

Abstract
Spinocerebellar ataxias (SCAs) are a heterogeneous group of autosomal-dominant neurodegenerative disorders involving the cerebellum and 23 different genes. We mapped SCA38 to a 56 Mb region on chromosome 6p in a SCA-affected Italian family by whole-genome linkage analysis. Targeted resequencing identified a single missense mutation (c.689G>T [p.Gly230Val]) in ELOVL5. Mutation screening of 456 independent SCA-affected individuals identified the same mutation in two further unrelated Italian families. Haplotyping showed that at least two of the three families shared a common ancestor. One further missense variant (c.214C>G [p.Leu72Val]) was found in a French family. Both missense changes affect conserved amino acids, are predicted to be damaging by multiple bioinformatics tools, and were not identified in ethnically matched controls or within variant databases. ELOVL5 encodes an elongase involved in the synthesis of polyunsaturated fatty acids of the ω3 and ω6 series. Arachidonic acid and docosahexaenoic acid, two final products of the enzyme, were reduced in the serum of affected individuals. Immunohistochemistry on control mice and human brain demonstrated high levels in Purkinje cells. In transfection experiments, subcellular localization of altered ELOVL5 showed a perinuclear distribution with a signal increase in the Golgi compartment, whereas the wild-type showed a widespread signal in the endoplasmic reticulum. SCA38 and SCA34 are examples of SCAs due to mutations in elongase-encoding genes, emphasizing the importance of fatty-acid metabolism in neurological diseases.
Main Text
Spinocerebellar ataxias (SCAs) are autosomal-dominant neurological disorders with an average prevalence of 5–7 in 100,000 individuals.1,2 SCAs are phenotypically characterized by gait ataxia and incoordination of eye movements, speech, and hand movements and are usually associated with cerebellar atrophy.3–6 Their genetic bases are highly heterogeneous and only partially known. More than 30 SCAs have been mapped, and 23 genes have been identified, but in most countries 40%–50% of families lack a genetic diagnosis.3,7 On the basis of the type of mutation, three main categories of SCAs can be identified: (1) disorders of CAG-coding polyglutamine repeat expansions, which are the most prevalent forms and include SCA1, SCA2, SCA3, SCA6, SCA7, and SCA17 (MIM 164400, 183090, 109150, 183086, 164500, and 607136, respectively); (2) noncoding repeat expansions, which include different type of repeats, such as ATTCT in SCA10 (MIM 603513), TGGAA in SCA31 (MIM 117210), and GGCCTG in SCA36 (MIM 614153); and (3) conventional mutations in genes encoding mostly unrelated proteins, except for ion channels in SCA13 (MIM 605259), SCA19 (also known as SCA22 [MIM 607346]), and episodic ataxia types 1 and 2 (MIM 160120 and 108500, respectively). SCA8 (MIM 608768) and SCA12 (MIM 604326) have been associated with a polyglutamine disorder or an untranslated-repeat disorder. Pathogenetic mechanisms that underlie neurodegeneration remain poorly understood and are triggered by a toxic gain of function in polyglutamine-expanded genes, an RNA effect in polyglutamine and/or noncoding repeat expansions, and/or a likely loss of function in almost all genetic entities.8 Compelling evidence to explain neuronal loss observed during the neurodegenerative process points to major etiological roles for interference with transcriptional regulation, protein aggregation and clearance, the ubiquitin-proteasome system, and alterations of calcium homeostasis. In the present study, we mapped SCA38, a form of SCA due to mutations in ELOVL fatty acid elongase 5 (ELOVL5 [MIM 611805]).
We collected a large Italian family with seven members affected by a pure form of cerebellar ataxia (family SCA38-01-BS, Figure 1A). The participants or an authorized representative provided written informed consent. The ethics committee of the Hospital of Brescia in Italy provided ethical approval.
Figure 1.
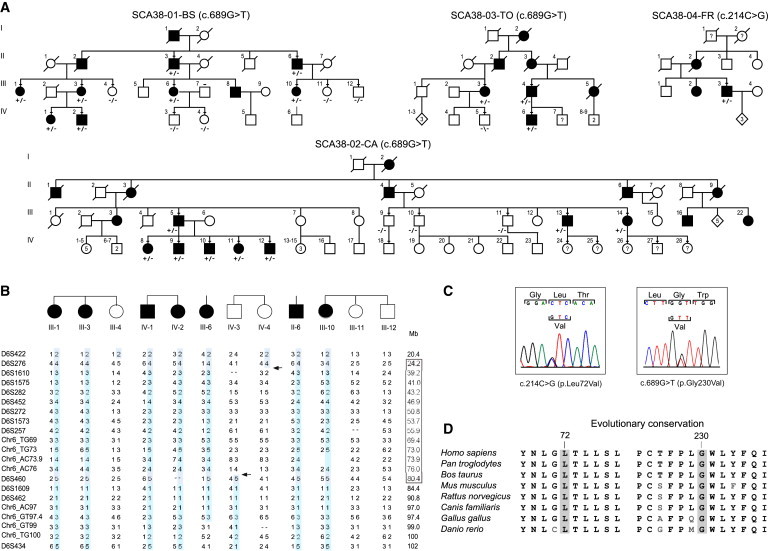
Family Trees, Haplotype Analysis, and ELOVL5 Mutation Analysis
(A) Pedigrees of SCA38-affected families. Open symbols indicate unaffected family members, and solid black symbols indicate affected members. Lines above the symbols indicate individuals for whom DNA was available. The genotype of the ELOVL5 mutations is indicated below each tested subject.
(B) Microsatellite genotypes with haplotype reconstruction of family SCA38-01-BS. Arrows indicate recombination events in healthy subjects IV-3 and IV-4; these events define the minimal region (boxed). On the right, distances of the markers from the top of chromosome 6p are indicated in Mb.
(C) Electropherograms of missense mutations c.214C>G (p.Leu72Val) and c.689G>T (p.Gly230Val).
(D) The amino acid sequence alignment of a portion of the human ELOVL5 in various orthologs. The two changed amino acids are conserved through vertebrates.
All clinical evaluations included a full medical history and examination, estimation of the age of onset, observation of additional neurological signs, electroneuromyographic studies, and whenever possible, brain MRI and/or fluorodesoxyglucose positron emission tomography (FDG-PET) scans. Epstein-Barr-virus-transformed lymphoblastoid cell lines were available for five affected individuals: subjects III-5 and III-10 from family SCA38-01-BS and subjects IV-8, IV-9, and IV-12 from family SCA38-02-CA.
Initially, CAG expansions within genes involved in SCA were excluded. Genome-wide linkage analysis was performed on seven affected individuals (II-6, III-1, III-3, III-6, III-10, IV-1, and IV-2) and three healthy relatives (III-4, III-11, and III-12) with the Illumina LINKAGE_12 microarray, containing 6,090 SNP markers with an average gap of 441 kb and 0.58 cM across the genome, according to the manufacturer’s protocol.
Three regions showed suggestive genetic linkage with Z > 1.5 (Figure S1A, available online): on chromosome 6, the region between rs560194 and rs2180419 (10.5–21.7 Mb) showed Zmax = +2.3 and the region between rs159988 and rs7776325 (25.1–99.9 Mb) showed Zmax = +2.8, and on chromosome 10, the region between rs10824792 and rs736594 (54.5–71.2 Mb) showed Zmax = +1.7. In addition, nine regions presented with uninformative multipoint LOD scores ranging from −1.9 to +0.1; two of them overlapped with SCA25- and SCA28-associated loci (Figure S1A).
To confirm linkage and refine mapping, we further genotyped the above affected individuals and four additional family members (II-3, III-7, IV-3, and IV-4) for 21 microsatellite markers located on chromosome 6 (UCSC Genome Browser hg19 and Table S1). Healthy status of subjects IV-3 and IV-4 was ascertained by neurologic examination and a FDG-PET scan when they were 40 years of age.
A significant multipoint LOD score (Zmax = +3.08) was found in a single region between markers D6S1610 and chr6_AC73.9 (Figure S1B). Haplotype analysis of the recombinant individuals restricted this locus, which we named SCA38 in agreement with the HUGO nomenclature, to a 56.2 Mb interval flanked by markers D6S276 (24.2 Mb) and D6S460 (80.4 Mb) on 6p22.2–q14.1 (Figure 1B).
Using custom array comparative genomic hybridization, we did not identify any deletions or duplications in this candidate region in the genomic DNA of subject III-7 from family SCA38-01-BS (custom design containing 3,700 probes between positions 50,854,000 and 76,107,000 Mb [hg19] on chromosome 6; ∼1 probe per 5.9 Kb; Agilent Technologies).
We performed targeted resequencing of all genes within the candidate region in subjects III-6 and III-10. Probes recognizing coding exons between positions 20,402,398 and 98,664,560 bp on chromosome 6, as well as 2 Kb upstream and downstream of the 5′ UTR and 3′ UTR of all genes, were designed with eArray (Agilent). They represented 3,307 regions (1.3 Mb), 2,837 of which had nonrepeated sequences and were finally captured in the DNA of subjects III-6 and III-10 of family SCA38-01-BS with a custom target-enrichment method (SureSelect Agilent array). The enriched samples were sequenced on the Applied Biosystems SOLiD platform. Reads of 50 bp (50,513,128 and 44,906,479 reads for affected individuals III-10 and III-6, respectively) were aligned to hg19 with Bioscope v.1.3 (Applied Biosystems) with default parameters. For subjects III-6 and III-10, approximately 49.2% and 47.8% of the sequences, respectively, matched the targeted region, and 85.4% and 85.5% of the targeted regions, respectively, were covered at least 30×; also, the average coverage was 170× and 147×, respectively, and a total of 9,669 and 10,341 variants, respectively, were identified. Sequences were then analyzed with the Gene Variant Analyzer (v.1.2), an in-house tool developed at the IBiSA Transcriptomics and Genomics facility of Marseille-Luminy (France). Variants not shared by both the affected individuals, homozygous variants, and variants reported as validated polymorphisms in human variation resources (dbSNP132, 1000 Genomes, NHLBI Exome Sequencing Project Exome Variant Server [EVS], or HapMap) were filtered out. Candidate genes were prioritized on the basis of potentially damaging variants.9
Among 91 heterozygous protein-altering variants shared by both subjects, we identified five missense variants with a database frequency < 0.1%. Two were excluded on the basis of their nonconservation in mammals and considered to be tolerated by SIFT and MutationTaster: c.821C>G (p.Pro274Arg) (RefSeq accession numb NM_005865.3) in protease, serine, 16 (thymus) (PRSS16 [MIM 607169]) and c.821A>G (p.His274Arg) (RefSeq NM_152880.3) in protein tyrosine kinase 7 (PTK7 [MIM 601890]). Two other variants, c.5682A>T (p.Glu1894Asp) (RefSeq NM_015153.3) in PHD finger protein 3 (PHF3 [MIM 607789]) and c.11901G>C (p.Leu3967Phe) (RefSeq NM_015548.4) in dystonin (DST [MIM 113810]), were not confirmed by Sanger sequencing. The last variant, c.689G>T (p.Gly230Val) (RefSeq NM_ 021814.4), was found in exon 7 of ELOVL5, which encodes an enzyme involved in the elongation of polyunsaturated fatty acids (PUFAs).10
Supporting ELOVL5 mutations as causative of SCA38, a mutation in its paralog, ELOVL4 (MIM 605512), has been recently described in a single family affected by an autosomal-dominant form of cerebellar ataxia and erythrokeratodermia (SCA34 [MIM 133190]).11 Mutations in ELOVL4 can also cause autosomal-dominant Stargardt disease 3 (MIM 600110)12,13 and ichthyosis, spastic quadriplegia, and mental retardation (MIM 614457).14
We screened ELOVL5 (conditions are reported in Table S2) in 456 index European individuals with most likely autosomal-dominant ataxia with an unknown genetic cause; these individuals were collected by the SPATAX network and other collaborators with approval from local ethics committees. We identified one heterozygous missense mutation (c.214C>G [p.Leu72Val] in exon 3) in a family of French origin and the c.689G>T (p.Gly230Val) mutation in two families of Italian origin (Figures 1A–1C). Two further changes were unlikely to be mutations (Table S3). We demonstrated segregation of the c.689G>T (p.Gly230Val) mutation with the disease in a total of 20 available affected subjects from families SCA38-01-BS, SCA38-02-CA, and SCA38-03-TO (Figure 1A). Both c.214C>G and c.689G>T were absent in dbSNP137, 1000 Genomes, and the NHLBI EVS (13,006 chromosomes) and in 800 chromosomes from healthy controls. The protein changes affect highly conserved amino acids in vertebrates and are predicted to be damaging by bioinformatics tools (Figure 1D; Table S4).
Haplotype comparison among the three Italian families carrying the c.689G>T (p.Gly230Val) substitution showed that it was associated with the same SNP haplotype covering 66.4 Kb (Table S5) (13% in the Italian TSI [Toscani in Italia] population). The haplotype—extended to 25.8 Mb with short-tandem-repeat markers—suggested a common ancestor in families SCA38-01-BS and SCA38-03-TO (Figures S1C and S2). For family SCA38-02-CA, sharing of the SNP haplotype might suggest a more ancient progenitor or two independent de novo c.689G>T mutations.
All affected individuals had common clinical features (Table 1). Initial symptoms were truncal ataxia and gait disturbance with an onset at about 40 years (up to 51 years in family SCA38-04-FR). Gaze-evoked nystagmus was always associated. Symptoms progressed very slowly. During the disease course, affected individuals had been able to walk until 50 years of age; later, they needed a walking stick. Dysarthria, as well as limb ataxia, was progressively evident. In two cases, diplopia was observed. Mild to moderate axonal neuropathy was diagnosed in five subjects. Mild sensory complains were reported in four affected individuals. Arterial hypertension and type 2 diabetes were present in one affected subject each. Cognitive disturbances were not reported, but individual II-2 (with c.214C>G [p.Leu72Val]) had psychiatric problems.
Table 1.
Clinics and Neuroradiology in SCA38-Affected Individuals
|
Family SCA38-01-BS |
Family SCA38-02-CA |
Family SCA38-04-FR |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| II-3 | III-1 | III-3 | III-6 | III-10 | IV-1 | IV-2 | IV-8 | II-2 | |
| Sex | male | female | female | female | female | female | male | female | female |
| Age at examination (years) | 81 | 77 | 79 | 59 | 60 | 47 | 45 | 40 | 63a |
| Age at onset of gait ataxia (years) | 45 | 40 | 34 | 45 | 38 | 38 | 38 | 35 | 51 |
| Disease duration (years) | 36 | 37 | 35 | 14 | 22 | 9 | 7 | 14 | 12 |
| Time from disease onset to wheelchair use (years) | 30 | NA | 25 | NA | NA | NA | NA | − | 10 |
| Initial symptom | walking difficulties | walking difficulties | walking difficulties | walking difficulties | walking difficulties | walking difficulties | walking difficulties | walking difficulties, nystagmus | walking difficulties |
| Neurology | |||||||||
| Gait ataxia | +++ | +++ | +++ | ++ | ++ | + | + | ++ | +++ |
| Dysarthria | ++ | + | + | − | + | − | − | + | ++ |
| Slow saccades | ++ | ++ | ++ | + | + | − | − | − | − |
| Nystagmus | + | +++ | +++ | +++ | +++ | ++ | ++ | + | − |
| Distal weakness | − | − | − | − | − | − | − | + | + |
| Myoclonus | − | − | − | − | − | − | − | − | − |
| Tremor | − | + | − | − | − | − | − | − | − |
| Peripheral neuropathy | ++ | NA | ++ | + | − | − | − | + | + |
| Extrapyramidal | − | − | − | − | − | − | − | − | − |
| Sensory loss | ++ | − | + | − | + | − | − | − | + |
| Brain MRI | |||||||||
| Vermian atrophy | +++ | NA | +++ | ++ | ++ | + | NA | + | ++ |
| Brainstem atrophy | − | NA | − | − | − | − | NA | − | − |
| Others Signs | |||||||||
| Cognitive impairment | − | − | − | − | − | − | − | − | psychiatric problems |
| Amino acid change | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Gly230Val | p.Leu72Val |
Abbreviations are as follows: −, none; +, mild; ++, moderate; +++, severe; and NA, not available.
Died at 65 years of age.
Structural neuroimaging (MRI) revealed cerebellar atrophy without obvious brainstem involvement in the moderate-severe stages (Figure 2A). In mid disease stages, MRI was within the normal range, but brain FDG-PET scans showed clear-cut cerebellar hypometabolism (Figure S3).
Figure 2.
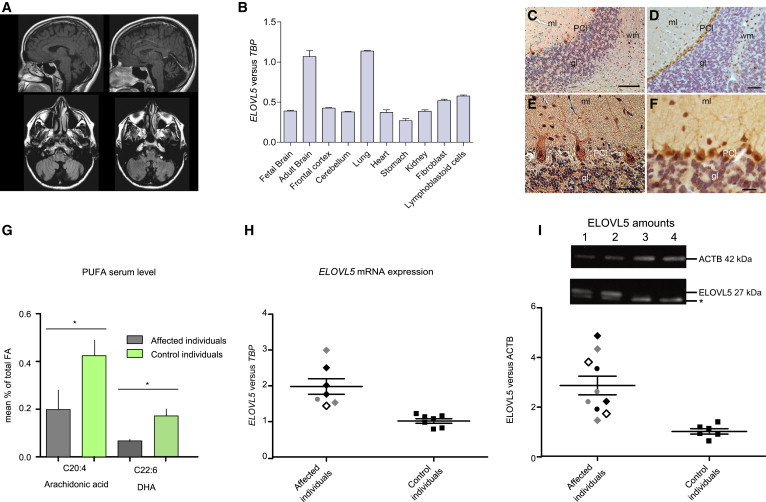
MRI Scans, ELOVL5 Expression, ELOVL5 Localization, and PUFA Serum Levels
(A) MRI scans of an affected individual (III-10 in family SCA38-01-BS). Coronal, midsagittal, and horizontal sections are reported. Cerebellar atrophy with vermis involvement is shown; the cortex and brainstem are preserved.
(B) The normal expression pattern of ELOVL5 in five human tissues, lymphoblastoid cells, and four brain areas (mean ± SD). ELOVL5 showed prominent expression in brain and lung. Gene expression was measured by quantitative PCR with FAM-labeled Universal ProbeLibrary LNA probes (Roche Diagnostics) (Table S6).
(C–F) Micrographs of sections of healthy human (C and E) and murine (D and F) cerebellar cortex immunostained with ELOVL5 antibody. Human cerebellar sections were obtained from paraffin-embedded tissues of postmortem brain derived from a healthy subject. Sections were incubated with ELOVL5 polyclonal antibody (C15621, diluted 1:400, AssaybIoTech) and counterstained with hematoxylin. Higher magnifications of sections of human and mouse cerebellar cortex are shown in (E) and (F), respectively. Note the similar labeling pattern of human and mouse Purkinje cells and the presence of numerous labeled cells in the white matter. Abbreviations are as follows: ml, molecular layer; PCl, Purkinje cell layer; gl, granular layer; and wm, white matter. Scale bars represent 200 μm (C), 80 μm (D), 50 μm (E), and 20 μm (F).
(G) Fatty-acid analysis in serum isolated from whole blood of four affected individuals (III-10 in family SCA38-01-BS, IV-8 and IV-9 in family SCA38-02-CA, and III-3 in family SCA38-03-TO). Fatty acids were analyzed in the serum from affected and control individuals by liquid chromatography-tandem mass spectrometry.15 Reduced AA and DHA were measured in affected individuals (mean ± SEM; AA, ∗p < 0.05; DHA,∗p < 0.05). Statistical analysis was performed with a one-tailed unpaired t test.
(H) Quantitative real-time PCR on the affected individuals’ cDNA showed an increase in ELOVL5 expression in comparison to the TBP reference (mean ± SEM; ∗∗∗p < 0.001, Mann Whitney test; ∗∗p < 0.01, two-tailed unpaired t test, Welch’s correction).
(I) ELOVL5 showed higher amounts in comparison to β-actin (ACTB) (mean ± SEM; ∗∗∗p < 0.001, Mann Whitney test; ∗∗p < 0.01, two-tailed unpaired t test, Welch’s correction). Total proteins were extracted from lymphoblasts of five affected individuals (III-6 and III-10 in family SCA38-01-BS and IV-8, IV-9, and IV-12 in family SCA38-02-CA) and five age- and origin-matched healthy controls. ELOVL5 amounts were normalized to ACTB. Different symbols indicate different affected individuals. The asterisk indicates an uncharacterized immunoblot signal as demonstrated in Elovl5-knockout mice (see Figure S3A). Lanes 1 and 2 are affected individuals IV-8 and IV-9, respectively, of family SCA38-02-CA; lanes 3 and 4 are two healthy controls.
ELOVL5 belongs to a multigenic family of genes encoding elongases. These enzymes are located in the endoplasmic reticulum (ER) and synthesize very-long-chain fatty acids with more than 16 carbons. Each elongase has its own substrate specificity, but there is some functional overlap.16 ELOVL5 is involved in the synthesis of PUFAs with more than 20 carbons of both the ω3 and ω6 series (double bond [C = C] at the third and sixth carbon atoms, respectively, from the end of the chain),17,18 as shown by Elovl5-knockout mice, which have decreased synthesis of arachidonic acid (AA, 20:4, ω6), eicosapentaenoic acid (EPA, 20:5, ω3), and docosahexaenoic acid (DHA, 22:6, ω3).10
ELOVL5 mRNA showed ubiquitous expression in different human tissues and prominent expression in adult brain and lung (Figure 2B). We explored the protein localization pattern in the cerebellum of human and mouse control samples. In Purkinje cells, the protein localized to the soma and the proximal portion of the dendritic tree (Figures 2C–2F), strengthening the likelihood that disruption of this gene plays a role in the pathogenesis of SCA38.
To verify ELOVL5 activity, we quantified very-long-chain fatty acids in the serum of four affected subjects with the c.689G>T (p.Gly230Val) mutation. We detected a significantly decreased level of two PUFAs directly synthesized by ELOVL5: AA and DHA (Figure 2G).
Lipid metabolism is strictly regulated by feedback loops that control the synthesis of enzymes whenever the amount of their products is altered. In Elovl5-knockout mice, EPA and DHA reduction induces an increase in the nuclear form of the transcription factor sterol regulatory element binding protein 1c (SREBP-1c),10 which is known to induce the expression of Elovl5 and several other lipogenic genes involved in fatty-acid and triglyceride synthesis. In lymphoblasts from affected individuals, ELOVL5 was significantly increased at both the mRNA level (2.0 ± 0.2, mean ± SEM, unpaired t test, p = 0.0011; Figure 2H; Table S6) and the protein level (2.85 ± 0.4, mean ± SEM, unpaired t test, p = 0.0023; Figure 2I; Figure S4A). Moreover, we demonstrated increased expression at the mRNA level of ELOVL6 (MIM 611546), a typical SREBP-1c target gene (2.5 ± 0.1, mean ± SEM, unpaired t test, p = 0.0012, data not shown).
This loop is responsible for an accumulation of lipids in the Elovl5-knockout mouse liver, leading to hepatic steatosis.10 Neither this feature nor other metabolic anomalies were evidenced in SCA38-affected individuals, suggesting that the wild-type ELOVL5 allele has a compensatory residual function. However, given the specific expression of ELOVL5 in Purkinje cells, a reduction in DHA and EPA synthesis might have an effect on the cerebellum, although Elovl5 ablation in mice does not display an overt neurologic phenotype, but this deserves further careful investigation.10
Lipid metabolism plays an important role in pre- and postnatal development and normal functioning of the CNS, given that this organ has the highest lipid content after adipose tissue. Altered levels of lipid molecules, such as fatty acids, are demonstrated in the neuronal system of individuals affected by neurodegenerative diseases, mental disorders, stroke, and trauma.
A variety of other neurological diseases, particularly hereditary spastic paraplegias19–23 and related diseases,24 also have altered lipid metabolism, which confirms the importance of lipid homeostasis in brain membranes. In particular, knocking down the biosynthesis of gangliosides affects the composition of membranes on other lipid components.25 Interestingly, activity of beta-glucosidase bile acid glucocerebrosidase was found to be altered in a complex phenotype characterized by spastic paraplegia with cerebellar ataxia (SPG46 [MIM 614409]).21,26
On the other hand, missense changes in ELOVL5 might lead to protein misfolding that alters the homeostasis of subcellular compartments. To verify ELOVL5 mislocalization, we prepared wild-type and mutant constructs containing the full-length human ELOVL5 cDNA in frame with a C-terminal HaloTag (pReceiver-M50, code U1147, GeneCopoeia). Point mutations c.214C>G and c.689G>T were introduced by PCR-induced mutagenesis (primers are listed in Table S7). Subcellular localization was studied by confocal microscopy in four different cell types (COS-7, SK-N-BE2, NIH/3T3, and HeLa cells) (Figure 3; Figures S4B, S5, and S6).
Figure 3.
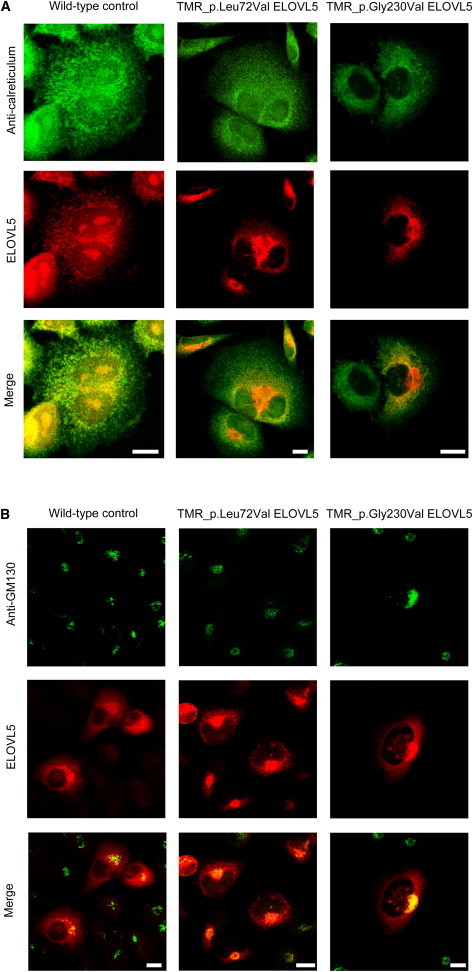
Subcellular Localization of Altered ELOVL5 in COS7 Cells
COS-7 cells transfected with wild-type or mutated (c.214C>G [p.Leu72Val] and c.689G>T [p.Gly230Val]) ELOVL5 cDNAs. The day before transfection, COS-7 (gray monkey kidney) cells were seeded at 90% confluence in 500 μl DMEM and 10% fetal bovine serum without antibiotic. In vitro transfections were performed with 500 ng of ScaI-digested linearized plasmid and 1 μl Lipofectamine 2000 according to the manufacturer’s (Life Technologies) instructions. After 24 hr, transfected cells were selected for 7 days with the use of G418 (Invivo Gen). Transfected COS-7 cells (5 × 104) were seeded on fibronectin-coated coverslips (24-well plates) and maintained with complete medium (DMEM, GIBCO) at 37°C. After 24 hr, cells were incubated with HaloTag TMR Direct Ligand at a final concentration of 1 μM (Promega) and the indicated primary antibodies. Samples were analyzed with an Olympus FV300 laser-scanning confocal microscope equipped with a blue argon (488 nm) laser, a green helium-neon (543 nm) laser, and FluoView 300 software (Olympus Biosystems). Cells were imaged with a 60× oil-immersion objective (1.4 NA). Images of optical sections (512 × 512 pixels) were digitally recorded and processed with BioView software (Center for Bio-Image Informatics).
(A) The ER is shown in green (rabbit anti-calreticulin; C4606, 1:400, Sigma-Aldrich), and ELOVL5 is shown in red (HaloTag TMR Direct Ligand). The two proteins completely colocalized in cells expressing wild-type ELOVL5, as shown by the merge row. This pattern was observed in 96% of cells (n = 49) expressing wild-type ELOVL5, 23% of cells (n = 42) with p.Leu72Val, and 17.5% of cells (n = 40) with p.Gly230Val (Fisher’s exact test performed between the wild-type and altered proteins, two-sided, p < 0.0001). Scale bars represent 10 μm.
(B) The Golgi is shown in green (mouse anti-GM130; 6108223, 1:400, BD Biosciences), and ELOVL5 is shown in red. Both p.Leu72Val and p.Gly230Val ELOVL5 were apparently reduced in the ER and mislocalized to the Golgi compartment, as shown by the merge row. This pattern was observed in 80% of cells (n = 40) expressing wild-type ELOVL5, 83% of cells (n = 47) with p.Leu72Val, and 71% of cells (n = 66) with p.Gly230Val. Experiments were performed at least three times. Scale bars represent 10 μm.
Wild-type ELOVL5 mainly localized in the ER compartment, as expected by published data.27 In contrast, p.Leu72Val and p.Gly230Val ELOVL5 had a less diffuse ER signal but did show perinuclear and polarized subcellular localization (Figure 3A; Figures S5A and S6A), consistent with an increase in the Golgi apparatus, as shown by the Golgi marker GM-130, a peripheral cytoplasmic protein tightly bound to the cis-Golgi membranes (Figure 3B; Figures S5B and S6B).28
The Golgi apparatus has a central role in the secretory pathway. It is expected that any alteration of the Golgi proteome would affect its homeostasis and consequently the anterograde or retrograde protein trafficking through it. The Golgi might also serve as the last quality-control checkpoint for correct protein conformation and activation of the unfolded protein response (UPR).29,30 In particular, the cis-Golgi might serve as a backup quality-control system to retain and send back misfolded proteins to the ER.31
Misfolded ELOVL5 might escape ER control, accumulate in the Golgi, and lead to chronic activation of the UPR, as hypothesized for infantile hypophosphatasia (MIM 241500), which is caused by a p.Asn153Asp substitution in alkaline phosphatase, tissue-nonspecific isozyme.32 The UPR induces apoptosis by distinct overlapping mechanisms, among which is the upregulation of the transcription factor C/EBP-homologous protein (CHOP) via the protein kinase RNA-like ER kinase.33 In p.Gly230Val-transfected COS-7 cells, the amount of CHOP was significantly increased, and it was slightly enhanced in p.Leu72Val-tranfected cells (Figure S7).
Overall, our data show that the p.Gly230Val substitution causes ELOVL5 mislocalization and might activate the UPR (although this point requires further investigation). The p.Leu72Val substitution might alter trafficking at the Golgi, in terms of both receiving material from the ER and recycling components back to the ER.34
On the basis of our screening, SCA38 is a rare hereditary ataxia with an estimated incidence of <1% among European individuals affected by an autosomal-dominant ataxia. This figure is in agreement with other SCAs due to conventional mutations, where higher frequencies are generally associated with founder effects.
Analogous with other SCAs caused by conventional mutations, SCA38 has a pure cerebellar phenotype with a progression slower than that of polyglutamine expansions3 and joins an emerging group of neurological diseases due to fatty-acids defects.
Acknowledgments
We are grateful to all family members who contributed to this study and to Fanny Mochel and Foudil Lamari for helpful discussions. We thank C. Castagnoli (Città della Salute e della Scienza, Torino) and G. Matullo (Hugef Foundation, Torino) for providing control cell lines and R. Sitia (Ospedale San Raffaele - Milano) for providing anti-calreticulum, anti-ERGIC-53/p58, and anti-GM130 antibodies. The Galliera Genetic Bank of the Telethon Network of Genetic Biobanks (project GTB12001), funded by Telethon Italy, provided us with specimens. This work was funded by the Associazione Italiana Sindromi Atassiche, the Associazione E.E. Rulfo per la Genetica Medica, PRIN 2010_2011 project 20108WT59Y (to A. Brusco), the GIS - Institut des Maladies Rares (A10021DS to G.S.), the Agence Nationale de la Recherche (SPATAX-Quest project to G.S.), the European Commission Seventh Framework Programme (E12009DD [Neuromics] and E04006DD [EUROSCA] to A. Brice), the program “Investissements d’Avenir” ANR-10-IAIHU-06 (to the Institut du Cerveau et de la Moelle Épinière), the Verum Foundation (to A. Brice and G.S.), and the Fondation Roger de Spoelberch (R12123DD to A. Brice). We thank Jay D. Horton (SouthWestern University, Dallas) for comments and critical discussion of the manuscript.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Haploview, http://www.broadinstitute.org/haploview
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
MutationAssesor, http://mutationassessor.org/
MutationTaster, http://www.mutationtaster.org/
Neuromuscular Disease Center, Washington University, http://neuromuscular.wustl.edu/ataxia/domatax.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PANTHER, http://www.pantherdb.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/index.php
SNPs&GO, http://snps.biofold.org/snps-and-go/pages/method.html
SPATAX Network, http://spatax.wordpress.com/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.van de Warrenburg B.P., Sinke R.J., Verschuuren-Bemelmans C.C., Scheffer H., Brunt E.R., Ippel P.F., Maat-Kievit J.A., Dooijes D., Notermans N.C., Lindhout D. Spinocerebellar ataxias in the Netherlands: prevalence and age at onset variance analysis. Neurology. 2002;58:702–708. doi: 10.1212/wnl.58.5.702. [DOI] [PubMed] [Google Scholar]
- 2.Craig K., Keers S.M., Archibald K., Curtis A., Chinnery P.F. Molecular epidemiology of spinocerebellar ataxia type 6. Ann. Neurol. 2004;55:752–755. doi: 10.1002/ana.20110. [DOI] [PubMed] [Google Scholar]
- 3.Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. doi: 10.1016/S1474-4422(10)70183-6. [DOI] [PubMed] [Google Scholar]
- 4.Manto M., Marmolino D. Cerebellar disorders—at the crossroad of molecular pathways and diagnosis. Cerebellum. 2009;8:417–422. doi: 10.1007/s12311-009-0142-4. [DOI] [PubMed] [Google Scholar]
- 5.van Gaalen J., Giunti P., van de Warrenburg B.P. Movement disorders in spinocerebellar ataxias. Mov. Disord. 2011;26:792–800. doi: 10.1002/mds.23584. [DOI] [PubMed] [Google Scholar]
- 6.Matilla-Dueñas A., Ashizawa T., Brice A., Magri S., McFarland K.N., Pandolfo M., Pulst S.M., Riess O., Rubinsztein D.C., Schmidt J. Consensus paper: pathological mechanisms underlying neurodegeneration in spinocerebellar ataxias. Cerebellum. 2014;13:269–302. doi: 10.1007/s12311-013-0539-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brusco A., Gellera C., Cagnoli C., Saluto A., Castucci A., Michielotto C., Fetoni V., Mariotti C., Migone N., Di Donato S., Taroni F. Molecular genetics of hereditary spinocerebellar ataxia: mutation analysis of spinocerebellar ataxia genes and CAG/CTG repeat expansion detection in 225 Italian families. Arch. Neurol. 2004;61:727–733. doi: 10.1001/archneur.61.5.727. [DOI] [PubMed] [Google Scholar]
- 8.La Spada A., Ranum L.P. Molecular genetic advances in neurological disease: special review issue. Hum. Mol. Genet. 2010;19(R1):R1–R3. doi: 10.1093/hmg/ddq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moon Y.A., Hammer R.E., Horton J.D. Deletion of ELOVL5 leads to fatty liver through activation of SREBP-1c in mice. J. Lipid Res. 2009;50:412–423. doi: 10.1194/jlr.M800383-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cadieux-Dion M., Turcotte-Gauthier M., Noreau A., Martin C., Meloche C., Gravel M., Drouin C.A., Rouleau G.A., Nguyen D.K., Cossette P. Expanding the clinical phenotype associated with ELOVL4 mutation: study of a large French-Canadian family with autosomal dominant spinocerebellar ataxia and erythrokeratodermia. JAMA Neurol. 2014;71:470–475. doi: 10.1001/jamaneurol.2013.6337. [DOI] [PubMed] [Google Scholar]
- 12.Barabas P., Liu A., Xing W., Chen C.K., Tong Z., Watt C.B., Jones B.W., Bernstein P.S., Križaj D. Role of ELOVL4 and very long-chain polyunsaturated fatty acids in mouse models of Stargardt type 3 retinal degeneration. Proc. Natl. Acad. Sci. USA. 2013;110:5181–5186. doi: 10.1073/pnas.1214707110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logan S., Agbaga M.P., Chan M.D., Kabir N., Mandal N.A., Brush R.S., Anderson R.E. Deciphering mutant ELOVL4 activity in autosomal-dominant Stargardt macular dystrophy. Proc. Natl. Acad. Sci. USA. 2013;110:5446–5451. doi: 10.1073/pnas.1217251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aldahmesh M.A., Mohamed J.Y., Alkuraya H.S., Verma I.C., Puri R.D., Alaiya A.A., Rizzo W.B., Alkuraya F.S. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am. J. Hum. Genet. 2011;89:745–750. doi: 10.1016/j.ajhg.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cermenati G., Abbiati F., Cermenati S., Brioschi E., Volonterio A., Cavaletti G., Saez E., De Fabiani E., Crestani M., Garcia-Segura L.M. Diabetes-induced myelin abnormalities are associated with an altered lipid pattern: protective effects of LXR activation. J. Lipid Res. 2012;53:300–310. doi: 10.1194/jlr.M021188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kihara A. Very long-chain fatty acids: elongation, physiology and related disorders. J. Biochem. 2012;152:387–395. doi: 10.1093/jb/mvs105. [DOI] [PubMed] [Google Scholar]
- 17.Leonard A.E., Bobik E.G., Dorado J., Kroeger P.E., Chuang L.T., Thurmond J.M., Parker-Barnes J.M., Das T., Huang Y.S., Mukerji P. Cloning of a human cDNA encoding a novel enzyme involved in the elongation of long-chain polyunsaturated fatty acids. Biochem. J. 2000;350:765–770. [PMC free article] [PubMed] [Google Scholar]
- 18.Moon Y.A., Shah N.A., Mohapatra S., Warrington J.A., Horton J.D. Identification of a mammalian long chain fatty acyl elongase regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2001;276:45358–45366. doi: 10.1074/jbc.M108413200. [DOI] [PubMed] [Google Scholar]
- 19.Tesson C., Nawara M., Salih M.A., Rossignol R., Zaki M.S., Al Balwi M., Schule R., Mignot C., Obre E., Bouhouche A. Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1051–1064. doi: 10.1016/j.ajhg.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boukhris A., Schule R., Loureiro J.L., Lourenço C.M., Mundwiller E., Gonzalez M.A., Charles P., Gauthier J., Rekik I., Acosta Lebrigio R.F. Alteration of ganglioside biosynthesis responsible for complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2013;93:118–123. doi: 10.1016/j.ajhg.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin E., Schüle R., Smets K., Rastetter A., Boukhris A., Loureiro J.L., Gonzalez M.A., Mundwiller E., Deconinck T., Wessner M. Loss of function of glucocerebrosidase GBA2 is responsible for motor neuron defects in hereditary spastic paraplegia. Am. J. Hum. Genet. 2013;92:238–244. doi: 10.1016/j.ajhg.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuurs-Hoeijmakers J.H., Geraghty M.T., Kamsteeg E.J., Ben-Salem S., de Bot S.T., Nijhof B., van de Vondervoort I.I., van der Graaf M., Nobau A.C., Otte-Höller I., FORGE Canada Consortium Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am. J. Hum. Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dick K.J., Eckhardt M., Paisán-Ruiz C., Alshehhi A.A., Proukakis C., Sibtain N.A., Maier H., Sharifi R., Patton M.A., Bashir W. Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35) Hum. Mutat. 2010;31:E1251–E1260. doi: 10.1002/humu.21205. [DOI] [PubMed] [Google Scholar]
- 24.Morgan N.V., Westaway S.K., Morton J.E., Gregory A., Gissen P., Sonek S., Cangul H., Coryell J., Canham N., Nardocci N. PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat. Genet. 2006;38:752–754. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sonnino S., Prinetti A. Gangliosides as Regulators of Cell Membrane Organization and Functions. In: Chalfant C., Del Poeta M., editors. Sphingolipids as Signaling and Regulatory Molecules. Landes Bioscience and Springer Science+Business Media; New York: 2010. pp. 165–184. [Google Scholar]
- 26.Hammer M.B., Eleuch-Fayache G., Gibbs J.R., Arepalli S.K., Chong S.B., Sassi C., Bouhlal Y., Hentati F., Amouri R., Singleton A.B. Exome sequencing: an efficient diagnostic tool for complex neurodegenerative disorders. Eur. J. Neurol. 2013;20:486–492. doi: 10.1111/j.1468-1331.2012.03883.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gregory M.K., Gibson R.A., Cook-Johnson R.J., Cleland L.G., James M.J. Elongase reactions as control points in long-chain polyunsaturated fatty acid synthesis. PLoS ONE. 2011;6:e29662. doi: 10.1371/journal.pone.0029662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J. Pharmacol. Sci. 2010;112:255–264. doi: 10.1254/jphs.09r03cr. [DOI] [PubMed] [Google Scholar]
- 29.Halliday M., Mallucci G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology. 2014;76(Pt A):169–174. doi: 10.1016/j.neuropharm.2013.08.034. [DOI] [PubMed] [Google Scholar]
- 30.Hurtley S.M., Helenius A. Protein oligomerization in the endoplasmic reticulum. Annu. Rev. Cell Biol. 1989;5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- 31.Hammond C., Helenius A. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 1994;126:41–52. doi: 10.1083/jcb.126.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito M., Amizuka N., Ozawa H., Oda K. Retention at the cis-Golgi and delayed degradation of tissue-non-specific alkaline phosphatase with an Asn153—>Asp substitution, a cause of perinatal hypophosphatasia. Biochem. J. 2002;361:473–480. doi: 10.1042/0264-6021:3610473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hetz C., Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- 34.Bexiga M.G., Simpson J.C. Human diseases associated with form and function of the Golgi complex. Int. J. Mol. Sci. 2013;14:18670–18681. doi: 10.3390/ijms140918670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.