

Abstract
The mimicry of protein tertiary structure by oligomers with unnatural backbones is a significant contemporary research challenge. Among common elements of secondary structure found in natural proteins, sheets have proven the most difficult to address. Here, we report the systematic comparison of different strategies for peptide backbone modification in β-sheets with the goal of identifying the best method for replacing a multi-stranded sheet in a protein tertiary fold. The most effective sheet modifications examined lead to native-like tertiary folding behavior with thermodynamic fold stability comparable to the prototype protein on which the modified backbones are based.
Introduction
Synthetic oligomers with the capacity to adopt discrete folded structures (“foldamers”)1 have received significant research attention,2 due in part to their ability to mimic natural peptide folding patterns. In more than two decades of work showing increasingly sophisticated structures from unnatural backbones, tertiary folds like those commonly found in proteins have proven difficult to recreate. Although significant progress has been made with helix-turn-helix targets,3 these represent only a small fraction of the diverse array of folds found in nature. Reproducing a wider selection of natural protein structural motifs with unnatural oligomers is an important goal because it opens the door to reproducing the full repertoire of functions enabled by those folds.
One design concept that shows promise in addressing the challenge of tertiary structure mimicry is the systematic backbone alteration of natural sequences. Folded proteins can tolerate diverse backbone modifications without compromising sequence-encoded folding.4 Bridging the gap between these observations and precedent on de novo foldamer design2 suggests an approach toward protein mimicry, in which a number of α-residues in a sequence with known folding behavior are replaced with various unnatural building blocks to generate heterogeneous backbones capable of adopting native-like folds. The versatility of the above method for mimicry of isolated α-helix5 and β-sheet6 secondary structures has recently been leveraged to simultaneously modify all the secondary structures in a small protein tertiary fold.7 A fundamental question that must be addressed for sequence-guided backbone alteration to be effective for the widest array of target folds is how to best apply chemical modification without disrupting sequence-encoded folding.
Among common protein secondary structures, sheet folds have proved more challenging targets than helices or turns for mimicry by unnatural oligomers. Building on pioneering work carried out largely in organic solvents,8 we have recently focused on developing strategies for the design of heterogeneous-backbone β-sheet mimics that fold in water.6 Hairpin model systems, widely used in fundamental studies on β-sheet formation in proteins,9 have proved valuable in assessing sheet propensity of unnatural building blocks. Unfortunately, the lessons learned in the hairpin context are not always applicable in a more complex protein tertiary fold. As an example, when incorporated in each strand of a hairpin-forming peptide, appropriately substituted β-amino acid residues (homologated analogues of α-residues) can maintain native-like folding,6a,6b but the same modifications abolish folding entirely when made in a four-stranded β-sheet in a small protein.7
Here, we report the side-by-side comparison of several different strategies for peptide backbone modification in β-sheet secondary structures with the goal of identifying the best method for replacing a multi-stranded sheet in a protein tertiary fold. The most effective sheet modifications examined lead to native-like tertiary folding behavior with thermodynamic fold stability comparable to the prototype protein on which the modified backbones are based.
Results and Discussion
Strategies Examined for Sheet Backbone Alteration
We compared three strategies for peptide backbone alteration in two different β-sheet forming host sequences – a two-stranded hairpin peptide and a four-stranded sheet in a small protein tertiary fold (Figure 1). α-Residues in each prototype sequence were replaced with N-methylated analogues (α→N-Me-α), (E)-vinylogous γ4-residues (α→γ4), or the cyclically constrained γ-residue Acc (α→γcyc). These three backbone modifications and some of the unanswered questions we sought to address about each are discussed in more detail below.
Fig. 1.

Summary of strategies examined for peptide backbone modification in β-sheets. The impact of three different types of α-residue replacement on folding was evaluated in two different structural contexts, a β-hairpin peptide and a four-stranded β-sheet in a small protein.
Methylation of backbone amide nitrogen atoms has been widely applied in small peptides10 and can be used to modify capping strands of sheet-forming sequences.11 We recently showed α→N-Me-α residue substitution is accommodated in a small bacterial protein, but it resulted in a degree of destabilization that was surprising given the subtle nature of the chemical change.7 One goal in the present work was to elucidate the molecular basis of this destabilization by systematically examining the site-dependent structural and thermodynamic effects of N-methylation on the folding of a small hairpin sequence.
In another effort toward sheet mimetics based on systematic modification of natural sequences, we recently reported that the cyclically constrained γ-residue (1R,3S)-3-aminocyclohexane carboxylic acid (Acc) can be incorporated into a hairpin-forming α-peptide sequence (α→γcyc substitution in each strand) without significantly altering the folded structure.6c Interestingly, the fold of the chimeric α/γcyc-peptide was actually more thermodynamically stable than that of the prototype α-peptide on which it was based.6c A second open question we wanted to address in this study is whether such α→γcyc substitutions are tolerated in the complex structural environment of a multi-stranded sheet in a protein tertiary fold.
A third strategy examined for backbone modification in β-sheets involved the replacement of α-residues with γ-residues bearing a side chain at Cγ and an (E)-double bond between Cα and Cβ. These vinylogous γ4-residue building blocks are known to be compatible with hairpin formation in organic solvent,8a,8f but their impact on folding in aqueous environments has not been reported. γ4-Residues offer an advantage over the γcyc residue Acc in that they can retain protein-derived side chains when they replace α-residues in a native sequence. We incorporated vinylogous γ4-residues (α→γ4 substitution in each strand) into both peptide hairpin and protein sheet contexts in order to ascertain the compatibility of these residues with the native folds.
Backbone Alteration in a Peptide β-Hairpin Host Sequence
Peptide hairpin 1 (Figure 2), derived from the C-terminal segment of the B1 domain of Streptococcal protein G,12 has proven a useful host sequence for exploring the sheet folding propensities of modified peptide backbones in aqueous solution.6b,6c Sequences 2–7 are variants of peptide 1 designed to systematically compare the impact of the different strategies for backbone alteration described above on the structure and stability of the sequence-encoded hairpin fold.
Fig. 2.

Sequences of peptides 1–7, key to α-residue replacements (Xxx indicates the side chain on the unnatural monomer is the same as the corresponding α-residue), and minimized average coordinates from the NMR solution structure of prototype peptide 1 in pH 6.3 phosphate buffer.
In peptide 2, α-residues Ala4 and Ala13 in 1 are replaced by the constrained γcyc-residue Acc (α→γcyc substitution). We have previously reported the synthesis and biophysical analysis of 2, and it is included here as a point of comparison.6c Peptide 3 has the same sites of backbone modification as 2, but the unnatural building blocks are vinylogous γ4-residues bearing the side chain of the replaced α-residues in 1 (α→γ4 substitution). In peptides 4–7, α-residues Trp3, Tyr5, Phe11, or Val13 from host sequence 1 are individually modified by N-methylation (α→N-Me-α). These four sites, all at non-hydrogen-bonding positions in the hairpin, were modified separately to determine how sequence context influences the thermodynamic impact of N-methylation on hairpin folded stability.
Peptides 1–7 were synthesized by Fmoc solid-phase peptide methods, purified by reverse-phase HPLC, and the identities of the purified oligomers confirmed by mass spectrometry. We compared the folding behavior of 1–7 by a series of multidimensional NMR experiments carried out in pH 6.3 phosphate buffer at 5 °C. Homonuclear 1H–1H COSY, TOCSY, and NOESY spectra were sufficient to enable full resonance assignment of each oligomer. As described in prior work, we used the chemical shift separation of the two diastereotopic Hα’s in Gly10 to quantify folded population and estimate folding free energy in the modified hairpins (Table 1).6b,6c,12–13
Table 1.
Folding thermodynamics of peptides 1–7 from NMR measurementsa
| Peptide | Δδ Gly10 Hα/Hα′ (ppm) | Fraction Folded (%) | ΔGfold (kcal mol−1) | ΔΔGfold vs. 1 (kcal mol−1) |
|---|---|---|---|---|
| 1 | 0.20 | 65 | −0.3 | |
|
| ||||
| 2 | 0.26 | 83 | −0.9 | −0.6 |
|
| ||||
| 3 | 0.12 | 39 | +0.2 | +0.5 |
|
| ||||
| 4 | 37 b | +0.3 | +0.6 | |
| 4trans (60%) | 0.19 | 61 | −0.3 | +0.0 |
| 4cis (40%) | 0.09 | |||
|
| ||||
| 5 | 19b | +0.8 | +1.1 | |
| 5trans (76%) | 0.09 | 30 | +0.5 | +0.8 |
| 5cis (24%) | 0.00 | |||
|
| ||||
| 6 | 29b | +0.5 | +0.8 | |
| 6trans (65%) | 0.12 | 38 | +0.3 | +0.6 |
| 6cis (35%) | 0.00 | |||
|
| ||||
| 7 | 55b | −0.1 | +0.2 | |
| 7trans (87%) | 0.20 | 63 | −0.3 | +0.0 |
| 7cis (13%) | 0.12 | |||
NMR carried out at 5 °C in pH 6.3 phosphate buffer. Assuming a 0.01 ppm uncertainty in measured Gly Hα/Hα′ separation, error propagation estimates uncertainties of 5% for fraction folded and ~0.2 kcal mol−1 for ΔGfold and ΔΔGfold.
Overall folded population calculated as product of the fraction of peptide in the trans amide configuration and fraction folded for trans isomer.
Comparison of the NMR data for peptides 2 and 3 suggests that the connectivity of the γ-residue incorporated into each strand of the hairpin has a significant effect on folding energetics. We previously showed that peptide 2, which contains two α→γcyc substitutions relative to 1, forms a more stable hairpin than the wild-type backbone.6c In contrast, the α→γ4 substitutions in peptide 3 measurably destabilized the fold (~0.6 kcal mol−1 relative to prototype 1 and ~1.1 kcal mol−1 relative to variant 2 with the Acc residues).
In considering the different impact of γcyc and γ4 residues on hairpin folded stability, we saw two possible origins: a significant change in the folded structure or altered backbone flexibility. In order to test the former hypothesis, we pursued a solution structure of 3 by simulated annealing with NMR-derived distance restraints and compared the resulting coordinates to the previously determined structure of γcyc-residue-containing variant 2. Due to the low folded population of 3, we synthesized a cyclic derivative for NMR structural analysis (3cyc), which has Cys residues appended to each terminus and linked together via a disulfide bond. α/γ4-Peptide 3cyc forms a β-hairpin fold very similar to that of α/γcyc-peptide 2 (Figure 3). The similarity among the solution structures of 2 and 3cyc suggest the different number of freely rotatable bonds in the two γ-residue classes (three for each unsaturated γ4 vs. two for each γcyc) is likely responsible for the different folded stabilities of the hairpins containing them.
Fig. 3.

Minimized average coordinates from NMR solution structures of α,γcyc-peptide 2 and α/γ4-peptide 3cyc in pH 6 phosphate buffer. Carbons are colored green for γ-residues and yellow for α-residues. Most side chains are omitted for clarity.
Analysis of the NMR data for peptides 4–7 reveals two important issues with respect to backbone N-methylation in sheet-forming sequences. First and most pronounced are complications arising from cis/trans amide isomerization.10,14 Each N-methyl hairpin showed signals for two distinct species by NMR, which we attributed to a mixture of cis and trans isomers at the tertiary amide introduced upon N-methylation. We calculated the isomer ratio by integrating well-resolved peaks in the TOCSY spectra. Population ratios varied among the four oligomers, but the trans amide was predominant in each case. We made this assignment based on analysis of the NOESY data, which showed close contacts between the backbone methyl group in the trans isomer and both the side-chain and Hα protons of the preceding residue. The consistently lower Gly Hα/Hα′ chemical shift separation indicates the presence of a cis amide in the chain destabilizes the hairpin fold considerably. This is reasonable considering how such a change would disrupt backbone direction and side chain contacts that enable parent sequence 1 to fold.
Separate from the issue of amide isomerization in N-Me-α-peptides 4–7 is the question of how the folded stability of the all-trans isomers compare to α-peptide 1. The answer depends on the positioning of the backbone methyl group relative to the hairpin turn. The presence of an N-methyl amide in a trans configuration at either Trp3 (peptide 4) or Val14 (peptide 7) led to a folded population identical within error to that of prototype sequence 1. By contrast, when the site of backbone methylation was closer to the turn (peptides 5 and 6), the folded state was measurably destabilized – even after taking into account the detrimental contribution of the cis isomer.
Computational studies have shown that the energetically accessible backbone conformations of N-Me-α-residues are more restricted than their non-methylated analogues.15 Notably, one region of the Ramachandran plot that becomes significantly disfavored energetically upon N-methylation corresponds to typical backbone dihedrals for strands from an antiparallel β-sheet.16 The above observations help to rationalize the observed destabilization of the hairpin folds of trans-amide isomers of N-Me-α-peptides 5 and 6 relative to α-peptide 1.
Backbone Alteration in a Protein β-Sheet Host Sequence
As a host sequence to examine sheet backbone modification in the context of a tertiary structure, we employed the full-length 56-residue B1 domain of Streptococcal protein G (8, Figure 4). This sequence, from which hairpin peptide 1 is derived, adopts a compact tertiary fold consisting of an α-helix packed against a four-stranded β-sheet.17 Of the three backbone alteration strategies above, one has previously been examined in GB1: α→N-Me-α substitution at terminal strands in the sheet (protein 9).7 We include data for protein 9 here for comparison. In proteins 10 and 11, α→γ residue substitutions are made in place of Ile6, Glu15, Thr44, and Thr53. Protein 10 incorporates constrained γcyc residues at these positions, while variant 11 bears vinylogous γ4-residues with side chains derived from the natural GB1 sequence. In both 10 and 11, the positioning of γ-residues is designed to create a stripe of unnatural residues along the center of the sheet if the modified backbones adopt a native-like tertiary fold. Protein 12 is a variant of GB1 with a completely natural backbone but mutations that remove three polar side-chain functional groups that are lost upon incorporation of Acc residues in protein 11. Finally, protein 13 is a variant of 11 with the same number of γcyc residues but incorporated at positions intended shift the stripe of unnatural monomers to a different region of the sheet.
Fig. 4.
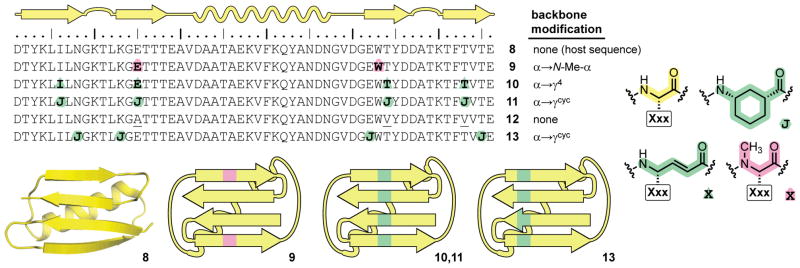
Sequences of proteins 8–13, key to α-residue replacements (Xxx indicates the side chain on the unnatural monomer is the same as the corresponding α-residue), crystal structure of wild-type GB1 8 (PDB 2QMT), and schematics showing the placement of unnatural residues in 9, 10, 11, and 13.
We compared the folding behavior of proteins 8–13 by circular dichroism (CD) spectroscopy in pH 7 phosphate buffer. CD scans (Figure 5A) for three of the four modified backbones (proteins 9, 11, and 13) showed shapes and magnitudes similar to wild-type GB1. These results suggest α→N-Me-α substitution and two different patterns of α→γcyc substitution are well tolerated in the tertiary fold. Protein 10, bearing four α→γ4 residue replacements, had a CD spectrum qualitatively different from all the other GB1 analogues examined. Its dissimilarity to typical random coil signatures and dependence on temperature (vide infra) argue against 10 existing as an unstructured chain. We cannot definitively say whether the spectrum of 10 is a result of an altered folded state or a change in the CD signature of the native-like tertiary fold due to the presence of four α,β-unsaturated amides in the backbone. In an effort to clarify this point, we attempted to obtain diffraction-quality crystals of protein 10 but were unsuccessful.
Fig. 5.

Circular dichroism scans at 25 °C (A) and thermal melts monitored at 220 nm (B) for proteins 8–13. Experiments were carried out on 40 μM concentration protein samples in 20 mM phosphate buffer, pH 7.
We performed thermal denaturation experiments to determine the impact of backbone substitutions on folded stability, monitoring the CD minima at 220 nm as a function of temperature (Figure 5B). All the proteins showed sharp sigmoidal unfolding transitions with cooperativities similar to wild-type GB1. The steep thermal unfolding transitions suggest the modified oligomers have well-ordered folded states. Comparing the midpoints of the thermal unfolding transitions (Tm) provides an estimate of the energetic impact of various backbone alterations on folding (Table 2).
Table 2.
Folding thermodynamics of proteins 8–13 from circular dichroism measurementsa
| Protein | Tm (°C)a | ΔΔGfold vs. 8 (kcal mol−1) | substitutions vs. 8 | ΔΔGfold per substitution (kcal mol−1) |
|---|---|---|---|---|
| 8 | 82.1 | |||
| 9 | 75.6 | +1.1 | 2 α→N-Me-α | 0.6 |
| 10 | 43.5 | +6.3 | 4 α→γ4 | 1.6 |
| 11 | 46.7 | +5.9 | 4 α→γcyc | 1.5 |
| 12 | 78.0 | +0.7 | 4 side chains | 0.2 |
| 13 | 74.3 | +1.3 | 4 α→γcyc | 0.3 |
CD experiments carried out in pH 7 phosphate buffer
As reported previously, protein 9 bearing two α→N-Me-α substitutions has a stability only slightly lower than wild-type GB1.7 In contrast, α→γ residue replacements in proteins 10 and 11 destabilize their folded states considerably. Consistent with their relative folding propensities in the hairpin host sequence, the γcyc Acc residues (protein 11) are superior to vinylogous γ4 residues (protein 10) in supporting the tertiary structure when incorporated at identical positions it the host sequence.
In considering the data for α/γ-hybrid protein 11, we found it striking that the γcyc residue Acc, which stabilized the hairpin secondary structure, was so destabilizing to the tertiary fold. One difference between protein 11 and wild-type 8 besides the altered backbone in the former is the loss of three functionalized side chains upon substitution of α-residues Glu15, Thr44, and Thr53 with Acc. Inspection of the crystal structure of wild-type GB1 shows three of these side chains are involved in inter-strand polar contacts that potentially stabilize the tertiary fold. The CD thermal stability observed for protein 12, which has a natural backbone but lacks polar groups necessary for these contacts, indicate that the lost side-chain functionality is at most a very small contributor to the difference in folding behavior between 11 and 8.
Another factor we considered as potentially responsible for destabilization of the γ-residue modified proteins is the increase in backbone length by two atoms with each α→γ residue substitution. When found in the core of the protein as in 10 and 11, this change may disrupt critical hydrophobic contacts between the sheet and helix necessary for folding. In order to test this hypothesis, we examined an analogue of GB1 (protein 13), bearing four Acc residues in a pattern that would create a stripe of γ-residues in the sheet as in 10 and 11 but further removed from the hydrophobic core of the protein in the folded state. We reasoned that the alteration in backbone length of the sheet would be better tolerated if it was not located in close proximity to key tertiary contacts. Supporting the above hypothesis, protein 13 showed a dramatically improved thermal stability compared to closely related analogue 11.
Conclusions
In summary, we have reported here the systematic comparison of three different strategies for peptide backbone modification in β-sheet secondary structures using two different host systems – a hairpin peptide and a small protein with a defined tertiary fold. Our results provide new insights into the design of heterogeneous backbones based on natural peptide sequences that encode for β-sheet folds. In the peptide hairpin host sequence, α→γcyc substitution was superior to α→γ4, which was better than backbone methylation (α→N-Me-α). Destabilization of the sheet fold by α→N-Me-α substitution appears to result primarily from population of an unproductive cis tertiary amide isomer at the methylation site, though the fold of the trans isomer is also destabilized relative to native due to local stereoelectronic effects. In the best case for hairpin modification (α→γcyc), the heterogeneous backbone had a more stable fold than the prototype α-peptide on which it was based.
When substitutions are applied to a central stripe of strand residues in the protein tertiary structure, the trend was significantly different than the hairpin host sequence: N-Me-α residue incorporation at capping strands of the sheet was best tolerated, followed by γcyc and vinylogous γ4 substitutions in all four strands. Optimization of the placement of γcyc residues, however, had a dramatic effect on the thermodynamic consequences of the modification. Shifting the position of the backbone expansion resulting from α→γ residue substitution away from the hydrophobic core of the protein led to a heterogeneous backbone with near wild-type folded stability. We anticipate these results will aid in ongoing efforts to recreate β-sheet folding patterns from heterogeneous backbones and open the way toward design of protein-mimetic oligomers with increasingly diverse tertiary folding topologies.
Experimental
Peptide and Protein Synthesis
Protected γ4-amino acids were prepared via the corresponding α-amino aldehydes18 according to published methods.19 Fmoc-Acc was prepared as previously described.6c Full experimental details and characterization data for new compounds are given in the Supporting Information (SI).
β-Hairpin peptides were synthesized using microwave-assisted Fmoc solid-phase synthesis techniques on a MARS microwave reactor (CEM) using NovaPEG Rink Amide resin. Couplings were carried out in NMP at 70 °C for 4 min using 4 equiv of Fmoc-protected amino acid, 4 equiv of HCTU, and 6 equiv DIEA. PyAOP was used in place of HCTU for the coupling of N-methylated residues and residues immediately following them. Deprotections were performed using an excess of 20% 4-methylpiperidine in DMF at 80 °C for 2 min. After each coupling or deprotection cycle, the resin was washed three times with DMF. Double couplings were performed at sequence positions following proline or N-methylated residues. Prior to cleavage, the resin was washed three times each with DMF, dichloromethane, and methanol, and then dried. Peptide cleavage was accomplished using 95% trifluoroacetic acid (TFA), 2.5% triisopropylsilane (TIS), and 2.5% water.
Peptide was precipitated from the cleavage solution by addition of diethyl ether and purified by preparative HPLC on a C18 column using gradients between 0.1% TFA in water and 0.1% TFA in acetonitrile. After purification, the linear precursor to peptide 3cyc was dissolved in 10 mM pH 8.9 phosphate buffer with 5% v/v DMSO, stirred until analytical HPLC and MS showed complete conversion to the cyclic disulfide (2 d), and then purified by HPLC to obtain 3cyc.
Protein GB1 and variants were synthesized on a PTI Tribute synthesizer using NovaPEG Rink Amide resin (70 μmol scale). Coupling reactions were performed by combining 3 mL of 0.4 M N-methylmorpholine in DMF with 7 equiv Fmoc-amino acid and 7 equiv HCTU. Following a two minute preactivation, the activated amino acid was added to the resin and vortexed for 45 min. Deprotection reactions were carried out twice with 3 mL of a 20% v/v solution of 4-methylpiperidine in DMF for 4 min. The resin was washed three times with 3 mL of DMF for 40 s between each cycle. After the final deprotection step, the resin was washed with 3 mL of dichloromethane followed by 3 mL of methanol. Resin was dried and subjected to cleavage by treatment with a solution of 94% TFA, 1% TIS, 2.5% water, and 2.5% ethanedithiol. Crude protein was precipitated by addition of cold diethyl ether. The solid was pelleted by centrifugation and dissolved in 6 M guanidinium chloride, 25 mM sodium phosphate, pH 6. This solution was subjected to purification by preparative C18 reverse-phase HPLC using gradients between 0.1% TFA in water and 0.1% TFA in acetonitrile. Each protein was subjected to a second round of purification by anion-exchange chromatography on a monoQ 5/50GL column (GE Healthcare) using 0.02 M Tris buffer at pH 8 and eluting with increasing concentrations of KCl.
All peptides and proteins used for biophysical analysis were >95% pure as determined by analytical HPLC on a C18 column. Identities were confirmed by mass spectrometry using a Voyager DE Pro MALDI-TOF instrument (Table S1).
NMR Sample Preparation, Data Collection, and Analysis
NMR samples were prepared by dissolving peptide in 750–850 μL of degassed 50 mM phosphate, 9:1 H2O/D2O, pH 6.3 (uncorrected for the presence of D2O) to a final concentration of 0.8–3 mM. 3-(Trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS, 50 mM in water) was added to a final concentration of 0.2 mM. Each solution was passed through a 0.2 μm syringe filter, and transferred to an NMR tube. The NMR tube headspace was purged with a stream of nitrogen prior to capping.
NMR experiments were performed on a Bruker Avance-700 spectrometer. Chemical shifts are reported relative to DSS (0 ppm). TOCSY, NOESY, and COSY pulse programs used excitation-sculpted gradient-pulse solvent suppression. For all 2D experiments, 2048 data points were collected in the direct dimension and 512 data points in the indirect dimension. The mixing times for TOCSY and NOESY were 80 ms and 200 ms, respectively. NMR measurements were performed at a temperature of 278 K for hairpin peptides 3–7 and at 293 K for cyclized hairpin peptide 3cyc. The Sparky software package (T. D. Goddard and D. G. Kneller, SPARKY 3, University of California, San Francisco) was used to analyze 2D NMR data. Backbone chemical shift assignments for peptides 3–7 are reported in Tables S2–7. Analysis of NMR data for 3–7 and estimation of folded populations followed previously published methods.6b,6c Tabulated NOEs for peptide 3cyc are reported in Table S8. These data were applied to calculate an NMR solution structure of 3cyc using the Crystallography and NMR system (CNS) software package20 according to published methods.6b,6c
Circular Dichroism Spectroscopy
CD measurements were performed on an Olis DSM17 Circular Dichroism Spectrometer in 2 mm quartz cells. Samples consisted of 40 μM protein in 20 mM sodium phosphate buffer, pH 7. Scans were carried out at 25 °C over the range of 200–260 nm in 1 nm increments with a 2 nm bandwidth. Scan data were smoothed by the Savitzky-Golay method. Melts were monitored at 220 nm over the range of 4 ºC to 98 ºC with 2 ºC increments, a dead band of 0.5 ºC, and a 2 min equilibration time at each temperature. All measurements were baseline corrected for blank buffer. Temperature-dependent CD data were fit to a two-state unfolding model to obtain melting temperature (Tm). The change in free energy of folding for each mutant relative to wild-type (ΔΔGfold) was estimated from the change in Tm (ΔTm) using the enthalpy of folding determined for GB1 by differential scanning calorimetry.21
Supplementary Material
Acknowledgments
Funding for this work was provided by the University of Pittsburgh and the National Institutes of Health (R01GM107161).
Footnotes
Electronic Supplementary Information (ESI) available: Figures S1–S3, Tables S1–S8, and experimental methods for the synthesis of unnatural amino acid monomers. See DOI: 10.1039/b000000x/
Notes and References
- 1.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 2.(a) Bautista AD, Craig CJ, Harker EA, Schepartz A. Curr Opin Chem Biol. 2007;11:685–692. doi: 10.1016/j.cbpa.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Horne WS. Expert Opin Drug Discov. 2011;6:1247–1262. doi: 10.1517/17460441.2011.632002. [DOI] [PubMed] [Google Scholar]; (d) Guichard G, Huc I. Chem Commun. 2011;47:5933–5941. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; (e) Pilsl LA, Reiser O. Amino Acids. 2011;41:709–718. doi: 10.1007/s00726-011-0894-2. [DOI] [PubMed] [Google Scholar]
- 3.(a) Cheng RP, DeGrado WF. J Am Chem Soc. 2002;124:11564–11565. doi: 10.1021/ja020728a. [DOI] [PubMed] [Google Scholar]; (b) Sharma GVM, Subash V, Narsimulu K, Sankar AR, Kunwar AC. Angew Chem Int Ed. 2006;45:8207–8210. doi: 10.1002/anie.200603084. [DOI] [PubMed] [Google Scholar]; (c) Delsuc N, Hutin M, Campbell VE, Kauffmann B, Nitschke JR, Huc I. Chemistry – A European Journal. 2008;14:7140–7143. doi: 10.1002/chem.200800988. [DOI] [PubMed] [Google Scholar]; (d) Lee BC, Chu TK, Dill KA, Zuckermann RN. J Am Chem Soc. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Petersson EJ, Schepartz A. J Am Chem Soc. 2008;130:821–823. doi: 10.1021/ja077245x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Price JL, Hadley EB, Steinkruger JD, Gellman SH. Angew Chem Int Ed. 2010;49:368–371. doi: 10.1002/anie.200904714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Lu W, Qasim MA, Laskowski M, Kent SBH. Biochemistry. 1997;36:673–679. doi: 10.1021/bi9625612. [DOI] [PubMed] [Google Scholar]; (b) Chapman E, Thorson JS, Schultz PG. J Am Chem Soc. 1997;119:7151–7152. [Google Scholar]; (c) Viles JH, Patel SU, Mitchell JBO, Moody CM, Justice DE, Uppenbrink J, Doyle PM, Harris CJ, Sadler PJ, Thornton JM. J Mol Biol. 1998;279:973–986. doi: 10.1006/jmbi.1998.1764. [DOI] [PubMed] [Google Scholar]; (d) Odaert B, Jean F, Melnyk O, Tartar A, Lippens G, Boutillon C, Buisine E. Protein Sci. 1999;8:2773–2783. doi: 10.1110/ps.8.12.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Arnold U, Hinderaker MP, Nilsson BL, Huck BR, Gellman SH, Raines RT. J Am Chem Soc. 2002;124:8522–8523. doi: 10.1021/ja026114n. [DOI] [PubMed] [Google Scholar]; (f) Deechongkit S, Nguyen H, Powers ET, Dawson PE, Gruebele M, Kelly JW. Nature. 2004;430:101–105. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]; (g) David R, Gunther R, Baumann L, Luhmann T, Seebach D, Hofmann HJ, Beck-Sickinger AG. J Am Chem Soc. 2008;130:15311–15317. doi: 10.1021/ja802453x. [DOI] [PubMed] [Google Scholar]; (h) Lee BC, Zuckermann RN. ACS Chem Biol. 2011;6:1367–1374. doi: 10.1021/cb200300w. [DOI] [PubMed] [Google Scholar]; (i) Valverde IE, Lecaille F, Lalmanach G, Aucagne V, Delmas AF. Angew Chem Int Ed. 2012;51:718–722. doi: 10.1002/anie.201107222. [DOI] [PubMed] [Google Scholar]
- 5.(a) Horne WS, Price JL, Gellman SH. Proc Natl Acad Sci USA. 2008;105:9151–9156. doi: 10.1073/pnas.0801135105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Horne WS, Boersma MD, Windsor MA, Gellman SH. Angew Chem Int Ed. 2008;47:2853–2856. doi: 10.1002/anie.200705315. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Horne WS, Johnson LM, Ketas TJ, Klasse PJ, Lu M, Moore JP, Gellman SH. Proc Natl Acad Sci USA. 2009;106:14751–14756. doi: 10.1073/pnas.0902663106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Lengyel GA, Frank RC, Horne WS. J Am Chem Soc. 2011;133:4246–4249. doi: 10.1021/ja2002346. [DOI] [PubMed] [Google Scholar]; (b) Lengyel GA, Horne WS. J Am Chem Soc. 2012;134:15906–15913. doi: 10.1021/ja306311r. [DOI] [PubMed] [Google Scholar]; (c) Lengyel GA, Eddinger GA, Horne WS. Org Lett. 2013;15:944–947. doi: 10.1021/ol4001125. [DOI] [PubMed] [Google Scholar]
- 7.Reinert ZE, Lengyel GA, Horne WS. J Am Chem Soc. 2013;135:12528–12531. doi: 10.1021/ja405422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Hagihara M, Anthony NJ, Stout TJ, Clardy J, Schreiber SL. J Am Chem Soc. 1992;114:6568–6570. [Google Scholar]; (b) Seebach D, Abele S, Gademann K, Jaun B. Angew Chem Int Ed. 1999;38:1595–1597. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1595::AID-ANIE1595>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; (c) Karle IL, Gopi HN, Balaram P. Proc Natl Acad Sci USA. 2001;98:3716–3719. doi: 10.1073/pnas.071050198. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martinek TA, Toth GK, Vass E, Hollosi M, Fulop F. Angew Chem Int Ed. 2002;41:1718–1721. doi: 10.1002/1521-3773(20020517)41:10<1718::aid-anie1718>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; (e) Langenhan JM, Guzei IA, Gellman SH. Angew Chem Int Ed. 2003;42:2402–2405. doi: 10.1002/anie.200350932. [DOI] [PubMed] [Google Scholar]; (f) Bandyopadhyay A, Mali SM, Lunawat P, Raja KMP, Gopi HN. Org Lett. 2011;13:4482–4485. doi: 10.1021/ol201840p. [DOI] [PubMed] [Google Scholar]
- 9.(a) Searle MS, Ciani B. Curr Opin Struct Biol. 2004;14:458–464. doi: 10.1016/j.sbi.2004.06.001. [DOI] [PubMed] [Google Scholar]; (b) Nowick JS. Acc Chem Res. 2008;41:1319–1330. doi: 10.1021/ar800064f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee J, Rechenmacher F, Kessler H. Angew Chem Int Ed. 2013;52:254–269. doi: 10.1002/anie.201205674. [DOI] [PubMed] [Google Scholar]
- 11.Spencer R, Chen KH, Manuel G, Nowick JS. Eur J Org Chem. 2013;2013:3523–3528. [Google Scholar]
- 12.Fesinmeyer RM, Hudson FM, Andersen NH. J Am Chem Soc. 2004;126:7238–7243. doi: 10.1021/ja0379520. [DOI] [PubMed] [Google Scholar]
- 13.Searle MS, Griffiths-Jones SR, Maynard AJ. J Mol Biol. 1999;292:1051–1069. doi: 10.1006/jmbi.1999.3119. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fischer G. Chem Soc Rev. 2000;29:119–127. [Google Scholar]; (b) Dugave C, Demange L. Chem Rev. 2003;103:2475–2532. doi: 10.1021/cr0104375. [DOI] [PubMed] [Google Scholar]
- 15.Manavalan P, Momany FA. Biopolymers. 1980;19:1943–1973. doi: 10.1002/bip.1980.360191103. [DOI] [PubMed] [Google Scholar]
- 16.Hovmoller S, Zhou T, Ohlson T. Acta Crystallogr Sect D Biol Crystallogr. 2002;58:768–776. doi: 10.1107/s0907444902003359. [DOI] [PubMed] [Google Scholar]
- 17.(a) Gronenborn AM, Filpula DR, Essig NZ, Achari A, Whitlow M, Wingfield PT, Clore GM. Science. 1991;253:657–661. doi: 10.1126/science.1871600. [DOI] [PubMed] [Google Scholar]; (b) Gallagher T, Alexander P, Bryan P, Gilliland GL. Biochemistry. 1994;33:4721–4729. [PubMed] [Google Scholar]; (c) Frericks Schmidt HL, Sperling LJ, Gao YG, Wylie BJ, Boettcher JM, Wilson SR, Rienstra CM. J Phys Chem B. 2007;111:14362–14369. doi: 10.1021/jp075531p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Fehrentz JA, Castro B. Synthesis. 1983:676–678. [Google Scholar]; (b) Guichard G, Briand JP, Friede M. Pept Res. 1993;6:121–124. [PubMed] [Google Scholar]
- 19.(a) Debaene F, Mejias L, Harris JL, Winssinger N. Tetrahedron. 2004;60:8677–8690. [Google Scholar]; (b) Mali SM, Bandyopadhyay A, Jadhav SV, Kumar MG, Gopi HN. Org Biomol Chem. 2011;9:6566–6574. doi: 10.1039/c1ob05732d. [DOI] [PubMed] [Google Scholar]
- 20.Brunger AT. Nat Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 21.Alexander P, Fahnestock S, Lee T, Orban J, Bryan P. Biochemistry. 1992;31:3597–3603. doi: 10.1021/bi00129a007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.