

Abstract
Individuals harboring germ-line DICER1 mutations are predisposed to a rare cancer syndrome, the DICER1 Syndrome or pleuropulmonary blastoma-familial tumor and dysplasia syndrome [online Mendelian inheritance in man (OMIM) #601200]. In addition, specific somatic mutations in the DICER1 RNase III catalytic domain have been identified in several DICER1-associated tumor types. Pituitary blastoma (PitB) was identified as a distinct entity in 2008, and is a very rare, potentially lethal early childhood tumor of the pituitary gland. Since the discovery by our team of an inherited mutation in DICER1 in a child with PitB in 2011, we have identified 12 additional PitB cases. We aimed to determine the contribution of germ-line and somatic DICER1 mutations to PitB. We hypothesized that PitB is a pathognomonic feature of a germ-line DICER1 mutation and that each PitB will harbor a second somatic mutation in DICER1. Lymphocyte or saliva DNA samples ascertained from ten infants with PitB were screened and nine were found to harbor a heterozygous germ-line DICER1 mutation. We identified additional DICER1 mutations in nine of ten tested PitB tumor samples, eight of which were confirmed to be somatic in origin. Seven of these mutations occurred within the RNase IIIb catalytic domain, a domain essential to the generation of 5p miRNAs from the 5′ arm of miRNA-precursors. Germ-line DICER1 mutations are a major contributor to PitB. Second somatic DICER1 “hits” occurring within the RNase IIIb domain also appear to be critical in PitB pathogenesis.
Keywords: DICER1, Pituitary blastoma, miRNA, Pediatric tumors
Introduction
Scheithauer et al. [24] described the first case of PitB occurring in a 13-month-old female Korean child presenting with Cushing's syndrome and diabetes insipidus (case 1 in this report). A sellar and suprasellar mass measuring 3.5 cm was identified on magnetic resonance imaging (MRI). Histopathologically, the excised tumor was distinct from other pediatric adenohypophyseal tumors, exhibiting primitive Rathke-type epithelium arranged in rosettes and glandular structures, small folliculo-stellate (FS) cells and a limited range of fully differentiated secretory cells. Ultrastructurally, the tumorous pituitary resembled that of 10–12 week embryonic-stage pituitary gland [24]. The name “blastoma” was chosen by Scheithauer to reflect the embryonic-primordial appearance and neonatal presentation of these tumors.
In 2012, Scheithauer and colleagues discussed six PitB cases [23] including the 2008 case [24], one recently reported PitB [28], one newly accessioned case, and three cases previously described as pituitary adenomas [15, 16, 20]. In addition, based on the description in the reports, it was considered that three further cases probably represented PitB [13, 14, 17]. The clinical presentations and tumor histopathology of each PitB were strikingly similar, but the main predisposing genetic factors were not known.
The DICER1 syndrome, or pleuropulmonary blastoma (PPB)-familial tumor and dysplasia syndrome [online Mendelian inheritance in man (OMIM) #601200], is caused by heterozygous germ-line mutations in the DICER1 gene, which encodes DICER1, a small RNA processing endoribonuclease that cleaves precursor microRNAs (miRNA) into mature miRNAs which, in turn, post-transcriptionally regulate messenger RNA expression [11]. The main manifestations of the DICER1 syndrome include PPB, cystic nephroma (CN), Sertoli–Leydig cell tumors (SLCT), multinodular goiter (MNG) and other rare childhood sarcomas and dysplasias [2, 5, 21, 25]. The majority of these known manifestations of the syndrome are fetal, pediatric or adolescent in onset.
In 2011, we identified an inherited DICER1 mutation in an infant with PitB who had a family history strongly suggestive of DICER1 syndrome (Supplementary Figure S1: case 3) [28]. We subsequently sought to determine the frequency of germ-line DICER1 mutations in other children with PitB, as well as to test PitB tumors for the presence of second somatic mutations in DICER1.
Materials and methods
The study was approved by the Institutional Review Board of the Faculty of Medicine of McGill University, Montreal, Quebec, Canada, no. A12-M117-11A. Participants were recruited to the study in compliance with the second edition of the Canadian Tri-Council Policy Statement of Ethical Conduct of Research involving Humans and, because of the ages of the participants, eligible relatives signed a consent form in accordance with the above-mentioned IRB protocol. All potential PitB tumors were reviewed by our central reference pathologists, Dr. Eva Horvath (EH) and Dr. Kalman Kovacs (KK).
Sample acquisition
We ascertained nine published cases (cases 1–5 and 7–10) and four unpublished cases (cases 6 and 11–13) (Table 1). The cases presented in this manuscript are numbered in the order in which they were ascertained by our group. As detailed in Fig. 1 and Supplementary Table S1, we collected PitB and non-tumor tissue samples as follows: formalinfixed paraffin-embedded (FFPE) tumor for ten cases, fresh frozen tumor for two cases and FFPE normal tissue for three cases. We obtained extracted tumor DNA from one case and were unable to obtain tumor tissue from two cases. No germ-line DNA samples were attainable from two cases. We obtained DNA extracted from blood lymphocytes from five cases. Genomic DNA was extracted from 3 to 5 ml of blood collected in EDTA blood collection tubes from one case using the gentra Puregene Blood kit (Qiagen, California, USA) following the manufacturer's instructions. The Gentra Puregene Blood kit was also used to extract genomic DNA from saliva samples collected from two cases using the OragenėDNA OG-250 DNA collection kit (DNA Genotek, Ottawa, Canada) (Supplementary Table S1). Therefore, of the thirteen cases, we had the ability to determine the origin of the identified mutations for ten cases as both germ-line and somatic samples were available (case 2, cases 4–6 and cases 8–13); for case 1, we had a tumor sample only; for case 3, only germ-line gDNA was available; and for case 7, we were unable to acquire a germ-line and/or somatic DNA sample (Fig. 1 and Supplementary Table S1).
Table 1.
Literature and case summary
| Case # | Sex | 1st Sx; age (months) | Age at Dx of PitB (months) | Endocrine function at presentation | Clinical outcome (time from 1st surgery to last F-U appointment) | Evidence of DICER1 syndrome? | Germ-line DICER1 mutation | Somatic DICER1 mutation | References |
|---|---|---|---|---|---|---|---|---|---|
| Case 1 | F | Cushing's, DI; 13 | 13 | ↑ ACTH; nl: PRL, GH; ↓:TSH, LH, FSH | Deceased 1.5 months post-surgery | None | Not available | c.5437 G>Aa | Scheithauer et al. [24] |
| Case 2 | F | Strabismus, ↓ visual acuity, short stature; 24 | 24 | ↑ ACTH; nl: “other markers” | Alive (7 years) | None | c.3277_3280delAACT | No RNase IIIa or IIIb mutation; No LOH | Minetal. [16]; Present case 3 in Scheithauer et al. [23] |
| Case 3 | M | Strabismus, proptosis, hypothyroidism; 9 | 9 | ↓ TSH; nl: bone age and growth; ↑ ACTH at 11 months | Alive (3.11 years) | Family: PPB, CN, RC,OSLCT | c.2379T>G (NMD of mutant) | Not available | Present case 1 in Scheithauer et al. [23], Wildi-Runge et al. [28] |
| Case 4 | F | ↑ Weight, fatigue; 6 | 23 | ↑ ACTH | Alive (7.5 years) | Family: thryoid nodule and hypothyroidism | c.3535_3538delTCTT | c.5125G>T | Moriarty et al. [17] |
| Case 5 | F | Ophthalmoplegia, ptosis; 13 | 13 | nl: TSH, PRL, somatomedin C. (ACTH not measured) | Alive (4.8 years) | None | c.l525C>T(de novo) | c.5425G>T | Present case 2 in Scheithauer et al. [23] |
| Case 6 | M | Strabismus; 5 | 7 | nl: Free T4, TSH; ↑ IGF2, IGFBP, blood AFP | Deceased 8 months post-surgery | None | c.4309_4312delGACT | c.5125G>A | Not previously published |
| Case 7 | M | Cushing's; 6 | 11 | ↑ Cortisol; nl: ACTH,TSH, GH, PRL; ↓ bone age | Deceased 26 months post-surgery—died of tumor | Bilateral lung cysts; bilateral RC | Not Available | Not Available | Levy etal. [12], Pullins et al. [20], Sumner et al. [26] |
| Case 8 | M | ↑ Weight, ↓ height (4); Cushing's (6) | 8 | ↑: ACTH, cortisol; nl: LH, FSH; ↓: TSH, PRL | Deceased at 0 months post-surgery (died post-op) | None | DNA quality too poor | DNA quality too poor | Miller etal. [15] |
| Case 9 | F | ↑ Weight, fatigue; 5 | 7 | ↑: ACTH, TSH; nl: PRL, GH, HCG | Alive (17.4 years) | None | c.2026C>T | c.5439G>T | List etal. [13] |
| Case 10 | F | DI, ↓ weight, short stature; 12 | 12 | ↑ Cortisol; ↓ bone age | Deceased 18 months post-surgery | None | c.2026C>T | c.5438A>T | Maederetal. [14] |
| Case 11 | F | Hydrocephalus; 7 | 7 | Not Knownb | surgery Alive (23 months) | Family: OSLCT, Thyroid nodule | c.l284delGA | c.5437 G>A | Not previously published |
| Case 12 | M | Cushing's; 8 | 8 | ↑: ACTH | Alive (21 months) | Multi-focal bilateral lung cysts & RC | c.5125G>C(de novo) | LOH | Not previously published |
| Case 13 | F | Strabismus, ↑ ICP, “plump” (Cushing's not suspected); 9 | 9 | ↑: Cortisol; ↓ TSH; nl: PRL, GH, FT4 | Alive (13.4 years) | None | Negative | LOH | Not previously published |
ACTH adrenocorticotropic hormone; AFP alpha-fetoprotein; CN cystic nephroma; CT computerized tomography; Cashing's Cushing's syndrome with cushingoid facies, truncal obesity, hyper-trichosis with or without acne vulgaris, hypertension and short stature; DI diabetes insipidus; Dx diagnosis; F female; FSH follicle stimulating hormone; F-U follow-up; GH growth hormone; HCG human chorionic gonadotropin; IGF insulin-like growth factor; IGFBP insulin-like growth factor binding protein; LH luteinizing hormone; LOH loss of heterozygosity; M male; Mo. months; nl normal; NMD nonsense-mediated decay; OSLCT ovarian Sertoli–Leydig cell tumor; post-op post-operatively; PPB pleuropulmonary blastoma; PRL prolactin; RC renal cysts; Sx symptoms; TSH thyroid stimulating hormone; ↑ increased; ↓ decreased
Mutation not confirmed to be somatic in origin
Due to precipitous admission and operation, pre-operative endocrine function test results are not available
Fig. 1.

Flowchart summarizing case identification, sample acquisition, molecular analysis and results of the study
Molecular screening of DICER1
In seven cases, screening for germ-line DICER1 mutations was conducted on DNA extracted from blood or saliva by PCR and sequencing of the region of interest [7, 22, 30]. In one case where no germ-line DICER1 mutation was identified by conventional sequencing, we screened for large deletions or duplications using our multiplex ligation-based probe amplification assay [22]. For four cases, germ-line DICER1 mutations were screened for using the Fluidigm access array system and next-generation sequencing (Supplementary Table S1). Fluidigm access array system involves an array-based PCR amplification of a specific region of interest (target enrichment). For our purpose, the exons of DICER1, all exon–intron boundaries and the 3′UTR were selectively targeted. Parallel amplification of 48 samples was carried out using custom design primers (Supplementary Table S2), designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) and to which CS1 and CS2 tails were added. Samples were barcoded during the targeted enrichment to allow for multiplexed sequencing and amplicons were tagged with adaptor sequences during the PCR amplification reaction. Next-generation sequencing was carried out using the Illumina MiSeq (McGill University and Genome Quebec Innovation Centre (MUGQIC)) and the Integrative Genomics Viewer software (IGV version 2.3; http://www.broadinstitute.org/igv/) was used to analyze the dataset. All variants identified were confirmed with Sanger sequencing.
Somatic “hotspot” mutations were identified by PCR amplification of gDNA derived from each PitB [29, 30] followed by Sanger sequencing (MUGQIC). Loss of heterozygosity (LOH) analysis in tumor samples was performed by amplifying tumor gDNA by PCR concurrently with the patient's germ-line gDNA, using primers specific to the region of interest. The ~150–200 base-pair PCR products were analyzed by direct Sanger sequencing and the relative intensity of the peaks at the position of the germ-line DICER1 mutation and/or single-nucleotide polymorphisms (SNPs) within the 3′UTR of the gene were compared between germ-line and tumor gDNA to determine whether LOH occurred (Supplementary Table S3). For one case, genotyping of the short tandem repeat (STR) markers, D14S265 and D14S1054, was performed using γ-P33 as previously described [27] to ascertain LOH in the absence of coding variants that could be interrogated using Sanger sequencing.
Pathological review
Suspected PitB cases were evaluated by EH and KK using light microscopy, immunohistochemistry, and transmission electron microscopy. For the histologic investigation, FFPE tissue was used. The electron microscopic study was performed on glutaraldehyde fixed and osmicated material.
Immunohistochemistry
Immunohistochemical staining for adrenocorticotrophic hormone (ACTH) and growth hormone (GH) was performed using the streptavidin–biotin–peroxidase complex method, using antibodies directed against ACTH and GH. Details of the methods including source and dilution of antibodies have been described previously [8, 10]. Immunostaining of Ki-67 and p53 was performed on formalinfixed, paraffin-embedded sections, 7 μm in thickness, that were de-paraffinized using xylol and rehydrated in serial aqueous dilutions of alcohol. Immunohistochemistry was performed after heat induced epitope antigen retrieval using citraconic acid at pH 7.4 and 45 min at 100 °C. Monoclonal mouse anti-human Ki-67 antigen antibody (Dako clone MIB-1) was used at a dilution of 1:100 and monoclonal mouse anti-human p53 antibody (Dako clone DO-7) was used at a dilution of 1:50. For both antibodies, immunostaining was performed using a DAKO Auto-stainer Link 48 and with DAKO EnVision™ Flex visualization system.
Results
We investigated 13 cases of PitB, the features of which are summarized in Table 1. Twelve of the 13 cases were reviewed by EH and KK, and the remaining case was included based on convincing published evidence [12, 20, 26]. Four cases were newly ascertained; nine have been previously reported between 1979 and 2012. Eight of the infants were females and five were males. Ages at notice of first symptom ranged from 7 to 24 months (median 8 months) and the ages at pathological diagnosis ranged from 7 to 24 months (median 9 months). Of the 13 infants, five died of disease within 0–26 months of the first surgery (median 8 months) and eight remain alive, with survival time from first surgery to last follow-up ranging from 21 months to 17.4 years (median 5.9 years). Two children with PitB had a personal medical history of other diseases associated with a DICER1 mutation; a further three children had a family history of diseases suggesting DICER1 syndrome; and in eight children, PitB was the only notable disease (Table 1). Family pedigrees are depicted in Supplementary Figures S1 to S6.
Cushing's syndrome and/or ophthalmoplegia were the most frequent presenting symptoms of PitB. Elevated blood ACTH levels were demonstrated in seven of the eight patients for which data are available (Table 1). Figure 2 presents the diagnostic MR images and mutation traces from case 12.
Fig. 2.

Case 12: a chest CT following IV contrast: multiple bilateral thin-walled air filled cysts evident within all lobes of the lungs; likely PPB Type I or Ir (not biopsied). b Coronal CT image of abdomen and pelvis. Liquid-filled cyst in lower pole of right kidney is typical of CN (not biopsied). Other CT images (not shown) also revealed a smaller cyst (likely CN, not biopsied) in the left kidney. c T1 midline sagittal MR image. d T2-weighted axial MR image just superior to the pituitary. In c and d, the pituitary tumor is indicated with arrows. e Panel I a mono-allelic germ-line DICER1 mutation, c.5125G>C [p.(Asp1709His)]. Panel II clear loss of heterozygosity at the position of the germ-line DICER1 mutation within the tumor (wild-type allele lost). f The proband (individual II-1) was diagnosed with PitB at the age of 8 months and was found to carry the de novo c.5125G>C germ-line DICER1 mutation
Morphologically, all cases showed the classic features of PitB as previously described by Scheithauer and colleagues [23, 24] (Fig. 3). This included a combination of Rathketype epithelial rosettes/glands, small primitive appearing cells and secretory cells, the latter of which were synaptophysin and chromogranin immunoreactive and always expressed ACTH in at least a subset of cells. Some cases also included a GH secreting subset (Fig. 3b, c-I, c-II), whereas ultrastructural features of FS cells were also commonly identified in a subset of tumor cells. However, differences in the proliferative and mitotic activity were evident in some cases. Case 12 was noted to have marked nuclear labelling for Ki-67 (Fig. 4a) and thus had a high proliferative fraction. The tumors of case 3, case 5 [23] and case 11 were similarly markedly proliferative. In contrast, the Ki-67 nuclear labelling in case 2 was estimated at 1.63 % (Fig. 4b), which is low despite the large tumor size detected at presentation. Nuclear expression of p53 protein was present in the cells that form the rosettes, but was scant elsewhere (Fig. 4c, d).
Fig. 3.

a case 13, T1-weighted post-contrast midline sagittal MR image showing pituitary region mass (red arrow). b case 4, hematoxylin and eosin (H&E) staining ×250: three enlarged follicles lined by stem cells. c Immunohistochemical staining I case 10, Growth hormone (GH) immunostaining ×400: enlarged GH/alpha subunit cells immunopositive for GH. II case 10, ACTH immunostaining ×400: small vessel surrounded by stem cells. Some cells display ACTH immunoreactivity
Fig. 4.

Immunohistochemical staining a case 12, Ki-67 labelling ×200: there is marked labelling for Ki-67, indicating a high proliferative fraction that is limited to the rosette-like epithelial structures. b Case 2, Ki-67 labelling ×100: Ki-67 labelling index estimated at 1.63 %, indicating low proliferative activity. c Case 12, p53 immunostaining ×400. d Case 2, p53 immunostaining ×100. For c and d, p53 expression is present in cells forming the rosette structures, but is scant elsewhere
DICER1 mutation data from each case are summarized in Table 1 and Fig. 5 with additional details including samples analyzed and mutation traces presented in Fig. 1, Supplementary Table S1 and Supplementary Table S4. We were able to obtain germ-line DNA from eleven children with PitB (ten of which were successfully screened for DICER1 mutations) and PitB tumor tissue from a different set of 11 cases. In one case reported in 1979, we were unable to extract reliable gDNA from FFPE to allow for molecular testing (Supplementary Table S1). Of the ten cases analyzed, nine children with PitB harbored a germline DICER1 mutation, among which, one germ-line mutation was predicted to affect the critical RNase IIIb catalytic site of DICER1. Nine of ten PitB tumor tissue specimens analyzed had DICER1 mutations, eight of which are confirmed to be somatic in origin where germ-line gDNA was available. Of the nine DICER1 mutations identified in the tumor samples, seven are predicted to affect the RNase IIIb catalytic site of DICER1 (Fig. 5 and Supplementary Table S1), consistent with the vast majority of other reported somatic mutations for DICER1-associated diseases (i.e., that they occur within exons encoding the RNase IIIb site). The remaining two somatic mutations identified were LOH of the wild-type allele within the tumor. Overall, we demonstrated DICER1 mutations in 11 of 12 PitB cases for which we had germ-line and/or tumor sample(s): both germ-line and somatic mutations were present in seven of 12 PitB cases; whereas a germ-line mutation only was found in two cases; a somatic mutation only in one case; one case harbored an RNase IIIb missense mutation in the tumor sample, but was not confirmed to be somatic in origin; and in one case, genetic screening of FFPE gDNA was not successful (Fig. 5 and Supplementary Table S4).
Fig. 5.
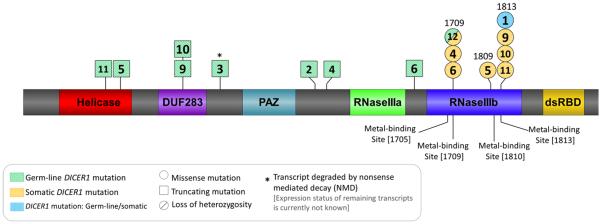
Graphic representation of the DICER1 protein structure (NP_001258211.1) indicating the approximate positions of the germ-line (green diagonal stripes) and somatic (orange horizontal stripes) DICER1 mutations observed in the 13 cases being reported. Mutation shaded with blue vertical stripes represent mutations that were identified within tumour gDNA, but are not confirmed to be somatic in origin. Case number indicated at the position of each mutation: case 1: somatic DICER1 amino acid change: p.(Glu1813Lys). Case 2: germ-line DICER1 amino acid change: p.(Asn1093*). Case 3: germ-line DICER1 amino acid change: p.(Tyr793*), (NMD of mutant). Case 4: germ-line DICER1 amino acid change: p.(Ser1179Thrfs*12); Somatic DICER1 amino acid change: p.(Asp1709Thr). Case 5: germ-line DICER1 amino acid change: p.(Arg509*); somatic DICER1 amino acid change: p.(Gly1809Trp). Case 6: germ-line DICER1 amino acid change: p.(Asp1437Metfs*16); somatic DICER1 amino acid change: p.(Asp1709Asn). Case 9: germ-line DICER1 amino acid change: p.(Arg676*); somatic DICER1 amino acid change: p.(Glu1813Asp). Case 10: germ-line DICER1 amino acid change: p.(Arg676*); somatic DICER1 amino acid change: p.(Glu1813Val). Case 11: germ-line DICER1 amino acid change: p.(Lys429Alafs*47); somatic DICER1 amino acid change: p.(Glu1813Lys). Case 12: germ-line DICER1 amino acid change: p.(Asp1709His); somatic DICER1 change: loss of heterozygosity
Discussion
The results presented here expand the spectrum of the DICER1 syndrome to include PitB as a highly characteristic phenotype with a high likelihood of germ-line and/or somatic DICER1 mutations. This tumor may now be considered a rare but pathognomonic manifestation of a germ-line DICER1 mutation. The penetrance appears to be well below 1 %. The clinical phenotype is noteworthy in that the primary manifestation is the onset of Cushing's syndrome during infancy, an exceedingly rare endocrinopathy in this age group. Even in the absence of a formal genetic diagnosis, this clinical presentation should motivate the clinician to look for other DICER1-related diseases in the patient and family and to consider PitB a strong diagnostic possibility.
The inclusion of PitB in the blastoma category was based on several factors delineated by Scheithauer [24]. The term “blastoma” implies that the tumor has the appearance of pituitary embryonic tissue and exhibits malignant potential, but questions have been raised as to whether PitB is indeed clinically malignant. Many of the PitBs in our study behave aggressively and were fatal in approximately 40 % of cases (Table 1). Death of these infants could be due to local tumor effects causing increased intracranial pressure and damage to surrounding tissues or due to excess serum cortisol causing severe, often lethal, Cushing's disease. One child had an aggressive recurrence with apparent intraventricular metastases, but histologic confirmation of the CNS disease was not possible. As noted in the results, cell proliferation, as measured by Ki-67 labelling index, within the PitBs studied was variable with four cases exhibiting high proliferative activity (case 3, 5 [23], 11 and 12) and one showing minimal proliferation (case 2) (Fig. 4). Three of the four children with PitBs showing high proliferative activity had recurrences and all four remain alive. These limited data suggest that the Ki-67 proliferation index has uncertain predictive value for PitB, although it should be studied further. The implications of p53 expression in the cells forming the rosettes similarly require further investigation. Perhaps both indolent and more active PitBs exist and as yet, we have only a limited understanding of the biologic behavior of this dysontogenic lesion.
There is also speculation surrounding the cell of origin of PitB. Histological studies suggest perturbation of pituitary stem cells could underlie the pathogenesis of these tumors and the recent advances in the identification of cancer stem cells in other lesions [3, 4] further substantiates this argument. The identification of DICER1 mutations in our cohort of PitBs (predicted to alter the miRNA profiles of the tumors) taken together with emerging data showing that miRNAs play a role in regulating stem cell markers [19] and the presence of FS “stem cells” within PitB [23] provides a possible explanation of pituitary dysontogenesis leading to PitB that could be explored.
Eight of the nine germ-line mutations identified are loss-of-function mutations that are predicted to inactivate one allele of DICER1, suggesting that germ-line mutation of DICER1 is a key predisposing genetic event. Five of the germ-line DICER1 mutations we identified are known to be inherited and two are de novo mutations (Fig. 2f and Supplementary Figure S1 to S6). Seven of the nine DICER1 mutations identified within the tumor samples (eight of which are confirmed to be somatic in origin) were localized within the sequence encoding the protein's RNase IIIb domain, thereby affecting highly conserved amino acid residues (Asp1709, Gly1809, Glu1813) (Fig. 5). Mutations affecting the metal ion-binding amino acid residues of DICER1, Glu1705, Asp1709, Asp1810 and Glu1813, have been shown to reduce the processing of mature 5p miRNA strands, shifting the mature miRNA expression within these tumors towards 3p-derived miRNAs, a shift in expression thought to contribute to tumorigenesis [1, 6]. The somatic mutation identified in case 13 was LOH of one allele evident at two STR markers, D14S1054 and D14S265 (Supplementary Figure S7). No germ-line mutation was found within the coding region of DICER1 in this patient, but we suspect that a non-coding mutation that deleteriously affects the expression of the transcript from the other allele may be present. In five cases where two DICER1 mutations were not identified, the genetic analysis of DICER1 in an exon-to-exon approach remains incomplete either due to difficulty amplifying DNA or because we did not have tissue available to us to complete the analysis. We predict that all cases will have two DICER1 mutations, hypothesizing that a second somatic “hit” (most often affecting an RNase IIIb metal ion-binding residue of DICER1) in the embryonic pituitary is required in addition to a loss-of-function germ-line DICER1 mutation to initiate development of an embryonic-appearing blastomatous tumor.
The germ-line and somatic DICER1 mutations identified in case 12 require further discussion. The de novo mutation, c.5125G>C [p.(Asp1709His)], is the third reported DICER1 mutation in lymphocyte gDNA affecting a metal ion-binding residue within an RNase III domain [9]. Approximately, 75 previously reported germ-line DICER1 mutations are distributed throughout the gene. In contrast, almost all reported somatic mutations in DICER1 syndrome diseases affect the metal-binding residues. The other two reports of de novo missense mutations directly affecting a metal ion binding site within an RNase III domain were determined by the researches to be mosaic mutations and were associated with overgrowth, bilateral Wilms tumor and bilateral lung cysts in both of their patients. There was no evidence of overgrowth in our case, but the clinical presentation in this child was particularly severe. In addition to the PitB, the infant has extensive multi-focal bilateral lung cysts (likely PPB Type I or Type Ir) and bilateral renal cystic masses (likely CN) which have not been biopsied (Fig. 2a, b). Furthermore, with only lymphocyte gDNA and PitB tumor tissue available to us, we were unable to determine whether the child is a mosaic for the observed p.(Asp1709His) mutation. The somatic “hit” in case 12 was identified to be loss of the wild-type allele within the tumor (Fig. 2e), which together with the germ-line mutation, would result in the absence of expression of any normal DICER1 within the affected cell lineages in the developing pituitary gland. We speculate that a germ-line DICER1 mutation affecting one of the metal ion-binding sites may result in especially severe clinical disease as in our case 12 and the two children previously reported [9]. In case 1, we identified an RNase IIIb mutation (c.5437G>A; p.[Glu1813Lys)] in FFPE tumor gDNA. In contrast to case 12, LOH was not evident within the tumor. Without a germ-line gDNA sample available, we are unable to confirm whether the identified mutation in case 1 is somatic in origin. Although consistent with somatic mutations most often identified within DICER1-related tumors, based on the severe clinical presentation of case 1 (Table 1), perhaps this RNase IIIb DICER1 mutation is in fact a germ-line mutation. As with case 12, the clinical presentation of case 7 was particularly severe (Table 1), but no tissue was available for molecular analysis.
The specific downstream miRNA perturbations of the germ-line and somatic DICER1 mutations identified and the mechanisms of tumorigenesis remain to be explored, but may be related to a relative excess of 3p-derived miRNAs as postulated [1, 6] and as recently reported in a case of PPB [18]. Further studies on this rare pediatric tumor should focus on mRNA, miRNA and gDNA profiling and will include explorations of the mechanistic implications of DICER1 mutations found in PitB. We encourage submission of any suspected PitB case for expert pathological review and intensive molecular study.
Conclusion
The results of this study suggest that a germ-line DICER1 mutation is the major and possibly sole predisposing genetic contributor to development of a PitB. The acquisition of a somatic mutation in DICER1 in the RNase IIIb domain also appears to be a critical second “hit” in PitB pathogenesis. The particularly, complex clinical situation surrounding children with PitB also requires communication between clinicians, neuropathologists and pediatric oncologists to help determine the most effective management of these infants.
Supplementary Material
Acknowledgements
We thank the families involved in this research for their consent to participation and all the clinicians for referring cases and providing samples. We thank Dr. Benoît Lhermitte, Dr. Alistair Lammie, Dr. Cynthia Andoniadou, Dr. Helen Spoudeas, Dr. Oh-Lyong Kim, Dr. Andrew Peet, Dr. Angela Hübner, Dr. Walter Miller, Claudia Retamal-Muñoz, Dr. Bénédict Rilliet, Dr. Ty W. Abel and Dr. Duncan MacGregor for their assistance with ascertainment and analysis of their respective cases. We thank the MUGQIC staff for assisting Pierre Lepage with the Fluidigm Access Array and next-generation sequencing. This research was made possible thanks to the support of the Lady Davis Institute/TD Bank Studentship Award, CCSRI Innovative grant to Dr. William D. Foulkes and K12 CA 090625 to Dr. Adam Esbenshade.
Footnotes
This paper is dedicated to Bernd W. Scheithauer, MD, with whom several of the co-authors have trained and/or collaborated. Bernd was a world-renowned neuropathologist with special expertise in pituitary disease. With his colleagues, Bernd described pituitary blastoma (PitB) in 2008 and he was an early participant in the work reported here. Just before his untimely death, he was thrilled to learn that several additional PitB cases had been accessioned and more so that DICER1 mutations explain PitB and its association with other childhood blastomas in this familial tumor predisposition syndrome.
Electronic supplementary material The online version of this article (doi:10.1007/s00401-014-1285-z) contains supplementary material, which is available to authorized users.
Conflict of interest The authors have no conflicts of interest to disclose.
References
- 1.Anglesio MS, Wang Y, Yang W, Senz J, Wan A, Heravi-Moussavi A, Salamanca C, Maines-Bandiera S, Huntsman DG, Morin GB. Cancer-associated somatic DICER1 hotspot mutations cause defective miRNA processing and reverse-strand expression bias to predominantly mature 3p strands through loss of 5p strand cleavage. J Pathol. 2013;229(3):400–409. doi: 10.1002/path.4135. doi:10.1002/path.4135. [DOI] [PubMed] [Google Scholar]
- 2.Bahubeshi A, Bal N, Rio Frio T, Hamel N, Pouchet C, Yilmaz A, Bouron-Dal Soglio D, Williams GM, Tischkowitz M, Priest JR, Foulkes WD. Germline DICER1 mutations and familial cystic nephroma. J Med Genet. 2010;47(12):863–866. doi: 10.1136/jmg.2010.081216. doi:10.1136/jmg.2010.081216. [DOI] [PubMed] [Google Scholar]
- 3.Baker M. Cancer stem cells tracked. Nature. 2012;488(7409):13–14. doi: 10.1038/488013a. doi:10.1038/488013a. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488(7412):522–526. doi: 10.1038/nature11287. doi:10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foulkes WD, Bahubeshi A, Hamel N, Pasini B, Asioli S, Baynam G, Choong CS, Charles A, Frieder RP, Dishop MK, Graf N, Ekim M, Bouron-Dal Soglio D, Arseneau J, Young RH, Sabbaghian N, Srivastava A, Tischkowitz MD, Priest JR. Extending the phenotypes associated with DICER1 mutations. Hum Mutat. 2011;32(12):1381–1384. doi: 10.1002/humu.21600. doi:10.1002/humu.21600. [DOI] [PubMed] [Google Scholar]
- 6.Gurtan AM, Lu V, Bhutkar A, Sharp PA. In vivo structure-function analysis of human Dicer reveals directional processing of precursor miRNAs. RNA. 2012;18(6):1116–1122. doi: 10.1261/rna.032680.112. doi:10.1261/rna.032680.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, Jarzembowski JA, Wikenheiser-Brokamp KA, Suarez BK, Whelan AJ, Williams G, Bracamontes D, Messinger Y, Goodfellow PJ. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325(5943):965. doi: 10.1126/science.1174334. doi:10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horvath E, Vidal S, Syro LV, Kovacs K, Smyth HS, Uribe H. Severe lymphocytic adenohypophysitis with selective disappearance of prolactin cells: a histologic, ultrastructural and immunoelectron microscopic study. Acta Neuropathol. 2001;101(6):631–637. doi: 10.1007/s004010000288. [DOI] [PubMed] [Google Scholar]
- 9.Klein S, Lee H, Ghahremani S, et al. Expanding the phenotype of mutations in DICER1: mosaic missense mutations in the RNase IIIb domain of DICER1 cause GLOW syndrome. J Med Genet. 2014;51:294–302. doi: 10.1136/jmedgenet-2013-101943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kovacs K, Lloyd R, Horvath E, Asa SL, Stefaneanu L, Killinger DW, Smyth HS. Silent somatotroph adenomas of the human pituitary. A morphologic study of three cases including immunocytochemistry, electron microscopy, in vitro examination, and in situ hybridization. Am J Pathol. 1989;134(2):345–353. [PMC free article] [PubMed] [Google Scholar]
- 11.Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. doi: 10.1038/nrg2843. doi:10.1038/nrg2843. [DOI] [PubMed] [Google Scholar]
- 12.Levy SR, Wynne CV, Jr, Lorentz WB., Jr Cushing's syndrome in infancy secondary to pituitary adenoma. Am J Dis Child. 1982;136(7):605–607. doi: 10.1001/archpedi.1982.03970430037010. [DOI] [PubMed] [Google Scholar]
- 13.List JV, Sobottka S, Huebner A, Bonk C, Koy J, Pinzer T, Schackert G. Cushing's disease in a 7-month-old girl due to a tumor producing adrenocorticotropic hormone and thyreotropin-secreting hormone. Pediatr Neurosurg. 1999;31(1):7–11. doi: 10.1159/000028824. [DOI] [PubMed] [Google Scholar]
- 14.Maeder P, Gudinchet F, Rillet B, Theintz G, Meuli R. Cushing's disease due to a giant pituitary adenoma in early infancy: CT and MRI features. Pediatr Radiol. 1996;26(1):48–50. doi: 10.1007/BF01403705. [DOI] [PubMed] [Google Scholar]
- 15.Miller WL, Townsend JJ, Grumbach MM, Kaplan SL. An infant with Cushing's disease due to an adrenocorticotropin-producing pituitary adenoma. J Clin Endocrinol Metab. 1979;48(6):1017–1025. doi: 10.1210/jcem-48-6-1017. doi:10.1210/jcem-48-6-1017. [DOI] [PubMed] [Google Scholar]
- 16.Min HS, Lee SJ, Kim SK, Park SH. Pituitary adenoma with rich folliculo-stellate cells and mucin-producing epithelia arising in a 2-year-old girl. Pathol Int. 2007;57(9):600–605. doi: 10.1111/j.1440-1827.2007.02145.x. doi:10.1111/j.1440-1827.2007.02145.x. [DOI] [PubMed] [Google Scholar]
- 17.Moriarty M, Hoe F. Cushing disease in a toddler: not all obese children are just fat. Curr Opin Pediatr. 2009;21(4):548–552. doi: 10.1097/MOP.0b013e32832d1f4f. doi:10.1097/MOP.0b013e32832d1f4f. [DOI] [PubMed] [Google Scholar]
- 18.Murray MJ, Bailey S, Raby KL, Saini HK, de Kock L, Burke GA, Foulkes WD, Enright AJ, Coleman N, Tischkowitz M. Serum levels of mature microRNAs in DICER1-mutated pleuropulmonary blastoma. Oncogenesis. 2014;3:e87. doi: 10.1038/oncsis.2014.1. doi:10.1038/onc sis.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peng C, Li N, Ng YK, Zhang J, Meier F, Theis FJ, Merkenschlager M, Chen W, Wurst W, Prakash N. A unilateral negative feedback loop between miR-200 microRNAs and Sox2/E2F3 controls neural progenitor cell-cycle exit and differentiation. J Neurosci. 2012;32(38):13292–13308. doi: 10.1523/JNEUROSCI.2124-12.2012. doi:10.1523/JNEURO SCI.2124-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pullins DI, Challa VR, Marshall RB, Davis CH., Jr ACTH-producing pituitary adenoma in an infant with cysts of the kidneys and lungs. Histopathology. 1984;8(1):157–163. doi: 10.1111/j.1365-2559.1984.tb02330.x. [DOI] [PubMed] [Google Scholar]
- 21.Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD. Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. J Med genet. 2012;49(7):417–419. doi: 10.1136/jmedgenet-2012-100898. doi:10.1136/jmedgenet-2012-100898. [DOI] [PubMed] [Google Scholar]
- 22.Sabbaghian N, Srivastava A, Hamel N, Plourde F, Gajtko-Metera M, Niedziela M, Foulkes WD. Germ-line deletion in DICER1 revealed by a novel MLPA assay using synthetic oligo-nucleotides. Eur J Hum Genet. 2013 doi: 10.1038/ejhg.2013.215. doi:10.1038/ejhg.2013.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheithauer BW, Horvath E, Abel TW, Robital Y, Park SH, Osamura RY, Deal C, Lloyd RV, Kovacs K. Pituitary blastoma: a unique embryonal tumor. Pituitary. 2012;15(3):365–373. doi: 10.1007/s11102-011-0328-x. doi:10.1007/s11102-011-0328-x. [DOI] [PubMed] [Google Scholar]
- 24.Scheithauer BW, Kovacs K, Horvath E, Kim DS, Osamura RY, Ketterling RP, Lloyd RV, Kim OL. Pituitary blastoma. Acta Neuropathol. 2008;116(6):657–666. doi: 10.1007/s00401-008-0388-9. doi:10.1007/s00401-008-0388-9. [DOI] [PubMed] [Google Scholar]
- 25.Slade I, Bacchelli C, Davies H, Murray A, Abbaszadeh F, Hanks S, Barfoot R, Burke A, Chisholm J, Hewitt M, Jenkinson H, King D, Morland B, Pizer B, Prescott K, Saggar A, Side L, Traunecker H, Vaidya S, Ward P, Futreal PA, Vujanic G, Nicholson AG, Sebire N, Turnbull C, Priest JR, Pritchard-Jones K, Houlston R, Stiller C, Stratton MR, Douglas J, Rahman N. DICER1 syndrome: clarifying the diagnosis, clinical features and management implications of a pleiotropic tumour predisposition syndrome. J Med Genet. 2011;48(4):273–278. doi: 10.1136/jmg.2010.083790. doi:10.1136/jmg.2010.083790. [DOI] [PubMed] [Google Scholar]
- 26.Sumner TE, Volberg FM. Cushing's syndrome in infancy due to pituitary adenoma. Pediatr Radiol. 1982;12(2):81–83. doi: 10.1007/BF00972437. [DOI] [PubMed] [Google Scholar]
- 27.Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, Li G, van Beers EH, Li L, Khalil T, Quenneville LA, Omeroglu A, Poll A, Lepage P, Wong N, Nederlof PM, Ashworth A, Tonin PN, Narod SA, Livingston DM, Foulkes WD. Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA. 2007;104(16):6788–6793. doi: 10.1073/pnas.0701724104. doi:10.1073/pnas.0701724104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wildi-Runge S, Bahubeshi A, Carret A-S, Crevier L, Robitaille Y, Kovacs K, Horvath E, Scheithauer BW, Foulkes WD, Deal C. New phenotype in the familial DICER1 tumor syndrome: pituitary blastoma presenting at age 9 months. 2011. pp. P1–P777. endocrine reviews 32(03_MeetingAbstracts) [Google Scholar]
- 29.Witkowski L, Mattina J, Schonberger S, Murray MJ, Choong CS, Huntsman DG, Reis-Filho JS, McCluggage WG, Nicholson JC, Coleman N, Calaminus G, Schneider DT, Arseneau J, Stewart CJ, Foulkes WD. DICER1 hotspot mutations in non-epithelial gonadal tumours. Br J Cancer. 2013;109(10):2744–2750. doi: 10.1038/bjc.2013.637. doi:10.1038/bjc.2013.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu MK, Sabbaghian N, Xu B, Addidou-Kalucki S, Bernard C, Zou D, Reeve AE, Eccles MR, Cole C, Choong CS, Charles A, Tan TY, Iglesias DM, Goodyer PR, Foulkes WD. Biallelic DICER1 mutations occur in Wilms tumours. J Pathol. 2013;230(2):154–164. doi: 10.1002/path.4196. doi:10.1002/path.4196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.