

Abstract
Stem cells divide asymmetrically to generate two progeny cells with unequal fate potential: a self-renewing stem cell and a differentiating cell. Given their relevance to development and disease, understanding the mechanisms that govern asymmetric stem cell division has been a robust area of study. Because they are genetically tractable and undergo successive rounds of cell division about once every hour, the stem cells of the Drosophila central nervous system, or neuroblasts, are indispensable models for the study of stem cell division. About 100 neural stem cells are located near the surface of each of the two larval brain lobes, making this model system particularly useful for live imaging microscopy studies. In this work, we review several approaches widely used to visualize stem cell divisions, and we address the relative advantages and disadvantages of those techniques that employ dissociated versus intact brain tissues. We also detail our simplified protocol used to explant whole brains from third instar larvae for live cell imaging and fixed analysis applications.
Keywords: Neuroscience, Issue 89, live imaging, Drosophila, neuroblast, stem cell, asymmetric division, centrosome, brain, cell cycle, mitosis
Introduction
Stem cells maintain a balance of differentiation and self-renewal to generate cellular diversity during early development and replace damaged cells in adult tissues. Regulation of this homeostasis prevents both the loss and over-expansion of the stem cell population, which can result in deleterious tissue degeneration or tumorigenesis.
Neuroblasts (NBs) are the widely studied neural stem cells of the Drosophila central nervous system (Figure 1A) that first form during embryonic stages1,2. NBs undergo repeated rounds of asymmetric cell division (ACD) to produce two unequally fated cells: a self-renewing stem cell and a differentiating cell. ACD is guided by centrosomes, the non-membranous organelles that act as the microtubule-organizing centers of most cells3. During mitosis, NB centrosomes organize and orient the bipolar mitotic spindle along the apical-basal polarity axis. Upon cleavage of the dividing NB, apical fate determinants that specify the stem cell fate, and basal fate determinants that specify differentiation, are segregated into the unequal daughter cells.
In the larval central brain, two types of NBs may be distinguished by their number, position, transcription factor expression, and cell lineage (Figure 1A)4-6. Type I NBs are the most abundant, and about 90 of them populate the anterior and posterior sides of each optic lobe of the brain7. These NBs express the transcription factor Asense (Ase), and they characteristically divide into one self-renewing NB and one smaller ganglion mother cell (GMC; Figure 1B). Each GMC undergoes a single terminal division to generate two neurons or glia (Figure 1B). In contrast, the eight Type II NBs that populate the posterior side of each optic lobe lack Ase expression5. They undergo ACD to produce one self-renewing NB and one intermediate neural progenitor (INP).
The INP, in turn, divides asymmetrically three to five times. Each of these divisions results in regeneration of the INP and the production of a single GMC4. Collectively, the specific NB identity and the temporal order of GMC birth gives rise to the astounding neuronal diversity of the adult central nervous system.
Understanding the cell biology that underlies NB ACD has been vastly improved through the use of live cell imaging techniques. Published protocols used by researchers to image live NBs vary widely. Overall, however, these methods may be grouped into two general categories distinguished by whether the larval brain is left intact or mechanically dissociated. Both techniques have distinct advantages and disadvantages depending on the researcher’s application.
Early reports revealing live NB cell divisions involve some degree of manual dissociation of the larval brain. These protocols detail smearing8 or teasing apart9 the brain in order to grow short-term primary cultures of the NBs on glass coverslips. To improve imaging, the round NBs are usually flattened on the coverslips with either glass slides8 or agarose pads9. Although flattened cells have improved optics, these techniques often lead to NB mitotic defects, including regression of the cleavage furrow and the inability to divide more than one time. Therefore, protocols that involve both manual dissociation and physical distortion of the larval brain tissue are generally only suited to very short-term (i.e., one cell cycle or less) applications. Likewise, semi-squashing or completely flattening intact brains between glass slides and coverslips limits the time one may spend imaging a given specimen to an approximately 30 min period10. Despite this limitation, this approach has been used successfully11-14.
Recent efforts to image live dissociated NBs limit physical distortion of the isolated cells. These techniques are particularly useful for applications where it is necessary to sustain cell cultures for multiple cell division cycles. For example, dispersed cells from manually dissociated brains may be partially embedded in a clot made from the mixture of fibrinogen and thrombin15 for long-term imaging16. Alternatively, explanted brains may be first chemically dissociated with the enzyme collagenase, next manually disrupted by repeated passage through a pipet tip, and subsequently plated on poly-L-lysine-coated glass bottom dishes17,18. Coating glass dishes with either fibrinogen or poly-L-lysine promotes the attachment and slight spreading of round cells, bringing them as close to the coverslip as possible without major physical distortion. In addition, these methods involve culturing the NBs in medium that may be readily exchanged for pharmacological perturbation experiments18. Overall, directly plating dispersed NBs improves imaging optics without sacrificing long-term imaging capabilities. Recently, a protocol describing the long-term culturing of dissociated NBs has been detailed19.
However, this approach is not without its limitations. There are several caveats to imaging live dissociated NBs. In a field of dispersed cells, for example, it is difficult to identify specific NB lineages that are spatially organized in the intact brain1. Likewise, distinguishing the Type I versus Type II NBs within dissociated tissue is also challenging without the expression of lineage tracers or subtype-specific transgenes, such as worniu-GAL4, asense-GAL8020. Although these transgenes are quite useful for some applications, they do limit researchers to those transgenes expressed under the control of the UAS enhancer element21 and require more complex genetic schemes. Studies that concern the spatial or temporal development of NBs, therefore, require in vivo imaging in the context of an intact tissue.
Moreover, studies of dissociated embryonic NBs indicate that the physical contact of adjacent cells is critical for the interphase localization of key polarity determinants22,23. These studies show completely isolated NBs no longer maintain the invariant mitotic spindle axis. Instead, these cells display a more randomized spindle axis and wide angles of separation between successive GMC buds, perhaps due to the loss of apical centrosome positioning22. Although the relationship between the larval NBs and their neighboring cells has not been extensively studied, it is worth considering that the larval stem cell microenvironment may impinge upon cell polarity or other behaviors. To maintain the physiological context of larval NBs, we image ACD from whole brains.
Given the inherent limitations associated with imaging dissociated NBs, our lab and others have established protocols to image successive rounds of NB ACD from whole larval brains. Techniques to image intact brains often replace the rigid surface of glass on one side of the culture chamber with a flexible membrane to prevent tissue distortion or damage and allow for gas exchange. Some methods involve mounting explanted brains in a mixture of insect culturing medium, serum, and ascorbic acid with the fat bodies isolated from ten larvae24. The isolated fat bodies are added because they secrete a mitogen known to stimulate NB cell division25. This protocol preserves the integrity of the tissue for over 3 hr and is, therefore, amenable to long-term imaging24,26-28. Recently, a protocol describing the long-term imaging of intact larval brains in the presence of ascorbic acid, serum, and fat bodies has been detailed29. Importantly, because the tissue is neither exposed nor immobilized, this approach is less useful for applications where culturing medium is exchanged (e.g., drug studies, immunodepletion, etc.).
Our lab has simplified the whole mount preparation of live larval brains for long-term imaging30,31. The main advantage to our protocol over others is its simplicity: our observations indicate neither serum, ascorbic acid, insulin, nor fat bodies are necessary additives to image successive rounds of NB ACD. Although additives, such as insulin, have been useful for the prolonged imaging of stem cell divisions32 and collective cell migration in Drosophila ovaries33, they appear to be dispensable for NB ACD, as our imaging medium consists of a simple mixture of standard insect cell culturing medium supplemented with antibiotic-antimycotic to prevent contamination. Through the use of reusable gas-permeable or glass bottom culture dishes, we have been able to sustain several rounds of successive NB ACDs. The same explanted samples can readily be used for immunofluorescence and other procedures with fixed tissue. In this protocol, we detail the dissection of intact larval brains, as well as the preparation of brains for both live and fixed analysis.
Protocol
1. Preparation of Materials Required to Explant Third Instar Larval Brains
- Prepare tools required for microdissection.
- Forge two dissecting tools (a scalpel and a hook) by first placing a single dissecting pin into each pin holder. Secure the pins tightly by hand or with pliers (Figure 1C).
- Use a pair of old forceps to bend one dissecting pin to an approximately 120° angle. Do this under a dissecting microscope to ensure that the top half of the pin lies flat when the pin holder is held in hand. This tool will be used as a scalpel to hold down or slice through tissue.
- Use a pair of old forceps to bend the second pin into a hook that will be used to hold and scrape away tissue.
- Cover each dissecting tool with a pipette tip to protect the tools from damage. With care, well-formed tools may be used for dissection indefinitely. Replace damaged or deformed pins as necessary.
- Prepare dissecting medium.
- In a tissue culture hood, dilute the antibiotic-antimycotic supplement into the Schneider’s culturing medium. Swirl the media to incorporate the supplement.
- Prepare several 5 ml aliquots of the supplemented media. Store all media at 4 °C until ready for use.
- Warm a 5 ml aliquot of supplemented medium to room temperature. Draw the medium into a sterile 5 ml syringe and screw on a sterile 0.2 µm syringe filter.
Select larvae for dissection. Use a dissecting probe to select crawling third instar larvae as the brain will be easiest to dissect. Optional: deposit larvae on a grape agar plate. Note: Do not use overly crowded vials or bottles with “wet food,” as these tend to force second or first instar larvae to crawl out prematurely. Note: Some genetic backgrounds will necessitate using younger larvae. This does not affect the protocol, but does make the dissection more difficult.
2. Dissection of the Central Nervous System
On a single glass slide, create two 50 ml pools of warm dissecting media separated by about 3 cm. One pool will be used for gross dissection and other used for fine dissection (Figure 1C).
Transfer several larvae to one of the media drops.
Move the coverslip with larvae to a dissecting microscope. Zoom into the pool containing the larvae such that the anterior and posterior ends of the larvae are discernible.
Use a pairs of forceps to grasp the mid-region of a single larva. Take a second pair of forceps in the other hand and grasp the larva close to the same region. Pull the two pairs of forceps apart to gently tear the larva in half. Repeat the above two steps for all larvae on the coverslip.
Dispose of the posterior halves of the larval body by transferring them to a tissue.
Zoom into a larva so that the anterior mouthhooks are clearly visible. Use both pairs of forceps to create a small incision, or notch, on opposite sides of the larval mouthhooks between the third thoracic and first anterior denticle bands.
Grasp the mouthhooks with the forceps in the non-dominant hand. Use the forceps in the other hand to gently peel away cuticle in an anterior-to-posterior direction. Continue to slowly peel away cuticle until the central nervous system is exposed.
Isolate the central nervous system from the rest of the larva by tearing tissues with the forceps. Use caution not to disturb the central nervous system. Critical step: Do not tug on the central nervous system! Discard damaged samples with a torn ventral nerve cord (Figure 2A) or distorted optic lobes (Figures 2B and 2B’).
Repeat the above two steps for all larvae on the coverslip. Transfer the healthy explanted tissue to the second (clean) pool of medium on the coverslip for the fine dissection stage.
Use the dissecting pin tools to clear away the peripheral tissues (e.g., eye, wing, and leg discs) from the brain. Slide the scalpel between the wanted (brain) and the unwanted tissue, pinning the connective tissue to the coverglass. Use the hook to gently tease away the undesired tissue while simultaneously pressing the scalpel to the coverslip in saw-like movements.
Critical step: Work slowly and deliberately to remove all discs from each brain. Use caution not to disturb the brain morphology. A healthy brain will have an intact ventral nerve cord and symmetric, round optic lobes (Figure 2C).
3. Mounting Explanted Brains for Live Imaging
Invert a 50 mm gas-permeable culture dish with the clear film facing up (Figure 2D). Depress the syringe to deposit a small drop (about 30 µl) of medium onto the center of the dish.
Transfer the isolated brains, one at a time, to the drop of medium on the dish. Use the hook tool to scoop up a brain underneath the optic lobes. Alternatively, use forceps to grasp axons projecting from the ventral nerve cord.
Collect 5-10 brains in the drop of medium. Submerge the brains by using the dissecting pin tools to push individual brains to the bottom of the drop of medium as many of them will be trapped at the meniscus (Figure 2E).
Orient the brains using the dissecting pin tools to align brains depending on the NBs to be imaged (Figure 3A). To image the dorsal NBs, place the ventral nerve cord facing down (along the membrane). To image the anterio-ventral NBs, place the brains with the ventral nerve cord facing upward (away from membrane). Align the brains such that all the ventral nerve cords point in the same direction within the x-y plane.
Use a syringe or plastic transfer pipette to place 4 halocarbon (HC) oil drops (about 30 µl each) onto the gas permeable membrane. The volume of each drop of oil should be equivalent to each other and to the drop of medium. Space the drops of oil to correspond to the four corners of a coverslip centered around the drop of culturing medium. Once complete, the five drops (four drops of oil and one drop of medium in the center) will resemble the five facets on Western-style dice (Figure 3B).
Lower a #1.5 22 mm coverslip on the assembly such that it hits all 5 drops at the same time. Maintaining an even pressure across the samples reduces the likelihood that they will be disrupted. Wait about 3 min as the oil and medium disperse under the weight of the coverslip. A cross-like shape of media will form (Figures 3C and 3D).
Critical step: Lower the coverslip onto the surface of the brains by removing excess media using a tissue paper wick (Figure 3C). Do this while watching the sample under the dissecting scope. As media is removed, the coverslip will lower. Stop once the coverslip touches the brains. Do not over-wick as this will cause tissue distortion or brain explosions.
If imaging dorsal NBs (Figure 3E), move to step 3.9. If imaging anterio-ventral NBs, the coverslip must be slightly moved (by gently nudging it with forceps) in the direction of the ventral nerve cord tips. This causes the brain to rotate, which brings the brain lobes in direct contact with the coverslip to facilitate imaging (Figure 3F).
Seal the chamber by encircling the coverslip with small amounts of halocarbon oil (Figure 3D). Excess oil oozing from the coverslip should be minimized and blotted away with a tissue.
4. Live Imaging of Central Brain Neuroblasts
Note: For this work, a Nikon Eclipse Ti spinning-disk confocal microscope (inverted microscope) was used for live imaging; details of our setup have been previously published31. Alternatively, the sample can be imaged using an upright system.
Add a drop of immersion oil to the coverslip just above the mounted brains. This will help center the objective directly above the sample.
Critical step: Maintain brains at a constant temperature of 25 °C with a stage incubator. Gently position the mounted sample into the stage incubator and cover the chamber with its lid.
Locate central brain NBs using transmitted light by focusing on the large, round cells that reside in the central and medial areas of the optic lobes; do not use epi-fluorescence. This becomes easier with practice. It is very important to expose the sample to the least amount of light as possible. Do not waste time imaging brains that are damaged.
- Once in place, take quick exposures using the confocal to determine proper location and depth. Limit depth to the first 10-15 µm. Adjust as needed, and take another test image.
- Optional step: Collect a test movie of the ventral nerve cord to ensure there is no x-y drift. If drift is detected, wait 15-30 min for the sample to settle. If the drift does not settle in 30 min, prepare a new sample. Major drift can usually be traced back to the addition of excess oil, or the unequal size of oil drops during mounting.
To eliminate axial drift (z-drift) use a continuous automatic focusing device that uses an infrared LED. Alternatively, manually correct the focal plane.
- Once a NB of interest is identified, proceed with imaging regimen. As with all live cell imaging, the amount of laser light, exposure time, camera binning, objective magnification, time and/or z-step interval must all be empirically optimized for a particular experiment to answer the question at hand. The goal is to minimize light damage while still collecting useful data. See discussion for additional guidance.
- Image the entire volume of the NB by collecting 12-14 images spaced by 1 µm in the z-axis. Adjust the time resolution to capture single or multiple cell cycles of the same NB. As a general guideline, use 10-30 sec intervals for single cell cycle imaging and 1-3 min intervals for multiple cell cycles.
Critical step: Confirm that the sample is healthy by monitoring the duration of mitosis. If mitosis takes more than 15 min, move on to another brain. A healthy brain will show many NBs within the same field of view undergoing several rounds of mitosis. Note that each cell cycle is slightly longer than the previous one due to light damage.
Once imaging is complete, disassemble the incubation chamber, and discard everything but the gas-permeable culturing dishes. Rinse the culture dish surface with 95% ethanol and wipe well with a tissue. Repeat two more times.
5. Preparation of Brains for Immunofluorescence
Collect 20 or more explanted brains into a 1.5 ml tube containing approximately 0.2 ml of supplemented medium.
Remove medium from the tube and rinse brains once with 0.5 ml PBSTx (Phosphate Buffered Saline, PBS, with 0.3% Triton X-100). Let brains settle to the bottom of the tube between all exchanges of liquid. The Triton X-100 detergent is used to permeabilize the tissue. Rinsing typically takes less than 30 sec; simply remove the PBSTx once brains have settled to the bottom of the tube.
Replace the solution with 0.5 ml of 9% electron microscopy grade paraformaldehyde diluted in PBSTx. Incubate with nutation for 15 min at room temperature.
Dispose of fixative according to hazardous waste regulations.
Wash the samples with 0.5 ml PBSTx for 15 min. Repeat the wash step with fresh PBSTx two more times.
Replace the solution with 0.5 ml PBT (PBS,1% Bovine Serum Albumin (BSA), and 0.1% Tween-20). Incubate with nutation for 1 hr at room temperature. This step blocks the nonspecific binding of antibody to the tissue.
Replace the solution with 0.5 ml primary antibody diluted in PBT supplemented with 4% normal goat serum (NGS). Incubate with nutation overnight at 4 °C.
Discard or save the diluted primary antibody.
Wash the samples with 0.5 ml PBT for 15 min. Repeat the wash step with fresh PBT two more times.
Replace the solution with 0.5 ml of modified PBT (PBS, 2% BSA, 0.1% Tween-20, and 4% NGS). Incubate with nutation for 1 hr at room temperature.
Replace the solution with 0.5 ml secondary antibody diluted into modified PBT. Incubate with nutation for 2 hr at room temperature.
Replace the solution with 0.5 ml PBST (PBS with 0.1% Tween-20). Incubate with nutation for 15 min at room temperature. Repeat the wash step with fresh PBST two more times.
6. Mounting Samples for Immunofluorescence
Carefully transfer the samples as a drop on a glass slide, restricting the drop towards the left of the center of slide.
Add a large drop (about 2 cm in diameter) of mounting medium (for example, Aqua-Poly/Mount) to the center of the slide. Avoid the pool of PBST that is toward the left.
Use forceps to transfer the brains from the pool of PBST to the pool of mounting medium. Adding mounting medium directly on top of the brains may disrupt their orientation and should be avoided.
Under a light microscope, arrange the samples in the mounting medium with the side that will be imaged facing up.
With the slide under the light microscope, slowly lower a #1.5 glass coverslip on top of the samples. Optional step: Dislodge minor air bubbles by lightly pushing on the coverslip.
Allow the mounting medium to polymerize for several hours, or overnight, by lying slides flat, light-protected, at room temperature.
Slides may be imaged the next day, or stored prior to imaging.
Representative Results
Live imaging has elucidated a number of mechanisms that regulate NB ACD. Prior to image acquisition, it is necessary to determine the optimal imaging conditions. It is necessary to balance the frame capture rate with the duration of live cell imaging. For fine cellular analysis, for example, increased temporal and optical resolution may be required. When visualizing a single NB cell cycle (Figure 4A), the rate at which images are captured may be increased without introducing photodamage. Typically, single cell division events may be monitored at approximately 10-30 sec time intervals. One way to increase the temporal resolution without damaging tissue is to modulate the image acquisition rate. For example, time-lapse imaging may be initiated at a lower frame rate, which is then increased upon observation of an interesting feature or time point (Movie 1).
Imaging successive rounds of cell division (Movie 2), on the other hand, is often done at reduced frame rates (Figure 4B). We typically image multiple (two or more) rounds of ACD through the entire NB cell volume at 1-3 min time intervals over a period of 2-3 hr. Both the temporal resolution and the imaging acquisition period may be improved by limiting the amount of light that hits the sample. For example, time-lapse imaging of successive NB divisions may be extended in excess of 8 hr if only a single optical plane is imaged. A selection of particularly useful transgenic lines for the study of NB ACD is listed (Table 1)16,31,34-38.
For analysis of large numbers of samples, it is often necessary to analyze fixed NBs. Our fixation protocol preserves the overall morphology of the NBs and is compatible with immunofluorescence assays (Figures 4C-4E). Whole mount fixed brains may be imaged at low magnification (Figure 4C) to visualize the overall brain size and shape or to detect NBs. To visualize NBs or their progeny GMCs in more detail, higher magnification must be used. At moderate magnification (Figure 4D), entire NB lineage clusters may be observed. For detailed cellular analysis, higher magnification is used (Figure 4E).
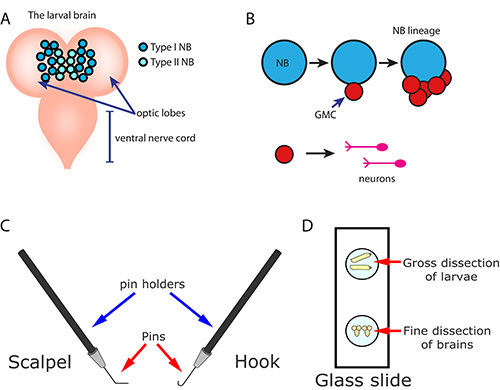 Figure 1. Explanting whole brains for live cell imaging of NB ACD. A) The larval central nervous system consists of two optic lobes and a ventral nerve cord. Two subtypes of NBs populate the optic lobe in stereotypical positions: Type I (dark blue) and Type II (light blue). B) NBs undergo ACD. With each cell division, the NB cleaves to give rise to one GMC (or INP, not shown) and regenerates the NB stem cell. GMCs will divide to give rise to two neurons (or glia, not shown). C) Two microdissection tools are assembled by inserting dissecting pins into pin holders. Bending the pins creates unique tools, a scalpel and a hook, which are used to sever, pierce, and displace unwanted tissue without damaging the underlying brain. D) Brains are explanted into one of two pools of dissecting media (blue circles). One pool of medium is used to remove the central nervous system from the larvae (gross dissection), and the other pool is used for fine microdissection of peripheral tissues from the explanted brains (fine dissection).
Figure 1. Explanting whole brains for live cell imaging of NB ACD. A) The larval central nervous system consists of two optic lobes and a ventral nerve cord. Two subtypes of NBs populate the optic lobe in stereotypical positions: Type I (dark blue) and Type II (light blue). B) NBs undergo ACD. With each cell division, the NB cleaves to give rise to one GMC (or INP, not shown) and regenerates the NB stem cell. GMCs will divide to give rise to two neurons (or glia, not shown). C) Two microdissection tools are assembled by inserting dissecting pins into pin holders. Bending the pins creates unique tools, a scalpel and a hook, which are used to sever, pierce, and displace unwanted tissue without damaging the underlying brain. D) Brains are explanted into one of two pools of dissecting media (blue circles). One pool of medium is used to remove the central nervous system from the larvae (gross dissection), and the other pool is used for fine microdissection of peripheral tissues from the explanted brains (fine dissection).
 Figure 2. Preservation of brain morphology during microdissection. Hasty brain dissection can induce severe tissue damage that includes (A) torn ventral nerve cords or (B) distorted optic lobes. Even more subtle tissue damage (B’) should be avoided for live cell imaging. (C) An example of a healthy tissue sample suitable for live imaging. D) An optically clear, gas permeable membrane. E) A collection of several healthy brains arranged in culturing medium on a gas-permeable dish. These brains are ready to be mounted for microscopy. Scale bar: (A-C), 100 µm; (D), 25 mm; (E) 1 mm.
Figure 2. Preservation of brain morphology during microdissection. Hasty brain dissection can induce severe tissue damage that includes (A) torn ventral nerve cords or (B) distorted optic lobes. Even more subtle tissue damage (B’) should be avoided for live cell imaging. (C) An example of a healthy tissue sample suitable for live imaging. D) An optically clear, gas permeable membrane. E) A collection of several healthy brains arranged in culturing medium on a gas-permeable dish. These brains are ready to be mounted for microscopy. Scale bar: (A-C), 100 µm; (D), 25 mm; (E) 1 mm.
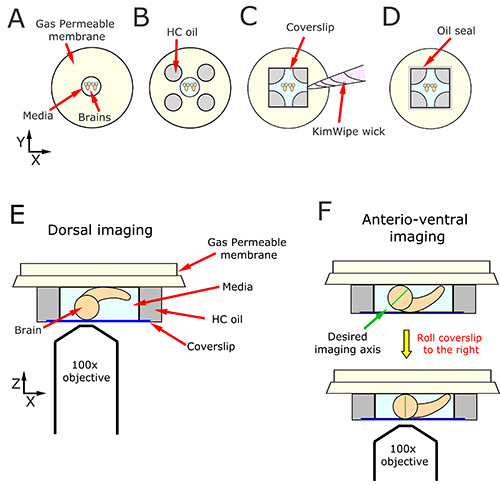 Figure 3. Mounting brains for live cell imaging. A) Brains are aligned in rows within a drop of culturing medium deposited in the center of the dish. B) The drop of medium is surrounded by drops of HC oil of approximately equal volume. The four drops of HC oil and the central drop of medium resemble the five facets of playing dice. After a coverslip is lowered onto the drops of oil, (C) a wick is fashioned out of KimWipe tissue and used to sop up excess fluid. D) The exterior edge of the coverslip is encircled by a thin layer of oil to stop the evaporation of the culturing medium. These brains are now ready for microscopy. E and F) The orientation of the brain relative to the coverslip determines which neuroblasts are accessible for live cell imaging. E) Dorsal neuroblasts are imaged by arranging the brains with the ventral side touching the gas permeable membrane. F) Anterior-ventral neuroblasts may be imaged by arranging the brains with the dorsal side touching the gas permeable membrane and then nudging the coverslip, and the underlying tissue, in the direction of the ventral nerve cord tips. This motion slightly tilts the optic lobes such that the anterio-dorsal surface contacts the coverslip, bringing the desired imaging axis into perfect alignment (green line).
Figure 3. Mounting brains for live cell imaging. A) Brains are aligned in rows within a drop of culturing medium deposited in the center of the dish. B) The drop of medium is surrounded by drops of HC oil of approximately equal volume. The four drops of HC oil and the central drop of medium resemble the five facets of playing dice. After a coverslip is lowered onto the drops of oil, (C) a wick is fashioned out of KimWipe tissue and used to sop up excess fluid. D) The exterior edge of the coverslip is encircled by a thin layer of oil to stop the evaporation of the culturing medium. These brains are now ready for microscopy. E and F) The orientation of the brain relative to the coverslip determines which neuroblasts are accessible for live cell imaging. E) Dorsal neuroblasts are imaged by arranging the brains with the ventral side touching the gas permeable membrane. F) Anterior-ventral neuroblasts may be imaged by arranging the brains with the dorsal side touching the gas permeable membrane and then nudging the coverslip, and the underlying tissue, in the direction of the ventral nerve cord tips. This motion slightly tilts the optic lobes such that the anterio-dorsal surface contacts the coverslip, bringing the desired imaging axis into perfect alignment (green line).
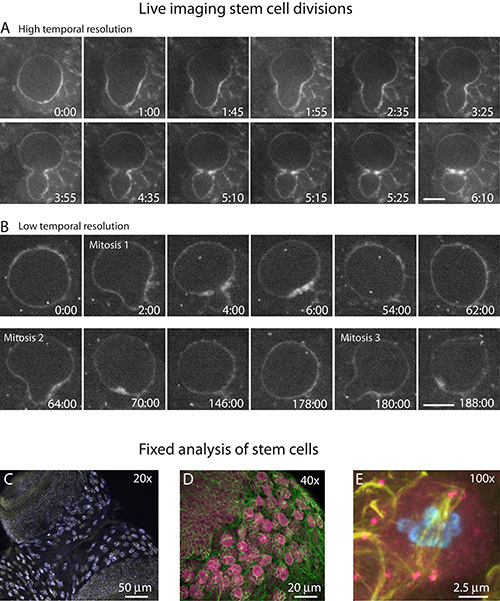 Figure 4. Representative results from live imaging of dividing neuroblasts. A and B) Live imaging of NB stem cell divisions in whole mount preparations using a 40X, 1.3 NA oil-immersion objective. Time is displayed as min:sec relative to the start of the time-lapse. A) Stills from time-lapse imaging of a single cell cycle from a NB expressing GFP-Moesin (GFP-Moe). The image acquisition rate of a single cell cycle may be increased to enhance temporal resolution without inducing photodamage. B) Stills from time-lapse imaging of successive cell division cycles from a NB expressing both GFP-Moe and a GFP-labeled centrosome marker. The prolonged period associated with multiple cell cycles necessitates reduced temporal resolution to avoid photodamage. Note that the length of time between each cell division increases during the course of imaging. C-E) Confocal projections of fixed stem cells from whole mount preparations. C)) To image all NBs located on both optic lobes, 20X magnification is used. This analysis is particularly useful to examine the overall brain morphology and to quantify NB numbers (light blue), etc. D) The majority of NBs (pink) from a single optic lobe may be visualized with 40X magnification. Microtubules, (green). E) For detailed cellular analysis of a few NBs, 100X magnification must be used. This mutant NB shows multiple centrosomes at each spindle pole, an observation that would be difficult to detect at lower magnification. Microtubules (yellow), centrosomes (anti-Asterless; pink), and DNA (DAPI; blue). Scale bars: (A and B) 5 µm; (C) 50 µm; (D) 20 µm; (E) 2.5 µm.
Figure 4. Representative results from live imaging of dividing neuroblasts. A and B) Live imaging of NB stem cell divisions in whole mount preparations using a 40X, 1.3 NA oil-immersion objective. Time is displayed as min:sec relative to the start of the time-lapse. A) Stills from time-lapse imaging of a single cell cycle from a NB expressing GFP-Moesin (GFP-Moe). The image acquisition rate of a single cell cycle may be increased to enhance temporal resolution without inducing photodamage. B) Stills from time-lapse imaging of successive cell division cycles from a NB expressing both GFP-Moe and a GFP-labeled centrosome marker. The prolonged period associated with multiple cell cycles necessitates reduced temporal resolution to avoid photodamage. Note that the length of time between each cell division increases during the course of imaging. C-E) Confocal projections of fixed stem cells from whole mount preparations. C)) To image all NBs located on both optic lobes, 20X magnification is used. This analysis is particularly useful to examine the overall brain morphology and to quantify NB numbers (light blue), etc. D) The majority of NBs (pink) from a single optic lobe may be visualized with 40X magnification. Microtubules, (green). E) For detailed cellular analysis of a few NBs, 100X magnification must be used. This mutant NB shows multiple centrosomes at each spindle pole, an observation that would be difficult to detect at lower magnification. Microtubules (yellow), centrosomes (anti-Asterless; pink), and DNA (DAPI; blue). Scale bars: (A and B) 5 µm; (C) 50 µm; (D) 20 µm; (E) 2.5 µm.
Movie 1. Live imaging of a single NB cell cycle. Time-lapse images of a single NB cell division from a brain expressing GFP-Moe to label the cell cortex. Images were acquired from a single optical plane at 40X magnification. In this movie, frames were captured every 15 sec. After about 7 min of acquisition, the rate was increased to one frame every 5 sec. Click here to view movie.
Movie 2. Live imaging successive rounds of NB ACD. Time-lapse images of a NB undergoing three rounds of cell division in a brain expressing GFP-Moe and a GFP-labeled centrosome marker. Images were acquired at 2 min time intervals at 60X magnification. Click here to view movie.
| Line | Labeled Cellular Structure | Fluorophore | Expression/Promoter | Citation |
| YFP-Asterless | Centriole | YFP | Ubiquitin | Rebollo, et al. (2007) Dev. Cell. 12: 467. |
| SAS6-mCherry | Centriole | mCherry | Endogenous | Rusan and Peifer (2007) J. Cell Biol. 177: 13. |
| GFP-SAS6 | Centriole | GFP | Endogenous (BAC) | Lerit and Rusan (2013) J. Cell Biol. 202: 1013. |
| GFP-PACT | Centriole | GFP | Ubiquitin | Martinez-Campos, et al. J. Cell Biol. 165: 673. |
| GFP-SAS4 | Centrosome | GFP | Ubiquitin | Dix and Raff (2007) Curr. Biol. 17: 1759. |
| GFP-Polo | Centrosome | GFP | Endogenous | Moutinho-Santos, et al. (1999) Biol Cell. 91: 585. |
| GFP-γ-Tubulin 23C | Centrosome | GFP | Ubiquitin | Lerit and Rusan (2013) J. Cell Biol. 202: 1013. |
| GFP-Centrosomin | Centrosome | GFP | UAS | Megraw, et al. (2002) J. Cell Sci. 115: 4707. |
| GFP-Spd-2 | Centrosome | GFP | Ubiquitin | Dix and Raff (2007) Curr. Biol. 17: 1759. |
| mCherry-α-Tubulin | Microtubules | mCherry | Ubiquitin | Rusan and Peifer (2007) J. Cell Biol. 177: 13. |
| GFP-α-Tubulin | Microtubules | GFP | Ubiquitin | Rebollo, et al. (2004) PLoS Biol. 2: E8. |
| Jupiter-GFP ("G147") | Microtubules | GFP | Endogenous | Morin, et al. (2001) PNAS. 98: 15050. |
| mCherry-Jupiter | Microtubules | mCherry | UAS | Cabernard and Doe (2009) Dev. Cell 17: 134. |
| H2AvD-mRFP | DNA | mRFP | Endogenous | Pandey, et al. (2005) J. Cell Sci. 118: 733. |
| H2AvD-GFP | DNA | GFP | Endogenous | Clarkson and Saint (1999) DNA Cell Biol. 18: 457. |
| Moesin-GFP | Cortical actin | GFP | Spaghetti squash | Kiehart, et al. (2000) J. Cell Biol. 149: 471. |
Table 1. A selection of useful transgenic lines for the study of ACD. These fusion constructs are particularly useful for studying cell division. They are routinely used in NB live imaging studies.
Discussion
Live cell imaging is an invaluable approach used to study the mechanisms that regulate ACD of stem cells, the differentiation of cell lineages, and the morphogenesis of complex tissues. Although research groups have historically employed a variety of techniques to visualize NB ACD, these approaches can be generally grouped into two categories that primarily differ with respect to whether the brain tissue is left intact or dissociated.
Imaging dissociated NBs has its advantages. For one, dissociated NBs are easier to image because they are especially easy to identify as the largest (about 12 µm in diameter) cells in a culturing dish. Moreover, nearly all stem cells that are cultured on a glass surface will be equally close to the microscope objective throughout the image acquisition period. The optical quality of time-lapse images is further improved with the use of either poly-L-lysine or fibrinogen coated glass to induce the semi-flattening of the round NBs16,17. Another advantage to imaging dissociated NBs grown in culture is the ease with which culturing medium can be exchanged. Once cells adhere to the coated glass surface, culturing medium containing any number of pharmacological agonists, antagonists, or immunoblocking antibodies, etc. may be added or removed. Finally, recent approaches to visualize dissociated NBs have demonstrated that this technique is, indeed, amenable to prolonged image acquisition18,19.
Nonetheless, the disadvantages to imaging dissociated NBs include the complexity of the protocol. Most methods detail lengthy (30-60 min) chemical digestion, followed by a long incubation step (60 min) to allow dispersed cells to settle and adhere to the culturing dish17,19.
In contrast, imaging NBs from an intact brain preserves both the physiological context and morphology of the tissue. The advantages to this approach include the ability to identify specific NB lineages that develop in stereotypical arrangements or at defined developmental times1. Therefore, our simplified protocol has utility that extends beyond studies of ACD, as it also allows for the live investigation of differentiation and morphogenesis events. One disadvantage to this method, however, is the inability to exchange culturing medium. Therefore, researchers interested in long-term imaging of successive stem cell divisions must consider the approach (dissociated primary culture versus whole mount) that best suits their application. Likewise, the necessity for culturing additives, such as insulin or serum, should be determined empirically based on the imaging conditions (duration of imaging, amount of light hitting the sample, etc.) and experimental setup.
In general, we recommend utilizing dissociated NBs for studies where it is necessary to assess the acute response of NBs to the introduction of small molecules (e.g., inhibitors, etc.), for high-throughput applications when fine microdissection of brains might be too cumbersome, and for advanced imaging studies that require the NB to be in physical contact with the coverslip. In contrast, we recommend utilizing intact brains for applications where it is useful to image a few NBs from the same brain, and especially for studies that concern cell-cell interactions, autonomy, clonal analysis, cell shape changes, and other investigations where the native environment of the cells is important.
To ensure the successful preparation of intact brains for live imaging, several critical steps must be carefully executed. Foremost, it is imperative to avoid exposing the tissue to unnecessary trauma. Specifically, explanting the brain and removing peripheral tissue should be performed with care. One should minimize direct contact between the dissecting tools and the brain. In addition, one should avoid tugging or pulling on tissue attached to the brain. Mishandling the tissue can readily result in torn or distorted brains. In our hands, NBs from damaged brains seldom divide. Individuals who are new to larval brain dissection often benefit from mastering the dissection before attempting to prepare samples for live imaging.
Another critical step in the protocol is the mounting of the brains prior to live cell imaging. The correct volumes of both culturing medium and HC oil are essential to complete a useful preparation. Although it is difficult to accurately measure the viscous HC oil with a pipet, one may approximate the correct volume (about 30 µl) by eye. Too much liquid prevents the coverslip from settling into place. Often, this results in the brains moving on the surface of the culturing dish. During live imaging, an unsettled coverslip is evident when the tissue drifts or the optic lobes roll. Samples that are not positioned in place should be avoided. On the contrary, adding too little culturing medium or HC oil results in catastrophic tissue damage, including burst optic lobes. There is a careful balance between using enough liquid medium and oil to handle the weight of the coverslip versus using too much liquid, which causes the coverslip to slide. This balance is best learned through practice. In general, it is best to only image those brains that have a healthy morphology, such as an intact ventral nerve cord and symmetrical, round optic lobes.
Once the sample is mounted with no detectable drift, keeping the sample alive becomes the next critical aspect of this protocol. This is best achieved by maintaining proper temperature and, most importantly, minimizing the amount of light that hits the sample. The microscope setup and the sample are the two variables that will govern the success of live cell imaging. High quality objectives, filters (~98% transmission), and cameras (back-thinned electron multiplying and complementary metal-oxide semiconductor) on the emission side will allow one to reduce the amount of light that hits the sample on the excitation side. It is well known that blue light (used to image GFP) is highly toxic to cells. One must determine how much total light (total exposure times) a given sample can tolerate and then spread that total time along the duration of the experiment. For example, if a sample can only tolerate 500 laser light exposures based on a particular microscope setup, then one could collect 50 x 10-slice stacks. These stacks could be spaced out by 1 min to collect a single NB cell cycle, or 2-3 min to collect ~3 cell cycles. In addition, optimizing settings for each genotype is critical for successful live imaging. During the optimization period, a good starting point for 488 nm laser power is 25-50 µW emanating from a 100X, 1.4 NA oil-immersion objective. Finally, one must determine what imaging condition will properly answer the question at hand, as it is not always the case that the most sophisticated imaging is needed.
For experiments with either live or fixed tissue, brains may be mounted so as to image specific NB lineages. For example, in order to image Type II NBs, which stereotypically reside in the posterior-medial region of the optic lobe7, one would mount the brains, as illustrated in Figure 3E. Because the spatial and temporal pattern of central brain NBs have been well documented1, one may use our protocol to image a variety of specific NB clusters while utilizing any number of the widely available transgenic constructs that are expressed under ubiquitous or, ideally, endogenous promoters (Table 1). Nonetheless, lineage-specific labeling technologies20 remain useful for a number of applications. With fixed analysis, brains with minor damage are generally acceptable. In addition, the complete removal of peripheral tissue can be postponed until just before embedding the brains in mounting medium. Studies with fixed tissue are particularly useful when large numbers of NBs must be imaged.
In conclusion, studies of NB ACD have greatly benefited from the use of both live cell imaging and detailed fixed analysis. The protocol outlined here details one approach to prepare Drosophila larvae for either live or fixed applications. We anticipate the continued investigation of NB ACD through detailed imaging studies will reveal novel and exciting results that will advance our understanding of stem cells in development and disease. The application of long-term live imaging of intact larval brains has the potential to uncover novel insights into the temporal sequence of differentiation and morphogenesis events that govern the development (and degeneration) of the central nervous system.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the division of intramural research at the National Institutes of Health/NHLBI (1ZIAHL006126) and a Lenfant Biomedical Postdoctoral Fellowship awarded to DAL.
References
- Truman JW, Bate M. Spatial and temporal patterns of neurogenesis in the central nervous system of Drosophila melanogaster. Developmental Biology. 1988;125:145–157. doi: 10.1016/0012-1606(88)90067-x. [DOI] [PubMed] [Google Scholar]
- Doe CQ. Molecular markers for identified neuroblasts and ganglion mother cells in the Drosophila central nervous system. Development. 1992;116:855–863. doi: 10.1242/dev.116.4.855. [DOI] [PubMed] [Google Scholar]
- Lerit DA, Smyth JT, Rusan NM. Organelle asymmetry for proper fitness, function, and fate. Chromosome research. 2013;21:271–286. doi: 10.1007/s10577-013-9350-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello BC, Izergina N, Caussinus E, Reichert H. Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural development. 2008;3 doi: 10.1186/1749-8104-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman SK, et al. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Developmental Cell. 2008;14:535–546. doi: 10.1016/j.devcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone JQ, Doe CQ. Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev Neurobiol. 2008;68:1185–1195. doi: 10.1002/dneu.20648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homem CC, Knoblich JA. Drosophila neuroblasts: a model for stem cell biology. Development. 2012;139:4297–4310. doi: 10.1242/dev.080515. [DOI] [PubMed] [Google Scholar]
- Savoian MS, Rieder CL. Mitosis in primary cultures of Drosophila melanogaster larval neuroblasts. Journal of Cell Science. 2002;115:3061–3072. doi: 10.1242/jcs.115.15.3061. [DOI] [PubMed] [Google Scholar]
- Fleming SL, Rieder CL. Flattening Drosophila cells for high-resolution light microscopic studies of mitosis in vitro. Cell Motil Cytoskeleton. 2003;56:141–146. doi: 10.1002/cm.10143. [DOI] [PubMed] [Google Scholar]
- Buffin E, Lefebvre C, Huang J, Gagou ME, Karess RE. Recruitment of Mad2 to the kinetochore requires the Rod/Zw10 complex. Current biology. 2005;15:856–861. doi: 10.1016/j.cub.2005.03.052. [DOI] [PubMed] [Google Scholar]
- Basto R, et al. Flies without centrioles. Cell. 2006;125:1375–1386. doi: 10.1016/j.cell.2006.05.025. [DOI] [PubMed] [Google Scholar]
- Lucas EP, Raff JW. Maintaining the proper connection between the centrioles and the pericentriolar matrix requires Drosophila centrosomin. J. Cell Biol. 2007;178:725–732. doi: 10.1083/jcb.200704081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, et al. Centrosome amplification can initiate tumorigenesis in flies. Cell. 2008;133:1032–1042. doi: 10.1016/j.cell.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit PT, Raff JW. Cnn dynamics drive centrosome size asymmetry to ensure daughter centriole retention in Drosophila neuroblasts. Curr. Biol. 2010;20:2187–2192. doi: 10.1016/j.cub.2010.11.055. [DOI] [PubMed] [Google Scholar]
- Forer A, Pickett-Heaps JD. Cytochalasin D and latrunculin affect chromosome behaviour during meiosis in crane-fly spermatocytes. Chromosome research. 1998;6:533–549. doi: 10.1023/a:1009224322399. [DOI] [PubMed] [Google Scholar]
- Rebollo E, et al. Functionally unequal centrosomes drive spindle orientation in asymmetrically dividing Drosophila neural stem cells. Dev. Cell. 2007;12:467–474. doi: 10.1016/j.devcel.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Januschke J, Llamazares S, Reina J, Gonzalez C. Drosophila neuroblasts retain the daughter centrosome. Nat. Commun. 2011;2 doi: 10.1038/ncomms1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Januschke J, et al. Centrobin controls mother-daughter centriole asymmetry in Drosophila neuroblasts. Nat Cell Biol. 2013;15:241–248. doi: 10.1038/ncb2671. [DOI] [PubMed] [Google Scholar]
- Homem CC, Reichardt I, Berger C, Lendl T, Knoblich JA. Long-Term Live Cell Imaging and Automated 4D Analysis of Drosophila Neuroblast Lineages. PLoS One. 2013;8 doi: 10.1371/journal.pone.0079588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumuller RA, et al. Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell. 2011;8:580–593. doi: 10.1016/j.stem.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Siegrist SE, Doe CQ. Extrinsic cues orient the cell division axis in Drosophila embryonic neuroblasts. Development. 2006;133:529–536. doi: 10.1242/dev.02211. [DOI] [PubMed] [Google Scholar]
- Broadus J, Doe CQ. Extrinsic cues, intrinsic cues and microfilaments regulate asymmetric protein localization in Drosophila neuroblasts. Curr. Biol. 1997;7:827–835. doi: 10.1016/s0960-9822(06)00370-8. [DOI] [PubMed] [Google Scholar]
- Siller KH, Serr M, Steward R, Hays TS, Doe CQ. Live imaging of Drosophila brain neuroblasts reveals a role for Lis1/dynactin in spindle assembly and mitotic checkpoint control. Molecular biology of the cell. 2005;16:5127–5140. doi: 10.1091/mbc.E05-04-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton JS, Edgar BA. Environmental control of the cell cycle in Drosophila: nutrition activates mitotic and endoreplicative cells by distinct mechanisms. Development. 1998;125:2149–2158. doi: 10.1242/dev.125.11.2149. [DOI] [PubMed] [Google Scholar]
- Siller KH, Doe CQ. Lis1/dynactin regulates metaphase spindle orientation in Drosophila neuroblasts. Developmental Biology. 2008;319:1–9. doi: 10.1016/j.ydbio.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabernard C, Doe CQ. Apical/basal spindle orientation is required for neuroblast homeostasis and neuronal differentiation in Drosophila. Dev. Cell. 2009;17:134–141. doi: 10.1016/j.devcel.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Januschke J, Gonzalez C. The interphase microtubule aster is a determinant of asymmetric division orientation in Drosophila neuroblasts. J. Cell Biol. 2010;188:693–706. doi: 10.1083/jcb.200905024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabernard C, Doe CQ. Live imaging of neuroblast lineages within intact larval brains in Drosophila. Cold Spring Harb Protoc. 2013;2013:970–977. doi: 10.1101/pdb.prot078162. [DOI] [PubMed] [Google Scholar]
- Rusan NM, Akong K, Peifer M. Putting the model to the test: are APC proteins essential for neuronal polarity, axon outgrowth, and axon targeting. J. Cell Biol. 2008;183:203–212. doi: 10.1083/jcb.200807079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerit DA, Rusan NM. PLP inhibits the activity of interphase centrosomes to ensure their proper segregation in stem cells. J. Cell Biol. 2013;202:1013–1022. doi: 10.1083/jcb.201303141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris LX, Spradling AC. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 2011;138:2207–2215. doi: 10.1242/dev.065508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad M, Jang AC, Starz-Gaiano M, Melani M, Montell DJ. A protocol for culturing Drosophila melanogaster stage 9 egg chambers for live imaging. Nat. Protoc. 2007;2:2467–2473. doi: 10.1038/nprot.2007.363. [DOI] [PubMed] [Google Scholar]
- Rusan NM, Peifer M. A role for a novel centrosome cycle in asymmetric cell division. J. Cell Biol. 2007;177:13–20. doi: 10.1083/jcb.200612140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Campos M, Basto R, Baker J, Kernan M, Raff JW. The Drosophila pericentrin-like protein is essential for cilia/flagella function, but appears to be dispensable for mitosis. J. Cell Biol. 2004;165:673–683. doi: 10.1083/jcb.200402130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix CI, Raff JW. Drosophila Spd-2 recruits PCM to the sperm centriole, but is dispensable for centriole duplication. Curr. Biol. 2007;17:1759–1764. doi: 10.1016/j.cub.2007.08.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutinho-Santos T, Sampaio P, Amorim I, Costa M, Sunkel CE. In vivo localisation of the mitotic POLO kinase shows a highly dynamic association with the mitotic apparatus during early embryogenesis in Drosophila. Biol. Cell. 1999;91:585–596. [PubMed] [Google Scholar]
- Megraw TL, Kilaru S, Turner FR, Kaufman TC. The centrosome is a dynamic structure that ejects PCM flares. J. Cell Sci. 2002;115:4707–4718. doi: 10.1242/jcs.00134. [DOI] [PubMed] [Google Scholar]