

Abstract
During cardiac thin-filament activation, the N-domain of cardiac troponin C (N-cTnC) binds to Ca2+ and interacts with the actomyosin inhibitory troponin I (cTnI). The interaction between N-cTnC and cTnI stabilizes the Ca2+-induced opening of N-cTnC and is presumed to also destabilize cTnI–actin interactions that work together with steric effects of tropomyosin to inhibit force generation. Recently, our in situ steady-state FRET measurements based on N-cTnC opening suggested that at long sarcomere length, strongly bound cross-bridges indirectly stabilize this Ca2+-sensitizing N-cTnC–cTnI interaction through structural effects on tropomyosin and cTnI. However, the method previously used was unable to determine whether N-cTnC opening depends on sarcomere length. In this study, we used time-resolved FRET to monitor the effects of cross-bridge state and sarcomere length on the Ca2+-dependent conformational behavior of N-cTnC in skinned cardiac muscle fibers. FRET donor (AEDANS) and acceptor (DDPM)-labeled double-cysteine mutant cTnC(T13C/N51C)AEDANS-DDPM was incorporated into skinned muscle fibers to monitor N-cTnC opening. To study the structural effects of sarcomere length on N-cTnC, we monitored N-cTnC opening at relaxing and saturating levels of Ca2+ and 1.80 and 2.2-μm sarcomere length. Mg2+-ADP and orthovanadate were used to examine the structural effects of noncycling strong-binding and weak-binding cross-bridges, respectively. We found that the stabilizing effect of strongly bound cross-bridges on N-cTnC opening (which we interpret as transmitted through related changes in cTnI and tropomyosin) become diminished by decreases in sarcomere length. Additionally, orthovanadate blunted the effect of sarcomere length on N-cTnC conformational behavior such that weak-binding cross-bridges had no effect on N-cTnC opening at any tested [Ca2+] or sarcomere length. Based on our findings, we conclude that the observed sarcomere length-dependent positive feedback regulation is a key determinant in the length-dependent Ca2+ sensitivity of myofilament activation and consequently the mechanism underlying the Frank-Starling law of the heart.
Introduction
Regulation of cardiac muscle contraction requires the Ca2+-dependent, integrated activity of highly synchronized and fine-tuned thin-filament protein-protein interactions, many of which involve the cardiac troponin (cTn) complex. To activate the thin filament at the beginning of systole, Ca2+ binds to the N-domain of cTnC (N-cTnC). This initiates a series of intra- and intermolecular structural changes in thin-filament proteins (1–3), including partially exposing a previously buried hydrophobic patch in N-cTnC (4–6), followed by the interaction between the hydrophobic patch and the switch region of cTnI (cTnI-Sr). This interaction switches the actomyosin-inhibitory C-terminal domain of cTnI away from interacting with actin toward interacting with N-cTnC (4,5,7–12) and further stabilizes Ca2+-induced N-cTnC opening (4,5,11,13). The detachment of cTnI from actin leads to tropomyosin shifting from the blocked toward the closed state of thin-filament regulation, a transition in which cTnT also plays a role (14). Ultimately, it is this shifting of tropomyosin toward the closed state that enables the strong attachment of cross-bridges to actin to generate force (15–20). (See Table 1 for Glossary of General Terms used in this article.)
Table 1.
Glossary of general abbreviations
| Term | Definition |
|---|---|
| BDM | 2, 3-butanedione monoxime |
| c | Cardiac muscle |
| DDPM | N-(4-dimethylamino-3,5-dinitrophenyl)maleimide |
| DTT | Dithiothreitol |
| EDTA | Ethylenediaminetetraacetic acid |
| EGTA | Ethylene glycol-bis-(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| FRET | Förster resonance energy transfer |
| AEDANS | 5-(io-doacetamidoethyl)aminonaphthelene-1-sulfonic acid |
| Tn | Troponin |
| TnC | Troponin C |
| TnI | Troponin I |
| TnT | Troponin T |
| S1 | Myosin subfragment-1 |
Interestingly, such force-generating strong actomyosin interactions induce further structural changes in the thin filament that sensitize it to Ca2+ in what is termed “positive feedback regulation” (21–24). This feedback activation pathway is thought to arise from the ability of strong myosin head binding to actin to shift tropomyosin toward the open state (25), which concomitantly disrupts cTnI–actin interactions even in the absence of Ca2+ (11,12). These structural changes indirectly stabilize N-cTnC opening through strengthening the N-cTnC–cTnI-Sr interaction (13,26), which is known to be important for stabilizing N-cTnC in the open conformation, as mentioned above (4,5).
Recently, a fourth state of thin-filament regulation has even been proposed to account for such Ca2+ independent activation of thin-filament regulatory units by myosin heads (27). In the fourth state, tropomyosin is in the open state of thin-filament regulation and cTnI-Sr can thus interact with N-cTnC, but Ca2+ is not bound to cTnC site II. To more fully understand the molecular mechanisms underlying these processes, our group has previously used in vitro fluorescence spectroscopy techniques to extensively study and characterize the distinct protein conformational changes that serve as the biophysical basis for thin-filament and positive feedback regulation (8–12,28–31). In these studies and in related work by others (20,32–36), a vast amount of information on how conformational changes in cTnC and cTnI participate in the molecular mechanism underlying cardiac thin-filament regulation has been uncovered using in vitro, reconstituted, thin filaments. An ongoing question is how well this information will hold in the context of the myofilament lattice found in myocardial sarcomeres (hereafter referred to as “in situ”).
While important information has been uncovered using reconstituted thin filaments, they lack important features of the myofilament lattice, including a loaded condition and lattice-associated geometric and mechanical constraints on protein structure and function. To study thin-filament regulation under a more-nearly native setting, we recently implemented steady-state Förster resonance energy transfer (ss-FRET) in detergent-skinned cardiac muscle fibers and used it to monitor in situ N-cTnC opening (37). Our results demonstrated that strong cross-bridge binding to actin leads to a stabilization of N-cTnC opening. Because stabilizing the open conformation of N-cTnC increases Ca2+ sensitivity (38–42), this finding provides a structural explanation for the thin-filament Ca2+-sensitizing effect of positive feedback regulation. Due to the fluorophore-concentration-dependent nature of our in situ ss-FRET technique, we did not further investigate an additional important mechanism to positive feedback regulation—one that further modulates thin-filament activation in vivo—namely, cardiac myofilament length-dependent activation (43–45).
Length-dependent activation is thought to underlie the Frank-Starling law of the heart, which describes the increase in stroke volume that occurs in response to an increase in diastolic ventricular filling (46). Increased filling stretches the myocardium, producing an increase in sarcomere length (SL) that leads to myofilament-length-dependent activation wherein the maximum tension developed by myofilaments is increased and their response to Ca2+ is enhanced. Accordingly, in this study we investigate the effect of changes in SL on cardiac thin-filament activation at the molecular level. Recent studies have shown that the maximum extent of ensemble-averaged N-cTnC opening within thin-filament regulatory units is positively correlated with thin-filament Ca2+ sensitivity (37,47), and as discussed above, strong actomyosin interactions lead to a stabilization of N-cTnC opening.
Additionally, recent small-angle x-ray diffraction evidence reported by Farman et al. (48) has suggested that increased SL leads to structural changes in thick-filament myosin heads that should promote increased interaction between strong-binding cross-bridges and actin. Thus, we hypothesized that a decrease in SL should produce decreased interaction between strong-binding cross-bridges and actin, which should consequently lead to decreased force and ensemble-averaged N-cTnC opening. Furthermore, we reasoned that changes in SL should produce no effect on N-cTnC opening when strong-binding cross-bridges are absent.
Although fluorescence intensity of a fluorophore is concentration-dependent, its fluorescence lifetime is not. Thus, to test our hypothesis that a decrease in SL would decrease N-cTnC opening, we have developed an in situ time-resolved FRET (tr-FRET) technique by retrofitting our Güth muscle research system (MRS) with time-correlated single-photon counting technology. This modification of the MRS allows us to perform fluorescence lifetime measurements of AEDANS-labeled cTnC incorporated into fiber samples to determine the effects of SL changes on in situ N-TnC opening. Accordingly, isometric tr-FRET experiments were performed on skinned fibers wherein endogenous cTnC was replaced with the double cysteine cTnC(T13C/N51C) mutant labeled with AEDANS and DDPM as FRET donor and acceptor, respectively. The roles that strong- and weak-binding cross-bridges play in Ca2+- and length-dependent activation were tested by promoting or inhibiting strong cross-bridge binding with Mg2+-ADP and sodium orthovanadate (Vi), respectively.
Fluorescence intensity decays yielded from these tr-FRET experiments were used to determine AEDANS-DDPM distance distributions as a function of cTnC site-II Ca2+ occupancy, cross-bridge states, and SL. AEDANS-DDPM-modified cTnC indicated that the ensemble-averaged extent of N-cTnC opening was not only dependent on Ca2+ occupancy and cross-bridge state, but also on SL. Specifically, reduction of SL attenuated the stabilizing effects of strong-binding cross-bridges on N-cTnC opening, and treatment with Vi eliminated SL-dependent changes in N-cTnC opening altogether. Thus, modulation of positive feedback regulation by SL was shown to be mechanistically involved in the length-dependent Ca2+ sensitivity associated with myofilament length-dependent activation.
Materials and Methods
Animal handling protocols
The handling of all the experimental animals followed the institutional guidelines and protocols approved by the Animal Care and Use Committee and the Office of Laboratory Animal Welfare, National Institutes of Health, Bethesda, MD. Our muscle-fiber study also followed the established guidelines of, and was approved by, the Washington State University Institutional Animal Care and Use Committee.
Experimental preparations
Methods based on previously reported protocols (37) were followed for tissue dissection; preparation of proteins, highly relaxing solution, pCa 9 and 4.3 solutions, and pCa solutions containing either Vi or ADP, and detergent-skinned fibers; cTnC extraction and reconstitution; and characterization of reconstitution efficiency using sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting. See the Supporting Material for further details.
Simultaneous measurement of isometric force and time-resolved fluorescence intensity in detergent-skinned cardiac muscle fibers
The same measurement protocol as described in our Supporting Material was used to perform simultaneous measurements of force and tr-FRET of N-cTnC opening as a function of cTnC site-II Ca2+ occupancy, SL, and cross-bridge state. Tested cross-bridge states included cycling cross-bridges; Mg2+-ADP-induced noncycling, strong-binding cross-bridges; or Vi-induced noncycling, weak-binding cross-bridges. There are some important experimental design considerations for using Vi to inhibit strong cross-bridge binding (see the Supporting Material for further details) (49–52), and we have previously demonstrated that the method works reliably under our experimental conditions (37). This was confirmed here by the fact that by following the tr-FRET measurement protocol and regardless of test condition, maximal force could be recovered on average to 88.0 ± 2.3%. Furthermore, no significant change in passive tension at pCa 9 was observed by following the tr-FRET measurement protocol conducted under any of the conditions tested.
Fig. S1 in the Supporting Material shows typical recordings of intensity decays (see Fig. S1 A) and isometric force traces (see Fig. S1 B) that were obtained using the protocol. As consistent with our prior ss-FRET observations (11,31,37), the fluorescence intensity decay of AEDANS observed in fibers containing donor-only-labeled cTnC(T13C/N51C)AEDANS, was insensitive to changes in Ca2+ concentration and cross-bridge attachment (see Fig. S1 A, inset), whereas the intensity decay from fibers containing cTnC(T13C/N51C)AEDANS-DDPM showed significant changes that were indicative of N-cTnC opening in response to elevated Ca2+ concentrations and strong cross-bridge binding (see Fig. S1 A). For example, in cTnC(T13C/N51C)AEDANS-DDPM-reconstituted fibers in the normal resting state (pCa 9.0), the AEDANS excited-state lifetime was appreciably reduced due to FRET with DDPM (see Fig. S1 A, gray dots without lines versus those with lines). Upon activation of the fibers with Ca2+ (pCa 4.3), the fluorescence lifetime was substantially recovered (see Fig. S1 A, dashed lines versus solid lines), which suggested that a Ca2+-induced reduction in FRET had been caused by increased ensemble-averaged separation between AEDANS and DDPM.
Decay traces were used to calculate the distribution of interprobe distances between AEDANS and DDPM using the equations described in our Supporting Material. The global analysis procedure described therein yields a distribution of intersite distances to which an observed AEDANS excited-state decay is attributed. Measurements of the anisotropy of AEDANS under each test condition were used to ensure that the average orientations of the probes attached to Cys-13 and Cys-51 did not change significantly between different test conditions (see Fig. S2). Fig. 1 shows a typical distribution produced by decay fitting (A) with residue and autocorrelation analysis and distance distributions (B) recovered from observations made at 2.2 μm SL. The distance at the peak of the distribution was taken as the mean distance (r) between donor and acceptor sites within the N-cTnC population being observed. The spread of the distance distribution is described by a half-width at half-maximum (HWHM). Fitted values of r and HWHM were compared with the use of Student’s t-test, with significance at the 95% confidence level (p < 0.05).
Figure 1.
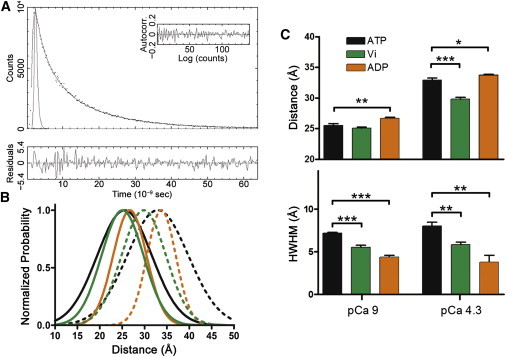
Comparison of N-cTnC Cys-13–Cys-51 distance distributions as a function of cTnC site-II Ca2+ occupancy and cross-bridge binding state when observed at long 2.2-μm SL in cTnC(T13C/N51C)AEDANS-DDPM-reconstituted myocardial fibers. (A) (Upper panel) Representative trace (black dots) of the in situ total fluorescence intensity decay of FRET donor AEDANS in the presence of nonfluorescent acceptor DDPM, which was measured at 2.2-μm SL in the presence of Ca2+ (pCa 4.3) under normal cross-bridge cycling. The decay profile was fit with Eq. 1 (gray line). (Inset and lower panel) Autocorrelation function and residuals associated with the fitting, respectively, which were used to judge goodness of fit. (B) Normalized distance distributions obtained using global-curve analysis of intensity decay profiles observed under the following conditions: pCa 9 + ATP (black solid line), pCa 4.3 + ATP (black dotted line), pCa 9 + ADP (orange solid line), pCa 4.3 + ADP (orange dotted line), pCa 9 + Vi (green solid line), and pCa 4.3 + Vi (green dotted line). (Area normalized versions of these distance distributions are shown in Fig. S4 in the Supporting Material.) (C) As a function of whether Ca2+ is bound to N-cTnC, this panel shows the statistical significance of changes in r and HWHM that occurred because of changes in pCa level or cross-bridge binding state. Parameter values are reported as the mean ± SE. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001.
Results
In situ time-resolved FRET measurements of the opening of the N-domain of cTnC and its analysis
To test our hypothesis that positive-feedback regulation is modulated by changes in SL, the in situ conformational behavior of N-cTnC was quantitatively characterized using tr-FRET measurements performed on cTnC(T13C/N51C)AEDANS- and cTnC(T13C/N51C)AEDANS-DDPM-reconstituted myocardial fibers. Densitometric analysis of Western blots using cTnC-specific antibodies (see Fig. S3) indicated that >85% of endogenous cTnC was replaced with fluorescently labeled cTnC constructs, which was similar to the reconstitution efficiency reported previously (37). Our tr-FRET experiments on these fibers were designed to evaluate the interrelationship between the effects of cTnC site-II Ca2+-occupancy, cross-bridge binding state, and SL on the conformational behavior of N-cTnC. The AEDANS intensity decay observed under each test condition was analyzed in terms of a Cys-13–Cys-51 distance distribution from which r and HWHM were derived (see Materials and Methods). Fitted distribution parameters recovered from analysis of intensity decays are summarized in Table 2 (normalized values are also given in Table S1 in the Supporting Material), wherein the r was taken from where the peak of the distribution fell. As described below, changes in r and HWHM were used to evaluate the effects of changes in pCa, cross-bridge binding state, or SL on the conformational behavior of N-cTnC.
Table 2.
Cys-13–Cys-51 distance distributions observed in cTnC(N13C/T51C)AEDANS-DDPM-reconstituted fibers under different biochemical conditions
| Cross-bridge state | pCa |
ra (Å) |
HWHMb (Å) |
||
|---|---|---|---|---|---|
| 1.8 μm SL | 2.2 μm SL | 1.8 μm SL | 2.2 μm SL | ||
| 5 mM ATP | 9.0 | 26.21 ± 0.27 | 25.53 ± 0.29 | 6.23 ± 0.64 | 7.19 ± 0.07 |
| 4.3 | 31.42 ± 0.42 | 32.97 ± 0.25 | 5.52 ± 0.42 | 8.05 ± 0.37 | |
| 1 mM Via | 9.0 | 25.46 ± 0.12 | 25.07 ± 0.10 | 5.11 ± 0.62 | 5.52 ± 0.20 |
| 4.3 | 29.97 ± 0.40 | 29.83 ± 0.31 | 5.15 ± 0.31 | 5.85 ± 0.26 | |
| 5 mM ADPb | 9.0 | 25.70 ± 0.39 | 26.72 ± 0.16 | 5.74 ± 0.45 | 4.39 ± 0.19 |
| 4.3 | 31.80 ± 0.14 | 33.78 ± 0.08 | 5.48 ± 0.56 | 3.78 ± 0.71 | |
| 0.5 mM Via | 9.0 | — | 25.75 ± 0.19 | — | 6.57 ± 0.12 |
| 4.3 | — | 32.03 ± 0.18 | — | 5.43 ± 0.43 | |
| 2.5 mM ADPb | 9.0 | — | 26.39 ± 0.26 | — | 5.18 ± 0.10 |
| 4.3 | — | 33.28 ± 0.22 | — | 6.41 ± 0.37 | |
Absolute parameter values are given as mean ± SE.
Vi solutions also contained 5 mM ATP.
ATP was absent in ADP solutions.
Because of the locations of Cys-13 and Cys-51 within the quaternary structure of N-cTnC, r is interpreted to represent the ensemble-averaged extent of N-cTnC hydrophobic patch exposure (37). Therefore, r can increase when a greater proportion of the N-cTnC ensemble transitions from the closed conformation of N-cTnC (53) to the open conformation (4). HWHM represents the breadth of the range of Cys-13–Cys-51 distances that occur as N-cTnC molecules transition between the conformational substates that form the conformation under discussion. A significantly broadened HWHM may thus indicate that observed N-cTnC molecules are in equilibrium between the closed and open conformations, which have significantly different extents of ensemble-averaged opening. As will be demonstrated in the following sections, a combined analysis of r and HWHM along these lines expanded upon previous insights into the effects of strong-binding cross-bridges on N-cTnC opening and provided three major findings on the effects of SL, each validating our hypothesis.
ADP stimulated more ensemble-averaged opening in Ca2+-bound N-cTnC than ATP at long sarcomere length
We will first focus on analyzing results from in situ tr-FRET experiments conducted at long, 2.2 μm SL (Fig. 1) because the results are directly comparable with observations from our prior in situ ss-FRET study (37). Changes in r were observed that were consistent with our prior ss-FRET observations, while changes in HWHM represented new information pertinent to changes in the protein dynamics of N-cTnC. We will now explore the changes in r that were observed before analyzing changes in HWHM. Fig. 1 B and Table 2 show that in the absence of cTnC site-II Ca2+ occupancy and presence of ATP, the N-cTnC ensemble exhibited a low r value. Introduction of Ca2+ resulted in a significant 7.44 ± 0.38 Å increase in r (Fig. 1, B and C; Table 2). This suggested that in the absence of Ca2+, the N-cTnC molecules were predominantly in the closed conformation (53). Ca2+-binding to N-cTnC and consequent strong actomyosin interactions then led to a considerable fraction of the N-cTnC ensemble transitioning to the open conformation of N-cTnC (4).
Subsequent treatment with Vi at pCa 4.3 to block strong cross-bridge attachment then significantly decreased r by 3.14 ± 0.40 Å (Fig. 1, B and C; Table 2). This strongly suggested that when strong actomyosin interactions became blocked in the presence of Ca2+, the N-cTnC ensemble ended up adopting a balanced mixture of N-cTnC molecules in the closed conformation with molecules in open conformation. Such a mixture suggests that the N-cTnC ensemble is in a conformational equilibrium between the closed (53) and open (4) conformations of N-cTnC, and that changes in r represent shifts in equilibrium position.
Experiments with ADP provided further evidence of an N-cTnC conformational equilibrium, and indicated that strong actomyosin interactions are required to maximally stabilize the in situ N-cTnC ensemble in the open conformation. Treatment with ADP at pCa 9 (with ATP having been removed) to produce strong-binding, noncycling cross-bridges stimulated a significant increase in r by 1.19 ± 0.33 Å. This was accompanied by Ca2+-independent active force generation that was 1.35 ± 0.10-fold higher than what was observed for the pCa 4.3 + ATP + 2.2 μm SL test condition (Table 3). These results indicated that even in the absence of Ca2+, maximal strong actomyosin interaction had led to a fraction of the N-cTnC ensemble transitioning from the closed to the open conformation. While this finding was consistent with our prior FRET work (31,37), an important fresh observation was that at pCa 4.3 and long SL and compared to cycling cross-bridges, noncycling strong-binding cross-bridges significantly further increased r by 0.81 ± 0.26 Å (Fig. 1, B and C; Table 2). Thus, even at saturating Ca2+ levels, the presence of weak-binding cross-bridge states during cross-bridge cycling results in a small fraction of N-cTnC molecules in the thick- and thin-filament overlap zone occupying the closed conformation. Taken together, these results with ATP, Vi, and ADP treatment at long SL strongly suggest that thin-filament regulation involves a N-cTnC conformational equilibrium that is not only highly sensitive to Ca2+, but also to positive feedback from strong-binding cross-bridges.
Table 3.
Force values (mean ± SE, n = 5–6) from cTnC(T13C/N51C)AEDANS-DDPM-reconstituted fibers observed at pCa 9 and 4.3 as a function of XB state and SL
| Cross-bridge state | pCa | Force (mN mm−2) |
|
|---|---|---|---|
| 1.8 μm SL | 2.2 μm SL | ||
| 5 mM ATP | 9.0 | 0.449 ± 0.377 | 8.62 ± 0.45 |
| 4.3 | 20.0 ± 1.1 | 34.3 ± 1.6 | |
| 1 mM Via | 9.0 | 1.03 ± 0.24 | 8.41 ± 0.26 |
| 4.3 | 1.51 ± 0.88 | 8.98 ± 2.01 | |
| 5 mM ADPb | 9.0 | 24.0 ± 2.2 | 43.5 ± 2.1 |
| 4.3 | 22.1 ± 3.7 | 40.7 ± 2.2 | |
| 0.5 mM Via | 9.0 | — | 8.63 ± 1.05 |
| 4.3 | — | 13.0 ± 1.9 | |
| 2.5 mM ADPb | 9.0 | — | 26.2 ± 2.4 |
| 4.3 | — | 33.6 ± 1.2 | |
Vi solutions also contained 5 mM ATP.
ATP was absent in ADP solutions.
Cycling cross-bridges caused increased N-cTnC conformational dynamics at long sarcomere length
In the form of HWHM values, tr-FRET provided information on N-cTnC protein dynamics that is inaccessible to ss-FRET. Thus, if an N-cTnC conformational equilibrium is present during thin-filament regulation and is highly sensitive to strong actomyosin interactions, HWHM values should reflect these facts. Interestingly, at long SL, the pCa-averaged HWHM value associated with cycling cross-bridges; noncycling, strong-binding cross-bridges; and noncycling, weak-binding cross-bridges was 7.62 ± 0.19, 4.09 ± 0.37, and 5.69 ± 0.16 Å, respectively, and these values were all significantly different from each other (p < 0.01). Thus, in the presence of cycling cross-bridges and regardless of cTnC site-II Ca2+ occupancy, our single Gaussian model suggested that the relative number of N-cTnC conformational substates was on average 1.86 ± 0.17-fold (p < 0.001) and 1.33 ± 0.05-fold (p < 0.001) greater than in the presence of noncycling strong and noncycling weak cross-bridges, respectively. However, no significant difference in HWHM value was noted for changes in cTnC site-II Ca2+ occupancy with respect to the same cross-bridge state.
These results indicated that the number of conformational substates being sampled by the N-cTnC ensemble was sensitive to cross-bridge state, but not cTnC site-II Ca2+ occupancy. Compared to Vi test conditions, ATP test conditions led to an increase in the number of N-cTnC conformational substates, whereas ADP test conditions tended to decrease them. As explored further in the Discussion below, this strongly suggests that the N-cTnC conformational equilibrium point is sensitive to the regulatory state of tropomyosin, which is itself highly sensitive to changes in strong actomyosin interaction. Cycling cross-bridges should accordingly produce the greatest variety of occupation of tropomyosin regulatory states, resulting in the highest HWHM values. In contrast, noncycling, strong-binding cross-bridges should maximally bias tropomyosin toward the open state of thin-filament regulation, leading to the lowest HWHM values.
To further evaluate the cross-bridge-state dependence of N-cTnC conformational dynamics, Cys-13–Cys-51 distance distributions were measured under varying concentrations of ADP and Vi (Fig. 2; Table 2). Martyn et al. (54) has suggested that the equilibrium of weak- and strong-binding cross-bridges during normal force generation can be altered by a change in [Vi], and changes in ADP concentration should similarly affect the stability of strong-binding states. Observed force values verified these expectations (Table 3), and the effects of the concomitant changes in strong actomyosin interaction were as follows: at both pCa 9.0 and 4.3, as Mg2+-ADP was increased from 0 to 2.5 and then 5 mM to increasingly bias cross-bridges toward the strong-binding state (Fig. 2 A), r was increasingly raised (p < 0.05) and HWHM was increasingly lowered (p < 0.05). We inferred that while tropomyosin was being increasingly stabilized in the open state of thin-filament regulation by strong actomyosin interactions, the N-cTnC ensemble was being increasingly stabilized in the open conformation.
Figure 2.
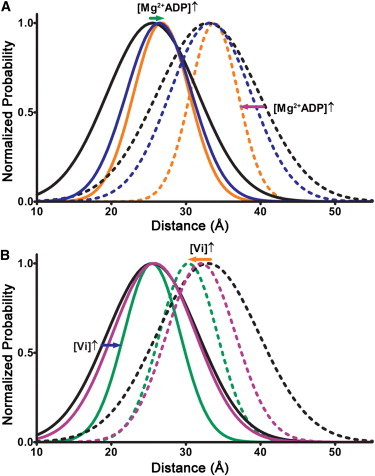
Cys-13–Cys-51 distance distributions calculated from intensity decays observed in cTnC(T13C/N51C)AEDANS- and cTnC(T13C/N51C)AEDANS-DDPM-reconstituted fibers set to 2.2 μm SL and exposed to intermediate and saturating concentrations of ADP and Vi. Similar to Fig. 1, the distance distributions were normalized against their peaks (area-normalized versions are shown in Fig. S5), and solid lines and dashed lines indicate measurements taken at pCa 9 and pCa 4.3, respectively. (A) Structural effects of 2.5 mM Mg2+-ADP (blue lines) versus 5 mM Mg2+-ADP (orange lines). For the purpose of visual comparison, distance distributions measured in presence of 5 mM ATP (black lines) are also plotted. (Green and purple arrows) How distance distributions change in response to increasing [Mg2+-ADP]. (B) Similarly to panel A, the structural effects of 0.5 mM Vi (purple lines) versus 1.0 mM Vi (0.5 mM, green lines) are shown. (Blue and orange arrows) How distance distributions change in response to increasing [Vi].
Furthermore, at pCa 4.3, as the concentration of Vi was increased from 0 to 0.5 and then 1 mM to increasingly disrupt strong cross-bridge attachment (Fig. 2 B), r was increasingly lowered (p < 0.05) and HWHM was increasingly lowered (p < 0.001). Accordingly, we inferred that while tropomyosin was being increasingly destabilized in the open state of thin-filament regulation, the N-cTnC ensemble was being increasingly transitioned toward the closed conformation of N-cTnC. Interestingly, at pCa 9, HWHM was increasingly lowered (p < 0.01), but no significant change in r was observed. This suggested that while cycling cross-bridges lead to increased sampling of the open state by the N-cTnC ensemble even at pCa 9, such sampling is not stable enough to cause a change in r that could be picked up by the single Gaussian model. Overall, the observed changes in HWHM further indicated that in situ N-cTnC is in a conformational equilibrium in which not only cTnC site-II Ca2+ occupancy but also strong actomyosin interactions are required to maximally stabilize N-cTnC in the open conformation.
Reduction of sarcomere length diminished the influence of strong-binding cross-bridges on N-cTnC conformational behavior
We next tested the effect of SL on the cross-bridge-state-dependent N-cTnC conformational behavior. Interestingly, the stabilizing effects of cycling cross-bridges on N-cTnC opening noted above for 2.2 μm SL test conditions (Fig. 1 B, black lines) were attenuated at short, 1.8 μm SL (Fig. 3 A, black lines). In the presence of ATP, both r and HWHM became significantly decreased at pCa 4.3 when the SL was changed from 2.2 to 1.8 μm (Fig. 3 D). Moreover, the normalized maximal active force generated by cycling cross-bridges was reduced by 23.9 ± 6.1% when SL was reduced from 2.2 to 1.8 μm (Table 3). Thus, when strong actomyosin interaction was decreased by SL reduction, the N-cTnC conformational equilibrium was concurrently shifted away from the open conformation toward the closed conformation, which resulted in reduced ensemble-averaged opening and fewer underlying conformational substates. This observation was the first major finding that validated our hypothesis. It was interpreted to indicate that a decrease in SL decreases the orientation and ordering of cross-bridges optimal for strong actomyosin interaction (48), which leads to a reduction in 1), steady-state force development and 2), N-cTnC opening through diminished NcTnC-cTnI-Sr interaction (37).
Figure 3.
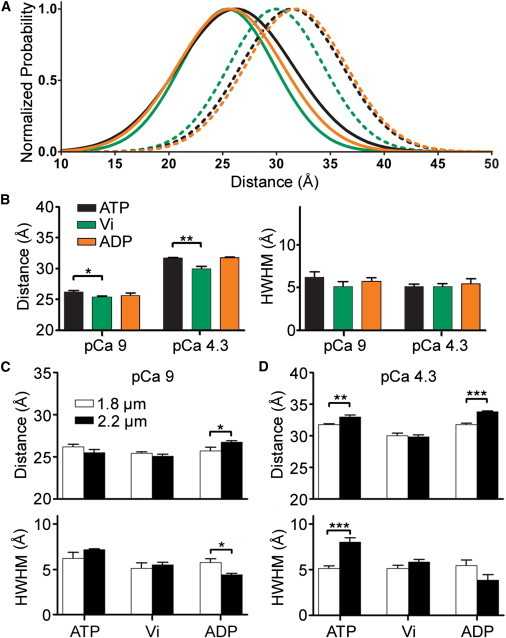
Comparison of N-cTnC Cys-13–Cys-51 distance distributions as a function of cTnC site-II Ca2+-occupancy and cross-bridge state when observed at short 1.8-μm SL in cTnC(T13C/N51C)AEDANS-DDPM-reconstituted myocardial fibers. (A) Normalized distance distributions obtained using global-curve analysis of intensity decay profiles observed under the following conditions: pCa 9 + ATP (black solid line), pCa 4.3 + ATP (black dotted line), pCa 9 + ADP (orange solid line), pCa 4.3 + ADP (orange dotted line), pCa 9 + Vi (green solid line), and pCa 4.3 + Vi (green dotted line). (Area-normalized versions of these distance distributions are shown in Fig. S6.) (B) As a function of whether Ca2+ is bound to N-cTnC and at 1.8 μm SL, this panel shows the statistical significance of differences in r and HWHM values that occurred because of changes in cross-bridge binding state. (C and D) Significance of differences in r and HWHM due to reduction of SL. The number of determinations was 5–6 for each group. ∗p < 0.05; ∗∗p < 0.005; ∗∗∗p < 0.001.
Based on this interpretation, we next predicted that a reduction in SL should also reduce the ability of noncycling, strong-binding cross-bridges to stabilize N-cTnC opening. Further testing demonstrated that at short SL, the replacement of ATP with ADP in pCa solutions had no significant effect on either r or HWHM (Fig. 3 B); hence, SL reduction from 2.2 to 1.8 μm caused the stabilizing effects of noncycling, strong-binding cross-bridges on N-cTnC opening to become significantly diminished (Fig. 3, C and D). Therefore, this observation also validated our hypothesis and further suggested that at short SL, the effects of positive feedback regulation on N-cTnC structure are significantly diminished. The consequent destabilization of the N-cTnC ensemble in the open conformation of N-cTnC provides a fresh structural insight into why reductions in Ca2+ sensitivity occur alongside decreases in active force generation as SL is decreased.
Treatment with vanadate eliminated sarcomere-length-dependent structural changes in N-cTnC
If SL-dependent modulation of positive feedback regulation is critically involved in effecting length-dependent Ca2+ sensitivity, then chemical prevention of strong cross-bridge attachment should eliminate SL-dependent changes in N-cTnC opening altogether (55). This prediction was verified by experiments wherein Vi was used to chemically inhibit the attachment of strong cycling cross-bridges to actin (Figs. 1 and 3). As expected, at pCa 4.3 and under Vi treatment, there was no statistical difference between the r and HWHM values obtained at 1.8 and 2.2 μm SL (Fig. 3, C and D). Moreover, the addition of 1 mM Vi to pCa 4.3 solutions reduced r and HWHM at both 1.8 and 2.2 μm SLs (Figs. 3 B and 1 C); however, the Vi-induced reduction in r observed at long SL was significantly greater than that observed at short SL. Therefore, the observed Vi-induced elimination of SL-modulation of N-cTnC opening represented additional evidence that implicated SL-dependent modulation of positive feedback regulation as mechanistically involved in length-dependent activation.
Discussion
The orientation and ordering of myosin heads relative to their binding sites on actin are key parameters for determining the probability that cross-bridges will form strong actomyosin interactions, and biophysical evidence has been accumulating that these parameters depend on SL (48,56–58). Results from our recent in situ ss-FRET study indicated that strong actomyosin interactions lead to a stabilization of ensemble-averaged N-cTnC opening (37), from which it was inferred that strong actomyosin interactions affect the associations of tropomyosin and cTnI with actin in a manner that indirectly leads to a stabilization of the Ca2+-sensitizing N-cTnC–cTnI-Sr interaction (10,24,37,59). Accordingly, we hypothesized in this study that SL-dependent modulation of strong actomyosin interactions leads to a concomitant modulation of the stabilizing effect of strong actomyosin interactions on N-cTnC opening.
Because our ss-FRET technique depends on the measurement of fluorescence intensity, it is technically challenging to test our hypothesis using this method because the fluorescence baseline is subject to change whenever the fiber being observed is stretched or slackened. To overcome this technical obstacle, we successfully modified our Güth MRS to perform in situ tr-FRET measurements, which take advantage of the fact that fluorescence lifetime is independent of fluorophore concentration. As demonstrated by our results, tr-FRET enabled us to characterize changes in the conformational behavior of N-cTnC caused by changes in cTnC site-II Ca2+ occupancy, cross-bridge binding state, or SL. We were also able to probe N-cTnC conformational dynamics because nanosecond timescale domain structural dynamics were embedded in the high-quality, time-resolved data. The implications of our major findings on our understanding of cardiac thin-filament regulation are discussed below. Note that the conformational changes discussed are considered as ensemble averages; we make no attempt to distinguish differences between cross-bridge overlapping and nonoverlapping regions of the thin filament.
N-cTnC is in a conformational equilibrium that is shifted toward the open conformation by positive feedback regulation
In our prior ss-FRET study (37), it was posited that the stabilizing effect of positive feedback from strong-binding cross-bridges on N-cTnC opening involves an N-cTnC conformational equilibrium shift (6,9,30,37). Supporting this argument, in situ FRET distance distributions obtained here at 2.2 μm SL under ATP conditions were dramatically broader than distributions observed under ADP or Vi test conditions (Fig. 1 and Table 2). Values of r obtained under various conditions also indicated that the conformational occupancy of the N-cTnC ensemble was mixed between the closed and open conformations. These results strongly suggested that the in situ N-cTnC ensemble is in equilibrium between the closed and open conformations of N-cTnC. Moreover, the equilibrium point, as represented by both r and HWHM, was observed to be highly sensitive not just to changes in cTnC site-II Ca2+ occupancy, but also cross-bridge state. Hence, Ca2+-binding to N-cTnC and positive feedback from strong actomyosin interactions together determine the fraction of the N-cTnC ensemble that occupies the open versus closed conformation at steady state (Fig. 4).
Figure 4.
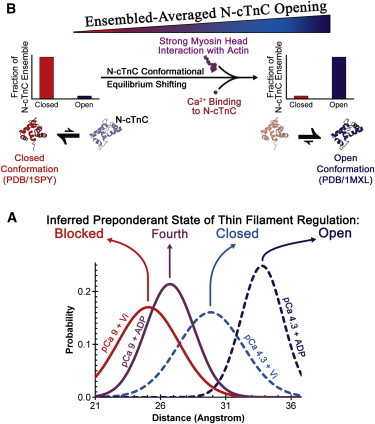
A schematic presenting our model for how changes in N-cTnC Ca2+ site-II occupancy and cross-bridge state lead to changes in ensemble-averaged N-cTnC opening at 2.2 μm SL. (A) Shown here are selected area-normalized distance distributions, which each represent a different a preponderant state of the four states of thin-filament regulation (27), as based on cTnC site-II Ca2+ occupancy and cross-bridge binding state. Hence, in terms of the preponderant state of thin-filament regulation, the extent of ensemble-averaged N-cTnC opening is ranked in the following order: Blocked < Fourth < Closed < Open. (B) We interpret the observed changes in ensemble-averaged N-cTnC opening as caused by shifts in an N-cTnC conformational equilibrium that is at work during thin-filament regulation. As explained further in the main text, Ca2+ binding to N-cTnC and strong cross-bridge binding to actin can each independently and allosterically lead to a shift in this equilibrium from the closed conformation of N-cTnC toward the open conformation. However, Ca2+ binding to N-cTnC and strong actomyosin interactions work in concert during physiological thin-filament regulation, and both are required to maximally activate the thin-filament and concomitantly shift the N-cTnC equilibrium toward the open conformation. The structures of N-cTnC presented in panel B were from PDB:1SPY (53) and PDB:1MXL (4), as indicated.
Because ensemble-averaged in situ N-cTnC opening is an excellent indicator of in situ thin-filament activation (37), the sensitivity of the N-cTnC conformational equilibrium to cross-bridge state strongly supports the importance of positive feedback regulation in cardiac thin-filament regulation. Our results indicated that Ca2+ alone was unable to fully activate the cardiac thin filament, just as argued previously by Wu et al. (60). In fact, based on ensemble-averaged N-cTnC opening at long SL, our results suggested that in the absence of strong actomyosin interactions and their effects on tropomyosin, cTnI, and cTnC structure, the thin filament is at most 52.3 ± 3.6% activated under Ca2+ saturation (Table 2 and see Table S1: 2.2 μm SL, pCa 4.3 + Vi vs. pCa 4.3 + ADP; calculation ignores role of overlap between thick and thin filaments). Furthermore, our previous ss-FRET Ca2+ titration results indicated that the Ca2+ sensitivity of the thin filament is depressed by ΔpCa50 = −0.26 ± 0.05 (mean ± standard deviation, n = 5) when positive feedback regulation is maximally inhibited by Vi (37). Therefore, without positive feedback regulation, the thin filament would achieve neither the level of activation in response to Ca2+ binding nor the Ca2+ sensitivity required for generating the correct amount of active force in response to normal systolic Ca2+ transients.
Myofilament length-dependent activation involves the length-dependent modulation of positive feedback regulation
Length-dependent activation is thought to be the basis of the Frank-Starling law of the heart. However, the identity of the complete mechanism underlying the SL-dependence of myocardial force production has proven complex and controversial (43,44). Contemporary hypotheses have variously posited that SL-dependent modulation of the probability of weak cross-bridge attachments (61,62), probability of strong cross-bridge attachments (55,63), extent to which myosin head orientation and ordering are favorable for the formation of strong actomyosin interactions (48), thin-filament-intrinsic cooperativity (62,64), structural changes in thin-filament proteins (65), titin strain (66), or thin-filament compliance (67–69) are important determinants in length-dependent activation. It is likely that all of these proposed mechanisms may contribute to length-dependent activation, but new insights are needed to clarify the cause-and-effect relationships that link these processes into a unified molecular mechanism. The biophysical data presented in this study help to address this need by contributing new insights into the effects of changes in SL and cross-bridge state on the conformational behavior of N-cTnC. We observed that increased SL leads to increased ensemble-averaged N-cTnC opening only when strong-binding cross-bridges are present. Because shifting of the N-cTnC ensemble toward the open conformation is known to lead to increased Ca2+ sensitivity (37–42), these observations represent a fresh structural insight into why increased SL leads to increased thin-filament Ca2+ sensitivity.
Our results provide additional support to the argument that length-dependent activation involves the modulation of strong cross-bridge binding and concomitant positive feedback regulation by changes in SL (24,45). This argument was recently advanced by small-angle x-ray diffraction measurements by Farman et al. (48) on the conformational behavior of cross-bridges in intact right ventricular trabeculae from rat. Their findings indicated that SL is positively correlated with an optimal average orientation and ordering of myosin heads for strong cross-bridge attachment to the thin filament. Consistent with this, we observed that decreased SL led to reduced strong actomyosin interaction, as implied by decreased force, concomitant with decreased ensemble-averaged N-cTnC opening. We interpret this N-cTnC structural effect as due to reduced positive feedback regulation.
In our model of positive feedback regulation (37), we infer that strong actomyosin interactions are allosterically transduced into stabilization of N-cTnC opening through changes in the associations of tropomyosin and cTnI with actin. Tropomyosin is shifted toward the open state and interactions between cTnI and actin/tropomyosin become disrupted. This is further inferred to indirectly promote increased interaction between cTnI-Sr and N-cTnC, which shifts the N-cTnC conformational equilibrium toward the open conformation of N-cTnC.
Finally, these structural changes are inferred to allosterically spread along the thin filament through tropomyosin-mediated couplings between regulatory units. In keeping with this model, because strong actomyosin interactions are influenced by SL, it is inferred that the allosteric structural changes they stimulate in tropomyosin, cTnI, and N-cTnC are consequently also influenced by SL. Thus, the proposed SL-dependent positive feedback regulation mechanism could explain why cTnI isoform, thin-filament cooperativity, and strong cross-bridge binding have all been implicated as determinants in the length-dependent activation mechanism. Titin may be inferred as important for determining how changes in SL modulate strong actomyosin interaction, and thin-filament compliance might be expected to modulate the protein-protein interactions involved in positive feedback regulation. Hence, complete characterization of the unified mechanism for length-dependent activation now not only seems possible, but inevitable.
Growing evidence that absence of strong cross-bridge binding results in the loss of the sarcomere-length-dependence of thin-filament Ca2+ sensitivity
Our Vi-associated results presented in this study contribute more-explicit structural evidence that Vi-mediated inhibition of strong-binding cross-bridge attachment to actin attenuates the effects of SL on thin-filament Ca2+ sensitivity. Smith et al. (55) previously used in situ linear dichroism to show that increasing levels of Vi resulted in decreasing extents of cross-bridge-dependent N-cTnC structural changes and decreasing dependencies of the Ca2+ sensitivity of contractile force on SL. Their results corroborated earlier linear dichroism measurements by Martyn and Gordon (26) showing that a saturating level of Vi eliminated SL-dependent changes in cTnC structure and the SL-dependence of Ca2+ sensitivity. Our observation here that 1 mM Vi treatment causes a loss of SL-induced changes in N-cTnC opening strongly indicates that the cTnC structural changes being observed by Smith et al. (55) and Martyn and Gordon (26) were associated with changes in N-cTnC opening and corresponding cross-bridge-dependent stabilization of the Ca2+-sensitizing N-cTnC–cTnI-Sr interaction (37).
Inhibitors of strong cross-bridge attachment other than Vi also appear to exert attenuating effects on the length dependence of Ca2+ sensitivity. Kurihara et al. (70) used aequorin-injected right ventricular papillary muscle from ferret to show that 10 mM BDM eliminated SL-dependent changes in Ca2+ binding to N-cTnC, and consistent with findings obtained using Vi, this effect of BDM treatment also leads to the conclusion that length-dependent activation involves positive feedback from the attachment of strong-binding cross-bridges. Farman et al. (62) reported that increasing concentrations of blebbistatin of up to 1 μM had a slightly steeper effect on the relationship between EC50 and maximal force at 2.0 μm SL than at 2.2 μm SL. However, the authors further found that this difference in steepness was statistically insignificant with p = 0.2, as determined by Student’s t-test. These findings suggest that an important experiment would be to directly compare the effects of Vi, BDM, and blebbistatin on the SL-dependence of N-cTnC opening. Unfortunately, such a direct comparison was not possible in this study, because the ultraviolet light required to excite AEDANS would rapidly inactivate blebbistatin (71). As of this writing, we are developing a FRET scheme relying on a longer-wavelength donor fluorophore that will make such a comparison possible in the future.
Conclusions
Our biophysical data presented here implicates positive feedback regulation as modulated by changes in SL, making it therefore mechanistically involved in myofilament length-dependent activation. In light of growing biophysical evidence that cooperativity is intrinsic to the thin filament such that strong actomyosin interactions may not be needed to achieve a steep relation between force and Ca2+ level (37,47,55), the uniquely high degree that cardiac thin-filament activation depends on strong cross-bridge binding (60) may therefore instead be an impressive evolutionary adaptation that helps facilitate length-dependent activation.
We note that much remains to be determined about the cause-and-effect relationships that link changes in SL, to changes in strong actomyosin interaction, to changes in N-cTnC opening. We expect that the posttranslational state of thin and thick filaments, titin strain, thin-filament cooperativity, thin-filament compliance, and protein isoform specific effects all play an important mechanistic role in these relationships. This is because all of these properties influence the allosteric interplay among cycling cross-bridges and tropomyosin, cTnI, and cTnC. Therefore, the reason why the mechanism underlying myofilament length-dependent activation has proven so difficult to establish is that many factors are working together in giving rise to the emergent property that is cardiac myofilament length-dependent activation.
Acknowledgments
We thank the laboratory of Jaak Panksepp for generously providing the rats that were used in this study. We are also grateful to Sheri Six for her assistance with animal handling.
This work was supported, in whole or in part, by National Institutes of Health grants No. R01 HL80186-5S1 (to W.-J.D.), No. R01 HL64035 (to R.J.S.), and No. R21 HL109693 (to W.-J.D.), and also by the M. J. Murdock Charitable Trust (to W.-J.D.). The study described in this manuscript was also supported by award No. T32GM008336 from the U.S. National Institute of General Medical Sciences.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
Supporting Material
Supporting Citations
Refs. (72–76) appear in the Supporting Material.
References
- 1.Tobacman L.S. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 2.Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 3.Wei B., Jin J.P. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch. Biochem. Biophys. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M.X., Spyracopoulos L., Sykes B.D. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 5.Dong W.J., Xing J., Cheung H.C. Conformation of the regulatory domain of cardiac muscle troponin C in its complex with cardiac troponin I. J. Biol. Chem. 1999;274:31382–31390. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- 6.Behrmann E., Müller M., Raunser S. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi T., Kobayashi M., Collins J.H. Inhibitory region of troponin I: Ca2+-dependent structural and environmental changes in the troponin-tropomyosin complex and in reconstituted thin filaments. Biochemistry. 2000;39:86–91. doi: 10.1021/bi991903b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong W.J., Robinson J.M., Cheung H.C. Ca2+-induced conformational transition in the inhibitory and regulatory regions of cardiac troponin I. J. Biol. Chem. 2003;278:8686–8692. doi: 10.1074/jbc.M212886200. [DOI] [PubMed] [Google Scholar]
- 9.Robinson J.M., Dong W.J., Cheung H.C. Switching of troponin I: Ca2+ and myosin-induced activation of heart muscle. J. Mol. Biol. 2004;340:295–305. doi: 10.1016/j.jmb.2004.04.046. [DOI] [PubMed] [Google Scholar]
- 10.Xing J., Chinnaraj M., Dong W.J. Structural studies of interactions between cardiac troponin I and actin in regulated thin filament using Förster resonance energy transfer. Biochemistry. 2008;47:13383–13393. doi: 10.1021/bi801492x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xing J., Jayasundar J.J., Dong W.J. Förster resonance energy transfer structural kinetic studies of cardiac thin filament deactivation. J. Biol. Chem. 2009;284:16432–16441. doi: 10.1074/jbc.M808075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Z., Li K.-L., Dong W.-J. Structural dynamics of C-domain of cardiac troponin I protein in reconstituted thin filament. J. Biol. Chem. 2012;287:7661–7674. doi: 10.1074/jbc.M111.281600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman R.M., Blumenschein T.M., Sykes B.D. An interplay between protein disorder and structure confers the Ca2+ regulation of striated muscle. J. Mol. Biol. 2006;361:625–633. doi: 10.1016/j.jmb.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 14.Potter J.D., Sheng Z., Zhao J. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J. Biol. Chem. 1995;270:2557–2562. doi: 10.1074/jbc.270.6.2557. [DOI] [PubMed] [Google Scholar]
- 15.Huxley H.E. Structural changes in the actin-and myosin-containing filaments during contraction. Cold Spring Harb. Symp. Quant. Biol. 1972;37:361–376. [Google Scholar]
- 16.Haselgrove J.C. X-ray evidence for a conformational change in the actin-containing filaments of vertebrate striated muscle. Cold Spring Harb. Symp. Quant. Biol. 1972;37:341–352. [Google Scholar]
- 17.Holmes K.C. The actomyosin interaction and its control by tropomyosin. Biophys. J. 1995;68:2S–7S. [PubMed] [Google Scholar]
- 18.Lehman W., Craig R., Vibert P. Ca2+-induced tropomyosin movement in Limulus thin filaments revealed by three-dimensional reconstruction. Nature. 1994;368:65–67. doi: 10.1038/368065a0. [DOI] [PubMed] [Google Scholar]
- 19.Lehman W., Hatch V., Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. J. Mol. Biol. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- 20.Poole K.J.V., Evans G., Holmes K.C. The effect of crossbridges on the calcium sensitivity of the structural change of the regulated thin filament. Biophys. J. 1995;68:A365. [Google Scholar]
- 21.Gordon A.M., Ridgway E.B. Extra calcium on shortening in barnacle muscle. Is the decrease in calcium binding related to decreased cross-bridge attachment, force, or length? J. Gen. Physiol. 1987;90:321–340. doi: 10.1085/jgp.90.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Güth K., Potter J.D. Effect of rigor and cycling cross-bridges on the structure of troponin C and on the Ca2+ affinity of the Ca2+-specific regulatory sites in skinned rabbit psoas fibers. J. Biol. Chem. 1987;262:13627–13635. [PubMed] [Google Scholar]
- 23.Hannon J.D., Martyn D.A., Gordon A.M. Effects of cycling and rigor crossbridges on the conformation of cardiac troponin C. Circ. Res. 1992;71:984–991. doi: 10.1161/01.res.71.4.984. [DOI] [PubMed] [Google Scholar]
- 24.Moss R.L., Razumova M., Fitzsimons D.P. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ. Res. 2004;94:1290–1300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 25.Miki M., Hai H., Wakabayashi T. Fluorescence resonance energy transfer between points on actin and the C-terminal region of tropomyosin in skeletal muscle thin filaments. J. Biochem. 2004;136:39–47. doi: 10.1093/jb/mvh090. [DOI] [PubMed] [Google Scholar]
- 26.Martyn D.A., Gordon A.M. Influence of length on force and activation-dependent changes in troponin c structure in skinned cardiac and fast skeletal muscle. Biophys. J. 2001;80:2798–2808. doi: 10.1016/S0006-3495(01)76247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lehrer S.S. The 3-state model of muscle regulation revisited: is a fourth state involved? J. Muscle Res. Cell Motil. 2011;32:203–208. doi: 10.1007/s10974-011-9263-8. [DOI] [PubMed] [Google Scholar]
- 28.Dong W.J., Xing J., Cheung H.C. Ca2+ induces an extended conformation of the inhibitory region of troponin I in cardiac muscle troponin. J. Mol. Biol. 2001;314:51–61. doi: 10.1006/jmbi.2001.5118. [DOI] [PubMed] [Google Scholar]
- 29.Dong W.J., Robinson J.M., Cheung H.C. Kinetics of conformational transitions in cardiac troponin induced by Ca2+ dissociation determined by Förster resonance energy transfer. J. Biol. Chem. 2003;278:42394–42402. doi: 10.1074/jbc.M304858200. [DOI] [PubMed] [Google Scholar]
- 30.Robinson J.M., Cheung H.C., Dong W. The cardiac Ca2+-sensitive regulatory switch, a system in dynamic equilibrium. Biophys. J. 2008;95:4772–4789. doi: 10.1529/biophysj.108.131318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong W.J., Jayasundar J.J., Cheung H.C. Effects of PKA phosphorylation of cardiac troponin I and strong crossbridge on conformational transitions of the N-domain of cardiac troponin C in regulated thin filaments. Biochemistry. 2007;46:9752–9761. doi: 10.1021/bi700574n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacchiocchi C., Lehrer S.S. Ca2+-induced movement of tropomyosin in skeletal muscle thin filaments observed by multi-site FRET. Biophys. J. 2002;82:1524–1536. doi: 10.1016/S0006-3495(02)75505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis J.P., Norman C., Tikunova S.B. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 2007;92:3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Wang H., Chalovich J.M., Marriott G. Structural dynamics of troponin I during Ca2+-activation of cardiac thin filaments: a multi-site Förster resonance energy transfer study. PLoS ONE. 2012;7:e50420. doi: 10.1371/journal.pone.0050420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akhter S., Zhang Z., Jin J.-P. The heart-specific NH2-terminal extension regulates the molecular conformation and function of cardiac troponin I. Am. J. Physiol. Heart C. 2012;302:H923–H933. doi: 10.1152/ajpheart.00637.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rieck D.C., Li K.-L., Dong W.-J. Structural basis for the in situ Ca2+ sensitization of cardiac troponin C by positive feedback from force-generating myosin cross-bridges. Arch. Biochem. Biophys. 2013;537:198–209. doi: 10.1016/j.abb.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gollapudi S.K., Mamidi R., Chandra M. The N-terminal extension of cardiac troponin T stabilizes the blocked state of cardiac thin filament. Biophys. J. 2012;103:940–948. doi: 10.1016/j.bpj.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holroyde M.J., Robertson S.P., Potter J.D. The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 1980;255:11688–11693. [PubMed] [Google Scholar]
- 40.Robertson I.M., Sun Y.B., Sykes B.D. A structural and functional perspective into the mechanism of Ca2+-sensitizers that target the cardiac troponin complex. J. Mol. Cell. Cardiol. 2010;49:1031–1041. doi: 10.1016/j.yjmcc.2010.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jayasundar J.J., Xing J., Dong W.J. Molecular dynamics simulations of the cardiac troponin complex performed with FRET distances as restraints. PLoS ONE. 2014;9:e87135. doi: 10.1371/journal.pone.0087135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D., Robertson I.M., Regnier M. Structural and functional consequences of the cardiac troponin C L48Q Ca2+-sensitizing mutation. Biochemistry. 2012;51:4473–4487. doi: 10.1021/bi3003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Tombe P.P., Mateja R.D., Irving T.C. Myofilament length dependent activation. J. Mol. Cell. Cardiol. 2010;48:851–858. doi: 10.1016/j.yjmcc.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cordina N.M., Liew C.K., Brown L.J. Effects of calcium binding and the hypertrophic cardiomyopathy A8V mutation on the dynamic equilibrium between closed and open conformations of the regulatory N-domain of isolated cardiac troponin C. Biochemistry. 2013;52:1950–1962. doi: 10.1021/bi4000172. [DOI] [PubMed] [Google Scholar]
- 45.Gillis T.E., Martyn D.A., Regnier M. Investigation of thin filament near-neighbor regulatory unit interactions during force development in skinned cardiac and skeletal muscle. J. Physiol. 2007;580:561–576. doi: 10.1113/jphysiol.2007.128975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lakatta E.G. Starling’s law of the heart is explained by an intimate interaction of muscle length and myofilament calcium activation. J. Am. Coll. Cardiol. 1987;10:1157–1164. doi: 10.1016/s0735-1097(87)80361-3. [DOI] [PubMed] [Google Scholar]
- 47.Shitaka Y., Kimura C., Miki M. The rates of switching movement of troponin T between three states of skeletal muscle thin filaments determined by fluorescence resonance energy transfer. J. Biol. Chem. 2005;280:2613–2619. doi: 10.1074/jbc.M408553200. [DOI] [PubMed] [Google Scholar]
- 48.Farman G.P., Gore D., de Tombe P.P. Myosin head orientation: a structural determinant for the Frank-Starling relationship. Am. J. Physiol. Heart Circ. Physiol. 2011;300:H2155–H2160. doi: 10.1152/ajpheart.01221.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campbell K.S. Impact of myocyte strain on cardiac myofilament activation. Pflugers Arch. 2011;462:3–14. doi: 10.1007/s00424-011-0952-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mocz G. Vanadate-mediated photocleavage of rabbit skeletal myosin. Eur. J. Biochem. 1989;179:373–378. doi: 10.1111/j.1432-1033.1989.tb14563.x. [DOI] [PubMed] [Google Scholar]
- 51.Fukuda N., Terui T., Kurihara S. Titin and troponin: central players in the Frank-Starling mechanism of the heart. Curr. Cardiol. Rev. 2009;5:119–124. doi: 10.2174/157340309788166714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Strauss J.D., Zeugner C., Rüegg J.C. Troponin replacement in permeabilized cardiac muscle. Reversible extraction of troponin I by incubation with vanadate. FEBS Lett. 1992;310:229–234. doi: 10.1016/0014-5793(92)81338-m. [DOI] [PubMed] [Google Scholar]
- 53.Spyracopoulos L., Li M.X., Sykes B.D. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 54.Martyn D.A., Smith L., Regnier M. The effects of force inhibition by sodium vanadate on cross-bridge binding, force redevelopment, and Ca2+ activation in cardiac muscle. Biophys. J. 2007;92:4379–4390. doi: 10.1529/biophysj.106.096768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith L., Tainter C., Martyn D.A. Cooperative cross-bridge activation of thin filaments contributes to the Frank-Starling mechanism in cardiac muscle. Biophys. J. 2009;96:3692–3702. doi: 10.1016/j.bpj.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDonald K.S., Moss R.L. Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circ. Res. 1995;77:199–205. doi: 10.1161/01.res.77.1.199. [DOI] [PubMed] [Google Scholar]
- 57.Fuchs F., Wang Y.-P. Sarcomere length versus interfilament spacing as determinants of cardiac myofilament Ca2+ sensitivity and Ca2+ binding. J. Mol. Cell. Cardiol. 1996;28:1375–1383. doi: 10.1006/jmcc.1996.0129. [DOI] [PubMed] [Google Scholar]
- 58.Farman G.P., Walker J.S., Irving T.C. Impact of osmotic compression on sarcomere structure and myofilament calcium sensitivity of isolated rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H1847–H1855. doi: 10.1152/ajpheart.01237.2005. [DOI] [PubMed] [Google Scholar]
- 59.Galińska-Rakoczy A., Engel P., Lehman W. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J. Mol. Biol. 2008;379:929–935. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu S., Liu J., Taylor K.A. Electron tomography of cryofixed, isometrically contracting insect flight muscle reveals novel actin-myosin interactions. PLoS ONE. 2010 doi: 10.1371/journal.pone.0012643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cremo C.R., Long G.T., Grammer J.C. Photocleavage of myosin subfragment 1 by vanadate. Biochemistry. 1990;29:7982–7990. doi: 10.1021/bi00486a029. [DOI] [PubMed] [Google Scholar]
- 62.Farman G.P., Allen E.J., de Tombe P.P. The role of thin filament cooperativity in cardiac length-dependent calcium activation. Biophys. J. 2010;99:2978–2986. doi: 10.1016/j.bpj.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuda N., Kajiwara H., Kurihara S. Effects of MgADP on length dependence of tension generation in skinned rat cardiac muscle. Circ. Res. 2000;86:E1–E6. doi: 10.1161/01.res.86.1.e1. [DOI] [PubMed] [Google Scholar]
- 64.Grammer J.C., Cremo C.R., Yount R.G. UV-induced vanadate-dependent modification and cleavage of skeletal myosin subfragment 1 heavy chain. 1. Evidence for active site modification. Biochemistry. 1988;27:8408–8415. doi: 10.1021/bi00422a017. [DOI] [PubMed] [Google Scholar]
- 65.Tachampa K., Wang H., de Tombe P.P. Cardiac troponin I threonine 144: role in myofilament length dependent activation. Circ. Res. 2007;101:1081–1083. doi: 10.1161/CIRCRESAHA.107.165258. [DOI] [PubMed] [Google Scholar]
- 66.Martyn D.A., Adhikari B.B., Yu L.C. Response of equatorial x-ray reflections and stiffness to altered sarcomere length and myofilament lattice spacing in relaxed skinned cardiac muscle. Biophys. J. 2004;86:1002–1011. doi: 10.1016/S0006-3495(04)74175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huxley H.E., Stewart A., Irving T. X-ray diffraction measurements of the extensibility of actin and myosin filaments in contracting muscle. Biophys. J. 1994;67:2411–2421. doi: 10.1016/S0006-3495(94)80728-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wakabayashi K., Sugimoto Y., Amemiya Y. X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys. J. 1994;67:2422–2435. doi: 10.1016/S0006-3495(94)80729-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Campbell K.S. Interactions between connected half-sarcomeres produce emergent mechanical behavior in a mathematical model of muscle. PLOS Comput. Biol. 2009;5:e1000560. doi: 10.1371/journal.pcbi.1000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurihara S., Saeki Y., Sudo N. Effects of length change on intracellular Ca2+ transients in ferret ventricular muscle treated with 2,3-butanedione monoxime (BDM) Jpn. J. Physiol. 1990;40:915–920. doi: 10.2170/jjphysiol.40.915. [DOI] [PubMed] [Google Scholar]
- 71.Sun Y.B., Lou F., Irving M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. J. Physiol. 2009;587:155–163. doi: 10.1113/jphysiol.2008.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fabiato A., Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J. Physiol. (Paris) 1979;75:463–505. [PubMed] [Google Scholar]
- 73.Chandra M., Tschirgi M.L., Campbell K.B. Interaction between myosin heavy chain and troponin isoforms modulate cardiac myofiber contractile dynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R1595–R1607. doi: 10.1152/ajpregu.00157.2007. [DOI] [PubMed] [Google Scholar]
- 74.Ford S.J., Mamidi R., Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. J. Mol. Cell. Cardiol. 2012;53:542–551. doi: 10.1016/j.yjmcc.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liao R., Wang C.K., Cheung H.C. Time-resolved tryptophan emission study of cardiac troponin I. Biophys. J. 1992;63:986–995. doi: 10.1016/S0006-3495(92)81685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dale R.E., Eisinger J., Blumberg W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.