

Abstract
The concentration of glutamate within the glutamatergic synapse is tightly regulated by the excitatory amino-acid transporters (EAATs). In addition to their primary role of clearing extracellular glutamate, the EAATs also possess a thermodynamically uncoupled Cl− conductance. Several crystal structures of an archaeal EAAT homolog, GltPh, at different stages of the transport cycle have been solved. In a recent structure, an aqueous cavity located at the interface of the transport and trimerization domains has been identified. This cavity is lined by polar residues, several of which have been implicated in Cl− permeation. We hypothesize that this cavity opens during the transport cycle to form the Cl− channel. Residues lining this cavity in EAAT1, including Ser-366, Leu-369, Phe-373, Arg-388, Pro-392, and Thr-396, were mutated to small hydrophobic residues. Wild-type and mutant transporters were expressed in Xenopus laevis oocytes and two-electrode voltage-clamp electrophysiology, and radiolabeled substrate uptake was used to investigate function. Significant alterations in substrate-activated Cl− conductance were observed for several mutant transporters. These alterations support the hypothesis that this aqueous cavity at the interface of the transport and trimerization domains is a partially formed Cl− channel, which opens to form a pore through which Cl− ions pass. This study enhances our understanding as to how glutamate transporters function as both amino-acid transporters and Cl− channels.
Introduction
Glutamate is the predominant excitatory neurotransmitter within the central nervous system and is important in development, memory, learning, and higher cognitive function (1). The concentration of glutamate within the glutamatergic synapse is tightly regulated by excitatory amino-acid transporters (EAATs). The transport of glutamate and aspartate into glia and neurones via the EAATs is coupled to the cotransport of three Na+ ions, one H+ ion, and the countertransport of one K+ ion. This coupling results in a net influx of two positive charges per transport cycle (Fig. 1 A) (2,3). The EAATs are particularly interesting because they function not only as glutamate transporters, but also as Cl− channels. This channel function requires the binding of substrate and Na+ for activation, but is thermodynamically uncoupled from substrate transport (4–6) and is mediated by molecular determinants distinct from those involved in substrate transport (7–9). In bipolar cells of the retina, Cl− permeation through EAAT5 generates hyperpolarization, which in turn prevents further glutamate release (10). In other cell types, it has been postulated that Cl− fluxes through the EAATs may play a role in ionic homeostasis (11).
Figure 1.
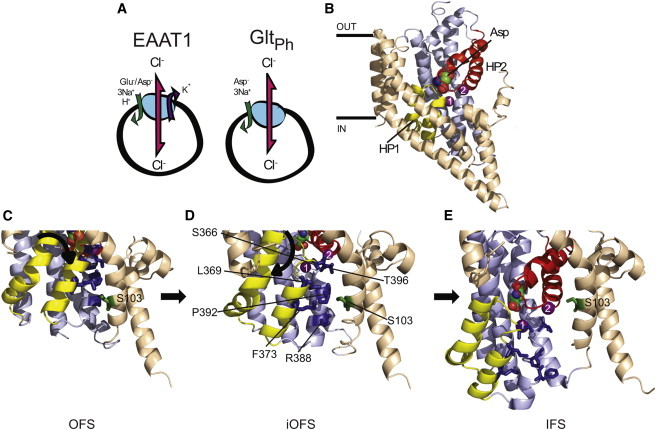
Transport stoichiometry of EAAT1 and GltPh and the putative glutamate transporter chloride channel. (A) Stoichiometry of ion-flux coupling for EAAT1 and GltPh. (B) The structure of a single protomer of GltPh in the outward-facing state (OFS; PDB:2NWX). (Space-filling representation) Bound aspartate; (purple spheres) two Na+ ions (the third Na+ ion is not shown). (Light brown) Trimerization domain; (light blue) transport domain; (yellow) HP1; (red) HP2. (C–E) Close-up of the region surrounding Ser-103 (Ser-65 in GltPh; shown in green) in the OFS (PDB:2NWX), the intermediate outward-facing state (iOFS; PDB:3V8G), and the IFS (PDB:3KBC). Residues investigated in this study are highlighted (dark blue) and labeled in panel D; TM5 has been removed for visual clarity. Figure was made using the software PYMOL (31).
In 2004, the first crystal structure of an archaeal EAAT homolog, GltPh, was determined (12). GltPh is a Na+-dependant aspartate transporter from Pyrococcus horikoshii and shares ∼37% amino-acid identity with the EAATs (13). It too has an uncoupled Cl− conductance, but is not coupled to the cotransport of H+ or the countertransport of K+ (Fig. 1 A) (14,15). Many residues that have been implicated in substrate and ion binding and/or translocation in the EAATs are conserved throughout the family (16–20). GltPh exists as a trimer, where three identical protomers come together to form an extracellular facing bowl-like structure that sits within the cell membrane. Each protomer consists of eight α-helical transmembrane domains (TMs 1–8) and two helical hairpin loops (HP1 and HP2), which cradle the substrate binding site, occluding it from both the intracellular and extracellular spaces (Fig. 1 B). The protomer can be divided into two functional domains: the trimerization domain (TM1, TM2, TM4 and TM5), which is responsible for maintaining interactions between protomers; and the transport domain (TM3, TM6, HP1, TM7, HP2, and TM8), which contains the binding sites for aspartate and Na+ ions (13,21). Each protomer has the ability to independently transport substrate and support a Cl− conductance (15,22–24).
Since 2004, several crystal structures of GltPh, at different stages of the transport cycle, have been solved (12,13,21,25). These structures demonstrate that transport of aspartate via GltPh, and presumably the EAATs, involves a large inward movement of the transport domain relative to the trimerization domain (Fig. 1, C–E). However, what these structures do not reveal is the location of the Cl− channel, which suggests that this channel is only transiently formed during the transport cycle. Several residues within TM2, TM5, and TM7 have been demonstrated in previous studies to be involved in EAAT-mediated Cl− permeation (26–29). The most interesting residue investigated to date is Ser-103. In 2004, we proposed that this serine residue directly interacts with permeating Cl− ions and may form part of the selectivity filter of the EAAT1 Cl− channel (29). Additionally, mutation of the equivalent residue in GltPh (Ser-65) reduces Cl− permeation (15). A recent structure of GltPh captures one of the protomers in a novel state termed the “intermediate outward facing state” (iOFS) (25). In this structure, we can see that these previously identified residues, including Ser-103, line a cavity located at the interface of the trimerization and transport domains (Fig. 1 D). This cavity is predicted to be accessible to the aqueous milieu, because cysteine residues in this region have been shown to be accessible to charged sulfhydryl reactive reagents (29). Also, molecular-dynamics simulations reveal that water can permeate the iOFS structure between the transport and trimerization domains in the vicinity of this cavity that is lined by HP1 and TM7 (30). In this study, we identify six residues in HP1 and TM7 that influence Cl− permeation through EAAT1. We propose that this intracellular facing cavity is a partially formed Cl− channel, and that during transitions of the transport domain toward the cytoplasm upon substrate transport, this cavity opens to form a channel through which Cl− ions permeate.
Materials and Methods
Site-directed mutagenesis
EAAT1 cDNA was subcloned into the plasmid oocyte transcription vector. Site-directed mutagenesis was performed with Velocity DNA polymerase (Bioline, Sydney, Australia) and oligonucleotide primers synthesized by Sigma Genosys (Sydney, Australia). The sequences of cDNA products were confirmed by the Australian Genome Research Facility (Sydney, Australia). Wild-type (WT) EAAT1 and mutant cDNAs were linearized with Spe1 (New England Biolabs, Ipswich, MA) and transcribed to cRNA by T7 RNA polymerase using the mMessage mMachine kit (Ambion, Austin, TX).
Harvesting and preparing oocytes
All chemicals were purchased from Sigma Chemical (Sydney, Australia) unless otherwise stated. Xenopus laevis frogs were obtained from NASCO (Fort Atkinson, WI). Stage V oocytes were harvested from Xenopus laevis, as previously described in Poulsen and Vandenberg (32) with all surgical procedures in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Defoliculated stage V oocytes were injected with 4.6 ng of cRNA (Drummond Nanoject; Drummond Scientific, Broomall, PA) and subsequently incubated at 16–18°C in standard frog Ringer’s solution (ND96: 96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, and 5 mM hemi-Na+-HEPES, pH 7.5) supplemented with 50 μg/mL gentamycin, 2.5 mM sodium pyruvate, 50 μg/mL tetracyclin, and 0.5 mM theophylline.
Electrophysiology
Two-to-four days after injections, current recordings were made using the two-electrode voltage-clamp technique with a Geneclamp 500 amplifier (Axon Instruments, Foster City, CA) interfaced with a PowerLab 2/20 chart recorder (ADInstruments, Sydney, Australia) and a Digidata 1322A (Axon Instruments) used in conjunction with LABCHART software (ADInstruments).
Current-voltage relationships for substrate-elicited conductances were determined by measuring substrate-elicited currents during 200-ms voltage pulses from −30 mV to membrane potentials ranging from −100 to +60/+80 mV at 10 mV steps. Background currents were eliminated by subtracting currents in the absence of substrate from substrate-elicited currents at corresponding membrane potentials. Current-voltage relationships were also recorded in NO3–-containing buffer (96 mM NaNO3, 2 mM KNO3, 1 mM Mg(NO3)2⋅6H2O, 1.8 mM Ca(NO3)2, and 5 mM hemi-Na+-HEPES, pH 7.5) All recordings were made with a bath grounded via a 3 M KCl/agar bridge linking to a 3 M KCl reservoir.
Substrate concentration responses were measured in ND96 and fitted by least-squares as a function of current at −60 mV (I) to a derivation of the Michaelis-Menten equation:
| (1) |
where Imax is the maximal current, [S] is the substrate concentration, and K0.5 is the concentration of substrate required to produce the half-maximal current.
Uptake of 3H-L-aspartate (PerkinElmer Life Sciences, Melbourne, Australia) under voltage-clamp was measured in oocytes expressing WT and mutant EAAT1 transporters. Oocytes were clamped at −100 mV and incubated with 30 μM 3H-L-aspartate for 1 min, then washed for 3 min with ND96. Oocytes were then removed from the bath and lysed with 1 M NaOH + SDS (1% w/v). After the addition of scintillant (Ultima Gold; Packard, Prospect, CT), counting was performed using a Microbeta TriLux scintillation counter (PerkinElmer Life Sciences).
Relative anion permeabilities (PX/PCl; X denoting Br−, I−, or NO3−), were determined by measuring the reversal potentials (Erev; Eq. 2) of substrate-elicited conductances in recording solution containing either 10 or 30 mM Na+ salts of Cl−, Br−, I−, or NO3− and either 86 or 66 mM Na+ gluconate (to obtain 96 mM Na+), 2 mM K+ gluconate, 1 mM Mg2+ gluconate, 1.8 mM Ca2+ gluconate, and 5 mM hemi-Na+-HEPES. A quantity of 1-mM permeant anion concentration was used to minimize anion loading of oocytes, which, unchecked, could affect reversal potential measurements. However, for some mutant transporters, reversal potential measurements could not be obtained in 10-mM permeant anion, in which case a permeant anion concentration of 30 mM was employed. Reversal potential measurements were then substituted into the Goldman-Hodgkin-Katz equation:
| (2) |
with R being the gas constant, T as the temperature in degrees Kelvin, z as the charge of the anion, and F as the Faraday’s constant. We assumed [Cl−]in = 41 mM (33) to calculate PX/PCl.
To examine the EAAT1 specific Na+-activated anion conductance, the nontransportable blocker TBOA (DL-threo-β-benzyloxyaspartate; Tocris, Bristol, UK), which blocks the Na+-activated anion conductance, was used. Steady-state currents were measured in the presence of 250 μM TBOA during 200-ms voltage pulses from −30 mV to membrane potentials ranging from −100 to +60 mV at 10-mV steps and were subtracted from currents in the absence of TBOA at corresponding membrane potentials.
Radiolabeled uptake
Uptake of 3H-L-glutamate (PerkinElmer Life Sciences) into oocytes expressing WT and mutant EAAT1 transporters was measured. Oocytes were incubated in ND96 containing 10 μM 3H-L-glutamate for 10 min. Uptake was terminated by washing oocytes three times in ice-cold ND96 followed by lysis and counting, as previously described.
Data analysis
Analysis of kinetic data was conducted using GRAPHPAD PRISM 5.0 (GraphPad Software, La Jolla, CA). All values presented are the mean ± SE of at least three cells. Analyses of variance were performed with a Dunnett’s post-hoc test. The p-values < 0.05 were interpreted as statistically significant and are indicated in figures as ∗, p < 0.05; ∗∗, p < 0.01; and ∗∗∗, p < 0.001.
The coupled transport component of the aspartate-elicited conductance presented in Fig. 2 A was modeled to an exponential function with an e-fold change in rate per 41 mV (29) using
| (3) |
where I is the current at a given membrane potential (V) and k is a normalization constant. The uncoupled Cl− component is assumed for simplicity to be Ohmic and to reverse at −22 mV (5,33). The net aspartate-activated conductance was then generated by the addition of these two components.
Results
Mutagenesis
The aim of this study was to investigate the role of residues in HP1 and TM7 in EAAT1-mediated Cl− permeation. These regions line an aqueous cavity located at the interface of the transport and trimerization domains (Fig. 1 D). This cavity is also lined by residues in TM2 and TM5 that have been shown to influence Cl− permeation through the EAATs and GltPh (15,26–29). Residues in HP1 (Ser-366, Leu-369,and Phe-373) and TM7 (Arg-388, Pro-392, and Thr-396) that line this cavity were mutated to hydrophobic residues including alanine, valine, and leucine to probe their involvement in EAAT1-mediated Cl− conduction. It is notable that these residues are conserved among GltPh, EAAT1, and other members of the glutamate transporter family with the exception of Phe-373, which is a methionine in GltPh and ASCT1, and Arg-388, which is a serine in GltPh.
Transporter activity
To examine the effects of the mutations on glutamate transporter function, 3H-L-glutamate uptake by oocytes expressing WT and mutant EAAT1 transporters was measured. All mutant transporters exhibit 3H-L-glutamate uptake levels above background (0.8 ± 0.2 fmol/oocyte/minute; Table 1), indicating an ability for all to transport glutamate. 3H-L-glutamate uptake by the L369A mutant transporter was ∼19% of WT EAAT1 levels (Table 1). In addition, the substrate-elicited currents of this transporter were below the measurable range, and thus no electrophysiological analysis of this mutant transporter could be performed. For WT EAAT1, the apparent affinities for L-glutamate and L-aspartate are 31 ± 2 μM and 18 ± 3 μM, respectively (Table 1). There was little or no change in apparent substrate affinities for mutant transporters compared to WT EAAT1 (Table 1), indicating that residues in this region are unlikely to be directly involved in substrate binding and transport.
Table 1.
Substrate transport and electrophysiological properties of WT and mutant EAAT1 transporters
| 3H-L-glutamate uptake (fmol/ oocyte/min) |
K0.5 (μM) |
Erev (mV) |
I at +60 mV (nA) |
||||||
|---|---|---|---|---|---|---|---|---|---|
| L-glutamate | L-aspartate | L-glutamate |
L-aspartate |
L-glutamate | L-aspartate | ||||
| Cl− | NO3− | Cl− | NO3− | ||||||
| EAAT1 | 113 ± 7 | 31 ± 2 | 18 ± 3 | 24 ± 1 | −30 ± 4 | 7 ± 1 | −50 ± 1 | 640 ± 80 | 1100 ± 100 |
| S366A | 112 ± 2 | 32 ±4 | 34 ± 5 | 18 ± 2 | −31 ± 1 | 7 ± 2 | −39 ± 1c | 1100 ± 80b | 1500 ± 10 |
| S366V | 77 ± 3 | 16 ± 2 | 14 ± 2 | 14 ± 1 | −44 ± 2a | 11 ± 1 | −49 ± 2 | 480 ± 50 | 690 ± 70 |
| L369A | 21 ± 1 | ND | ND | ND | ND | ND | ND | ND | ND |
| L369V | 84 ± 4 | 25 ± 2 | 10 ± 2 | 50 ± 7c | −18 ± 3a | 33 ± 5c | −36 ± 1c | 700 ± 100 | 1000 ± 100 |
| F373V | 82 ± 2 | 15 ± 2 | 13 ± 2 | −1 ± 3c | −35 ± 2 | −8 ± 1c | −39 ± 1c | 1550 ± 90c | 1700 ± 100 |
| R388A | 89 ± 1 | 25 ± 3 | 8 ± 2 | 16 ± 2 | −51 ± 1c | 10 ± 1 | −51 ± 1 | 2400 ± 200c | 2700 ± 300c |
| R388L | 82 ± 2 | 12 ± 1 | 14 ± 2 | 22 ± 3 | −50 ± 3b | 19 ± 3 | −50 ± 1 | 1600 ± 300c | 2000 ± 300a |
| P392A | 78 ± 7 | 8 ± 1 | 4.7 ± 0.6 | 59 ± 4c | −46 ± 3c | 11 ± 2 | −66 ± 2c | 600 ± 50 | 1340 ± 70 |
| P392V | 85 ± 3 | 23 ± 3 | 20 ± 3 | 0 ± 3c | −56 ± 3c | −13 ± 2c | −60 ± 2c | 4100 ± 300c | 6700 ± 500c |
| T396A | 90 ± 2 | 27 ± 2 | 9 ± 2 | 64 ± 2c | −8 ± 2c | 58 ± 3c | −14 ± 1c | 39 ± 3a | 67 ± 11c |
| T396V | 73 ± 4 | 40 ± 2 | 14.7 ± 0.4 | 44 ± 3b | −18 ± 2a | 37 ± 4c | −38 ± 1c | 190 ± 14a | 300 ± 20b |
3H-L-glutamate uptake was performed for 10 min with oocytes expressing WT and mutant EAAT1 transporters. Background (uninjected oocytes) 3H-L-glutamate uptake of 0.8 fmol/oocyte/min was subtracted from the data presented. Apparent substrate affinity (K0.5) values were determined from concentration responses conducted at −60 mV. Reversal potentials (Erev) were obtained from 30 μM substrate-elicited currents in 96 mM Cl−- or NO3−-based buffers. Raw current (I) at +60 mV in NO3−-containing buffer are shown for comparison. ND indicates that no electrophysiological data could be obtained.
Data shown represents the mean ± SE (n ≥ 3) with p < 0.05 compared to WT EAAT1.
Data shown represents the mean ± SE (n ≥ 3) with p < 0.01 compared to WT EAAT1.
Data shown represents the mean ± SE (n ≥ 3) with p < 0.001 compared to WT EAAT1.
Alterations in anion conductance
When oocytes expressing WT EAAT1 are exposed to L-glutamate or L-aspartate under physiological conditions, a net current that is comprised of a coupled transport component and an uncoupled Cl− component is generated (Fig. 2 A). The coupled transport component is inwardly directed at membrane potentials up to +60 mV, and for L-aspartate can be fit to an exponential function with an e-fold change in rate per 41 mV (29). On the other hand, the uncoupled Cl− component has a reversal potential of ∼−22 mV (4,5). Therefore, at negative membrane potentials (e.g., −100 mV), the net current produced is a combination of both components, and at positive membrane potentials, (e.g., +60 mV), the majority of current is due to the uncoupled Cl− component. The reversal potential of the net current will be determined by the relative proportions of the two components. Because all of the mutant transporters have comparable transport properties to that of WT EAAT1 (Table 1), any changes in reversal potential or the current amplitude at +60 mV will reflect changes in anion conductance properties.
Figure 2.
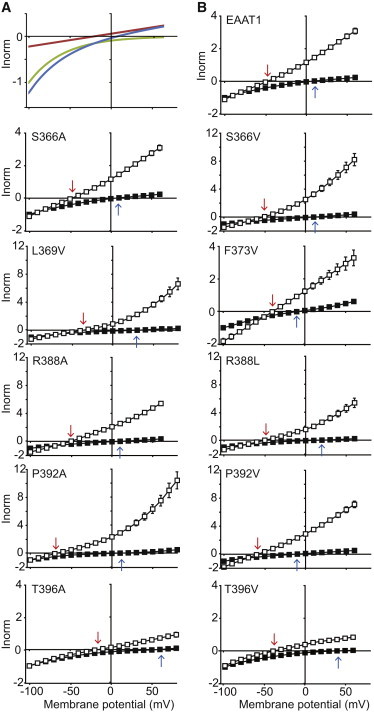
Substrate-activated current-voltage relationships for WT and mutant EAAT1 transporters. (A) Schematic current-voltage relationship showing the two components of the substrate-activated conductance of EAAT1 expressed in oocytes. The net aspartate-activated conductance (blue/solid line) is composed of the coupled transport component (green/dotted line) and the uncoupled Cl− component (red/dashed line). See Materials and Methods. (B) Current-voltage relationships for wild-type EAAT1 and mutant transporters elicited by 30 μM L-aspartate in either 96 mM Cl−-based buffer (solid squares) or 96 mM NO3−-based buffer (open squares). Currents were normalized (Inorm) to the substrate-elicited current at −100 mV in Cl−. (Arrows) Reversal potential of the substrate-activated conductance in Cl−-based buffer (blue/open arrow) and NO3−-based buffer (red/solid arrows). Reversal potentials are also listed in Table 1. Note that different scales are used for various mutant transporters. Where error bars cannot be seen, they lie within the symbol. Data shown represents the mean ± SE (n ≥ 3). To see this figure in color, go online.
Although Cl− is the physiological anion, a range of anions can permeate through EAAT1 (6). NO3− is ∼10 times more permeant through this channel than Cl−, and thus, when extracellular Cl− is replaced with NO3−, the substrate-elicited current at +60 mV is significantly larger and the reversal potential is shifted to a membrane potential that is more negative. For mutations that increase anion conductance, the reversal potentials will be shifted to potentials that are more negative and current amplitudes at +60 mV will be greater when compared to WT EAAT1. Conversely, for mutations that decrease the anion conductance, reversal potentials will be shifted to potentials that are more positive, with smaller current amplitudes at +60 mV compared to WT EAAT1. We have measured current-voltage relationships in both Cl− and NO3−-containing buffers to examine anion conductance properties. Data presented in Fig. 2 B is normalized to the current generated at −100 mV for each transporter. This normalization procedure accounts for any variability in expression levels of the different transporters, and allows us to measure changes in the anion conductance as a proportion of the total substrate-activated conductance.
Analysis of current-voltage relationships for mutant transporters revealed several alterations in reversal potential measurements and current amplitudes at +60 mV compared to WT EAAT1 (Table 1, Figs. 2 B and 3 A). For example, the reversal potential of the substrate-elicited conductance for T396A shifted to a more positive potential and the current amplitude at +60 mV was reduced, indicating that this mutation decreased EAAT1-mediated anion conduction. On the other hand, the reversal potential of the P392V conductance was shifted to a membrane potential that was more negative, and the current at +60 mV was increased, demonstrating that this mutation increased anion conduction. Interestingly, for several mutant transporters, these shifts in reversal potentials and changes in current amplitudes at +60 mV were either anion-dependent and/or substrate-dependent (Table 1, Figs. 2 B and 3 A). This data suggests that these mutations affect the relative permeabilities of Cl− and NO3−, and reveal that these mutations alter the aspartate- and glutamate-activated channels differently, highlighting the subtle differences in the properties of the channel when it is activated by glutamate or aspartate (29).
Figure 3.
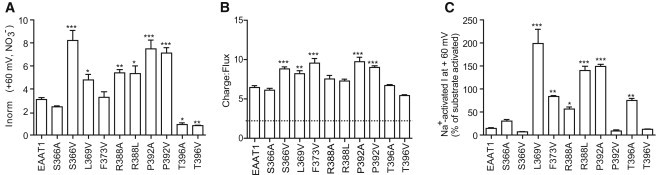
Anion conductance properties of WT and mutant EAAT1 transporters. (A) Currents at +60 mV elicited by 30 μM L-aspartate were measured in 96 mM NO3−-based buffer and normalized to the substrate-elicited current at −100 mV in 96 mM NO3−-based buffer. (B) 30 μM 3H-L-aspartate in 96 mM Cl−-based buffer was applied for 1 min to oocytes expressing WT and mutant transporters clamped at −100 mV. Charge transfer and 3H-L-aspartate uptake were measured and used to obtain a C/F ratio. (Dashed line) C/F ratio of 2, which is the charge transfer associated with coupled substrate transport; the remainder results from the uncoupled Cl− component. (C) Na+-activated anion currents blocked by 250 μM TBOA at +60 mV were measured in 96 mM NO3−-based buffer and are shown as a percentage of the current elicited by 300 μM L-glutamate at +60 mV in 96 mM NO3−-based buffer. Data shown represents the mean ± SE (n ≥ 3) with ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared to WT EAAT1.
Substrate transport via EAAT1 results in the net influx of two positive charges per transport cycle (Fig. 1 A) (3). Any additional charge transfer observed during transport is due to the uncoupled anion conductance. We can measure the amount of charge moving through the transporter by determining the charge/flux (C/F), where we monitor the current (charge) generated during the application of 3H-L-aspartate. WT EAAT1 has a C/F ratio of 6.9 ± 0.5 at −100 mV measured in a Cl− buffer (Fig. 3 B). This means the number of charges transferred per molecule of aspartate transported is ∼7 and thus ∼9 Cl− ions exit the cell with every transport cycle (as two positive charges enter per substrate transport cycle). Among the various EAAT1 mutant transporters analyzed in this study, S366V, L369V, F373V, P392A, and P392V demonstrated significant increases in C/F ratios compared to WT EAAT1. Additionally, T396V presented a reduced C/F ratio of 5.4 ± 0.1 mV (Fig. 3 B). Although this reduction is not statistically significant, it represents approximately seven Cl− ions exiting per transport cycle as opposed to the nine observed in WT EAAT1. As we have previously demonstrated these mutant transporters have similar transport properties to WT EAAT1 (Table 1), these changes in C/F ratios are indicative of a change in the number of Cl− ions passing through the channel with each transport cycle.
For most mutant transporters where there is an increase in the C/F ratio there is also an increase in current at +60 mV (e.g., S366V, L369V, P392A, and P392V; Fig. 3, A and B). However, it should be noted that the relationship does not always hold due to differences in experimental conditions and properties of the mutant transporters. The C/F experiment is conducted in a Cl− buffer on cells that are voltage-clamped at −100 mV. These conditions give us the best opportunity to measure the uncoupled anion component while still effectively measuring substrate transport. In contrast, changes in the current measured at +60 mV in a NO3−-based buffer allow us to isolate changes in the uncoupled anion component. In the cases of T396A, T396V, R388A, and R388L, significant changes in current at +60 mV were observed without corresponding changes in the C/F ratio. On the other hand, F373V displays a significant increase in the C/F ratio but no change in the current at +60 mV. These differential results in the NO3−- or Cl−-based buffers may be explained by changes in the relative permeability of NO3− compared to Cl− within these mutant transporters or when compared to wild-type EAAT1 (Table 1 and 2). In addition, the mutations may have changed the voltage-dependence of the activation of the uncoupled anion conductance, which could also contribute to the variability observed in the different experimental conditions.
Table 2.
Relative anion permeabilities of WT and mutant EAAT1 transporters
| PBr−/PCl− | PI−/PCl− | PNO3−/PCl− | |
|---|---|---|---|
| EAAT1 | 2.8 ± 0.2 | 11.4 ± 0.5 | 13.1 ± 0.4 |
| S366A | 3.7 ± 0.4 | 28 ± 3c | 27 ± 2c |
| S366V | 1.64 ± 0.04b | 8.5 ± 0.5 | 12.5 ± 8 |
| L369A | ND | ND | ND |
| L369V | 4.0 ± 0.7b | 17.5 ± 0.9 | 19 ± 1 |
| F373V | 2.8 ± 0.1 | 13.7 ± 0.7 | 18.3 ± 0.7 |
| R388A | 7.05 ± 0.09c | 39 ± 1c | 38 ± 2c |
| R388L | 4.76 ± 0.05c | 24.0 ± 0.6c | 25.0 ± 0.9b |
| P392A | 11.5 ± 0.6c | 74 ± 5c | 82 ± 7c |
| P392V | 2.51 ± 0.07 | 9.2 ± 0.5 | 11 ± 1 |
| T396A | 1.20 ± 0.05c | 3.6 ± 0.2b | 4.1 ± 0.3b |
| T396V | 1.38 ± 0.06c | 4.0 ± 0.2a | 5.0 ± 0.3a |
Reversal potentials (Erev) of 100 μM L-aspartate-elicited currents were obtained in buffers containing either 10 mM or 30 mM permeant anion salts. ND indicates that no electrophysiological data could be obtained. Relative anion permeabilities were calculated using the Goldman-Hodgkin-Katz equation (Eq. 2).
Data shown represents the mean ± SE (n ≥ 3) with p < 0.05 compared to WT EAAT1.
Data shown represents the mean ± SE (n ≥ 3) with p < 0.01 compared to WT EAAT1.
Data shown represents the mean ± SE (n ≥ 3) with p < 0.001 compared to WT EAAT1.
Relative anion permeabilities
The alterations in the C/F ratios, reversal potentials, and current amplitudes at +60 mV described above indicate an altered capacity for these mutant transporters to support the anion conductance. To further probe the role of these residues in anion conduction through EAAT1, we investigated relative anion permeabilities. The minimum pore diameter of the EAAT1 anion channel has been estimated to be 5 Å and permits the permeation of a range of anions. These anions have a permeability sequence of NO3− > I− > Br− > Cl−, where NO3− is the most permeant anion (6). Changes in the order or magnitude of this sequence as a result of mutagenesis suggest changes in interactions between the anions and protein (29). When measuring the relative permeabilities of Br−, I−, and NO3− compared to that of Cl−, we used L-aspartate as the substrate because the proportion of current due to the anion conductance is greater for L-aspartate than for L-glutamate, and thus yields more-consistent data. Significant changes in the relative permeabilities of various anions were observed for several mutant transporters (Table 2). For example, in R388A, R388L, and P392A, the relative permeabilities for Br−, I−, and NO3− were significantly increased compared to WT EAAT1 (Table 2). Conversely, in T396A and T396V, the relative permeabilities of Br−, I−, and NO3− were significantly decreased compared to WT EAAT1 (Table 2). Because no changes in the order of the relative anion permeability sequence were observed, we cannot confirm that these residues form part of the selectivity filter. However, these results support the hypothesis that residues within HP1 and TM7 are involved in anion permeation and/or channel open time.
The Na+-activated anion conductance
In addition to the substrate-activated anion conductance, the EAATs also permit a leak anion conductance. This leak requires only Na+ binding to be activated and thus is referred to as the Na+-activated anion conductance (6,11). In WT EAAT1, this Na+-activated anion conductance has a magnitude of 15 ± 1% of the anion conductance activated by saturating glutamate concentrations (Fig. 3 C). In oocytes expressing some mutant transporters, we noticed that whole-cell conductances in the absence of substrate were considerably greater compared to WT EAAT1, indicating that these mutations may also alter the nature of this Na+-activated anion conductance. To investigate this conductance, the EAAT1 competitive blocker, TBOA, was employed. TBOA binds to the substrate-binding site and traps the transporter in an open-to-out conformation (13), and is known to block the Na+-activated anion conductance (29).
This provides a tool to isolate this conductance from other conductances endogenous to the oocyte. In mutant transporters where the side chain was smaller than that of the native residue in WT EAAT1 (L369V, F373V, R388A, R388L, P392A, and T396A), the amplitude of Na+-activated anion conductance was significantly increased (Fig. 3 C). In some instances, the Na+-activated anion conductance was even greater than or equal to the substrate-activated anion conductance. On the other hand, in mutant transporters where the side chain was similar in size to that of the native residue in WT EAAT1 (S366A, S366V, P392V, and T396V), no significant changes in Na+-activated anion conductances were observed (Fig. 3 C). These results suggest that the bulk of residues within the cavity between the transport and trimerization domains influence the amplitude of the Na+-activated anion conductance.
Discussion
Several crystal structures of GltPh at different stages of the transport cycle have been solved (12,13,21,25). Although these structures have provided a comprehensive understanding of substrate transport by GltPh and the EAATs, the structural basis for anion conductance via these transporters is yet to be fully understood. Here we have used site-directed mutagenesis to demonstrate that residues in HP1 and TM7, which line a cavity at the interface of the transport and trimerization domains of EAAT1, affect anion permeation with little or no effect on glutamate transport properties. Given the strong sequence conservation of these residues in this region among members of the glutamate transporter family, it is likely that this domain interface serves a similar role across all members of the family. Although there are subtle changes in apparent substrate affinities for some of the mutant transporters (Table 1) that can result in shifts in reversal potential measurements and current amplitudes at +60 mV, we see no correlation between these measurements and the apparent substrate affinities observed. In addition, changes in apparent substrate affinity cannot account for the changes in relative anion permeabilities observed (Table 2).
Previous studies by our group and others (15,26–29) have identified residues involved in glutamate-transporter-mediated anion permeation. When we map these residues, and the believed-novel residues identified in this study, onto the iOFS and inward-facing state (IFS) of GltPh (Fig. 4 A), a hypothesis for anion channel formation emerges. As the transport domain shuttles toward the cytoplasm during transport, a structural state between the iOFS and the IFS will arise where residues involved in anion permeation will align (Fig. 4 A). Taken together with the high sequence conservation of these residues and that this region has been shown to line an anion-selective aqueous cavity in GltPh and EAAT1 (25,29,30), we propose that residues in TM2, TM5, HP1, and TM7 that line this domain interface contribute to the formation of an anion channel during substrate transport. A recent study by Shabaneh et al. (34) demonstrated that cross-linking between introduced cysteines designed to trap the transporter in the iOFS decreased glutamate transport but still allowed activation of the anion conductance. Because there is not an open pore through the protein in the iOFS structure (25), we propose that some further movement of the transport domain is required to activate the Cl− conductance, although the extent and exact nature of this movement remains to be resolved (Fig. 4 A).
Figure 4.
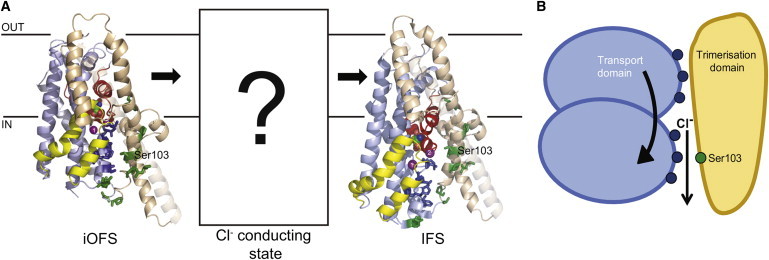
The transport domain forms both the gate and pore of the glutamate transporter anion channel. (A) The proposed anion-conducting state arises between the iOFS (PDB:3V8G) and the IFS (PDB:3KBC) of GltPh (colored as in Fig. 1). (Dark blue) Residues investigated in this study. (Green) Those previously established to be involved in the EAAT-mediated anion conductance including Gln-93, Pro-98, Ser-102, Ser-103, Thr-106, Asp-112, Lys-114, Asp-272, Lys-384, and Arg-385 (26–29). Serine 103 is labeled for reference. TM5 is shown in partial transparency for visual clarity. Figure was made using the software PYMOL (31). (B) We propose that the opening of the EAAT1 anion channel is associated with large conformational changes of the transport domain (light blue) relative to the trimerization domain (light brown). Upon movement (indicated by the curved arrow) of the transport domain during substrate translocation, the residues identified in this study (represented by the dark blue circles) line the anion channel pore, where they are involved in anion permeation. The trimerization domain, which contains Ser-103 (green circle), remains static during the transport process.
In comparison with traditional anion channels, the magnitude of Cl− conduction through EAAT1 is relatively small; from noise analysis it has been estimated to be ∼1 fS (6). For this reason, it is not possible to perform single-channel recordings, even in the presence of the highly permeant anion NO3−. Without this tool, it is difficult to distinguish between effects on channel gating and anion permeation as both effects prevail as changes in anion conductance properties. From the results obtained in this study, as well as other mutagenesis, crystallographic, and molecular-dynamic studies (12,13,15,21,25–29,35,36), we propose that the mechanism of gating of the EAAT1 anion conductance may be different from traditional channel gating, which usually relies on the movement or rotation of a few residues (Fig. 4 B). In this proposed mechanism, the anion channel pore is located at the interface of the trimerization and transport domains. The gating of this channel is associated with large conformational changes of the transport domain initiated during substrate translocation. This is supported by previous studies that have demonstrated a close relationship between the activation kinetics of substrate translocation and the uncoupled anion conductance (37,38). Because of this large downward movement of the transport domain, it is likely that the region forming the gate translates to form part of the anion permeation pathway. For this reason, when discussing effects of mutagenesis on gating and permeation of the EAAT1 anion channel, use of the two terms independently becomes somewhat redundant as the two components may not be separate entities, but rather inherently linked.
The relative anion permeability sequence of the Na+-activated anion conductance is I− > NO3− > Br− > Cl−, whereas that of the substrate-activated anion conductances is NO3− > I− > Br− > Cl− (29). This variance indicates that substrate binding alters the interactions between anions and the channel, and thus the channels for the two conductances are not identical. Until now it has been unclear whether the Na+-activated anion channel is entirely distinct from the substrate-activated channel, or if they are subtly distinct conformations of the same channel (11). Here we have identified a series of residues in HP1 and TM7 that are involved in both Na+- and substrate-activated anion permeation. So, although the permeation pathways for the two conductances are not identical, this observations supports the hypothesis that both the Na+- and substrate-activated anion conductances are mediated by distinct conformations of the same channel.
Conclusion
Mutagenesis, crystallographic, and MD studies are continuously expanding our understanding of glutamate transporter dynamics. In this report, we have identified a series of residues located in an intracellular facing cavity at the interface between the transport and trimerization domains that are involved in anion permeation. We propose a mechanism for anion channel formation in which the downward movement of the transport domain during substrate transport allows this intracellular-facing cavity to open and form a pore through which anions can permeate. This study provides greater clarity of the structural basis that enables glutamate transporters to carry out their dual functions as both amino-acid transporters and anion channels.
Acknowledgments
The authors thank Cheryl Handford for her expert technical assistance and all who maintain the University of Sydney Xenopus laevis colony. We thank members of the Transporter Biology Group at the University of Sydney, O. Boudker and G. Verdon, for helpful discussions.
This work was supported by a National Health and Medical Research Council (Australia) project grant No. APP1048784. R.J.C. is supported by an Australian Postgraduate Award, a University of Sydney Alumni scholarship, and a John Lamberton scholarship. R.M.R. is supported by a National Health and Medical Research Council (Australia) Career Development Fellowship (No. 571093).
References
- 1.Vandenberg R.J., Ryan R.M. Mechanisms of glutamate transport. Physiol. Rev. 2013;93:1621–1657. doi: 10.1152/physrev.00007.2013. [DOI] [PubMed] [Google Scholar]
- 2.Levy L.M., Warr O., Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
- 4.Fairman W.A., Vandenberg R.J., Amara S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 5.Wadiche J.I., Amara S.G., Kavanaugh M.P. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 6.Wadiche J.I., Kavanaugh M.P. Macroscopic and microscopic properties of a cloned glutamate transporter/chloride channel. J. Neurosci. 1998;18:7650–7661. doi: 10.1523/JNEUROSCI.18-19-07650.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borre L., Kavanaugh M.P., Kanner B.I. Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J. Biol. Chem. 2002;277:13501–13507. doi: 10.1074/jbc.M110861200. [DOI] [PubMed] [Google Scholar]
- 8.Ryan R.M., Vandenberg R.J. Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J. Biol. Chem. 2002;277:13494–13500. doi: 10.1074/jbc.M109970200. [DOI] [PubMed] [Google Scholar]
- 9.Seal R.P., Shigeri Y., Amara S.G. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc. Natl. Acad. Sci. USA. 2001;98:15324–15329. doi: 10.1073/pnas.011400198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veruki M.L., Mørkve S.H., Hartveit E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006;9:1388–1396. doi: 10.1038/nn1793. [DOI] [PubMed] [Google Scholar]
- 11.Vandenberg R.J., Huang S., Ryan R.M. Slips, leaks and channels in glutamate transporters. Channels (Austin) 2008;2:51–58. doi: 10.4161/chan.2.1.6047. [DOI] [PubMed] [Google Scholar]
- 12.Yernool D., Boudker O., Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- 13.Boudker O., Ryan R.M., Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- 14.Ryan R.M., Compton E.L., Mindell J.A. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J. Biol. Chem. 2009;284:17540–17548. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryan R.M., Mindell J.A. The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 2007;14:365–371. doi: 10.1038/nsmb1230. [DOI] [PubMed] [Google Scholar]
- 16.Bendahan A., Armon A., Kanner B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 2000;275:37436–37442. doi: 10.1074/jbc.M006536200. [DOI] [PubMed] [Google Scholar]
- 17.Kavanaugh M.P., Bendahan A., Kanner B.I. Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J. Biol. Chem. 1997;272:1703–1708. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- 18.Rosental N., Kanner B.I. A conserved methionine residue controls the substrate selectivity of a neuronal glutamate transporter. J. Biol. Chem. 2010;285:21241–21248. doi: 10.1074/jbc.M109.087163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teichman S., Kanner B.I. Aspartate-444 is essential for productive substrate interactions in a neuronal glutamate transporter. J. Gen. Physiol. 2007;129:527–539. doi: 10.1085/jgp.200609707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandenberg R.J., Arriza J.L., Kavanaugh M.P. Constitutive ion fluxes and substrate binding domains of human glutamate transporters. J. Biol. Chem. 1995;270:17668–17671. doi: 10.1074/jbc.270.30.17668. [DOI] [PubMed] [Google Scholar]
- 21.Reyes N., Ginter C., Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grewer C., Balani P., Rauen T. Individual subunits of the glutamate transporter EAAC1 homotrimer function independently of each other. Biochemistry. 2005;44:11913–11923. doi: 10.1021/bi050987n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koch H.P., Brown R.L., Larsson H.P. The glutamate-activated anion conductance in excitatory amino acid transporters is gated independently by the individual subunits. J. Neurosci. 2007;27:2943–2947. doi: 10.1523/JNEUROSCI.0118-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leary G.P., Stone E.F., Kavanaugh M.P. The glutamate and chloride permeation pathways are colocalized in individual neuronal glutamate transporter subunits. J. Neurosci. 2007;27:2938–2942. doi: 10.1523/JNEUROSCI.4851-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verdon G., Boudker O. Crystal structure of an asymmetric trimer of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 2012;19:355–357. doi: 10.1038/nsmb.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hotzy J., Machtens J.P., Fahlke C. Neutralizing aspartate 83 modifies substrate translocation of excitatory amino acid transporter 3 (EAAT3) glutamate transporters. J. Biol. Chem. 2012;287:20016–20026. doi: 10.1074/jbc.M112.344077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang S., Vandenberg R.J. Mutations in transmembrane domains 5 and 7 of the human excitatory amino acid transporter 1 affect the substrate-activated anion channel. Biochemistry. 2007;46:9685–9692. doi: 10.1021/bi700647f. [DOI] [PubMed] [Google Scholar]
- 28.Kovermann P., Machtens J.P., Fahlke C. A conserved aspartate determines pore properties of anion channels associated with excitatory amino acid transporter 4 (EAAT4) J. Biol. Chem. 2010;285:23676–23686. doi: 10.1074/jbc.M110.126557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryan R.M., Mitrovic A.D., Vandenberg R.J. The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J. Biol. Chem. 2004;279:20742–20751. doi: 10.1074/jbc.M304433200. [DOI] [PubMed] [Google Scholar]
- 30.Li J., Shaikh S.A., Tajkhorshid E. Transient formation of water-conducting states in membrane transporters. Proc. Natl. Acad. Sci. USA. 2013;110:7696–7701. doi: 10.1073/pnas.1218986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrodinger L.L.C. The PYMOL Molecular Graphics System, Ver. 1.3r1. 2010. http://www.pymol.org/
- 32.Poulsen M.V., Vandenberg R.J. Niflumic acid modulates uncoupled substrate-gated conductances in the human glutamate transporter EAAT4. J. Physiol. 2001;534:159–167. doi: 10.1111/j.1469-7793.2001.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barish M.E. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J. Physiol. 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shabaneh M., Rosental N., Kanner B.I. Disulfide cross-linking of transport and trimerization domains of a neuronal glutamate transporter restricts the role of the substrate to the gating of the anion conductance. J. Biol. Chem. 2014;289:11175–11182. doi: 10.1074/jbc.M114.550277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akyuz N., Altman R.B., Boudker O. Transport dynamics in a glutamate transporter homologue. Nature. 2013;502:114–118. doi: 10.1038/nature12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stolzenberg S., Khelashvili G., Weinstein H. Structural intermediates in a model of the substrate translocation path of the bacterial glutamate transporter homologue GltPh. J. Phys. Chem. B. 2012;116:5372–5383. doi: 10.1021/jp301726s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao Z., Grewer C. Cooperation of the conserved aspartate 439 and bound amino acid substrate is important for high-affinity Na+ binding to the glutamate transporter EAAC1. J. Gen. Physiol. 2007;129:331–344. doi: 10.1085/jgp.200609678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watzke N., Bamberg E., Grewer C. Early intermediates in the transport cycle of the neuronal excitatory amino acid carrier EAAC1. J. Gen. Physiol. 2001;117:547–562. doi: 10.1085/jgp.117.6.547. [DOI] [PMC free article] [PubMed] [Google Scholar]