

Abstract
The high prevalence and mortality of lung cancer, together with a poor 5-year survival of only approximately 15%, emphasize the need for prognostic and predictive factors to improve patient treatment. C4.4A, a member of the Ly6/uPAR family of membrane proteins, qualifies as such a potential informative biomarker in non-small cell lung cancer. Under normal physiological conditions, it is primarily expressed in suprabasal layers of stratified squamous epithelia. Consequently, it is absent from healthy bronchial and alveolar tissue, but nevertheless appears at early stages in the progression to invasive carcinomas of the lung, i.e., in bronchial hyperplasia/metaplasia and atypical adenomatous hyperplasia. In the stages leading to pulmonary squamous cell carcinoma, expression is sustained in dysplasia, carcinoma in situ and invasive carcinomas, and this pertains to the normal presence of C4.4A in squamous epithelium. In pulmonary adenocarcinomas, a fraction of cases is positive for C4.4A, which is surprising, given the origin of these carcinomas from mucin-producing and not squamous epithelium. Interestingly, this correlates with a highly compromised patient survival and a predominant solid tumor growth pattern. Circumstantial evidence suggests an inverse relationship between C4.4A and the tumor suppressor LKB1. This might provide a link to the prognostic impact of C4.4A in patients with adenocarcinomas of the lung and could potentially be exploited for predicting the efficacy of treatment targeting components of the LKB1 pathway.
Keywords: LYPD3, Non-small cell lung cancer, Prognosis, Solid growth pattern, Liver kinase B1, Precursor lesions, Atypical adenomatous hyperplasia, Metaplasia, Squamous differentiation, Ly6/Urokinase-type plasminogen activator receptor
Core tip: C4.4A is a new biomarker with potential prognostic value in pulmonary adenocarcinoma. High levels of protein expression correlate with poor patient survival and a histological growth pattern of the solid type. Recent data suggest that C4.4A is negatively regulated by the tumor suppressor liver kinase B1 (LKB1), which is inactivated in a fraction of adenocarcinomas of the lung. Such an inverse association between C4.4A and LKB1 could possibly render C4.4A a candidate predictive biomarker for therapeutic intervention targeting components of the LKB1 pathway, such as mammalian target of rapamycin.
INTRODUCTION
Lung cancer is the most prevalent and most mortal form of cancer, with an estimated 1.6 million new cases and 1.375 million deaths every year[1]. Survival is highly dependent on stage at presentation, decreasing drastically from localized (52%) to regional (25%) and distant disseminated disease (4%), yielding an overall relative 5-year survival of only approximately 15%[2]. Despite these discouraging statistics, there have, with the advent of personalized medicine, been some improvements in the treatment of lung cancer[3]. This pertains predominantly to an improved knowledge on tumor biology, which has uncovered a molecular rationale for different treatment efficacies. In this context, molecular and histologic parameters for prediction of drug responsiveness, in particular the identification of subsets of lung cancer harboring distinct targetable oncogenic driver mutations, have guided choice of therapy.
A striking example can be seen with small tyrosine kinase inhibitors (TKIs), for which activating mutations in exons 19 and 21 of the epidermal growth factor receptor (EGFR) gene are predictive of therapeutic efficacy[4-7]. Consequently, activating EGFR mutations are now a validated biomarker for decisions regarding first-line treatment of advanced non-small cell lung cancer (NSCLC)[4,8]. Response to the anaplastic lymphoma kinase (ALK)-TKI crizotinib similarly depends on the presence of the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion gene in a subset of pulmonary adenocarcinoma (AC)[9,10]. In addition to EGFR and ALK tests, the potential of other biomarkers is being validated in clinical trials, e.g., ROS1, human epidermal growth factor receptor 2/neu (HER2), BRAF and mesenchymal-epidermal transition proto-oncogene (MET) primarily in AC, and fibroblast growth factor receptor 1 (FGFR1) and discoidin domain receptor 2 (DDR2) in squamous cell carcinoma (SCC)[11]. Regarding NSCLC histology, there have been tolerance issues coupled to the inhibitor of angiogenesis, bevacizumab, which can cause life-threatening hemorrhage and hemoptysis in patients with SCC, and is thus contra-indicated in this subgroup[12,13]. Another drug, the antifolate pemetrexed, inhibits three enzymes of the folate metabolism, thymidylate synthase (TS), dihydrofolate reductase, and glycinamide ribonucleotide formyltransferase, the consequence of which is a reduction in de novo purine and pyrimidine synthesis, thus interfering with DNA and RNA synthesis. Overexpression of TS might be associated with a reduced efficacy of pemetrexed, and this can explain why it is more efficient in advanced AC than in SCC[14,15]. The fact that tumor histology and molecular features can influence the choice of treatment implies that different NSCLC histologic subtypes should be considered as distinct disease entities, which in turn comprise distinct molecular subsets that should be managed individually for successful outcome.
Earlier detection of lung cancer, as well as assessment of the malignant potential of a resected tumor for decisions regarding adjuvant treatment, represent other promising avenues for reducing the high mortality of the disease. Continuously increasing our knowledge on the mechanisms involved in pathogenesis is crucial to further improve the rational targeting strategy that has been adopted for lung cancer treatment. New biomarkers of early disease, prognosis and prediction of response to targeted therapy are relevant in this context, to ameliorate patient survival[16-18]. The protein C4.4A is a potential new biomarker in NSCLC.
C4.4A – A UROKINASE RECEPTOR (uPAR) PROTEIN HOMOLOG
C4.4A is a membrane protein anchored to the cell surface via a glycosylphosphatidylinositol (GPI) moiety, showing predicted structural homology to the other multidomain members of the Ly6/uPAR (LU) protein family, i.e., the urokinase-type plasminogen activator receptor (uPAR), Haldisin, TEX101, CD177 and LYPD4[19-23]. The genes encoding these proteins are clustered in a small region of chromosome 19q13 (Figure 1A). After posttranslational processing, C4.4A consists of 278 amino acids distributed in 2 N-terminal LU domains and a serine, threonine, proline-rich region C-terminally (Figure 1B).
Figure 1.
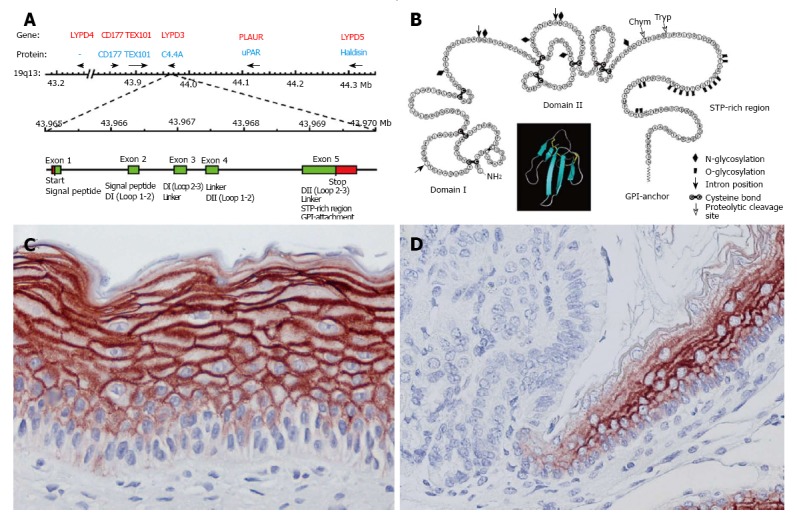
Structure and expression of the protein C4.4A. A: Schematic outline of the gene cluster encompassing multidomain members of the LU protein family, highlighting the gene encoding C4.4A (LYPD3) and its exon composition. Modified from Kriegbaum et al[20,24], 2011 and Jacobsen et al[20,24]; B: Cartoon of the structure of C4.4A. The insert represents the three-finger fold characteristic of LU domains (made in PyMOL, DeLano Scientific, using PDB coordinates 1NEA). Modified from Hansen et al[19]; C and D: C4.4A expression in suprabasal layers of squamous epithelium, exemplified by tissue-engineered human epidermis (C) and the transition between the glandular and non-glandular portions of the rodent stomach (D). C4.4A is detected by a polyclonal C4.4A antibody and visualized by NovaRED chromogen. D is reproduced with permission from BestPractice Onkologi, Denmark[89].
C4.4A IN NORMAL DIFFERENTIATION PROCESSES
Under normal physiological conditions, C4.4A is predominantly expressed in suprabasal layers of stratified squamous epithelia such as those of the skin, hair follicles, esophagus, oral and nasal cavity, vagina and cornea (Figure 1C). It is furthermore found in the cuboidal epithelium of human term placenta and sweat ducts, in the pigmented epithelium of the retina and in Hassall’s corpuscle in the thymus[19,24]. Remaining epithelia, including the alveolar and bronchial compartments of the healthy lung (Figures 2A and 3A; Table 1), are devoid of C4.4A, suggesting a tight regulation of its expression, possibly by the CCAAT/enhancer binding protein β (C/EBPβ) or estrogen[25,26]. This is clearly visualized at transition zones such as those present in the rodent stomach and at the ano-rectal junction, where the distinct C4.4A expression at the squamous side is abruptly terminated at the columnar side (Figure 1D). The stringent membrane-associated expression pattern of C4.4A would be in line with a putative role of this protein in cell adhesion[19,27,28], but its biological function is still to be delineated.
Figure 2.
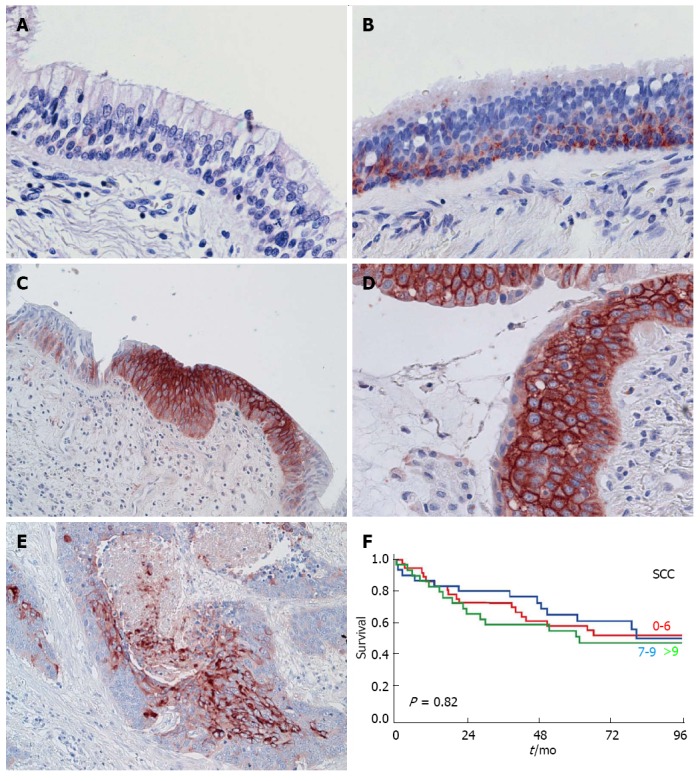
C4.4A in pulmonary squamous cell carcinoma. Panels A-E: C4.4A expression as detected by immunohistochemistry with a polyclonal antibody in normal bronchial epithelium (A), hyperplasia (B), metaplasia (C), dysplasia (D) and invasive squamous cell carcinoma (SCC) (E). A, B, D and C, E are reproduced with permission from Jacobsen et al[34], 2012 and BestPractice Onkologi, Denmark[89], respectively. Panel F: Kaplan-Meier curves for the survival of SCC patients, which is independent of C4.4A scores, here stratified by tertiles (red: Lowest level of C4.4A; blue: Intermediate level of C4.4A; green: Highest level of C4.4A). Modified from Jacobsen et al[33], 2013.
Figure 3.
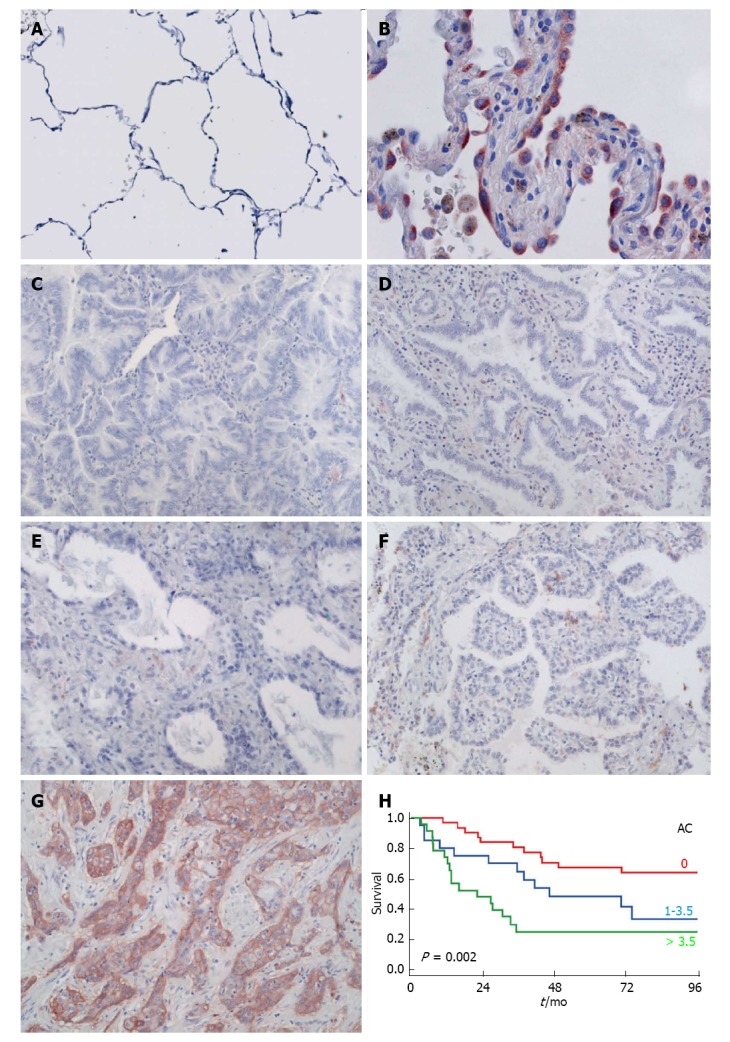
C4.4A in pulmonary adenocarcinoma. Panels A-E: C4.4A expression as detected by immunohistochemistry with a polyclonal antibody in normal alveoli (A; reproduced with permission from Jacobsen et al[34], 2012), atypical adenomatous hyperplasia (B), invasive AC with predominant mucinous lepidic (C), non-mucinous lepidic (D), acinar (E), papillary (F) and solid (G) pattern. Panel H: Kaplan-Meier estimates for the survival of AC patients, which is correlated with C4.4A scores, here stratified by tertiles (red: Lowest level of C4.4A; blue: Intermediate level of C4.4A; green: Highest level of C4.4A). Modified from Jacobsen et al[33], 2013.
Table 1.
Expression and prognostic significance of C4.4A in pulmonary squamous cell carcinoma and adenocarcinoma
| C4.4A reactivity | Multivariate survival analysis4 | Ref. | ||||||
| Premalignant lesions | 01 | + | ++ | +++ | [34] | |||
| SCC | Normal | 100% | Not available | |||||
| Hyperplasia | 59% | 41% | ||||||
| Metaplasia | 29% | 71% | ||||||
| Dysplasia | 20% | 80% | ||||||
| AC | Normal | 100% | ||||||
| AAH | 17% | 37% | 37% | 10% | ||||
| Invasive cancer | Median2 | Range | Lower quartile | Upper quartile | HR (95%CI) | P-value | [33] | |
| SCC | 8 | 0-16 | 5 | 10 | 0.82 | |||
| AC | 13 | 0-16 | 0 | 6 | 1.65 (1.24-2.19) | 0.0005 | ||
0: Negative; +: Weakly/focally positive; ++: Positive; +++: Strongly positive;
Refers to product of C4.4A intensity (0-4, where 0 = negative, 1 = very weak, 2 = weak, 3 = moderate and 4 = strong staining) and frequency (0-4, where 0 = 0%, 1 = 0%-25%, 2 = 26%-50%, 3 = 51%-75% and 4 ≥ 76% positive tumor cells);
Median value for non-solid and solid AC is 0 and 4, respectively;
Performed by the Cox proportional hazards model. SCC: Squamous cell carcinoma; AC: Adenocarcinoma.
C4.4A IN PATHOLOGICAL CONDITIONS
C4.4A was originally identified by two independent differential antigen screens as a candidate metastasis-associated protein[29,30], the first in a rat pancreatic adenocarcinoma cell line, and the second in an in vitro urothelial wound response model. Rösel et al[29] furthermore reported on the capability of C4.4A-positive but not C4.4A-negative tumor cells to penetrate a matrigel, in the absence but not in the presence of a monoclonal C4.4A antibody. C4.4A has indeed been implicated in a range of human cancers, including lung[31-35], esophageal[27], bladder[30] and colorectal[36], as evaluated by immunohistochemistry, in situ hybridization, PCR, Northern blotting or microarray screening. In colorectal cancer[36], esophageal squamous cell carcinomas[27] and bladder transitional cell carcinomas (Jacobsen et al[33], manuscript in preparation), C4.4A is upregulated at the tumor invasive front as compared to the tumor core, suggesting a possible association of C4.4A to the invasive process. Whether this can be further translated to a direct involvement in the ability of a tumor cell to metastasize as initially proposed has not been verified experimentally in vivo, but the expression of C4.4A seen in primary tumors is at least in the case of the esophagus and the lung recapitulated in corresponding lymph node metastases[27,33].
One of the most well-studied diseases regarding C4.4A expression is NSCLC[31,33,34]. The role of C4.4A in the progression to pulmonary SCC and AC is described in the following.
C4.4A IN PULMONARY SQUAMOUS CELL CARCINOMA
Progression to malignancy through well-described premalignant lesions
Lung cancer is divided into two main histological types, where small cell lung cancer comprises around 15% of cases and NSCLC the remaining 85% of cases. The latter is further subdivided into AC, SCC and large cell carcinoma (LCC), of which AC has become the most frequent type[37-39].
Pulmonary SCCs most often originate in the bronchial compartment. There is a high degree of consensus concerning the stages that transform normal, pseudostratified columnar bronchial epithelium into invasive SCC[40]. After an excessive proliferation phase resulting in hyperplasia of basal cells, metaplasia entails the transdifferentiation of bronchial cells resulting in the conversion of columnar epithelium to squamous epithelium. Dysplasia represents the first true premalignant stage and is followed by carcinoma in situ (CIS), which again can develop into malignant carcinoma. Morphologically, epithelial thickness, cell size and mitotic figures increase throughout this progression, which is also characterized by pleomorphism and loss of epithelial polarity[37]. Molecularly, activation of oncogenes and inactivation of tumor suppressor genes occur, along with allelic losses[41-43].
Morphologically normal bronchial epithelium is devoid of C4.4A (Figure 2A), but expression appears already at the stage of hyperplasia (Figure 2B) and becomes prominent upon metaplastic (Figure 2C) and dysplastic conversion (Figure 2D, Table 1)[34]. The protein is consistently present in invasive SCC (Figure 2E), with high levels detected in approx. 75% of patients in two different cohorts[31,33].
C4.4A expression mirroring normal squamous differentiation
Given that hyperplasia and metaplasia are not true premalignant lesions, but rather regarded as reactive changes primarily caused by chronic irritation such as cigarette smoking, they do not by themselves indicate an increased risk of developing into a manifest carcinoma, as contrasted to severe dysplasia and CIS[43-45]. A consequence of the very early appearance of C4.4A in basal cell hyperplasia/early metaplasia is that it cannot reflect the subsequent malignant transformation per se and therefore cannot be used as an early biomarker for pulmonary SCC. It thus reports on the differentiation status of the cells rather than on their malignant potential, and this provides an explanation for the presence of C4.4A in nearly all cases of invasive SCC[31,33,34]. With consistent high levels of C4.4A in both premalignant and malignant lesions, C4.4A cannot differentiate between individual cases, and this is furthered into a lack of prognostic information in SCC patients (Figure 2F)[31,33].
The detection of C4.4A in squamous metaplasia and dysplasia aligns excellently with the normal expression of C4.4A, which is closely linked to squamous differentiation of epithelium, as exemplified by murine embryogenesis, where C4.4A and the differentiation-specific cytokeratin 10 appear simultaneously in the nasal cavity (day E13.5) and in the interfollicular epidermis of the vibrissae (day E14.5)[24]. Due to the stringent regulation of C4.4A in squamous epithelia, as convincingly shown at squamo-columnar junctions (Figure 1D), the appearance of C4.4A in basal cell hyperplasia in the bronchial compartment is on the other hand an unexpected finding. This could suggest that C4.4A reports on the induction of the metaplastic conversion process, where pseudostrafied columnar epithelium is replaced by stratified squamous epithelium, even before the emerging squamous differentiation is evident morphologically. It has indeed been demonstrated that genetic aberrations such as mutations, deletions and overexpression of p53, and loss of heterozygosity (LOH) at chromosomes 3p and 9p (p16INK4a locus) occur in histologically normal respiratory mucosa of smokers and lung cancer patients[40,42,45-47]. Respiratory basal cells have been reported to be progenitors of squamous metaplastic cells and presumably preneoplastic epithelium[37,40,42,48,49]. Delineating the function of C4.4A might give an indication of its role in the transdifferentiation process occurring in the bronchial mucosa, setting the stage for ensuing neoplastic transformation. This would increase our knowledge on the very early pathogenesis of pulmonary SCC. Considering the complete lack of C4.4A in normal bronchial epithelium, it might be used in conjunction with other histopathological, molecular and genetic markers in risk assessment for identifying malignant clones of the bronchial mucosa before these are manifest morphologically[40], which could suggest a need for a closer patient follow-up.
C4.4A IN PULMONARY ADENOCARCINOMA
From atypical adenomatous hyperplasia to invasive adenocarcinomas
Most ACs of the lung originate peripherally, which in contrast to the central airways is much more challenging to image, rendering a delineation of the various stages of transformation from normal alveolar epithelium to invasive AC corresponding to the development to invasive SCC difficult[37,40,46]. On the basis of clinicopathologic and molecular studies, atypical adenomatous hyperplasia (AAH) has nevertheless been identified as a precursor lesion, which subsequently transforms into adenocarcinoma in situ (AIS), formerly known as bronchioloalveolar carcinoma (BAC)[38,40,42,50-56]. AAH and AIS thus represent the counterparts of squamous dysplasia and CIS, respectively. Evidence for the stepwise development from AAH to AC comes from longitudinal case studies by low-dose computed tomography imaging[57,58], the similarity of molecular aberrations[40,59-62] and a common expression pattern of markers of the suggested progenitor cells, Clara cells and type II pneumocytes, in AAH and AC[40,42,63] as well as mouse models of pulmonary AC, targeting KRAS/p53[64] or EGFR[65,66].
With a view to the preferential expression of C4.4A in squamous epithelium, as described above, one would not expect C4.4A expression in the alveolar compartment. This is indeed the case for normal alveoli (Figure 3A), but unexpectedly, the protein is present in approx. 25% of investigated cases of AC, albeit at much lower expression levels than in SCC, when scoring C4.4A semi-quantitatively as a product of intensity (0-4) and frequency (0-4) of immunohistochemical staining (median of 1 vs 8 on a scale up to 16) (Figure 3C-G, Table 1)[31,33]. Interestingly, a small fraction of atypical type II pneumocytes in AAH lesions is also positive for C4.4A (Figure 3B)[34]. This might be coupled to the cuboidal nature of these cells, as C4.4A also is found in other cuboidal epithelia such as the placenta and sweat ducts[19] ( and Kriegbaum, unpublished).
Prognostic impact of C4.4A in adenocarcinomas
As compared to known prognostic factors, such as stage, performance status, completeness of resection, tumor differentiation grade, nodal status, age and gender, the translational value of proposed lung tumor markers and gene expression signatures has been disappointing[16,67]. It is therefore of interest to find new biomarkers or a combination thereof that could allow a more accurate survival prediction and aid in the decision making regarding adjuvant therapy. The prognostic impact of C4.4A in NSCLC has been investigated by a specific and reproducible semi-quantitative immunohistochemical protocol with substantial inter-observer agreement in two independent patient cohorts (40 ACs/56 SCCs from Denmark[31] and 88 ACs/104 SCCs from Germany[33]). These studies showed that increasingly higher levels of C4.4A in ACs correlated with a decreasing patient survival, as evaluated by Kaplan-Meier analysis, where C4.4A scores were grouped according to tertiles (Figure 3H). Univariate and multivariate analysis of overall survival including C4.4A scores and the clinical covariates pathological stage, performance status, gender, age and treatment, likewise revealed C4.4A to be a significant prognostic factor (Table 1), together with pathological stage. The validation of the observations obtained in the first study in a second, larger patient population emphasizes the robust correlation between C4.4A levels and prognosis of AC patients. This has been further substantiated by another study based on quantitative real-time PCR, where the gene encoding C4.4A, LYPD3, was selected as one of 91 signature genes for survival prediction of pulmonary AC patients[68]. It could be interesting to use patient material from previously conducted large clinical trials to retrospectively test for superiority of C4.4A to current prognostic factors, with a view to future validation in a prospective, randomized trial.
Considering the sequential development from AAH to AC, it is tempting to speculate whether a C4.4A-positive AAH eventually could develop into a C4.4A-positive AC with an ensuing compromised patient survival. If this were the case, this would entail that there are subpopulations of C4.4A-positive AAH cells with higher malignant potential than the C4.4A-negative counterparts, with the possibility to be used as an early marker of a supposedly more aggressive subtype of AC. In addition, considering that only a minority of AAHs progresses to AC, it would be interesting to assess whether C4.4A-positive AAH cases are more prone to this malignant progression than C4.4A-negative AAH. Addressing these questions would, however, require prospective, longitudinal studies and advanced imaging techniques not yet available for proper visualization of both AAH lesions and C4.4A, or alternatively following the various stages of malignant progression in a suitable mouse lung cancer model[69].
Correlation of C4.4A expression with solid growth type
Pulmonary ACs encompass a histologically, molecularly and genetically very heterogeneous group of tumors. The new multidisciplinary classification suggests that AC should be described according to histological subtype, which can be mucinous or non-mucinous lepidic, acinar, papillary, micropapillary or solid[38]. Gene expression profiling clusters the different AC types according to morphological and molecular characteristics, which emphasizes that ACs cannot be classified in one homogeneous group[70,71]. Interestingly, patient survival also differs significantly with subtype, with 5-year relative survival rates of 86%-90% and 39%-70% when the lepidic and solid components, respectively, are predominant[72,73].
Detailed analysis of C4.4A in the fraction of positive ACs, with focus on histological subtypes, has revealed that C4.4A expression is tightly correlated with the solid growth pattern[33]. Whereas the protein is almost absent in cases with a predominant mucinous/non-mucinous lepidic, acinar or papillary pattern (Figure 3C-F), the median value in cases with solid growth is 4 on a scale ranging from 0 to 16. This places the majority of these patients in the upper tertile of C4.4A scores in the AC group (> 3.5), with an ensuing poor prognosis as illustrated by Kaplan-Meier survival curves (Figure 3H). Given that a solid component in itself predicts poor patient survival[72,73], it could be assumed that this clear interaction between C4.4A and solid growth would explain the prognostic impact of C4.4A. This is, however, refuted by multivariate overall survival analysis using the Cox proportional hazards model including growth pattern and the C4.4A/solid interaction in addition to classical clinical factors, where C4.4A and pathological stage are the only significant independent parameters. Despite the interaction between C4.4A and solid growth pattern, C4.4A is thus a stronger prognostic factor than solid growth[33]. Given that AC patients with a predominant solid growth pattern seem to benefit from adjuvant radiotherapy, this might be exploited in clinical decision-making regarding this form of treatment[74].
By comparing gene expression profiles of independent lung cancer patient populations obtained by DNA microarray analysis, Hayes et al[75] identified three AC subtypes that were termed bronchioid, squamoid and magnoid, with reference to their resemblance to bronchioalveolar carcinoma, SCC and LCC, respectively. AC tumors in a differentiation state close to the SCC phenotype thus seem to exist. This is further substantiated by a study of ACs of mainly the solid pattern with signet-ring morphology, which co-express thyroid transcription factor 1 (TTF-1) and p63, being markers of AC and SCC, respectively[76]. In the WHO classification from 2004, carcinomas with histological and immunohistochemical evidence of double differentiation are termed adenosquamous carcinomas[37]. The squamoid ACs are characterized by moderate or poor differentiation, solid growth, invasion, and a poor patient survival[75,77]. The fact that C4.4A is primarily expressed in AC of the solid type, coupled to its close association to squamous differentiation and prognostic impact in AC would suggest that the C4.4A-positive ACs according to this classification are of the squamoid type.
Regulation of C4.4A by the tumor suppressor LKB1
The normal stringent control of C4.4A expression as seen in squamous epithelia is apparently disrupted in pulmonary AC. Whether the unexpected expression of C4.4A in pulmonary AC has functional implications in the pathogenesis of AC remains unknown. It might also be reporting on an erroneous regulation of a signaling pathway that when activated in the lung results in highly malignant tumors. Circumstantial evidence suggests the involvement of the tumor suppressor liver kinase B1 (LKB1). Firstly, C4.4A is negatively regulated by LKB1 in esophageal cancer cell lines, where LYPD3 was upregulated upon loss of LKB1. The migratory and invasive potential of these cells was reduced when LYPD3 subsequently was knocked down[78]. Secondly, LKB1 reconstitution in the LKB1-deficient AC cell line H2126 downregulates C4.4A at the mRNA level[79]. Thirdly, 17%-54% of pulmonary AC patients present with LKB1 inactivating mutations, of which the majority is found in poorly differentiated cases[80-84]. Fourthly, LKB1 is lost in a fraction of AAH lesions[85]. If there is an inverse relationship between LKB1 and C4.4A, C4.4A-positivity with its ensuing worsened patient prognosis in pulmonary AC might be explained by LKB1 inactivation in the given cases. This would imply that C4.4A potentially could be used as a biomarker for the efficacy of treatment targeting the LKB1 pathway, including mammalian target of rapamycin (mTOR) signaling, which is activated upon LKB1 inactivation[86]. The potential of mTOR inhibitors as therapeutic agents in NSCLC is currently investigated in a range of clinical trials[87,88].
One of the most recent genetic murine lung cancer models exploits a combination of LKB1 inactivation and KRAS oncogene activation to obtain metastatic tumors of a mixed phenotype, including AC, SCC and LCC[79]. Crossing a C4.4A-deficient mouse with the KRAS/LKB1 model would allow a simultaneous investigation of the consequences of the absence or presence of C4.4A in the progression of both AC and SCC, revealing if C4.4A is functionally involved in or is a reporter of a molecular mechanism that could be targeted therapeutically.
CONCLUSION
C4.4A induction is an early event in the progression of two very distinct histological subtypes of lung cancer, i.e., AC and SCC, where expression is seen in the presumed precursor lesions AAH and hyperplasia/metaplasia, respectively. Nevertheless, the differential prognostic impact suggests that the protein plays distinct roles in the pathogenesis of these two entities, and this underscores the high heterogeneity of lung cancer and the concept of AC and SCC as two different diseases.
In the progression to SCC, C4.4A is inherently linked to the phenotype that arises following the transdifferentiation process that converts pseudostratified columnar respiratory epithelium to squamous epithelium, completely in line with its normal expression. The appearance already in basal cell hyperplasia is interesting from a biological point of view, indicating that C4.4A is such an early marker of squamous differentiation that it is present even before the phenotype is morphologically manifest. However, given that C4.4A consistently is expressed in reactive lesions even preceding true premalignant lesions, is sustained in the subsequent stages of malignant progression and is invariably present in invasive carcinomas, it cannot possibly provide any information on the survival of SCC patients.
In sharp contrast to this, C4.4A is a strong, independent prognostic indicator in AC patients, and it therefore has clinical relevance in this lung cancer subtype, especially in cases with a solid growth pattern, for which C4.4A is a surrogate marker. As such, whereas C4.4A follows a normal differentiation pattern in SCC, its expression in AC reflects an abnormal differentiation program. Clarifying whether this has implications for AC pathogenesis or is reporting on a dysregulated signaling pathway awaits the delineation of the biological function of C4.4A. Either way, it is tempting to speculate that there is a link between C4.4A-positive AAH lesions and ACs with a solid pattern, which would make C4.4A a very early biomarker for a particularly malignant form of AC. It is, however, at present unclear if AAH actually develops into overt solid AC[38,46,53]. It is furthermore of interest to investigate the putative inverse correlation between LKB1 inactivation and high C4.4A expression and the hypothetical candidacy of C4.4A as a predictive biomarker for therapy aimed at components of the LKB1 pathway. This is in line with the goal of supplementing traditional NSCLC management according to the TNM classification with personalized medicine[18], which depends on predictive biomarkers reporting on biological and molecular tumor characteristics that can identify the patient populations benefitting from a given targeted treatment.
ACKNOWLEDGMENTS
The authors would like to thank John Post (Finsen Laboratory) for skilled assistance in figure preparation. Dr. Eric Santoni-Rugiu participates in Pfizer's Nordic Board for ALK in NSCLC, has received speaker's fees from Pfizer, Roche, and Novartis, and his institution receives research funding from Pfizer and Roche.
Footnotes
Supported by Copenhagen University Hospital (Rigshospitalets Forskningspuljer), The Danish National Research Foundation (Danish-Chinese Centre for Proteases and Cancer), Harboefonden, Torben og Alice Frimodts Fond, Fabrikant Einar Willumsens Mindelegat, Holger Rabitz and hustrus Legat, The Lundbeck Foundation
P- Reviewer: Bugaj AM, Ren T, Scaggiante B, Wang X, Zoller M S- Editor: Wen LL L- Editor: A E- Editor: Lu YJ
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Kaneda H, Yoshida T, Okamoto I. Molecularly targeted approaches herald a new era of non-small-cell lung cancer treatment. Cancer Manag Res. 2013;5:91–101. doi: 10.2147/CMAR.S32973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gridelli C, De Marinis F, Di Maio M, Cortinovis D, Cappuzzo F, Mok T. Gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer with activating epidermal growth factor receptor mutation: Review of the evidence. Lung Cancer. 2011;71:249–257. doi: 10.1016/j.lungcan.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 5.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 6.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 7.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roengvoraphoj M, Tsongalis GJ, Dragnev KH, Rigas JR. Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non-small cell lung cancer: focus on epidermal growth factor receptor mutation testing and mutation-positive patients. Cancer Treat Rev. 2013;39:839–850. doi: 10.1016/j.ctrv.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 9.Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, Riely GJ, Solomon B, Ou SH, Kim DW, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–1019. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minuti G, D’Incecco A, Cappuzzo F. Targeted therapy for NSCLC with driver mutations. Expert Opin Biol Ther. 2013;13:1401–1412. doi: 10.1517/14712598.2013.827657. [DOI] [PubMed] [Google Scholar]
- 12.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab (Avastin) plus Carboplatin and Paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12:713–718. doi: 10.1634/theoncologist.12-6-713. [DOI] [PubMed] [Google Scholar]
- 13.Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, Langer CJ, DeVore RF, Gaudreault J, Damico LA, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22:2184–2191. doi: 10.1200/JCO.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 14.Ceppi P, Volante M, Saviozzi S, Rapa I, Novello S, Cambieri A, Lo Iacono M, Cappia S, Papotti M, Scagliotti GV. Squamous cell carcinoma of the lung compared with other histotypes shows higher messenger RNA and protein levels for thymidylate synthase. Cancer. 2006;107:1589–1596. doi: 10.1002/cncr.22208. [DOI] [PubMed] [Google Scholar]
- 15.Scagliotti G, Hanna N, Fossella F, Sugarman K, Blatter J, Peterson P, Simms L, Shepherd FA. The differential efficacy of pemetrexed according to NSCLC histology: a review of two Phase III studies. Oncologist. 2009;14:253–263. doi: 10.1634/theoncologist.2008-0232. [DOI] [PubMed] [Google Scholar]
- 16.Brundage MD, Davies D, Mackillop WJ. Prognostic factors in non-small cell lung cancer: a decade of progress. Chest. 2002;122:1037–1057. doi: 10.1378/chest.122.3.1037. [DOI] [PubMed] [Google Scholar]
- 17.Ettinger DS, Akerley W, Bepler G, Blum MG, Chang A, Cheney RT, Chirieac LR, D’Amico TA, Demmy TL, Ganti AK, et al. Non-small cell lung cancer. J Natl Compr Canc Netw. 2010;8:740–801. doi: 10.6004/jnccn.2010.0056. [DOI] [PubMed] [Google Scholar]
- 18.Tanoue LT. Staging of non-small cell lung cancer. Semin Respir Crit Care Med. 2008;29:248–260. doi: 10.1055/s-2008-1076745. [DOI] [PubMed] [Google Scholar]
- 19.Hansen LV, Gårdsvoll H, Nielsen BS, Lund LR, Danø K, Jensen ON, Ploug M. Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem J. 2004;380:845–857. doi: 10.1042/BJ20031478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobsen B, Ploug M. The urokinase receptor and its structural homologue C4.4A in human cancer: expression, prognosis and pharmacological inhibition. Curr Med Chem. 2008;15:2559–2573. doi: 10.2174/092986708785909012. [DOI] [PubMed] [Google Scholar]
- 21.Fujihara Y, Tokuhiro K, Muro Y, Kondoh G, Araki Y, Ikawa M, Okabe M. Expression of TEX101, regulated by ACE, is essential for the production of fertile mouse spermatozoa. Proc Natl Acad Sci USA. 2013;110:8111–8116. doi: 10.1073/pnas.1222166110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gårdsvoll H, Kriegbaum MC, Hertz EP, Alpízar-Alpízar W, Ploug M. The urokinase receptor homolog Haldisin is a novel differentiation marker of stratum granulosum in squamous epithelia. J Histochem Cytochem. 2013;61:802–813. doi: 10.1369/0022155413501879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korkmaz B, Kuhl A, Bayat B, Santoso S, Jenne DE. A hydrophobic patch on proteinase 3, the target of autoantibodies in Wegener granulomatosis, mediates membrane binding via NB1 receptors. J Biol Chem. 2008;283:35976–35982. doi: 10.1074/jbc.M806754200. [DOI] [PubMed] [Google Scholar]
- 24.Kriegbaum MC, Jacobsen B, Hald A, Ploug M. Expression of C4.4A, a structural uPAR homolog, reflects squamous epithelial differentiation in the adult mouse and during embryogenesis. J Histochem Cytochem. 2011;59:188–201. doi: 10.1369/0022155410394859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fries F, Nazarenko I, Hess J, Claas A, Angel P, Zöller M. CEBPbeta, JunD and c-Jun contribute to the transcriptional activation of the metastasis-associated C4.4A gene. Int J Cancer. 2007;120:2135–2147. doi: 10.1002/ijc.22447. [DOI] [PubMed] [Google Scholar]
- 26.Hardman MJ, Ashcroft GS. Estrogen, not intrinsic aging, is the major regulator of delayed human wound healing in the elderly. Genome Biol. 2008;9:R80. doi: 10.1186/gb-2008-9-5-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hansen LV, Laerum OD, Illemann M, Nielsen BS, Ploug M. Altered expression of the urokinase receptor homologue, C4.4A, in invasive areas of human esophageal squamous cell carcinoma. Int J Cancer. 2008;122:734–741. doi: 10.1002/ijc.23082. [DOI] [PubMed] [Google Scholar]
- 28.Paret C, Bourouba M, Beer A, Miyazaki K, Schnölzer M, Fiedler S, Zöller M. Ly6 family member C4.4A binds laminins 1 and 5, associates with galectin-3 and supports cell migration. Int J Cancer. 2005;115:724–733. doi: 10.1002/ijc.20977. [DOI] [PubMed] [Google Scholar]
- 29.Rösel M, Claas C, Seiter S, Herlevsen M, Zöller M. Cloning and functional characterization of a new phosphatidyl-inositol anchored molecule of a metastasizing rat pancreatic tumor. Oncogene. 1998;17:1989–2002. doi: 10.1038/sj.onc.1202079. [DOI] [PubMed] [Google Scholar]
- 30.Smith BA, Kennedy WJ, Harnden P, Selby PJ, Trejdosiewicz LK, Southgate J. Identification of genes involved in human urothelial cell-matrix interactions: implications for the progression pathways of malignant urothelium. Cancer Res. 2001;61:1678–1685. [PubMed] [Google Scholar]
- 31.Hansen LV, Skov BG, Ploug M, Pappot H. Tumour cell expression of C4.4A, a structural homologue of the urokinase receptor, correlates with poor prognosis in non-small cell lung cancer. Lung Cancer. 2007;58:260–266. doi: 10.1016/j.lungcan.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 32.Heighway J, Knapp T, Boyce L, Brennand S, Field JK, Betticher DC, Ratschiller D, Gugger M, Donovan M, Lasek A, et al. Expression profiling of primary non-small cell lung cancer for target identification. Oncogene. 2002;21:7749–7763. doi: 10.1038/sj.onc.1205979. [DOI] [PubMed] [Google Scholar]
- 33.Jacobsen B, Muley T, Meister M, Dienemann H, Christensen IJ, Santoni-Rugiu E, Lærum OD, Ploug M. Ly6/uPAR-related protein C4.4A as a marker of solid growth pattern and poor prognosis in lung adenocarcinoma. J Thorac Oncol. 2013;8:152–160. doi: 10.1097/JTO.0b013e318279d503. [DOI] [PubMed] [Google Scholar]
- 34.Jacobsen B, Santoni-Rugiu E, Illemann M, Kriegbaum MC, Laerum OD, Ploug M. Expression of C4.4A in precursor lesions of pulmonary adenocarcinoma and squamous cell carcinoma. Int J Cancer. 2012;130:2734–2739. doi: 10.1002/ijc.26305. [DOI] [PubMed] [Google Scholar]
- 35.Würfel J, Seiter S, Stassar M, Claas A, Kläs R, Rösel M, Marhaba R, Savelyeva L, Schwab M, Matzku S, et al. Cloning of the human homologue of the metastasis-associated rat C4.4A. Gene. 2001;262:35–41. doi: 10.1016/s0378-1119(00)00515-1. [DOI] [PubMed] [Google Scholar]
- 36.Konishi K, Yamamoto H, Mimori K, Takemasa I, Mizushima T, Ikeda M, Sekimoto M, Matsuura N, Takao T, Doki Y, et al. Expression of C4.4A at the invasive front is a novel prognostic marker for disease recurrence of colorectal cancer. Cancer Sci. 2010;101:2269–2277. doi: 10.1111/j.1349-7006.2010.01674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Travis WD, Brambilla E, Mller-Hermelink HK, Harris CC, eds . World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2004. [Google Scholar]
- 38.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol. 2011;6:244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wahbah M, Boroumand N, Castro C, El-Zeky F, Eltorky M. Changing trends in the distribution of the histologic types of lung cancer: a review of 4,439 cases. Ann Diagn Pathol. 2007;11:89–96. doi: 10.1016/j.anndiagpath.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu Rev Pathol. 2006;1:331–348. doi: 10.1146/annurev.pathol.1.110304.100103. [DOI] [PubMed] [Google Scholar]
- 41.Hirsch FR, Franklin WA, Gazdar AF, Bunn PA. Early detection of lung cancer: clinical perspectives of recent advances in biology and radiology. Clin Cancer Res. 2001;7:5–22. [PubMed] [Google Scholar]
- 42.Lantuéjoul S, Salameire D, Salon C, Brambilla E. Pulmonary preneoplasia--sequential molecular carcinogenetic events. Histopathology. 2009;54:43–54. doi: 10.1111/j.1365-2559.2008.03182.x. [DOI] [PubMed] [Google Scholar]
- 43.Wistuba II, Behrens C, Milchgrub S, Bryant D, Hung J, Minna JD, Gazdar AF. Sequential molecular abnormalities are involved in the multistage development of squamous cell lung carcinoma. Oncogene. 1999;18:643–650. doi: 10.1038/sj.onc.1202349. [DOI] [PubMed] [Google Scholar]
- 44.Bota S, Auliac JB, Paris C, Métayer J, Sesboüé R, Nouvet G, Thiberville L. Follow-up of bronchial precancerous lesions and carcinoma in situ using fluorescence endoscopy. Am J Respir Crit Care Med. 2001;164:1688–1693. doi: 10.1164/ajrccm.164.9.2012147. [DOI] [PubMed] [Google Scholar]
- 45.Jeanmart M, Lantuejoul S, Fievet F, Moro D, Sturm N, Brambilla C, Brambilla E. Value of immunohistochemical markers in preinvasive bronchial lesions in risk assessment of lung cancer. Clin Cancer Res. 2003;9:2195–2203. [PubMed] [Google Scholar]
- 46.Wistuba II. Genetics of preneoplasia: lessons from lung cancer. Curr Mol Med. 2007;7:3–14. doi: 10.2174/156652407779940468. [DOI] [PubMed] [Google Scholar]
- 47.Wistuba II, Mao L, Gazdar AF. Smoking molecular damage in bronchial epithelium. Oncogene. 2002;21:7298–7306. doi: 10.1038/sj.onc.1205806. [DOI] [PubMed] [Google Scholar]
- 48.Leube RE, Rustad TJ. Squamous cell metaplasia in the human lung: molecular characteristics of epithelial stratification. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;61:227–253. doi: 10.1007/BF02890425. [DOI] [PubMed] [Google Scholar]
- 49.Stosiek P, Kasper M, Moll R. Changes in cytokeratin expression accompany squamous metaplasia of the human respiratory epithelium. Virchows Arch A Pathol Anat Histopathol. 1992;421:133–141. doi: 10.1007/BF01607046. [DOI] [PubMed] [Google Scholar]
- 50.Niho S, Yokose T, Suzuki K, Kodama T, Nishiwaki Y, Mukai K. Monoclonality of atypical adenomatous hyperplasia of the lung. Am J Pathol. 1999;154:249–254. doi: 10.1016/S0002-9440(10)65271-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koga T, Hashimoto S, Sugio K, Yonemitsu Y, Nakashima Y, Yoshino I, Matsuo Y, Mojtahedzadeh S, Sugimachi K, Sueishi K. Lung adenocarcinoma with bronchioloalveolar carcinoma component is frequently associated with foci of high-grade atypical adenomatous hyperplasia. Am J Clin Pathol. 2002;117:464–470. doi: 10.1309/CHXA-3MH0-B7FD-JGUL. [DOI] [PubMed] [Google Scholar]
- 52.Maeshima AM, Tochigi N, Yoshida A, Asamura H, Tsuta K, Tsuda H. Clinicopathologic analysis of multiple (five or more) atypical adenomatous hyperplasias (AAHs) of the lung: evidence for the AAH-adenocarcinoma sequence. J Thorac Oncol. 2010;5:466–471. doi: 10.1097/JTO.0b013e3181ce3b73. [DOI] [PubMed] [Google Scholar]
- 53.Noguchi M. Stepwise progression of pulmonary adenocarcinoma--clinical and molecular implications. Cancer Metastasis Rev. 2010;29:15–21. doi: 10.1007/s10555-010-9210-y. [DOI] [PubMed] [Google Scholar]
- 54.Miller RR, Nelems B, Evans KG, Müller NL, Ostrow DN. Glandular neoplasia of the lung. A proposed analogy to colonic tumors. Cancer. 1988;61:1009–1014. doi: 10.1002/1097-0142(19880301)61:5<1009::aid-cncr2820610525>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 55.Chapman AD, Kerr KM. The association between atypical adenomatous hyperplasia and primary lung cancer. Br J Cancer. 2000;83:632–636. doi: 10.1054/bjoc.2000.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mori M, Chiba R, Takahashi T. Atypical adenomatous hyperplasia of the lung and its differentiation from adenocarcinoma. Characterization of atypical cells by morphometry and multivariate cluster analysis. Cancer. 1993;72:2331–2340. doi: 10.1002/1097-0142(19931015)72:8<2331::aid-cncr2820720808>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 57.Min JH, Lee HY, Lee KS, Han J, Park K, Ahn MJ, Lee SJ. Stepwise evolution from a focal pure pulmonary ground-glass opacity nodule into an invasive lung adenocarcinoma: an observation for more than 10 years. Lung Cancer. 2010;69:123–126. doi: 10.1016/j.lungcan.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 58.Soda H, Nakamura Y, Nakatomi K, Tomonaga N, Yamaguchi H, Nakano H, Nagashima S, Anami M, Hayashi T, Tsukamoto K, et al. Stepwise progression from ground-glass opacity towards invasive adenocarcinoma: long-term follow-up of radiological findings. Lung Cancer. 2008;60:298–301. doi: 10.1016/j.lungcan.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 59.Sakamoto H, Shimizu J, Horio Y, Ueda R, Takahashi T, Mitsudomi T, Yatabe Y. Disproportionate representation of KRAS gene mutation in atypical adenomatous hyperplasia, but even distribution of EGFR gene mutation from preinvasive to invasive adenocarcinomas. J Pathol. 2007;212:287–294. doi: 10.1002/path.2165. [DOI] [PubMed] [Google Scholar]
- 60.Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, Kerr KM, Laird-Offringa IA. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS One. 2011;6:e21443. doi: 10.1371/journal.pone.0021443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takamochi K, Ogura T, Suzuki K, Kawasaki H, Kurashima Y, Yokose T, Ochiai A, Nagai K, Nishiwaki Y, Esumi H. Loss of heterozygosity on chromosomes 9q and 16p in atypical adenomatous hyperplasia concomitant with adenocarcinoma of the lung. Am J Pathol. 2001;159:1941–1948. doi: 10.1016/S0002-9440(10)63041-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westra WH. Early glandular neoplasia of the lung. Respir Res. 2000;1:163–169. doi: 10.1186/rr28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mori M, Tezuka F, Chiba R, Funae Y, Watanabe M, Nukiwa T, Takahashi T. Atypical adenomatous hyperplasia and adenocarcinoma of the human lung: their heterology in form and analogy in immunohistochemical characteristics. Cancer. 1996;77:665–674. [PubMed] [Google Scholar]
- 64.Jackson EL, Olive KP, Tuveson DA, Bronson R, Crowley D, Brown M, Jacks T. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 65.Ji H, Li D, Chen L, Shimamura T, Kobayashi S, McNamara K, Mahmood U, Mitchell A, Sun Y, Al-Hashem R, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. doi: 10.1016/j.ccr.2006.04.022. [DOI] [PubMed] [Google Scholar]
- 66.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Subramanian J, Simon R. Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen G, Kim S, Taylor JM, Wang Z, Lee O, Ramnath N, Reddy RM, Lin J, Chang AC, Orringer MB, et al. Development and validation of a quantitative real-time polymerase chain reaction classifier for lung cancer prognosis. J Thorac Oncol. 2011;6:1481–1487. doi: 10.1097/JTO.0b013e31822918bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Seranno S, Meuwissen R. Progress and applications of mouse models for human lung cancer. Eur Respir J. 2010;35:426–443. doi: 10.1183/09031936.00124709. [DOI] [PubMed] [Google Scholar]
- 70.Motoi N, Szoke J, Riely GJ, Seshan VE, Kris MG, Rusch VW, Gerald WL, Travis WD. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am J Surg Pathol. 2008;32:810–827. doi: 10.1097/PAS.0b013e31815cb162. [DOI] [PubMed] [Google Scholar]
- 71.Takeuchi T, Tomida S, Yatabe Y, Kosaka T, Osada H, Yanagisawa K, Mitsudomi T, Takahashi T. Expression profile-defined classification of lung adenocarcinoma shows close relationship with underlying major genetic changes and clinicopathologic behaviors. J Clin Oncol. 2006;24:1679–1688. doi: 10.1200/JCO.2005.03.8224. [DOI] [PubMed] [Google Scholar]
- 72.Russell PA, Wainer Z, Wright GM, Daniels M, Conron M, Williams RA. Does lung adenocarcinoma subtype predict patient survival?: A clinicopathologic study based on the new International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary lung adenocarcinoma classification. J Thorac Oncol. 2011;6:1496–1504. doi: 10.1097/JTO.0b013e318221f701. [DOI] [PubMed] [Google Scholar]
- 73.Yoshizawa A, Motoi N, Riely GJ, Sima CS, Gerald WL, Kris MG, Park BJ, Rusch VW, Travis WD. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod Pathol. 2011;24:653–664. doi: 10.1038/modpathol.2010.232. [DOI] [PubMed] [Google Scholar]
- 74.Warth A, Muley T, Meister M, Stenzinger A, Thomas M, Schirmacher P, Schnabel PA, Budczies J, Hoffmann H, Weichert W. The novel histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society classification system of lung adenocarcinoma is a stage-independent predictor of survival. J Clin Oncol. 2012;30:1438–1446. doi: 10.1200/JCO.2011.37.2185. [DOI] [PubMed] [Google Scholar]
- 75.Hayes DN, Monti S, Parmigiani G, Gilks CB, Naoki K, Bhattacharjee A, Socinski MA, Perou C, Meyerson M. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J Clin Oncol. 2006;24:5079–5090. doi: 10.1200/JCO.2005.05.1748. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida A, Tsuta K, Watanabe S, Sekine I, Fukayama M, Tsuda H, Furuta K, Shibata T. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer. 2011;72:309–315. doi: 10.1016/j.lungcan.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 77.Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, Miller CR, Socinski MA, Parsons AM, Thorne LB, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS One. 2012;7:e36530. doi: 10.1371/journal.pone.0036530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gu Y, Lin S, Li JL, Nakagawa H, Chen Z, Jin B, Tian L, Ucar DA, Shen H, Lu J, et al. Altered LKB1/CREB-regulated transcription co-activator (CRTC) signaling axis promotes esophageal cancer cell migration and invasion. Oncogene. 2012;31:469–479. doi: 10.1038/onc.2011.247. [DOI] [PubMed] [Google Scholar]
- 79.Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 80.Conde E, Suarez-Gauthier A, García-García E, Lopez-Rios F, Lopez-Encuentra A, García-Lujan R, Morente M, Sanchez-Verde L, Sanchez-Cespedes M. Specific pattern of LKB1 and phospho-acetyl-CoA carboxylase protein immunostaining in human normal tissues and lung carcinomas. Hum Pathol. 2007;38:1351–1360. doi: 10.1016/j.humpath.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 81.Gao Y, Ge G, Ji H. LKB1 in lung cancerigenesis: a serine/threonine kinase as tumor suppressor. Protein Cell. 2011;2:99–107. doi: 10.1007/s13238-011-1021-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD, et al. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007;26:5911–5918. doi: 10.1038/sj.onc.1210418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- 84.Sanchez-Cespedes M. The role of LKB1 in lung cancer. Fam Cancer. 2011;10:447–453. doi: 10.1007/s10689-011-9443-0. [DOI] [PubMed] [Google Scholar]
- 85.Ghaffar H, Sahin F, Sanchez-Cepedes M, Su GH, Zahurak M, Sidransky D, Westra WH. LKB1 protein expression in the evolution of glandular neoplasia of the lung. Clin Cancer Res. 2003;9:2998–3003. [PubMed] [Google Scholar]
- 86.Carretero J, Medina PP, Blanco R, Smit L, Tang M, Roncador G, Maestre L, Conde E, Lopez-Rios F, Clevers HC, et al. Dysfunctional AMPK activity, signalling through mTOR and survival in response to energetic stress in LKB1-deficient lung cancer. Oncogene. 2007;26:1616–1625. doi: 10.1038/sj.onc.1209951. [DOI] [PubMed] [Google Scholar]
- 87.Ekman S, Wynes MW, Hirsch FR. The mTOR pathway in lung cancer and implications for therapy and biomarker analysis. J Thorac Oncol. 2012;7:947–953. doi: 10.1097/JTO.0b013e31825581bd. [DOI] [PubMed] [Google Scholar]
- 88. Available from: http: //clinicaltrials.gov/2013.
- 89.Kriegbaum MC, Jacobsen B, Santoni-Rugiu E, Ploug M. C4.4A er en ny biomarkør i ikke-småcellet lungekræft. BestPractice. 2012;5:58–61. [Google Scholar]