

Abstract
Alcohol consumption is associated with an increased risk of breast cancer, increasing linearly even with a moderate consumption and irrespectively of the type of alcoholic beverage. It shows no dependency from other risk factors like menopausal status, oral contraceptives, hormone replacement therapy, or genetic history of breast cancer. The precise mechanism for the effect of drinking alcohol in mammary cancer promotion is still far from being established. Studies by our laboratory suggest that acetaldehyde produced in situ and accumulated in mammary tissue because of poor detoxicating mechanisms might play a role in mutational and promotional events. Additional studies indicated the production of reactive oxygen species accompanied of decreases in vitamin E and GSH contents and of glutathione transferase activity. The resulting oxidative stress might also play a relevant role in several stages of the carcinogenic process. There are reported in literature studies showing that plasmatic levels of estrogens significantly increased after alcohol drinking and that the breast cancer risk is higher in receptor ER-positive individuals. Estrogens are known that they may produce breast cancer by actions on ER and also as chemical carcinogens, as a consequence of their oxidation leading to reactive metabolites. In this review we introduce our working hypothesis integrating the acetaldehyde and the oxidative stress effects with those involving increased estrogen levels. We also analyze potential preventive actions that might be accessible. There remains the fact that alcohol drinking is just one of the avoidable causes of breast cancer and that, at present, the suggested acceptable dose for prevention of this risk is of one drink per day.
Keywords: Alcohol, Ethanol, Acetaldehyde, Free radicals, Mammary cancer, Oxidative stress, Estrogens, Polyphenols
Core tip: Excessive alcohol drinking of any type of alcoholic beverage is known to linearly increase the risk of breast cancer. The precise mechanism involved to produce this effect is still far from being established. In this review we introduce our present working hypothesis integrating the participation in cancer initiation and promotion of: in situ accumulation of the acetaldehyde metabolite, the local promotion of oxidative stress and the effects of the increased levels of estrogen occurring during alcohol drinking. Some potential preventive alternatives were also analyzed.
EPIDEMIOLOGY OF ALCOHOL DRINKING INDUCED BREAST CANCER
Alcohol drinking is causally related to an increased risk of cancer of the upper aero-digestive tract, liver, colorectum, and female breast[1-4]. Of particular concern is the case of breast cancer promotion by chronic alcohol consumption. In effect, breast cancer is an extremely relevant cause of disease and death in women and alcohol intake is one of the few modifiable risk factors for breast cancer. The International Agency for Research on Cancer recently reported that by 2010 more than 100 epidemiological studies have evaluated the association between the consumption of alcoholic beverages and the risk of breast cancer[4]. Further, combined analysis of data from 53 studies around the world showed a clear dose-response relationship between alcohol consumption and increased risk of breast cancer[2]. This study showed 9% increase in risk per 10 g intake of alcohol per day. In fact, other recently epidemiological studies in a total of 1280296 middle-aged women in the United Kingdom reported that even drinking women consuming an average in only 10 g of alcohol (one drink) per day showed 12% increased risk of breast cancer[1]. In addition, a detailed prospective study in 254870 women, made in eight European countries, reported that 5% of the female breast cancer was attributable to alcohol consumption[5].
Unfortunately, there is limited information regarding a possible mechanism for this effect and about positive modulatory effects of dietary factors if alcohol drinking is not avoided. That issue was of particular interest in the studies made by our laboratory and by other workers[6-9]. In this review we are going to describe our working hypothesis on the pathogenesis of alcohol drinking induced mammary cancer and its potential prevention.
ACETALDEHYDE FORMATION, ACCUMULATION AND EFFECTS
Despite the relevance of the problem, the mechanism for ethanol-increased risk of breast cancer remains unknown.
Several lines of evidence indicate that acetaldehyde, a product of alcohol metabolism, might play a role in alcohol-related carcinogenic effects in different target tissues[10,11]. Animal experiments have clearly shown that acetaldehyde is an established mutagenic and carcinogenic chemical[12-14]. In addition, other factors such as oxidative stress, altered methyl transfer, abnormal metabolism of vitamin A and retinoic acid and perturbed levels of hormones might be of particular relevance according to the target tissue involved[3,8].
Concerning the specific case of mammary tissue, the knowledge of the acetaldehyde concentrations was of particular interest to our laboratory. In general terms, the concentration of acetaldehyde in any tissue, and also in the mammary, depends of its ability to produce it in situ, plus the additional arriving to the organ via blood supply and in the capacity of the given organ to degrade it[15].
The ability of mammary tissue to produce acetaldehyde in situ was studied in our laboratory. The available information was scarce to null at that time. Two different pathways of bioactivation of ethanol to acetaldehyde were reported by our laboratory to be present in the rat mammary tissue. One is in the cytosolic fraction and the other is in microsomes[16,17]. Both were preliminary characterized and showed to be susceptible to inhibitory effects by chemicals present in food (this last subject will be analyzed ahead when discussing the preventive potential of findings).
The enzyme involved in the cytosolic pathway was evidenced to be xanthine oxidoreductase (XOR) because of its susceptibility to inhibitory effects of allopurinol and by the ability of the process to occur only when the presence of NAD+ was accompanied by substrates of the XOR form of the enzyme such as hypoxanthine, xanthine, caffeine, theobromine, theophylline or 1,7-dimethylxanthine[16].
Moreover, it is also known that during acute alcohol intoxication, there is an increased purine degradation and hyperuricemia[18,19]. The enhanced supply of purines resulting from this process could also provide an extra amount of cofactors for the XOR-mediated pathway of metabolism of ethanol to acetaldehyde (and also free radicals) in the mammary tissue.
The presence of XOR in mammary tissue is well known[20,21], and past studies from our laboratory evidenced their presence in high amounts in the rat mammary tissue epithelial cells[22]. Interestingly, the activity of this cytosolic pathway significantly increased after repetitive alcohol drinking through a Lieber and De Carli diet for 28 d[22].
The contribution of enzymes present in cytosolic fraction of mammary tissue, other than XOR, to the activation of ethanol to acetaldehyde, for example, alcohol dehydrogenase (ADH) may be more limited. On one hand, previous studies[23] showed that no ADH activity was found in homogenates of rat mammary tissue. More recently, our laboratory reported traces of ADH activity in the cytosolic fraction of mammary tissue that was about 16 times smaller than in the liver[15]. By the other hand, Triano et al[24] reported that human mammary tissue contains a class of ADH having a limited potential to transform alcohol to acetaldehyde.
In addition to the mammary tissue cytosolic pathway of ethanol oxidation to acetaldehyde described above, our laboratory reported the presence of another one occurring in the microsomal fraction of that tissue[17].
In our earlier studies of that pathway it was established that the enzymatic transformation involved was oxygen and NADPH-dependent, but that the cytochrome P450 was not involved because it was not inhibited by CO:O2 (80:20 v/v) or by SFK525A[17].
Interestingly, this microsomal transformation of alcohol to acetaldehyde was strongly inhibited by diphenyleneiodonium (DPI), sodium diethyldithiocarbamate, sodium azide, nordihydroguaiaretic acid but not by dapsone, aminotriazole or indomethacin. Those results suggested us the potential participation in this biotransformation of an oxidase or a peroxidase but not of lactoperoxidase or cyclooxygenase[17]. We were unable to detect the formation of either hydroxyl or 1-hydroxyethyl radicals in those studies. In the course of following studies performed at the opportunity in rats exposed to a standard Lieber and De Carli diet for 28 d, we observed the induction not only of XOR cytosolic activation pathways but also of the microsomal one[22].
That was of particular significance, since we showed in the course of additional recent work that the enhancing effect is not due to the participation of CYP2E1 after chronic alcohol drinking as it is known for the liver microsomal fraction[25].
Further, acetone, another inducer of microsomal CYP2E1-mediated alcohol metabolism in liver microsomes, failed to enhance ethanol bioactivation and CYP2E1 enzymatic activity in the microsomal rat mammary tissue[25]. To ensure that CYP2E1 enzymatic activity in the microsomal rat mammary tissue was not present or was very low, we also included in those studies determination of chlorzoxazone hydroxylase activity. This activity was considered in literature as having a significant response to the presence of CYP2E1 in a given tissue[25]. We were not able to detect CYP2E1-mediated metabolism of chlorzoxazone in the mammary tissue microsomal fraction despite the fact we employed a particularly sensitive procedure developed in our laboratory, where the formation of 6-hydroxychlorzoxazone metabolite could be determined by HPLC with coulometric detection[25]. That further excluded the participation of CYP2E1 in this microsomal pathway of alcohol metabolism in the mammary tissue and encouraged us to challenge the possibility that a peroxidase or a lipoxygenase was involved in that process instead. That hypothesis was originally coined because of the potent inhibitory effect of nordihydroguaiaretic acid. This polyphenol is a known inhibitor of lipoxygenases[26,27]. We also envisaged the possibility that the potent inhibitory effect of DPI could be suggesting the additional participation of a NADPH oxidase enzyme as a supplier of hydrogen peroxide. Under this view, the role of NADPH oxidase would be the generation of the necessary co-substrate required by lipoxygenase to exert its activity against xenobiotics[8]. On behalf of this hypothesis is the fact that the specific inhibitory effect of DPI on NADPH oxidase is well established[28]. That hypothesis visualizes the overall process of microsomal ethanol oxidation to acetaldehyde in rat mammary tissue as a cooperative mechanism between NADPH oxidase and lipoxygenase (Figure 1).
Figure 1.
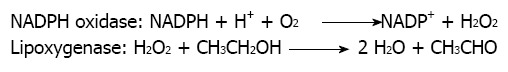
Cooperative mechanism between NADPH oxidase and lipoxygenase in the oxidation of ethanol to acetaldehyde in microsomes.
The concentration of acetaldehyde in different tissues depends on the production and degradation of acetaldehyde and on the amount of it arriving to the given tissue via blood supply. In a detailed study from our laboratory, we attempted to evaluate how much acetaldehyde accumulates in mammary tissue after single doses of ethanol and also the levels of it arriving via blood at different periods of time. The studies were performed at three different dose levels (high, medium and low). Values were compared with the equivalent occurring in liver[15].
The levels of acetaldehyde in mammary tissue were higher than in plasma and lower than in liver for at least 15 h for the higher dose tested or six hours for the medium dose or two hours for the case of the lower one. The shape of the curve concentration of acetaldehyde in mammary tissue vs time after p.o. administration always mimicked that observed in liver. But, more important, the levels of acetaldehyde in plasma were similar for the three ethanol doses given[15]. These results suggest that acetaldehyde present in the mammary tissue (and in the liver) reflects the balance between the ability of these tissues to generate acetaldehyde and the one to metabolize and excretes it. The liver is able to get rid of the acetaldehyde formed with the participation of aldehyde dehydrogenase (ALDH) and glutathione transferase (GST)[19]. In the case of mammary tissue the situation appears to be different. On one hand, ADH activity is about 16 times smaller than in the liver, but perhaps more important, ALDH activity present in three subcellular fractions tested in our study were in all of them at least ten times smaller than in the liver[15].
The overall conclusion of those results was that acetaldehyde is able to accumulate during significantly relevant periods of time in mammary tissue mainly as result of its ability to oxidize ethanol to acetaldehyde in situ and to a less important extent to the arrival of it via blood and produced elsewhere (for example, in the liver)[15].
Which might be the expected consequences of acetaldehyde accumulation in a given tissue for long lasting periods of time?
It is important to point out that acetaldehyde is a reactive chemical that proved to be toxic, mutagenic and carcinogenic and able to interact with many cellular constituents, including DNA, proteins (nuclear proteins), lipids (including nuclear lipids), glutathione and others[19,29,30].
The reactions of acetaldehyde with DNA are of particular concern because they clearly suggest that it acts as a tumor initiator and mutagenic compound. Structures of the identified DNA adducts of acetaldehyde were recently reviewed in literature[31-33]. Representative structures are shown in Figure 2.
Figure 2.
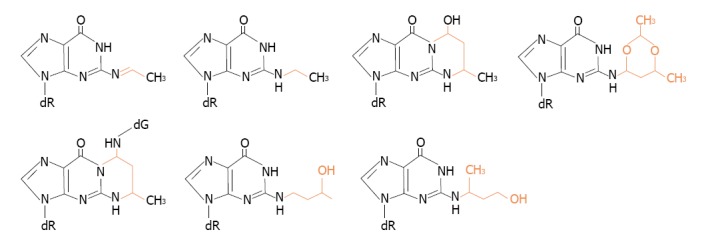
Structures of some of acetaldehyde-guanine adducts.
DNA adducts may cause polymerase errors and induce mutations in critical genes. Furthermore, they may lead to mutations that activate proto-oncogenes and inactivate tumor suppressor genes in replicating cells[34,35].
Notwithstanding, there are known DNA repair enzymes that can modify DNA damage caused by acetaldehyde, removing its adducts from DNA. The relation between adducts formation and their repair would be relevant to molecular epidemiology of cancer, particularly in replicating cells[35].
It is of particular interest in this respect that studies by Freudenheim et al[36] reported results that were consistent with an increased likelihood of tumors with p53 mutations for pre-menopausal breast cancer with increased alcohol intake 10 or 20 years previous. For intakes of 16 or more drinks per month in the period of 20 years before the interview compared with non drinkers, the OR was 5.25 (95%, 1.48-18.58)[36].
Acetaldehyde is also known to bind to proteins and amino acids and the subject might also be relevant to potential carcinogenic effects of alcohol drinking in different organs.
However, non specific studies related to breast cancer and alcohol drinking and the potential participation of acetaldehyde protein binding are available at present in literature. Notwithstanding, it is well known that acetaldehyde binds to reactive lysine residues, to some aromatic amino acids, cysteine, or to free alpha-amino groups, such as the terminal valine of hemoglobin. The subject was thoroughly reviewed by Niemelä[37], who also pointed the relevance of those interactions in different pathologies occurring in several tissues other than the breast.
In this respect, it might be relevant to take in consideration that acetaldehyde was shown to bind covalently to liver nuclear proteins[30]. In the case of liver, the biotransformation of ethanol to acetaldehyde may occur not only in the cytoplasm or in the endoplasmic reticulum but also in the nearby outer nuclear membrane[30,38]. Acetaldehyde, despite being a reactive molecule, does not have the equivalent reactivity to the one of a low molecular weight free radical (for example, trichloromethyl or 1-hydroxyethyl). This allows it to travel from cellular distant sites (cytoplasm or endoplasmic reticulum) to the nucleus and interact with DNA and nuclear proteins or lipids when acetaldehyde accumulates for long periods of time as those observed to occur in mammary tissue after alcohol drinking.
In the case of other liver carcinogenic chemicals, like carbon tetrachloride, a good correlation was observed between the covalent binding of its reactive metabolites to nuclear proteins and carcinogenicity. The interaction involved histone and non-histone proteins. Acidic and residual nuclear proteins were the favorite targets of that interaction and showed a good correlation between those covalent interactions and carcinogenicity[39].
In addition, is important to take in mind that ethanol is a well known inducer of CYP2E1-mediated metabolisms[19] and that this enhancing effect was observed in the outer nuclear membrane of the liver[40,41].
Far reaching consequences may also be expected from alterations in nuclear proteins resulting from an attack by acetaldehyde. In effect, it is known that nuclear proteins are critical for cell division, growth, differentiation and apoptosis[42-44]. A clear example of relevant nuclear proteins is the one of those involved in the cell cycle clock. They include cyclins, cyclin-dependent kinases; protein coded by proto-oncogenes and tumor suppressor genes (such as the myc and Bcl proteins or the pRB or p53 and others); enzymes required for DNA synthesis and for DNA repair[45,46].
Critical evidence that some of these activities may be affected by acetaldehyde produced during ethanol consumption was provided by several authors, who showed that chronic ethanol consumption results in inhibition of the DNA alkylation repair by O6-methylguanine transferase (O6-MeGT), which removes alkyl groups from the O6-position of guanine. Acetaldehyde has been shown to inhibit O6-MeGT[10,47-49]. Further, exposure to ethanol was reported in recent studies to interfere with the cell division machinery by perturbing the key G2 cycling protein[50].
Concerning the covalent binding to nuclear lipids, it should be noted that all lipid fractions were involved in our studies performed in the case of liver. Part of the covalent binding was acid labile, but a significant part was not. The labile portion might be attributable to the presence of Schiff base adducts of acetaldehyde with amino groups from phospholipids[30]. In effect, previous studies from other laboratories reported that amino-containing phospholipids like phosphatidylserine and phosphatidylethanolamine formed Schiff base adducts[51,52].
The fraction of covalent binding resistant to acid hydrolysis might be attributed in part to addition reactions of 1-hydroxyethyl on unsaturated fatty acid double bonds or on cholesterol moieties and on nitrogen-containing phospholipids. That behavior was previously reported for the case of other carbon-centered free radicals, for example the trichloromethyl radicals[53,54].
Another part of the covalent binding to lipids resistant to mild acid media might also be attributed to the formation of ethyl esters of fatty acids. It is known that liver microsomes and cytosol contain enzyme systems able to esterify free fatty acids or transesterify other esters to ethyl esters[55-57]. Whether nuclear preparations from mammary tissue have similar ability remains to be established. Irrespectively of the present lack of knowledge about the structure of the reaction products formed, it is not unexpected to envisage that the alteration of nuclear membranes by their reaction with either acetaldehyde or 1-hydroxyethyl or resulting from ester formation with ethanol, might lead to profound alterations in liver nuclear functions. It is well established that ethanol also strikingly alters other liver membranes and their fluidity[58]. It is known that nuclear lipids may be part of an intracellular signaling system modulating protein kinase C, an enzyme involved in phosphorylation of nuclear proteins[59]. The covalent binding to nuclear phospholipids deserves special interest since they have a known ability to regulate gene function, nucleosome structure, RNA synthesis and activation of DNA polymerase alpha[59-65]. Past studies from the laboratory reported correlations between alterations in nuclear lipids resulting from a free radical attack and the different response of the rat strains employed to the carcinogenic effects of CCl4[54].
FREE RADICALS, OXIDATIVE STRESS AND EFFECTS
Oxidative stress was also considered to be a potential factor involved in the alcohol drinking promoted carcinogenic effect on mammary tissue. Notwithstanding this, no evidence of its occurrence in mammary tissue were reported until our recent studies where we showed that oxidative stress can be observed in this tissue after an experimental protocol of 28 d of alcohol drinking through the Lieber and De Carli diet[15,22,25]. It is relevant at this point to have present what “oxidative stress” is all about. Oxidative stress has been defined as an imbalance between oxidants and antioxidants in favor of the former, resulting in an overall increase in cellular levels of reactive oxygen species (ROS). Many pathways play a role in how ethanol induces oxidative stress in the liver (the issue has exercised the interest of numerous researches for years) and is reviewed in Cederbaum et al[66]. Ethanol-induced oxidative stress in the liver included the ability to generate free radicals able to initiate the process (for example, 1-hydroxyethyl and hydroxyl radicals) and formation of hydroperoxides, peroxides, superoxide, H2O2 and other. In the case of liver exposed to alcohol, the evidence included the formation of products derived from the attack of ROS to relevant target molecules (for example, DNA, proteins or lipids) like 8-oxodeoxyguanosine, protein carbonyls, decreases in protein sulfhydryls, lipid hydroperoxides and other products such as malondialdehyde or 4-hydroxy-2-nonenal (4HNE)[66].
The evidence that cellular antioxidant defenses were overwhelmed might include determinations of the variety of enzymatic and non-enzymatic mechanisms that have evolved to protect cells against ROS such as superoxide dismutase, catalase, glutathione peroxidase, GST and other enzymes. Also are relevant low molecular weight antioxidants, such as glutathione itself, vitamin E, ascorbate, vitamin A and others. In summary, toxicity induced by ROS reflects the balance between the rates of production of ROS compared to the rates of removal of ROS plus repair of damage to cellular macromolecules[66].
After these introductory remarks learnt from free radical cell injury and from the effects of alcohol on liver, let us to introduce what kind of evidence we obtained for the case of mammary tissue.
Initially we began to consider the possibility that during alcohol drinking an oxidative stress process occurred in mammary tissue because in our hands repetitive alcohol drinking exposure led to increased XOR and lipoxygenase activities in the rat mammary tissue[22]. In effect, increased XOR and lipoxygenase activities by themselves would lead to not only higher generation of ROS, but also when occurring in the presence of ethanol, to increased generation of free radicals. The formation of hydroxyl radicals was detected during XOR mediated cytosolic alcohol metabolism[16].
Other reason leading to a similar potential effect was derived from our observation that acetaldehyde accumulates in mammary tissue during repetitive alcohol drinking[15]. It is well known that the generation of increased level of acetaldehyde may provoke decreases in GSH as it was respectively observed in liver[66]. Further, GSH and GSH-dependent enzymes such as glutathione peroxidase, glutathione reductase and glutathione transferase are a vital first line defense against oxidative stressful conditions[66].
The initial observation indicating that alcohol drinking could provoke oxidative stress that we obtained was derived from our studies in the t-butylhydroperoxide-induced chemiluminescence in mammary tissue homogenates from rats exposed to repetitive alcohol drinking when compared to those from control animals[15].
The samples from alcohol treated animals had a completely different response to the t-butylhydroperoxide challenge and the shape of their response curve compared to that observed in the control samples clearly suggested that animals exposed to alcohol have diminished defenses against t-butylhydroperoxide challenge[15]. However, that experiment did not give indication about the nature of the defensive process that was compromised. Further, it did not show which target molecule involved the potential oxidative process that was occurring.
To answer those questions, other studies were performed and we found that after repetitive alcohol drinking, the levels of lipid hydroperoxides in mammary tissue were significantly increased. Further, the protein sufhydryl and vitamin E content in the alcohol-treated animals decreased. However, in those experiments we did not observe protein carbonyl enhancement or increased formation of 8-hydroxyguanine[22]. One reason to explain that different response to alcohol drinking-provoked oxidative stress might be that much longer periods of expose to alcohol could be necessary. That is in our work we were detecting early manifestations of alcohol drinking induced oxidative stress in mammary tissue.
If that was the case, after more prolonged periods of alcohol drinking, other relevant effects might occur derived from generation of harmful reactive aldehydes produced from lipid peroxidation such as malondialdehyde and 4HNE. They are able to react with proteins, lipids and with sufhydryl-containing molecules of low molecular weight[33,66]. See Figure 3 for their structures.
Figure 3.
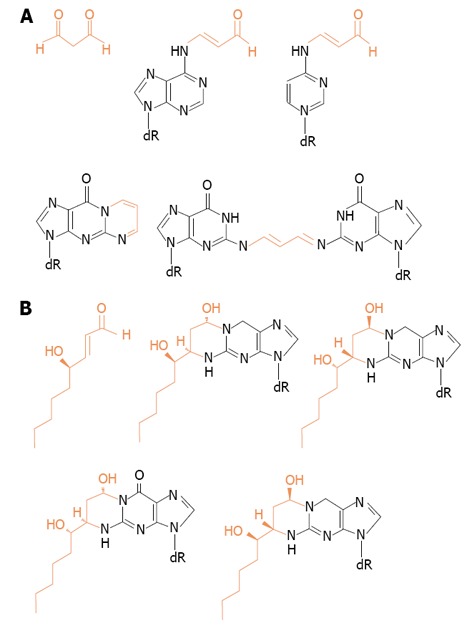
Structures of adducts formed between DNA bases and (A) malondialdehyde or (B) 4-hydroxy-2-nonenal.
The reaction with sufhydryl groups that we observed might be of significance considering that many sufhydryl-containing enzymes play a key role in cell functioning and also in cell signaling processes[67-70].
Free radicals not only are relevant in relation to the carcinogenesis initiation step. The progress of human breast cancer to the metastasis stage was linked to hydroxyl radical-induced DNA damage[71].
These findings should be of some significance in light of the well established correlation between oxidative stress and tumor promotion and cancer[72-74].
INCREASED LEVELS OF ESTROGENS AND CONSEQUENCES
Increased plasmatic levels of estrogen by alcohol drinking have been clearly demonstrated in controlled feeding studies in human female volunteers[75-84]. Further, the hypothesis about the role of estrogens in alcohol drinking-induced breast cancer is strongly supported by the fact that their higher risk was related to estrogen receptor ER-positive rather than to ER-negative mammary tumors[85-88]. However, estrogens may exert their carcinogenic effects in mammary tissue not only via ER, but also by direct damage to DNA[89]. Metabolic activation of estrogens to catechol-3,4-quinones is an example. These reactive metabolites are able to interact with DNA to give mutational events[90-93]. Those increased levels of estrogen after alcohol drinking would arise because alcohol increases aromatase activity and that leads to enhanced conversion of testosterone to estrogen, resulting in decreased testosterone and increased estrogen level[94].
These considerations might be of particular relevance, since there are available in literature clear evidences that high levels of estrogen in blood are associated with an increased risk of breast cancer[94].
One major mechanism of action of estrogens is that by which estrogen stimulates cell proliferation through nuclear ER-mediated signaling pathways, thus resulting in an increased risk of genomic mutations during DNA replications[95-97].
Another pathway involves estrogen metabolism that is mediated by cytochrome P450 1B1, which generates 2- and 4-catechol estrogens that easily autoxidize to the respective quinones, even without the need for enzymatic or metal ion catalysis[98]. At this point it is particularly relevant to do mention that alcohol consumption was reported to significantly increase the rate of NADPH-dependent oxidative metabolism of estrogens to quinones in females. CYP1B1 had a distinct, selective activity for the 4-hydroxylation of estradiol and estrone[99]. This inductive effect of alcohol drinking might increase chances of participation of cytochrome-mediated pathway in relation to other related to direct interactions of the estrogens with ER.
It is our idea that, under conditions where oxidative stress or peroxidizing conditions occur (like those reported by our laboratory to occur during exposure of mammary tissue to alcohol), the transformation of one to the other might be even further facilitated.
In addition, o-quinones are also potent redox-active compounds[98,100]. For example, these quinones can be reductively detoxified by a quinone reductase in a two-electron transfer process[101]. However, they can also undergo redox cycling with the semiquinone form of P450 reductase (a one-electron process) and generate superoxide radicals, that in the presence of iron or other transition metal give hydroxyl radicals[100] (Figure 4).
Figure 4.
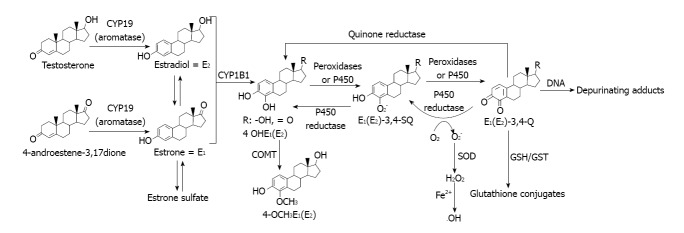
Estrogen oxidation to reactive metabolites that bind to DNA to generate depurinating adducts.
By the other hand, the estrogen quinones can directly damage DNA leading to genotoxic effects[102,103]. DNA adducts of estrogen quinones have been detected in the mammary glands of ACI rats treated with 4-hydroxyestradiol or its quinones[103]. Further, recently developed highly sensitive LC/MS-MS procedures were used to analyze DNA from human breast tumors and their adjacent tissue and the DNA adducts formed from estrogen quinones were detected[104].
All these results suggest that these mechanisms might be relevant for the carcinogenic effects of estrogens by themselves. However, there is not available in literature evidence of their formation in mammary tissue from alcohol drinking animals or humans at present.
As part of our present working hypothesis about the contribution of estrogens in the alcohol drinking effects in mammary tissue, we assumed that these estrogen quinones might be formed (Figure 4). The reasons for that hypothesis are going to be described ahead and are related to some of our results.
It is relevant to understand our “views” about the cooperative effects between alcohol metabolism to acetaldehyde, its ability to promote oxidative stress and the participation of induced levels of estrogen in carcinogenic effects in mammary tissue to describe what happens with the catechol estrogens and the estrogen quinones after their formation.
The catechol estrogens may be further metabolized by O-methylation, by reaction with glutathione, and also by glucuronidation and sufation[105,106].
Interestingly, catechol estrogen metabolites display a less intense ER-mediated estrogenic activity, implying that catechol estrogens are attributed to have both, direct genotoxic effects as well as ER-mediated tumor promoter actions[107]. Further, sulfate and glucuronide conjugates seem to play a role for free estrogens and, in general, it is at present a controversy about conjugation metabolism, if they may mitigate the catechol estrogen-mediated carcinogenic properties[107].
Notwithstanding, being the conjugated estrogens more water-soluble they seemed to be more readily excreted than the lipophilic parent estrogens[107]. These suggest that the conjugation pathway is considered as a protection mechanism against damage caused by reactive metabolites of estrogens[105].
An important aspect of the mechanism of generation of estrogen quinones from catechol estrogens concerns the resulting formation of ROS during the process. In effect, ROS are generated via the redox cycling between catechol estrogens and their quinone analogs[108-110] (Figure 4).
Catechol estrogens can be oxidized by any oxidative enzyme or metal ions such as Cu2+ or Fe3+ to give rise to semiquinones and o-quinones[108-110]. Is our hypothesis that it might very well be that this activating pathway of the catechol estrogens was stimulated during alcohol drinking by the inductive effect of alcohol on mammary tissue XOR and lipoxygenase or even more likely, by the alcohol drinking promoted oxidative stress that was observed[15,22,25].
The reduction of o-quinones back to semiquinones and catechols by P450 reductase provides an opportunity to generate ROS, including superoxide and hydroxyl radicals[107].
In our experiments on the effect of alcohol drinking on rat mammary tissue we were able to detect the formation of hydroxyl radicals and lipid hydroperoxides during the metabolism of ethanol in this tissue[16,22].
In those studies we also reported that repetitive alcohol drinking provoked the depletion of defenses against oxidative stress insult due to significantly reduced levels of glutathione, α-tocopherol and defensive enzymes such as glutathione transferase and glutathione reductase[25]. Besides the consequences that those effects have on the oxidative stress process itself and in the promotion of the carcinogenic process[73], this might have additional relevant consequences for the contribution of estrogens to the mammary carcinogenic process. In effect, glutathione is also able to react with the estrogen quinones and semiquinones to give glutathione conjugates in a reaction catalyzed by glutathione transferase[107]. Further, the lowered levels of glutathione would also decrease the capacity of mammary tissue to destroy hydroperoxides since it is the necessary cofactor for glutathione peroxidase to operate even when the levels of this enzyme were not decreased[25].
Decreased levels of two critical antioxidants like vitamin E and glutathione that we observed in mammary tissue after alcohol drinking might be not only of relevance for the promotion of oxidative stress[25], but also to understand that their depletion might be a potential factor of the genotoxic effect of estrogens, via their oxidative metabolism that might be enhanced[107] (Figure 4).
The increased generation of ROS produced by alcohol toxicity[66] and the effects of estrogen[107] might explain the decreased levels of α-tocopherol observed[25].
In addition, the significantly decreased levels of GST observed in our experiments on the effect of alcohol drinking on mammary tissue might also be explained as attributable to the potent inhibitory effects that specific glutathione-estrogen metabolites have on the GST molecule[111-113]; of course that remain to be proved. It might of particular relevance in light of previous observations reported by Zheng et al[114] in epidemiological studies, that alcohol consumption increased breast cancer risk in women among those who carry susceptible GST genotypes.
Not only the GSH/GST detoxicating pathway of the estrogen metabolites could be negatively modulated by alcohol drinking. Other might be the carboxymethyl transferase (COMT) pathway of catechol estrogens methylation (Figure 5). Several points of the cycle depicted in that process are already known that might be altered during alcohol drinking. For example, the S-adenosylmethionine (SAM) involved in the methyltransferase step of catechol estrogens methylation is known to be diminished because of SAM decreased hepatic biosynthesis. In alcoholic liver disease methionine adenosyltransferase capacity is affected. In fact, chronic alcoholics have hypermethioninemia and impaired methionine clearance[115]. That leads to decreased SAM biosynthesis and in turn to impact on methylation capacity of the cell and decreased antioxidant defenses[115].
Figure 5.
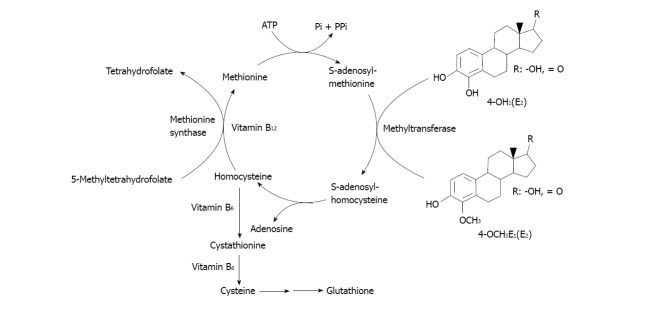
Pathways for catechol-estrogen methylation by carboxymethyl transferase.
Further, chronic exposure to ethanol has been shown not only to decrease hepatic SAM levels, but also to increase hepatic concentrations of S-adenosylhomocysteine and decrease plasma concentrations of folate in animals and humans[115,116]. In turn, homocysteine may exert pathogenic effects largely through metabolic accumulation of SAH, which is a strong non-competitive inhibitor of COMT-mediated methylation metabolisms of various catechol substrates, including those arising from estrogens[117].
In addition to depleting folate concentrations, chronic ethanol exposure decreases the activity of methionine synthase, which is required to catalyze the transfer of a methyl group from folate to homocysteine to form methionine[116,118,119].
All these facts might decrease the ability of COMT to methylate harmful catechol estrogens during chronic alcohol drinking as we describe in our working hypothesis about the concerted action of alcohol drinking, the formation of deleterious acetaldehyde, free radicals, oxidative stress and increased levels of estrogens.
WORKING HYPOTHESIS ABOUT HOW ALCOHOL DRINKING MIGHT PROMOTE CARCINOGENIC EFFECTS IN MAMMARY TISSUE
Considering the growing evidence about the relevance of acetaldehyde participation in the alcohol drinking-carcinogenic effects in target organs (for example, the aerodigestive tract) we consider that its in situ formation and accumulation in mammary tissue should play a role. Contribution of acetaldehyde arriving via blood supply should play some but a minor role.
Taking into account that oxidative stress promotion was observed and that free radical generation systems are present in mammary tissue exposed to ethanol during alcohol drinking we felt that it was relevant to consider oxidative stress as a relevant participant in the alcohol drinking promoted carcinogenic effects on mammary tissue. It is well known that it plays a role in promotion and initiation of tumors induced by other chemicals acting in other tissues as well as in mammary tissue.
Finally, alcohol drinking is also known to increase estrogen blood levels in animals and human beings and it is an established carcinogen for mammary tissue[95,120]. In women, Eriksson et al[121] observed an estrogen associated acetaldehyde elevation after alcohol intake.
Alcohol is able also to provoke even on culture studies proliferative and cell transforming effects, not only via estrogen receptors but also as a “chemical carcinogen”, mediated by products of their oxidative metabolism[87,122,123].
In light of the remarkable susceptibility of mammary tissue to alcohol drinking, clearly evidenced in epidemiological studies, we coined the working hypothesis that the resulting deleterious carcinogenic effects might arise from a sort of collaborative participation of these three components, making some of them more susceptible to the effect of the other. Some of those possibilities are going to be described at the time of proposing potential preventive treatments that we postulate based on existing linked literature.
Only the challenge of the hypothesis will tell whether is valid or not. Lack of hypothesis does lead to new experiments. New experiments may show the need of a new one. The present one in our hands is shown in Figure 6.
Figure 6.
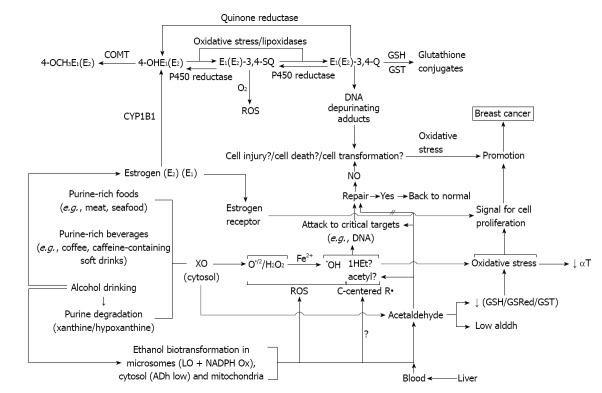
Working hypothesis about the mechanism of the promotion of mammary cancer by alcohol drinking.
POTENTIAL PREVENTIVE TREATMENTS AGAINST ALCOHOL DRINKING DELETERIOUS EFFECTS IN MAMMARY TISSUE
The first point to mention is that being alcohol drinking one of the few causes of breast cancer promotion a wise suggestion might be not to drink. Notwithstanding we feel that epidemiology shows that the real success of that advice may not be easily achievable. The present suggested tolerance for alcohol drinking by women is of one daily drink[4]. The analysis of the potential preventive strategies for women deciding to moderately exceed that suggested tolerated levels will be analyzed in light of the existing knowledge and of our working hypothesis.
The preventive alternatives to be analyzed include: (1) Blockade of acetaldehyde generation and accumulation in mammary tissue; (2) Prevention of alcohol and estrogen-induced oxidative stress in mammary tissue by antioxidants; (3) Treatments increasing defenses against estrogen provoked effects; and (4) Treatments able to be preventive in different aspects of the problem.
Inhibitory effects on acetaldehyde generating enzymes present in mammary tissue
Considering that most of the acetaldehyde accumulating in mammary tissue was generated in situ[15], we considered of particular interest to pay attention to inhibitors of the two pathways already evidenced by our laboratory, one present in cytosolic fraction and mediated by XOR and other, located at the microsomal fraction being linked to a lipoxygenase activity[8,16,17,124-126].
It was of our especial interest the search for compounds present in food or drinks or available as dietary supplements, having low toxicity, potent inhibitory effects and ideally acting on both, the cytosolic plus microsomal pathways of ethanol metabolism to acetaldehyde[127,128].
Several phytochemicals of polyphenolic nature tested appeared as potential candidates exhibiting those interesting characteristics and deserve our future attention. The most potent inhibitors among those polyphenols were flavonols quercetin, myricetin and kaempferol; also the flavones apigenin, luteolin and the polyphenols nordihydroguaiaretic acid and ellagic acid. Their structures are shown in Figure 7.
Figure 7.

Structures of some of the compounds with the most powerful ability to inhibit the oxidation of ethanol to acetaldehyde in rat mammary tissue microsomal and cytosolic fractions.
It might be important to consider at this point that several polyphenols were found to be inhibitory of carcinogenesis in laboratory investigations[129-131].
The case of ellagic acid particularly attracted our interest because it is present in several fruit sources[132]. This compound was able to diminish estrogen-mediated mammary tumorigenesis in ACI rats[133]. Other authors reported anti-proliferative activities in vitro with several cell types[134]. And more, it also exhibited beneficial anticancer effects based in a chemical study in male prostate cancer patients[135,136].
We also found that folic acid behaved a highly potent inhibitor of acetaldehyde formation in the cytosolic fraction of mammary tissue[124]. That suggests a preventive effect mediated not only by acetaldehyde depletion but also because it is a critical component of the overall methylation processes. Folate was found to be relevant in the prevention of breast cancer[137].
Removal of acetaldehyde accumulated in mammary tissue
Past studies from our laboratory evidenced that acetaldehyde accumulates in mammary tissue during long periods of time, even after single doses of ethanol given orally[15].
Removal of acetaldehyde from mammary tissue appears far more difficult than in liver. In effect, the ALDH activity in mammary tissue subcellular fractions (cytosol, mitochondria and microsomes) was at least ten times lower than in the liver[15]. In addition, the relevant mitochondrial ALDH activity might become irreversibly inhibited by the lipid peroxidation byproduct 4HNE, which is a potent inhibitor as well as a substrate for ALDH[138,139]. Lipid peroxidation might occur during the oxidative stress produced by ethanol acting on mammary tissue[22]. Further, the other potential GSH/GST system able to get rid of acetaldehyde is significantly decreased in mammary tissue after repetitive alcohol drinking[25].
Other alternative to eliminate acetaldehyde accumulation in mammary tissue might arise from cysteine administration. Cysteine is able to ameliorate the toxicity of acetaldehyde by forming a stable adduct, the 2-methylthiazolidine-4-carboxylic acid[140] (Figure 8). Its efficiency to lower the acetaldehyde accumulation in mammary tissue remains to be established. This alternative was suitably employed to prevent acetaldehyde accumulation in the oral cavity during alcohol drinking[141,142].
Figure 8.
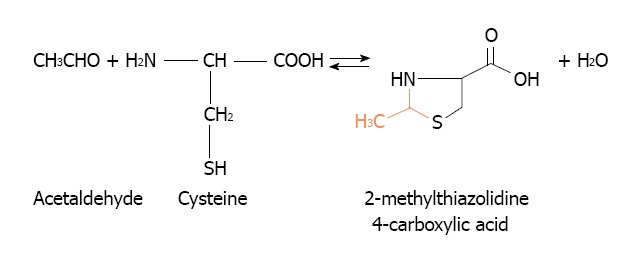
Reaction between acetaldehyde and cysteine to form the adduct 2-methylthiazolidine-4-carboxylic acid.
In the case of mammary tissue, we consider N-acetylcysteine (NAC) as having more likely potential preventive action. NAC might be less effective than cysteine in extracellular environment (e.g., saliva), but can be given at higher doses and during longer periods of time than cysteine because of its lower toxicity and because it is also a precursor of cysteine and glutathione[143]. In a number of studies since 1984, NAC also proved to have the potential to prevent cancer and other mutation-related diseases[143].
Prevention of estrogen oxidation or enhancement of detoxication of estrogen reactive metabolites
The invoking hypothesis displayed in Figure 6 assumes that a significant component in the alcohol drinking promotion of mammary cancer involves de formation of catechol estrogens that might be oxidized to their quinones, which can react with DNA.
The contribution of alcohol drinking to this deleterious pathway might involve the inductive effect on CYP1B1 and provision of relevant oxidizing conditions for the generation of the estrogen reactive metabolites and the decreased detoxication of those reactive moieties.
The contribution of antioxidants if that hypothesis was likely appears attractive to prevent this factor of the alcohol-drinking via the oxidative estrogen metabolism component. Recent studies[144] evidenced that the antioxidant N-acetylcysteine, a precursor of cysteine and of intracellular GSH[145] is able to block the initial step in the genotoxicity caused by estrogen quinones.
The authors concluded that N-acetylcysteine preventive action included multiple protective mechanisms, including nucleophilicity, antioxidant activity and inhibition of DNA adduct formation[146]. Preventive properties were also observed in human breast epithelial cells and mouse mammary epithelial cells[147].
Further, GSH, an ubiquitous antioxidant is able to react non-enzymatically with the catechol estrogen quinones or more efficiently, with the catalytic activation of GST[101,148].
Resveratrol, a natural antioxidant present in grapes and different plant products[149,150] was also effective in inhibiting the formation of estrogen-DNA adducts[146] and is also known to exert diverse anticarcinogenic effects in vitro and in vivo[149,151].
Dihydrolipoic acid, which is formed in vivo when lipoic acid is administered, can also inhibit the formation of depurinating estrogen-DNA adducts[146]. Resveratrol achieved the highest level of inhibitory effect and NAC or dihydrolipoic acid a moderate action[146].
Interestingly, our laboratory reported that alcohol-drinking decreased levels of GSH and α-tocopherol, and GST in mammary tissue and enhanced oxidative stress in mammary tissue[25].
These alcohol drinking-induced deleterious effects should decrease the detoxicating pathway of glutathione conjugates formation and give to the estrogen 3,4-quinones more opportunity to react with DNA to produce depurinating adducts.
Other harmful effect of alcohol drinking may also occur at the level of the COMT detoxicating pathway operating at the level of the catechol estrogens, especially the 4-hydroxylated metabolite of 17β-estradiol. In effect, the COMT pathway to proceed requires the participation of SAM, folic acid, vitamins B6 and B12 (Figure 5).
The SAM requiring methyl transferase then methylates the 4-OH group of the catechol estrogens to give the 4-OCH3 derivative having less toxic effects[98,107]. In the SAM-mediated transmethylation process, S-adenosylhomocysteine (SAH) is generated as a product and is hydrolyzed afterwards by SAH hydrolase to form homocysteine and adenosine. SAH is a potent competitive inhibitor of the methylation reaction and it is important to remove adenosine and homocysteine to prevent accumulation of SAH. Re-methylation of homocysteine requires folic acid and vitamin B12. Homocysteine can also form cysteine via enzymatic processes requiring vitamin B6. Cysteine is, in turn, the rate determining precursor for GSH synthesis via a two step enzymatic process. All these processes are better reviewed by several authors[115,116,119,152]. See Figure 5 for a summary of the SAM-mediated methylation process of catechol estrogens.
The reason for briefly introducing this matter is that this COMT detoxicating process also offers possibilities of preventive treatments.
First it is important to summarize which are the effects of alcohol drinking on the components of the above described participating molecules of the transmethylation process.
For example, it has been shown that chronic ethanol exposure decrease hepatic concentration of SAM, increasing plasma concentration of homocysteine, increase hepatic concentration of SAH and decrease plasma concentration of folate, in animal and human studies[116].
Conversely, exogenous administration of SAM has been shown to attenuate deleterious effects of alcohol drinking in animals. SAM administration is known to restore hepatic concentrations of GSH depleted by alcohol drinking[116].
That is, SAM treatment might also be of help to prevent alcohol drinking effects on mammary induced cancer by improving the COMT pathway of detoxication.
CONCLUSION
Our reviewed considerations about the pathogenesis of alcohol-drinking induced mammary cancer led to the working hypothesis that: acetaldehyde accumulation and its harmful effects, induced generation of ROS and oxidative deleterious effects on increased estrogen levels might cooperatively be involved in cancer promotion. That remains to be proved.
If assumptions were even partially correct, some useful preventive alternatives might be available: inhibition of acetaldehyde forming metabolic pathways, destruction of accumulated acetaldehyde, treatments with powerful and safe antioxidants and also enhancement of estrogen quinones detoxicating pathways. Further, cocktails having mixtures of components from each type might be envisaged.
Notwithstanding, it is always important to consider that alcohol drinking induced mammary cancer is one of the very few avoidable causes of cancer and it might be possible to avoid alcohol drinking at all or more probably acceptable to drink just one drink per day (12 g of ethanol per day only).
Footnotes
P- Reviewer: Duryee MJ S- Editor: Ma YJ L- Editor: A E- Editor: Liu SQ
References
- 1.Allen NE, Beral V, Casabonne D, Kan SW, Reeves GK, Brown A, Green J, Million Women Study Collaborators. Moderate alcohol intake and cancer incidence in women. J Natl Cancer Inst. 2009;101:296–305. doi: 10.1093/jnci/djn514. [DOI] [PubMed] [Google Scholar]
- 2.Hamajima N, Hirose K, Tajima K, Rohan T, Calle EE, Heath CW, Coates RJ, Liff JM, Talamini R, Chantarakul N, et al. Alcohol, tobacco and breast cancer--collaborative reanalysis of individual data from 53 epidemiological studies, including 58,515 women with breast cancer and 95,067 women without the disease. Br J Cancer. 2002;87:1234–1245. doi: 10.1038/sj.bjc.6600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seitz HK, Pelucchi C, Bagnardi V, La Vecchia C. Epidemiology and pathophysiology of alcohol and breast cancer: Update 2012. Alcohol Alcohol. 2012;47:204–212. doi: 10.1093/alcalc/ags011. [DOI] [PubMed] [Google Scholar]
- 4.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Alcohol consumption and ethyl carbamate. IARC Monogr Eval Carcinog Risks Hum. 2010;96:3–1383. [PMC free article] [PubMed] [Google Scholar]
- 5.Schütze M, Boeing H, Pischon T, Rehm J, Kehoe T, Gmel G, Olsen A, Tjønneland AM, Dahm CC, Overvad K, et al. Alcohol attributable burden of incidence of cancer in eight European countries based on results from prospective cohort study. BMJ. 2011;342:d1584. doi: 10.1136/bmj.d1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stewart BW, Kleihues P. WHO - International Agency for Research on Cancer. World Cancer Report. Lyon: IARC press; 2003. pp. 29–32. [Google Scholar]
- 7.Dumitrescu RG, Shields PG. The etiology of alcohol-induced breast cancer. Alcohol. 2005;35:213–225. doi: 10.1016/j.alcohol.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Castro GD, Castro JA. Metabolism of ethanol to acetaldehyde in the rat mammary tissue. Inhibitory effects of plant polyphenols and folic acid. In: Watson RR, Preedy VR, Zibadi S, editors. Alcohol, Nutrition and Health Consequences. Nutrition and Health. New York: Springer Science Business Media; 2013. pp. 145–154. [Google Scholar]
- 9.Castro GD, Quintans LN, Maciel ME, Castro JA. Preventive effects of plant polyphenols in the promotion of mammary cancer and testicular damage induced by alcohol drinking. In: Watson RR, Preedy VR, Zibadi S, editors. Polyphenols in Human Health and Disease. San Diego: Elsevier-Academic Press; 2014. pp. 1181–1190. [Google Scholar]
- 10.Garro AJ, Lieber CS. Alcohol and cancer. Annu Rev Pharmacol Toxicol. 1990;30:219–249. doi: 10.1146/annurev.pa.30.040190.001251. [DOI] [PubMed] [Google Scholar]
- 11.Seitz HK, Stickel F. Acetaldehyde as an underestimated risk factor for cancer development: role of genetics in ethanol metabolism. Genes Nutr. 2010;5:121–128. doi: 10.1007/s12263-009-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dellarco VL. A mutagenicity assessment of acetaldehyde. Mutat Res. 1988;195:1–20. doi: 10.1016/0165-1110(88)90013-9. [DOI] [PubMed] [Google Scholar]
- 13.Woutersen RA, Appelman LM, Feron VJ, Van der Heijden CA. Inhalation toxicity of acetaldehyde in rats. II. Carcinogenicity study: interim results after 15 months. Toxicology. 1984;31:123–133. doi: 10.1016/0300-483x(84)90004-0. [DOI] [PubMed] [Google Scholar]
- 14.Woutersen RA, Appelman LM, Van Garderen-Hoetmer A, Feron VJ. Inhalation toxicity of acetaldehyde in rats. III. Carcinogenicity study. Toxicology. 1986;41:213–231. doi: 10.1016/0300-483x(86)90201-5. [DOI] [PubMed] [Google Scholar]
- 15.Castro GD, Delgado de Layño AM, Fanelli SL, Maciel ME, Díaz Gómez MI, Castro JA. Acetaldehyde accumulation in rat mammary tissue after an acute treatment with alcohol. J Appl Toxicol. 2008;28:315–321. doi: 10.1002/jat.1281. [DOI] [PubMed] [Google Scholar]
- 16.Castro GD, Delgado de Layño AM, Costantini MH, Castro JA. Cytosolic xanthine oxidoreductase mediated bioactivation of ethanol to acetaldehyde and free radicals in rat breast tissue. Its potential role in alcohol-promoted mammary cancer. Toxicology. 2001;160:11–18. doi: 10.1016/s0300-483x(00)00433-9. [DOI] [PubMed] [Google Scholar]
- 17.Castro GD, Delgado de Layño AM, Costantini MH, Castro JA. Rat breast microsomal biotransformation of ethanol to acetaldehyde but not to free radicals: its potential role in the association between alcohol drinking and breast tumor promotion. Teratog Carcinog Mutagen. 2003;Suppl 1:61–70. doi: 10.1002/tcm.10060. [DOI] [PubMed] [Google Scholar]
- 18.Fam AG. Gout: excess calories, purines, and alcohol intake and beyond. Response to a urate-lowering diet. J Rheumatol. 2005;32:773–777. [PubMed] [Google Scholar]
- 19.Lieber CS. Alcohol metabolism: general aspects. In: Watson RR, Preedy VR, editors. Comprehensive Handbook of Alcohol Related Pathology, Volume 1. London: Elsevier Science Ltd-Academic Press; 2005. pp. 15–26. [Google Scholar]
- 20.Jarasch ED, Grund C, Bruder G, Heid HW, Keenan TW, Franke WW. Localization of xanthine oxidase in mammary-gland epithelium and capillary endothelium. Cell. 1981;25:67–82. doi: 10.1016/0092-8674(81)90232-4. [DOI] [PubMed] [Google Scholar]
- 21.Kooij A, Frederiks WM, Gossrau R, Van Noorden CJ. Localization of xanthine oxidoreductase activity using the tissue protectant polyvinyl alcohol and final electron acceptor Tetranitro BT. J Histochem Cytochem. 1991;39:87–93. doi: 10.1177/39.1.1983876. [DOI] [PubMed] [Google Scholar]
- 22.Castro GD, de Castro CR, Maciel ME, Fanelli SL, de Ferreyra EC, Gómez MI, Castro JA. Ethanol-induced oxidative stress and acetaldehyde formation in rat mammary tissue: potential factors involved in alcohol drinking promotion of breast cancer. Toxicology. 2006;219:208–219. doi: 10.1016/j.tox.2005.11.019. [DOI] [PubMed] [Google Scholar]
- 23.Guerri C, Sanchis R. Alcohol and acetaldehyde in rat’s milk following ethanol administration. Life Sci. 1986;38:1543–1556. doi: 10.1016/0024-3205(86)90493-5. [DOI] [PubMed] [Google Scholar]
- 24.Triano EA, Slusher LB, Atkins TA, Beneski JT, Gestl SA, Zolfaghari R, Polavarapu R, Frauenhoffer E, Weisz J. Class I alcohol dehydrogenase is highly expressed in normal human mammary epithelium but not in invasive breast cancer: implications for breast carcinogenesis. Cancer Res. 2003;63:3092–3100. [PubMed] [Google Scholar]
- 25.Fanelli SL, Maciel ME, Díaz Gómez MI, Delgado de Layño AM, Bietto FM, Castro JA, Castro GD. Further studies on the potential contribution of acetaldehyde accumulation and oxidative stress in rat mammary tissue in the alcohol drinking promotion of breast cancer. J Appl Toxicol. 2011;31:11–19. doi: 10.1002/jat.1555. [DOI] [PubMed] [Google Scholar]
- 26.O’Brien PJ. Peroxidases. Chem Biol Interact. 2000;129:113–139. doi: 10.1016/s0009-2797(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 27.Natarajan R, Nadler J. Role of lipoxygenases in breast cancer. Front Biosci. 1998;3:E81–E88. doi: 10.2741/a369. [DOI] [PubMed] [Google Scholar]
- 28.Doussiere J, Gaillard J, Vignais PV. The heme component of the neutrophil NADPH oxidase complex is a target for aryliodonium compounds. Biochemistry. 1999;38:3694–3703. doi: 10.1021/bi9823481. [DOI] [PubMed] [Google Scholar]
- 29.Fang JL, Vaca CE. Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis. 1997;18:627–632. doi: 10.1093/carcin/18.4.627. [DOI] [PubMed] [Google Scholar]
- 30.Díaz Gómez MI, Fanelli SL, Castro GD, Costantini MH, Castro JA. A liver nuclear ethanol metabolizing system. Formation of metabolites that bind covalently to macromolecules and lipids. Toxicology. 1999;138:19–28. doi: 10.1016/s0300-483x(99)00072-4. [DOI] [PubMed] [Google Scholar]
- 31.Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Identification of DNA adducts of acetaldehyde. Chem Res Toxicol. 2000;13:1149–1157. doi: 10.1021/tx000118t. [DOI] [PubMed] [Google Scholar]
- 32.Balbo S, Hashibe M, Gundy S, Brennan P, Canova C, Simonato L, Merletti F, Richiardi L, Agudo A, Castellsagué X, et al. N2-ethyldeoxyguanosine as a potential biomarker for assessing effects of alcohol consumption on DNA. Cancer Epidemiol Biomarkers Prev. 2008;17:3026–3032. doi: 10.1158/1055-9965.EPI-08-0117. [DOI] [PubMed] [Google Scholar]
- 33.Yu HS, Oyama T, Isse T, Kitagawa K, Pham TT, Tanaka M, Kawamoto T. Formation of acetaldehyde-derived DNA adducts due to alcohol exposure. Chem Biol Interact. 2010;188:367–375. doi: 10.1016/j.cbi.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 34.Matsuda T, Yabushita H, Kanaly RA, Shibutani S, Yokoyama A. Increased DNA damage in ALDH2-deficient alcoholics. Chem Res Toxicol. 2006;19:1374–1378. doi: 10.1021/tx060113h. [DOI] [PubMed] [Google Scholar]
- 35.Lorenti Garcia C, Mechilli M, Proietti De Santis L, Schinoppi A, Kobos K, Palitti F. Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat Res. 2009;662:3–9. doi: 10.1016/j.mrfmmm.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 36.Freudenheim JL, Bonner M, Krishnan S, Ambrosone CB, Graham S, McCann SE, Moysich KB, Bowman E, Nemoto T, Shields PG. Diet and alcohol consumption in relation to p53 mutations in breast tumors. Carcinogenesis. 2004;25:931–939. doi: 10.1093/carcin/bgh088. [DOI] [PubMed] [Google Scholar]
- 37.Niemelä O. Distribution of ethanol-induced protein adducts in vivo: relationship to tissue injury. Free Radic Biol Med. 2001;31:1533–1538. doi: 10.1016/s0891-5849(01)00744-4. [DOI] [PubMed] [Google Scholar]
- 38.Castro GD, Delgado de Layño AM, Castro JA. Liver nuclear ethanol metabolizing systems (NEMS) producing acetaldehyde and 1-hydroxyethyl free radicals. Toxicology. 1998;129:137–144. doi: 10.1016/s0300-483x(98)00076-6. [DOI] [PubMed] [Google Scholar]
- 39.Castro GD, Díaz Gómez MI, Castro JA. Species differences in the interaction between CCl4 reactive metabolites and liver DNA or nuclear protein fractions. Carcinogenesis. 1989;10:289–294. doi: 10.1093/carcin/10.2.289. [DOI] [PubMed] [Google Scholar]
- 40.Gómez MI, Valles E, Fanelli SL, de Layño AM, Castro GD, Castro JA. Alcohol induction of liver nuclear ethanol and N-nitrosodimethylamine metabolism to reactive metabolites. Teratog Carcinog Mutagen. 2002;22:139–145. doi: 10.1002/tcm.10009. [DOI] [PubMed] [Google Scholar]
- 41.Díaz Gómez MI, Fanelli SL, Delgado de Layño AM, Bietto FM, Castro JA, Castro GD. Deleterious effects induced by oxidative stress in liver nuclei from rats receiving an alcohol-containing liquid diet. Toxicol Ind Health. 2008;24:625–634. doi: 10.1177/0748233708101207. [DOI] [PubMed] [Google Scholar]
- 42.Dasso M. The role of the Ran GTPase pathway in cell cycle control and interphase nuclear functions. Prog Cell Cycle Res. 1995;1:163–172. doi: 10.1007/978-1-4615-1809-9_13. [DOI] [PubMed] [Google Scholar]
- 43.He D, Zeng C, Brinkley BR. Nuclear matrix proteins as structural and functional components of the mitotic apparatus. Int Rev Cytol. 1995;162B:1–74. doi: 10.1016/s0074-7696(08)62614-5. [DOI] [PubMed] [Google Scholar]
- 44.Martelli AM, Bareggi R, Bortul R, Grill V, Narducci P, Zweyer M. The nuclear matrix and apoptosis. Histochem Cell Biol. 1997;108:1–10. doi: 10.1007/s004180050140. [DOI] [PubMed] [Google Scholar]
- 45.Oliff A, Gibbs JB, McCormick F. New molecular targets for cancer therapy. Sci Am. 1996;275:144–149. doi: 10.1038/scientificamerican0996-144. [DOI] [PubMed] [Google Scholar]
- 46.Weinberg RA. How cancer arises. Sci Am. 1996;275:62–70. doi: 10.1038/scientificamerican0996-62. [DOI] [PubMed] [Google Scholar]
- 47.Garro AJ, Espina N, Farinati F, Salvagnini M. The effects of chronic ethanol consumption on carcinogen metabolism and on O6-methylguanine transferase-mediated repair of alkylated DNA. Alcohol Clin Exp Res. 1986;10:73S–77S. doi: 10.1111/j.1530-0277.1986.tb05184.x. [DOI] [PubMed] [Google Scholar]
- 48.Espina N, Lima V, Lieber CS, Garro AJ. In vitro and in vivo inhibitory effect of ethanol and acetaldehyde on O6-methylguanine transferase. Carcinogenesis. 1988;9:761–766. doi: 10.1093/carcin/9.5.761. [DOI] [PubMed] [Google Scholar]
- 49.Mufti SI, Salvagnini M, Lieber CS, Garro AJ. Chronic ethanol consumption inhibits repair of dimethylnitrosamine-induced DNA alkylation. Biochem Biophys Res Commun. 1988;152:423–431. doi: 10.1016/s0006-291x(88)80731-9. [DOI] [PubMed] [Google Scholar]
- 50.Mikami K, Haseba T, Ohno Y. Ethanol induces transient arrest of cell division (G2 + M block) followed by G0/G1 block: dose effects of short- and longer-term ethanol exposure on cell cycle and cell functions. Alcohol Alcohol. 1997;32:145–152. doi: 10.1093/oxfordjournals.alcalc.a008248. [DOI] [PubMed] [Google Scholar]
- 51.Kenney WC. Acetaldehyde adducts of phospholipids. Alcohol Clin Exp Res. 1982;6:412–416. doi: 10.1111/j.1530-0277.1982.tb05000.x. [DOI] [PubMed] [Google Scholar]
- 52.Kenney WC. Formation of Schiff base adduct between acetaldehyde and rat liver microsomal phosphatidylethanolamine. Alcohol Clin Exp Res. 1984;8:551–555. doi: 10.1111/j.1530-0277.1984.tb05728.x. [DOI] [PubMed] [Google Scholar]
- 53.Fanelli SL, Castro GD, Castro JA. Cholesterol interaction with free radicals produced from carbon tetrachloride or bromotrichloromethane by either catalytic decomposition or via liver microsomal activation. Chem Biol Interact. 1995;98:223–236. doi: 10.1016/0009-2797(95)03648-2. [DOI] [PubMed] [Google Scholar]
- 54.Fanelli SL, Castro JA. Covalent binding of carbon tetrachloride reactive metabolites to liver microsomal and nuclear lipid and phospholipid classes from Sprague Dawley and osborne Mendel male rats. Teratog Carcinog Mutagen. 1995;15:155–166. doi: 10.1002/tcm.1770150402. [DOI] [PubMed] [Google Scholar]
- 55.Goodman DS, Deykin D. Fatty acid ethyl ester formation during ethanol metabolism in vivo. Proc Soc Exp Biol Med. 1963;113:65–67. doi: 10.3181/00379727-113-28277. [DOI] [PubMed] [Google Scholar]
- 56.Lange LG. Nonoxidative ethanol metabolism: formation of fatty acid ethyl esters by cholesterol esterase. Proc Natl Acad Sci USA. 1982;79:3954–3957. doi: 10.1073/pnas.79.13.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laposata M. Fatty acid ethyl esters: ethanol metabolites which mediate ethanol-induced organ damage and serve as markers of ethanol intake. Prog Lipid Res. 1998;37:307–316. doi: 10.1016/s0163-7827(98)00013-7. [DOI] [PubMed] [Google Scholar]
- 58.Sun GY, Sun AY. Ethanol and membrane lipids. Alcohol Clin Exp Res. 1985;9:164–180. doi: 10.1111/j.1530-0277.1985.tb05543.x. [DOI] [PubMed] [Google Scholar]
- 59.Sylvia VL, Joe CO, Norman JO, Curtin GM, Busbee DL. Phosphatidylinositol-dependent activation of DNA polymerase alpha. Biochem Biophys Res Commun. 1986;135:880–885. doi: 10.1016/0006-291x(86)91010-7. [DOI] [PubMed] [Google Scholar]
- 60.Herlan G, Giese G, Wunderlich F. Influence of nuclear membrane lipid fluidity on nuclear RNA release. Exp Cell Res. 1979;118:305–309. doi: 10.1016/0014-4827(79)90155-1. [DOI] [PubMed] [Google Scholar]
- 61.Cocco L, Gilmour RS, Ognibene A, Letcher AJ, Manzoli FA, Irvine RF. Synthesis of polyphosphoinositides in nuclei of Friend cells. Evidence for polyphosphoinositide metabolism inside the nucleus which changes with cell differentiation. Biochem J. 1987;248:765–770. doi: 10.1042/bj2480765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Venkatraman JT, Lefebvre YA, Clandinin MT. Diet fat alters the structure and function of the nuclear envelope: modulation of membrane fatty acid composition, NTPase activity and binding of triiodothyronine. Biochem Biophys Res Commun. 1986;135:655–661. doi: 10.1016/0006-291x(86)90043-4. [DOI] [PubMed] [Google Scholar]
- 63.Venkatraman JT, Clandinin MT. Ribonucleic acid efflux from isolated mouse liver nuclei is altered by diet and genotypically determined change in nuclear envelope composition. Biochim Biophys Acta. 1988;940:33–42. doi: 10.1016/0005-2736(88)90005-3. [DOI] [PubMed] [Google Scholar]
- 64.Manzoli FA, Martelli AM, Capitani S, Maraldi NM, Rizzoli R, Barnabei O, Cocco L. Nuclear polyphosphoinositides during cell growth and differentiation. Adv Enzyme Regul. 1989;28:25–34. doi: 10.1016/0065-2571(89)90061-7. [DOI] [PubMed] [Google Scholar]
- 65.Chapkin RS, Davidson LD, Davidson LA. Phospholipid molecular species composition of mouse liver nuclei. Influence of dietary n-3 fatty acid ethyl esters. Biochem J. 1992;287(Pt 1):237–240. doi: 10.1042/bj2870237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cederbaum AI, Lu Y, Wu D. Role of oxidative stress in alcohol-induced liver injury. Arch Toxicol. 2009;83:519–548. doi: 10.1007/s00204-009-0432-0. [DOI] [PubMed] [Google Scholar]
- 67.Boyer PD. Sulfhydryl and Disulfide Groups of Enzymes. In: Boyer PD, Lardy H, Myrback K, editors. The Enzymes, 2nd ed. New York: Academic Press; 1959. pp. 512–588. [Google Scholar]
- 68.Johnstone RM. Sulfhydryl Agents: Arsenicals. In: Hochster RM, Quastel JH, editors. Metabolic Inhibitors, volume II. London: Academic Press; 1963. pp. 99–118. [Google Scholar]
- 69.Peters R. Inhibitors of enzymes containing thiol groups and toxicity. In: Biochemical Lesions and Lethal Synthesis., editor. New York: Pergamon Press; 1963. pp. 74–87. [Google Scholar]
- 70.Dixon M, Webb EC. Enzymes. New York: Academic Press; 1979. pp. 301–307. [Google Scholar]
- 71.Malins DC, Polissar NL, Gunselman SJ. Progression of human breast cancers to the metastatic state is linked to hydroxyl radical-induced DNA damage. Proc Natl Acad Sci USA. 1996;93:2557–2563. doi: 10.1073/pnas.93.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 73.Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- 74.Klaunig JE, Wang Z, Pu X, Zhou S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol. 2011;254:86–99. doi: 10.1016/j.taap.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 75.Reichman ME, Judd JT, Longcope C, Schatzkin A, Clevidence BA, Nair PP, Campbell WS, Taylor PR. Effects of alcohol consumption on plasma and urinary hormone concentrations in premenopausal women. J Natl Cancer Inst. 1993;85:722–727. doi: 10.1093/jnci/85.9.722. [DOI] [PubMed] [Google Scholar]
- 76.Ginsburg ES, Mello NK, Mendelson JH, Barbieri RL, Teoh SK, Rothman M, Gao X, Sholar JW. Effects of alcohol ingestion on estrogens in postmenopausal women. JAMA. 1996;276:1747–1751. doi: 10.1001/jama.1996.03540210055034. [DOI] [PubMed] [Google Scholar]
- 77.Purohit V. Moderate alcohol consumption and estrogen levels in postmenopausal women: a review. Alcohol Clin Exp Res. 1998;22:994–997. doi: 10.1111/j.1530-0277.1998.tb03694.x. [DOI] [PubMed] [Google Scholar]
- 78.Sarkola T, Mäkisalo H, Fukunaga T, Eriksson CJ. Acute effect of alcohol on estradiol, estrone, progesterone, prolactin, cortisol, and luteinizing hormone in premenopausal women. Alcohol Clin Exp Res. 1999;23:976–982. [PubMed] [Google Scholar]
- 79.Sarkola T, Fukunaga T, Mäkisalo H, Peter Eriksson CJ. Acute effect of alcohol on androgens in premenopausal women. Alcohol Alcohol. 2000;35:84–90. doi: 10.1093/alcalc/35.1.84. [DOI] [PubMed] [Google Scholar]
- 80.Sarkola T, Adlercreutz H, Heinonen S, von Der Pahlen B, Eriksson CJ. The role of the liver in the acute effect of alcohol on androgens in women. J Clin Endocrinol Metab. 2001;86:1981–1985. doi: 10.1210/jcem.86.5.7486. [DOI] [PubMed] [Google Scholar]
- 81.Coutelle C, Höhn B, Benesova M, Oneta CM, Quattrochi P, Roth HJ, Schmidt-Gayk H, Schneeweiss A, Bastert G, Seitz HK. Risk factors in alcohol associated breast cancer: alcohol dehydrogenase polymorphism and estrogens. Int J Oncol. 2004;25:1127–1132. [PubMed] [Google Scholar]
- 82.Mahabir S, Baer DJ, Johnson LL, Dorgan JF, Campbell W, Brown E, Hartman TJ, Clevidence B, Albanes D, Judd JT, et al. The effects of moderate alcohol supplementation on estrone sulfate and DHEAS in postmenopausal women in a controlled feeding study. Nutr J. 2004;3:11. doi: 10.1186/1475-2891-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sierksma A, Sarkola T, Eriksson CJ, van der Gaag MS, Grobbee DE, Hendriks HF. Effect of moderate alcohol consumption on plasma dehydroepiandrosterone sulfate, testosterone, and estradiol levels in middle-aged men and postmenopausal women: a diet-controlled intervention study. Alcohol Clin Exp Res. 2004;28:780–785. doi: 10.1097/01.alc.0000125356.70824.81. [DOI] [PubMed] [Google Scholar]
- 84.Seitz HK, Maurer B. The relationship between alcohol metabolism, estrogen levels, and breast cancer risk. Alcohol Res Health. 2007;30:42–43. [PubMed] [Google Scholar]
- 85.Singletary KW, Gapstur SM. Alcohol and breast cancer: review of epidemiologic and experimental evidence and potential mechanisms. JAMA. 2001;286:2143–2151. doi: 10.1001/jama.286.17.2143. [DOI] [PubMed] [Google Scholar]
- 86.Gavaler JS, Van Thiel DH. Hormonal status of postmenopausal women with alcohol-induced cirrhosis: further findings and a review of the literature. Hepatology. 1992;16:312–319. doi: 10.1002/hep.1840160206. [DOI] [PubMed] [Google Scholar]
- 87.Etique N, Flament S, Lecomte J, Grillier-Vuissoz I. Ethanol-induced ligand-independent activation of ERalpha mediated by cyclic AMP/PKA signaling pathway: an in vitro study on MCF-7 breast cancer cells. Int J Oncol. 2007;31:1509–1518. [PubMed] [Google Scholar]
- 88.Sarkar DK, Liehr JG, Singletary KW. Role of estrogen in alcohol promotion of breast cancer and prolactinomas. Alcohol Clin Exp Res. 2001;25:230S–236S. doi: 10.1097/00000374-200105051-00037. [DOI] [PubMed] [Google Scholar]
- 89.Roy D, Liehr JG. Estrogen, DNA damage and mutations. Mutat Res. 1999;424:107–115. doi: 10.1016/s0027-5107(99)00012-3. [DOI] [PubMed] [Google Scholar]
- 90.Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D. Estrogens as endogenous genotoxic agents--DNA adducts and mutations. J Natl Cancer Inst Monogr. 2000;(90):75–93. doi: 10.1093/oxfordjournals.jncimonographs.a024247. [DOI] [PubMed] [Google Scholar]
- 91.Cavalieri EL, Rogan EG. A unifying mechanism in the initiation of cancer and other diseases by catechol quinones. Ann N Y Acad Sci. 2004;1028:247–257. doi: 10.1196/annals.1322.029. [DOI] [PubMed] [Google Scholar]
- 92.Cavalieri EL, Rogan EG. Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer. J Steroid Biochem Mol Biol. 2011;125:169–180. doi: 10.1016/j.jsbmb.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 94.Fernandez SV. Estrogen, alcohol consumption, and breast cancer. Alcohol Clin Exp Res. 2011;35:389–391. doi: 10.1111/j.1530-0277.2010.01355.x. [DOI] [PubMed] [Google Scholar]
- 95.Feigelson HS, Henderson BE. Estrogens and breast cancer. Carcinogenesis. 1996;17:2279–2284. doi: 10.1093/carcin/17.11.2279. [DOI] [PubMed] [Google Scholar]
- 96.Henderson BE, Feigelson HS. Hormonal carcinogenesis. Carcinogenesis. 2000;21:427–433. doi: 10.1093/carcin/21.3.427. [DOI] [PubMed] [Google Scholar]
- 97.Flötotto T, Djahansouzi S, Gläser M, Hanstein B, Niederacher D, Brumm C, Beckmann MW. Hormones and hormone antagonists: mechanisms of action in carcinogenesis of endometrial and breast cancer. Horm Metab Res. 2001;33:451–457. doi: 10.1055/s-2001-16936. [DOI] [PubMed] [Google Scholar]
- 98.Bolton JL, Thatcher GR. Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol. 2008;21:93–101. doi: 10.1021/tx700191p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhu BT, Lee AJ. NADPH-dependent metabolism of 17beta-estradiol and estrone to polar and nonpolar metabolites by human tissues and cytochrome P450 isoforms. Steroids. 2005;70:225–244. doi: 10.1016/j.steroids.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 100.Bolton JL, Trush MA, Penning TM, Dryhurst G, Monks TJ. Role of quinones in toxicology. Chem Res Toxicol. 2000;13:135–160. doi: 10.1021/tx9902082. [DOI] [PubMed] [Google Scholar]
- 101.Gaikwad NW, Yang L, Rogan EG, Cavalieri EL. Evidence for NQO2-mediated reduction of the carcinogenic estrogen ortho-quinones. Free Radic Biol Med. 2009;46:253–262. doi: 10.1016/j.freeradbiomed.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R, Muti P, Rogan E, Russo J, Santen R, et al. Catechol estrogen quinones as initiators of breast and other human cancers: implications for biomarkers of susceptibility and cancer prevention. Biochim Biophys Acta. 2006;1766:63–78. doi: 10.1016/j.bbcan.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 103.Li KM, Todorovic R, Devanesan P, Higginbotham S, Köfeler H, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL. Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis. 2004;25:289–297. doi: 10.1093/carcin/bgg191. [DOI] [PubMed] [Google Scholar]
- 104.Embrechts J, Lemière F, Van Dongen W, Esmans EL, Buytaert P, Van Marck E, Kockx M, Makar A. Detection of estrogen DNA-adducts in human breast tumor tissue and healthy tissue by combined nano LC-nano ES tandem mass spectrometry. J Am Soc Mass Spectrom. 2003;14:482–491. doi: 10.1016/S1044-0305(03)00130-2. [DOI] [PubMed] [Google Scholar]
- 105.Raftogianis R, Creveling C, Weinshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Cancer Inst Monogr. 2000;(27):113–124. doi: 10.1093/oxfordjournals.jncimonographs.a024234. [DOI] [PubMed] [Google Scholar]
- 106.Hachey DL, Dawling S, Roodi N, Parl FF. Sequential action of phase I and II enzymes cytochrome p450 1B1 and glutathione S-transferase P1 in mammary estrogen metabolism. Cancer Res. 2003;63:8492–8499. [PubMed] [Google Scholar]
- 107.Chang M. Dual roles of estrogen metabolism in mammary carcinogenesis. BMB Rep. 2011;44:423–434. doi: 10.5483/BMBRep.2011.44.7.423. [DOI] [PubMed] [Google Scholar]
- 108.Zhang F, Bolton JL. Synthesis of the equine estrogen metabolites 2-hydroxyequilin and 2-hydroxyequilenin. Chem Res Toxicol. 1999;12:200–203. doi: 10.1021/tx980189g. [DOI] [PubMed] [Google Scholar]
- 109.Roy D, Bernhardt A, Strobel HW, Liehr JG. Catalysis of the oxidation of steroid and stilbene estrogens to estrogen quinone metabolites by the beta-naphthoflavone-inducible cytochrome P450 IA family. Arch Biochem Biophys. 1992;296:450–456. doi: 10.1016/0003-9861(92)90596-o. [DOI] [PubMed] [Google Scholar]
- 110.Markides CS, Roy D, Liehr JG. Concentration dependence of prooxidant and antioxidant properties of catecholestrogens. Arch Biochem Biophys. 1998;360:105–112. doi: 10.1006/abbi.1998.0934. [DOI] [PubMed] [Google Scholar]
- 111.Abel EL, Lyon RP, Bammler TK, Verlinde CL, Lau SS, Monks TJ, Eaton DL. Estradiol metabolites as isoform-specific inhibitors of human glutathione S-transferases. Chem Biol Interact. 2004;151:21–32. doi: 10.1016/j.cbi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 112.Yao J, Chang M, Li Y, Pisha E, Liu X, Yao D, Elguindi EC, Blond SY, Bolton JL. Inhibition of cellular enzymes by equine catechol estrogens in human breast cancer cells: specificity for glutathione S-transferase P1-1. Chem Res Toxicol. 2002;15:935–942. doi: 10.1021/tx020018i. [DOI] [PubMed] [Google Scholar]
- 113.Chang M, Shin YG, van Breemen RB, Blond SY, Bolton JL. Structural and functional consequences of inactivation of human glutathione S-transferase P1-1 mediated by the catechol metabolite of equine estrogens, 4-hydroxyequilenin. Biochemistry. 2001;40:4811–4820. doi: 10.1021/bi002513o. [DOI] [PubMed] [Google Scholar]
- 114.Zheng T, Holford TR, Zahm SH, Owens PH, Boyle P, Zhang Y, Zhang B, Wise JP, Stephenson LP, Ali-Osman F. Glutathione S-transferase M1 and T1 genetic polymorphisms, alcohol consumption and breast cancer risk. Br J Cancer. 2003;88:58–62. doi: 10.1038/sj.bjc.6600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shukla SD, Velazquez J, French SW, Lu SC, Ticku MK, Zakhari S. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32:1525–1534. doi: 10.1111/j.1530-0277.2008.00729.x. [DOI] [PubMed] [Google Scholar]
- 116.Purohit V, Abdelmalek MF, Barve S, Benevenga NJ, Halsted CH, Kaplowitz N, Kharbanda KK, Liu QY, Lu SC, McClain CJ, et al. Role of S-adenosylmethionine, folate, and betaine in the treatment of alcoholic liver disease: summary of a symposium. Am J Clin Nutr. 2007;86:14–24. doi: 10.1093/ajcn/86.1.14. [DOI] [PubMed] [Google Scholar]
- 117.Zhu BT. On the mechanism of homocysteine pathophysiology and pathogenesis: a unifying hypothesis. Histol Histopathol. 2002;17:1283–1291. doi: 10.14670/HH-17.1283. [DOI] [PubMed] [Google Scholar]
- 118.Kharbanda KK. Alcoholic liver disease and methionine metabolism. Semin Liver Dis. 2009;29:155–165. doi: 10.1055/s-0029-1214371. [DOI] [PubMed] [Google Scholar]
- 119.Zakhari S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013;35:6–16. doi: 10.35946/arcr.v35.1.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J Natl Cancer Inst. 1998;90:814–823. doi: 10.1093/jnci/90.11.814. [DOI] [PubMed] [Google Scholar]
- 121.Eriksson CJ, Fukunaga T, Sarkola T, Lindholm H, Ahola L. Estrogen-related acetaldehyde elevation in women during alcohol intoxication. Alcohol Clin Exp Res. 1996;20:1192–1195. doi: 10.1111/j.1530-0277.1996.tb01110.x. [DOI] [PubMed] [Google Scholar]
- 122.Etique N, Grillier-Vuissoz I, Lecomte J, Flament S. Crosstalk between adenosine receptor (A2A isoform) and ERalpha mediates ethanol action in MCF-7 breast cancer cells. Oncol Rep. 2009;21:977–981. doi: 10.3892/or_00000311. [DOI] [PubMed] [Google Scholar]
- 123.Izevbigie EB, Ekunwe SI, Jordan J, Howard CB. Ethanol modulates the growth of human breast cancer cells in vitro. Exp Biol Med (Maywood) 2002;227:260–265. doi: 10.1177/153537020222700406. [DOI] [PubMed] [Google Scholar]
- 124.Maciel ME, Castro GD, Castro JA. Inhibition of the rat breast cytosolic bioactivation of ethanol to acetaldehyde by some plant polyphenols and folic acid. Nutr Cancer. 2004;49:94–99. doi: 10.1207/s15327914nc4901_13. [DOI] [PubMed] [Google Scholar]
- 125.Maciel ME, Castro JA, Castro GD. Inhibition of rat mammary microsomal oxidation of ethanol to acetaldehyde by plant polyphenols. Hum Exp Toxicol. 2011;30:656–664. doi: 10.1177/0960327110377522. [DOI] [PubMed] [Google Scholar]
- 126.Hong J, Smith TJ, Ho CT, August DA, Yang CS. Effects of purified green and black tea polyphenols on cyclooxygenase- and lipoxygenase-dependent metabolism of arachidonic acid in human colon mucosa and colon tumor tissues. Biochem Pharmacol. 2001;62:1175–1183. doi: 10.1016/s0006-2952(01)00767-5. [DOI] [PubMed] [Google Scholar]
- 127.Cai Y, Luo Q, Sun M, Corke H. Antioxidant activity and phenolic compounds of 112 traditional Chinese medicinal plants associated with anticancer. Life Sci. 2004;74:2157–2184. doi: 10.1016/j.lfs.2003.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cos P, Ying L, Calomme M, Hu JP, Cimanga K, Van Poel B, Pieters L, Vlietinck AJ, Vanden Berghe D. Structure-activity relationship and classification of flavonoids as inhibitors of xanthine oxidase and superoxide scavengers. J Nat Prod. 1998;61:71–76. doi: 10.1021/np970237h. [DOI] [PubMed] [Google Scholar]
- 129.Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis by dietary polyphenolic compounds. Annu Rev Nutr. 2001;21:381–406. doi: 10.1146/annurev.nutr.21.1.381. [DOI] [PubMed] [Google Scholar]
- 130.Lambert JD, Hong J, Yang GY, Liao J, Yang CS. Inhibition of carcinogenesis by polyphenols: evidence from laboratory investigations. Am J Clin Nutr. 2005;81:284S–291S. doi: 10.1093/ajcn/81.1.284S. [DOI] [PubMed] [Google Scholar]
- 131.Collins-Burow BM, Burow ME, Duong BN, McLachlan JA. Estrogenic and antiestrogenic activities of flavonoid phytochemicals through estrogen receptor binding-dependent and -independent mechanisms. Nutr Cancer. 2000;38:229–244. doi: 10.1207/S15327914NC382_13. [DOI] [PubMed] [Google Scholar]
- 132.Valentovic M, Santanam N. Nutrition, oxidative stress and cancer. In: Claudio PP, Niles RM. Nutrition and Cancer from Epidemiology to Biology. Bentham E Books; 2012. pp. 77–86. [Google Scholar]
- 133.Aiyer HS, Srinivasan C, Gupta RC. Dietary berries and ellagic acid diminish estrogen-mediated mammary tumorigenesis in ACI rats. Nutr Cancer. 2008;60:227–234. doi: 10.1080/01635580701624712. [DOI] [PubMed] [Google Scholar]
- 134.Losso JN, Bansode RR, Trappey A, Bawadi HA, Truax R. In vitro anti-proliferative activities of ellagic acid. J Nutr Biochem. 2004;15:672–678. doi: 10.1016/j.jnutbio.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 135.Bell C, Hawthorne S. Ellagic acid, pomegranate and prostate cancer -- a mini review. J Pharm Pharmacol. 2008;60:139–144. doi: 10.1211/jpp.60.2.0001. [DOI] [PubMed] [Google Scholar]
- 136.Pantuck AJ, Leppert JT, Zomorodian N, Aronson W, Hong J, Barnard RJ, Seeram N, Liker H, Wang H, Elashoff R, et al. Phase II study of pomegranate juice for men with rising prostate-specific antigen following surgery or radiation for prostate cancer. Clin Cancer Res. 2006;12:4018–4026. doi: 10.1158/1078-0432.CCR-05-2290. [DOI] [PubMed] [Google Scholar]
- 137.Larsson SC, Giovannucci E, Wolk A. Folate and risk of breast cancer: a meta-analysis. J Natl Cancer Inst. 2007;99:64–76. doi: 10.1093/jnci/djk006. [DOI] [PubMed] [Google Scholar]
- 138.Deitrich RA, Petersen D, Vasiliou V. Removal of acetaldehyde from the body. In: Novartis Foundation Symposium 285., editor. Acetaldehyde-related pathology: bridging the trans-disciplinary divide. Chichester: Wiley; 2007. pp. 23–51. [DOI] [PubMed] [Google Scholar]
- 139.Doorn JA, Hurley TD, Petersen DR. Inhibition of human mitochondrial aldehyde dehydrogenase by 4-hydroxynon-2-enal and 4-oxonon-2-enal. Chem Res Toxicol. 2006;19:102–110. doi: 10.1021/tx0501839. [DOI] [PubMed] [Google Scholar]
- 140.Cederbaum AI, Rubin E. Protective effect of cysteine on the inhibition of mitochondrial functions by acetaldehyde. Biochem Pharmacol. 1976;25:963–973. doi: 10.1016/0006-2952(76)90323-3. [DOI] [PubMed] [Google Scholar]
- 141.Salaspuro VJ, Hietala JM, Marvola ML, Salaspuro MP. Eliminating carcinogenic acetaldehyde by cysteine from saliva during smoking. Cancer Epidemiol Biomarkers Prev. 2006;15:146–149. doi: 10.1158/1055-9965.EPI-05-0248. [DOI] [PubMed] [Google Scholar]
- 142.Salaspuro V. Pharmacological treatments and strategies for reducing oral and intestinal acetaldehyde. In: Novartis Foundation Symposium 285., editor. Acetaldehyde-related pathology: bridging the trans-disciplinary divide. Chichester: Wiley; 2007. pp. 145–157. [DOI] [PubMed] [Google Scholar]
- 143.De Flora S, Izzotti A, D’Agostini F, Balansky RM. Mechanisms of N-acetylcysteine in the prevention of DNA damage and cancer, with special reference to smoking-related end-points. Carcinogenesis. 2001;22:999–1013. doi: 10.1093/carcin/22.7.999. [DOI] [PubMed] [Google Scholar]
- 144.Zahid M, Saeed M, Ali MF, Rogan EG, Cavalieri EL. N-acetylcysteine blocks formation of cancer-initiating estrogen-DNA adducts in cells. Free Radic Biol Med. 2010;49:392–400. doi: 10.1016/j.freeradbiomed.2010.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Reed DJ. Glutathione: toxicological implications. Annu Rev Pharmacol Toxicol. 1990;30:603–631. doi: 10.1146/annurev.pa.30.040190.003131. [DOI] [PubMed] [Google Scholar]
- 146.Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL. Inhibition of depurinating estrogen-DNA adduct formation by natural compounds. Chem Res Toxicol. 2007;20:1947–1953. doi: 10.1021/tx700269s. [DOI] [PubMed] [Google Scholar]
- 147.Venugopal D, Zahid M, Mailander PC, Meza JL, Rogan EG, Cavalieri EL, Chakravarti D. Reduction of estrogen-induced transformation of mouse mammary epithelial cells by N-acetylcysteine. J Steroid Biochem Mol Biol. 2008;109:22–30. doi: 10.1016/j.jsbmb.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gaikwad NW, Rogan EG, Cavalieri EL. Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones. Free Radic Biol Med. 2007;43:1289–1298. doi: 10.1016/j.freeradbiomed.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aziz MH, Kumar R, Ahmad N. Cancer chemoprevention by resveratrol: in vitro and in vivo studies and the underlying mechanisms (review) Int J Oncol. 2003;23:17–28. [PubMed] [Google Scholar]
- 150.Jang M, Cai L, Udeani GO, Slowing KV, Thomas CF, Beecher CW, Fong HH, Farnsworth NR, Kinghorn AD, Mehta RG, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 151.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 152.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]