

Abstract
Pulmonary alveolar proteinosis (PAP) is an orphan disease characterized by the accumulation of excess of surfactant within alveoli and bronchioles. The primary form of PAP (P-PAP; also referred to as idiopathic or autoimmune) is the most common form. It is mediated through a circulating neutralizing antibody against granulocyte-macrophage colony-stimulating factor. Secondary PAP (S-PAP) can be induced by a host of inciting agents and is far more liable to progress to terminal respiratory failure. We describe a rare case of S-PAP occurring in a renal transplant recipient due to mycophenolate and cyclosporine combination-therapy, which resolved spontaneously following withdrawal of these drugs.
Keywords: Alveolar proteinosis, cyclosporin, mycophenolate, renal transplant, immunosuppression
INTRODUCTION
The final common pathway in the pathogenesis of pulmonary alveolar proteinosis (PAP) appears to be an excess of surfactant in the alveoli owing to failure of scavenging mechanisms, rather than an abnormal augmentation of surfactant production.[1] On the basis of clinical, histopathological and pathologic differences, PAP is now classified into primary, secondary and congenital. The primary PAP (P-PAP) accounts for approximately 90% of all cases and appears to be mediated through a circulating neutralizing antibody (anti granulocyte-macrophage colony-stimulating factor (GM-CSF) immunoglobulin neutralizing immunoglobulin G antibody).[2] Since GM-CSF normally plays a vital role in the catabolism of surfactant by alveolar macrophages, its functional deficiency allows surfactant to accumulate.
A variety of inciting agents, by reducing alveolar macrophage number or function, can induce the more heterogenous entity of Secondary PAP (S-PAP). Hematogenous malignancies account for the most part, but inhalational agents, certain infections, cancers and rare immunological disturbances have been implicated. PAP that occurs as a consequence of immunosuppressive agents also falls into this category. Although cyclosporin-A has been causally linked with PAP[3] and rare cases of alveolar proteinosis have been reported due to sirolimus,[2] a Medline/PubMed search revealed no occurrence of PAP in renal transplant patients that occurred as a consequence of cyclosporine-mycophenolate combination therapy.
CASE REPORT
This is a case report of a 36-year-old renal transplant recipient who presented with worsening dry cough and breathlessness on exertion since 1 year, which progressed to a point that she could walk no more than a few yards on level ground.
5 years ago, she had been diagnosed with chronic kidney failure. At that time, imaging (ultrasound and abdominal CT scans) had shown bilaterally shrunken kidneys. Standard work-up including serum anti-nuclear antibody (ANA) was negative; 3 years ago she underwent renal transplantation, with “triple immunosuppression” (prednisolone, mycophenolate and cyclosporine). A year ago she had developed breathlessness on exertion. There was no history of exposure to birds or animals, molds, organic or inorganic dusts, or to chemical fumes.
On examination
There was no pedal edema. Jugular venous pulse was not raised. A few crackles were audible at the lung bases. Cardiac auscultation was normal.
Investigations
Routine biochemistry was normal. Resting oxygen saturation was 86-88%, falling further on effort. The chest X-ray showed a bilateral perihilar and lower zone infiltrates [Figure 1]. A chest CT scan showed bilateral diffuse ground-glass haziness with superimposed interlobular septal thickening [Figure 2]. Serology for cytomegalovirus, human immunodeficiency virus, ANA, cytoplasmic antineutrophil cytoplasmic antibodies and perinuclear anti-neutrophil cytoplasmic antibodies was negative. Bronchoscopic alveolar lavage (BAL) revealed no gross inflammation. Silver methenamine stain and stains for acid fast bacteria (AFB) were negative. BAL fluid cultures (pyogenic and AFB) were negative. Transbronchial lung biopsies showed dilated alveoli [Figure 3a] filled with PAS-positive granular eosinophic material along with deeply eosinophilic structures; inflammatory cells were notably absent. The eosinophilic material was resistant to decolorization with diastase[4] [Figure 3b], which is conclusive of alveolar proteinosis. Anti GM-CSF antibodies are not useful in S-PAP.[1]
Figure 1.
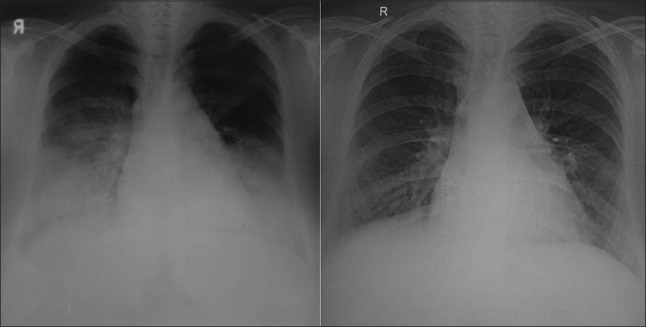
The panel on the left shows the chest radiograph at presentation showing bilateral consolidation without cardiomegaly or effusions. The panel on the right shows the chest radiograph 2 years after discontinuation of cyclosporine and mycophenolate. There is near-total resolution of the pulmonary parenchymal abnormality
Figure 2.
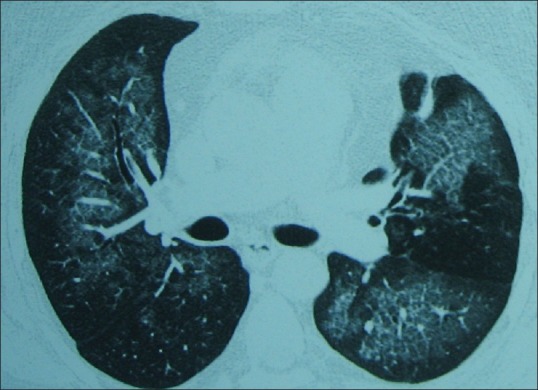
Computed tomography scan showing bilateral diffuse ground-glass haziness with superimposed interlobular septal thickening
Figure 3.
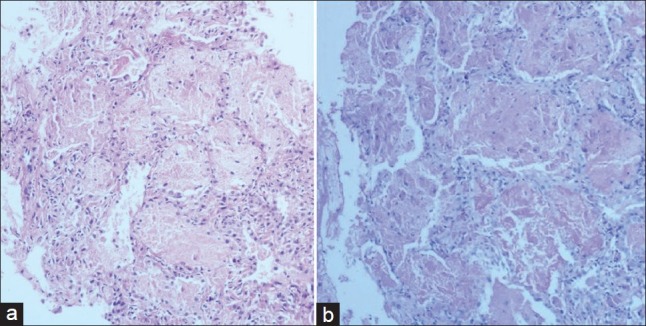
(a) Transbronchial lung biopsies shows preserved lung architecture. Alveoli are filled with granular eosinophiic material that noticeably lacks inflammatory cells; (b) periodic acid-Schiff stained tissue after diastase treatment
Course of illness
The immunosuppressive regimen was changed to azathioprine (75 mg/day). Prednisolone was continued (5 mg/day) and continuous oxygen advised. Patient declined a whole-lung lavage. 2 years after the diagnosis, the patient's breathlessness had improved significantly and she no longer required oxygen support. A chest film showed near-total resolution.
DISCUSSION
PAP is an orphan disease that predominantly affects non-smokers. Major advances have been made in unraveling the mechanisms underlying P-PAP, but the pathogenesis of S-PAP remains ill-understood. A failure of scavenging mechanisms lies at the heart of both forms. The prognosis of S-PAP can be unpredictable[2] and the outcome is generally regarded as much worse than that of the autoimmune variety, with a median survival time of around 20 months.[5]
A bilateral perihilar infiltrate (“butterfly distribution”) involving the lower lobes, but sparing the costophrenic angles in the relevant setting pattern is suggestive of PAP,[6] but pulmonary edema or Pneumocystis jirovecii pneumonia can cast a similar pattern on the X-ray (pleural effusions, unlike in cardiogenic pulmonary edema, are absent in PAP). On CT chest, a branching pattern of linear reticulations arranged in distinctive geometric shapes and overlying ground-glass opacities (“crazy-pavement distribution”) has been regarded as characteristic of PAP,[3] though it occasionally occurs in other conditions (e.g. broncho-alveolar carcinoma); this pattern, however, rarely manifests in S-PAP[5,6] as was the case in this patient. Anti-GM-CSF antibodies have an almost 100% sensitivity for P-PAP. However, in S-PAP their concentrations are close to undetectable.[1] The test, in developing countries, is not widely available and is expensive.
Tissue biopsy is very effective at ruling out other mimicking conditions and should still be considered the gold standard for the diagnosis of PAP.[1] The presence of intra-alveolar eosinophilic material resistant to decolorization by diastase is diagnostic.[4]
The appearance of the symptoms after a few months of the commencement of immunotherapy is very suggestive. This patient progressively worsened while on mycophenolate and cyclosporine and the improvement began only when these drugs were withdrawn. Cyclosporin-A has been causally linked with PAP[3] as has sirolimus.[2,7]
Without therapeutic lung lavage, the prognosis of P-PAP is unpredictable; though with therapeutic lavage, at 5-year survival rises to almost 95%.[8] Recently, results with GM-CSF for P-PAP have been encouraging.[9] The prognosis for S-PAP, though, is generally regarded as poor,[4,5] and palpably worse than that of the primary form.[10] This is especially true of cytotoxic-induced PAP.[3] In respect of this patient, the cessation of the offending drugs was the only therapeutic “intervention,” and no other therapy was given.
CONCLUSION
In the presence of bilateral diffuse alveolar infiltrates, due consideration should be given to S-PAP in the setting of immunosuppressant use. Lung biopsy still remains the gold standard for the diagnosis of S-PAP,[9] though antibody testing is now important in the primary form of the disease. Withdrawal of offending agents is vital and in an exceptional case such as this, may of itself suffice in securing the desired improvement.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349:2527–39. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 2.Kadikoy H, Paolini M, Achkar K, Suki W, Gaber AO, Anwar N, et al. Pulmonary alveolar proteinosis in a kidney transplant: A rare complication of sirolimus. Nephrol Dial Transplant. 2010;25:2795–8. doi: 10.1093/ndt/gfq265. [DOI] [PubMed] [Google Scholar]
- 3.Bacigalupo A, Frassoni F, van Lint MT, Raffo MR, Vitale V, Corbetta G, et al. Cyclosporin A in marrow transplantation for leukemia and aplastic anemia. Exp Hematol. 1985;13:244–8. [PubMed] [Google Scholar]
- 4.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: Progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–35. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 5.Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: Pre-mortem cases in Japan. Eur Respir J. 2011;37:465–8. doi: 10.1183/09031936.00092910. [DOI] [PubMed] [Google Scholar]
- 6.Rossi SE, Erasmus JJ, Volpacchio M, Franquet T, Castiglioni T, McAdams HP. "Crazy-paving" pattern at thin-section CT of the lungs: Radiologic-pathologic overview. Radiographics. 2003;23:1509–19. doi: 10.1148/rg.236035101. [DOI] [PubMed] [Google Scholar]
- 7.Feagans J, Victor D, Moehlen M, Florman SS, Regenstein F, Balart LA, et al. Interstitial pneumonitis in the transplant patient: Consider sirolimus-associated pulmonary toxicity. J La State Med Soc. 2009;161:166, 168–72. [PubMed] [Google Scholar]
- 8.Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177:752–62. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonfield TL, Kavuru MS, Thomassen MJ. Anti-GM-CSF titer predicts response to GM-CSF therapy in pulmonary alveolar proteinosis. Clin Immunol. 2002;105:342–50. doi: 10.1006/clim.2002.5301. [DOI] [PubMed] [Google Scholar]
- 10.Bonella F, Bauer PC, Griese M, Ohshimo S, Guzman J, Costabel U. Pulmonary alveolar proteinosis: New insights from a single-center cohort of 70 patients. Respir Med. 2011;105:1908–16. doi: 10.1016/j.rmed.2011.08.018. [DOI] [PubMed] [Google Scholar]