

Abstract
Metabolic syndrome (MS) is a collection of cardiometabolic risk factors that includes obesity, insulin resistance, hypertension, and dyslipidemia. Although there has been significant debate regarding the criteria and concept of the syndrome, this clustering of risk factors is unequivocally linked to an increased risk of developing type 2 diabetes and cardiovascular disease. Regardless of the true definition, based on current population estimates, nearly 100 million have MS. It is often characterized by insulin resistance, which some have suggested is a major underpinning link between physical inactivity and MS. The purpose of this review is to: (i) provide an overview of the history, causes and clinical aspects of MS, (ii) review the molecular mechanisms of insulin action and the causes of insulin resistance, and (iii) discuss the epidemiological and intervention data on the effects of exercise on MS and insulin sensitivity.
Introduction
One earliest references to metabolic syndrome (MS) can be traced back to Camus in 1966 (97). However, in 1988 Gerald Reaven gave the Banting Lecture at the American Diabetes Association national meeting and introduced the concept of what he called “Syndrome X,” an aggregation of independent, coronary heart disease (CHD) risk factors in the same individual. The risk factors included in the syndrome were insulin resistance, defined as the inability of insulin to optimally stimulate the transport of glucose into the body’s cell (hyperinsulinemia or impared glucose tolerance) (note: for purposes of this review, we will use the terms insulin resistance and insulin sensitivity interchangeably), hypertension, hypertriglyceridemia, and low, high-density lipotrotein cholesterol (HDL) (522). The following year Kaplan (333) called it “the deadly quartet” and Foster (205) described it as “a secret killer.” None of these acronyms described the point made by Reaven in his Banting Lecture that insulin resistance/hyperinsulinemia might be the underlying cause of the syndrome. Reaven also suggested that insulin resistance/hyperinsulinemia was an underlying risk factor for T2D, which, at the time, was referred to as noninsulin-dependent diabetes mellitus. In 1991, Ferrannini et al. (194) published an article entitled “Hyperinsulinemia: the key feature of a cardiovascular and metabolic syndrome,” terms that better reflected Reaven’s point of view. Furthermore, use of the term MS acknowledges that this array of factors is associated with abnormal carbohydrate and lipid metabolism. These authors emphasized that insulin resistance was the underlying factor and, once acquired, those with a genetic predisposition would develop all the other aspects of the disorder. However, Ferrannini et al. pointed out that dietary intake and exercise could reduce insulin resistance, suggesting that the final phenotypic expression involves both genetic and acquired influences. Additionally, Haffner et al. (249) coined the term “insulin resistance syndrome” for the disorder to highlight the fact that insulin resistance preceded other aspects of the syndrome. Some individuals still use the term insulin resistance syndrome but now the term “metabolic syndrome” is more commonly used to describe the aggregation of multiple CHD and T2D risk factors. Insulin sensitivity/resistance is closely related to MS and the major manifestation of MS is coronary artery disease (CAD). In the Insulin Resistance Atherosclerosis Study (IRAS), a multi-ethnic cohort with variable glucose tolerance, the two lowest quintiles of insulin sensitivity, as estimated by frequently sampled intravenous glucose tolerance tests (FSIGT), had ORs of 2.4 and 4.7 for CAD compared with the highest quintile (535).
In addition to the factors mentioned by Reaven, Kaplan suggested that upper-body or visceral obesity needs to be considered as part of the syndrome and as a major risk factor for CHD and T2D, independent of overall obesity. Subsequently, many studies confirmed that visceral obesity (553) was correlated with the MS and its individual components, as reviewed by Despres and Lemieux (155, 156). As more studies were conducted, additional CHD risk factors were added to the syndrome. Landin et al. (380) suggested that elevated serum levels of fibrinogen and tissue plasminogen activator inhibitor were related to metabolic factors for CHD. Small dense particles of low-density lipoprotein (LDL) were found to have increased atherogenicity compared to large, less dense particles (604). Subsequently Barakat et al (46) and Reaven et al. (526) reported that small dense LDL particles were associated with insulin resistance, obesity, and T2D. Thus, small dense LDL was added to the list of factors associated with the MS. The combination of elevated serum triglycerides (TG), depressed HDL, and elevated small-dense LDL particles is commonly referred to as dyslipidemia.
Other aspects of atherosclerosis development and myocardial infarction that have been linked with the MS include endothelial dysfunction (663), inflammation (395, 718), and oxidative stress (661). In an animal model of diet-induced MS (50), Reil et al. (528) reported defective bradykinin and acetylcholine-induced relaxation with a normal response to nitroprusside in isolated artery strips. When the animals were switched from the high-fat, sucrose diet back to a low-fat, starch diet for 4 weeks, the endothelium-dependent defects were eliminated. In a recent study of 819 subjects, Suzuki et al. (621) reported that those subjects with the MS (N = 377) had lower flow-mediated dilation, which is a measure of conduit artery endothelial function, compared to that of subjects without MS.
High-sensitivity C-reactive protein (CRP), a marker for systemic inflammation, is commonly used clinically to evaluate risk in primary and secondary prevention of vascular disease (648). Not surprisingly, CRP has been associated with the MS. Obesity is now recognized as a state of chronic, low-grade inflammation and is associated with increased serum markers of inflammation and oxidative stress (196, 345), and MS has been linked with inflammation and oxidative stress (196, 254). However, as pointed out by Despres and Lemieux (155) in their review, not all obese individuals have the MS. In a recent study, Van Guilder et al. (661) studied three groups of subjects: normal weight, obese without MS, and obese with MS. Plasma CRP was significantly elevated in both obese groups compared to the normal weight group but was significantly elevated in the obese with MS compared to obese without MS. Other markers of inflammation including tumor necrosis factor-alpha (TNFα), interleukin (IL)-6 and IL-18 were only elevated in the obese with MS. Ox-LDL was measured as a marker of oxidative stress and was found to be elevated in both obese groups; the obese with MS was significantly higher that the obese without MS. These results suggest that increased oxidative stress and inflammation might be involved in the development of the MS. In addition, inflamed adipose tissue, characterized by increased monocyte infiltration and cytokine production, (24, 300, 563) has recently been associated with MS.
What Causes the Metabolic Syndrome?
When Reaven introduced the concept in 1988, he suggested that insulin resistance/hyperinsulinemia was the underlying cause. His suggestion was based on cross-sectional data showing associations between hyperinsulinemia and the other aspects of the syndrome in patients as well as experimental studies on rodents fed diets high in sucrose or fructose. Support for the role of hyperinsulinemia in the development of the syndrome came in 1992 when Haffner et al. (249) reported 8 years prospective data from 2217 subjects in the San Antonio Heart Study showing that fasting hyperinsulinemia preceded the development of other aspects of the syndrome including hypertension, hypertriglyceridemia, and depressed HDL-C, as well as the development of T2D. After adjusting for baseline obesity and fat distribution, as well as weight gain over the period of observation, significant relationships between insulin and the other factors remained present. Further support for the importance of hyperinsulinemia came from an animal study by Barnard et al. (50). Feeding rodents a high-fat sucrose diet resulted in skeletal muscle insulin resistance and hyperinsulinemia within a few weeks before any change in body fat or abdominal fat cell size. The animals subsequently developed hypertriglyceridemia, enhanced clotting and hypertension, that is, the MS.
Through his Banting Lecture, Reaven suggested some mechanisms to explain how insulin resistance/ hyperinsulinemia might cause the other aspects of the MS. He pointed out that hypertension was associated with elevated levels of plasma catecholamines and suggested enhanced sympathetic nervous system activity as a contributing mechanism. He also cited studies reporting that insulin caused the kidney to promote sodium reabsorption and increase plasma volume. For the increase in TG associated with the syndrome, he cited a study reporting that in perfused rat livers insulin increased very low density lipoprotein (VLDL) TG production. The San Antonio group provided further support for these mechanisms in two subsequent papers (148, 248). They pointed out that although some data had shown that acutely, insulin could be a vasodilator [as reviewed by reference (54)], prolonged insulin resistance and hyperinsulinemia was associated with hypertension and could be due to several mechanisms including an overactive sympathetic nervous system, sodium retention, altered membrane ion transport, and proliferation of vascular smooth muscle cells. They also stated that hyperinsulinemia would increase liver production of VLDL to increase serum TG, while at the same time reduce high-density lipoprotein production and serum HDL-C. These data all support Reaven’s suggestion that insulin resistance/hyperinsulinemia is the primary factor responsible for the MS. Using the insulin/glucose clamp technique, DeFronzo and Ferrannini (148) demonstrated that cellular resistance to insulin action subtends hyperinsulinemia. The important question, however, is what causes the insulin resistance. Reaven suggested that elevated plasma-free fatty acids were involved in the development of insulin resistance, as originally suggested by Randle et al. (518), and presented some experimental data to support his claim (522).
Based on the 1947 observation of Vague (657), women with upper body obesity were far more likely to get heart disease and T2D compared to women with lower body obesity. Kaplan (333), in 1989, suggested that obesity, especially abdominal obesity, was the primary factor that induced hyper-insulinemia and subsequently the MS. Today, a well-accepted theory on a cause of insulin resistance and the MS is excess abdominal fat. Many studies using waist-to-hip ratio, computed tomography, or similar measures have shown that abdominal fat, especially visceral fat, correlates best with the MS and CHD risk (155). In 1990, Bjorntrop (69) proposed that abdominal or “portal adipose tissue” would release excess free fatty acid that would go directly to the liver, increasing TG formation while also suppressing insulin clearance, resulting in hyperinsulinemia. He further stated that the lipid mobilizing capacity of portal adipose tissue is pronounced in men and abdominally obese women because of an abundance of β-adrenergic receptors with little α-adrenergic inhibition. In their review, Despres and Lemieux (155) discuss other factors associated with abdominal obesity that might be involved in the MS, including increased inflammatory cytokine production by fat cells along with reduced adiponectin release. However, they point out that while an abundance of data show that excess visceral fat is associated with both atherogenic and diabetogenic risk factors, an important question is whether visceral fat is a causal factor or simply a marker for the MS. The prospective studies discussed earlier suggest that it is a marker as opposed to being the true underlying cause.
In an editorial, Landsberg (381) stated, “the most important environmental cause of insulin resistance is central obesity, but a list would also include sedentary lifestyle and high-fat intake.” We would reverse the order to state that the most important cause of insulin resistance is a high-fat, refined-carbohydrate diet and physical inactivity, which are exacerbated by genetic predispositions, such as the development of abdominal obesity, as was suggested in a review by Barnard and Wen published in 1994 (51). Two early studies from the Reaven group reported that feeding rodents diets high in sucrose or fructose resulted in insulin resistance and hyperinsulinemia in as little as two weeks while on the diets (527, 722). A series of studies from the Barnard laboratory (49, 50, 52, 53, 237, 238, 543, 715) demonstrated that when rodents were placed on a high-fat and/or refined-carbohydrate diet compared to a low-fat, starch diet, skeletal muscle insulin resistance with elevated serum insulin developed in a few weeks, prior to differences in body weight or body fat. The high-fat, refined-carbohydrate diet eventually led to hypertension, hypertriglyceridemia, and enhanced clotting as well as obesity, which are characteristics of the MS (49, 50). The observation of diet-induced skeletal muscle insulin resistance is significant, as skeletal muscle is the most important target tissue for insulin action, is the primary site for glucose disposal following a meal (147), and shows a major defect in insulin-resistant T2D patients (147). The diet-induced insulin resistance in the rodent model was associated with a decrease in insulin receptor autophosphorylation and tyrosine kinase activity similar to the defects observed in muscle from type 2 diabetic patients (53, 575, 714). The mechanisms underlying these and other defects that lead to insulin resistance are discussed in detail later.
Clinical Aspects of the Metabolic Syndrome
Since Reaven introduced the concept in 1988, thousands of papers related to MS have been published. A search of PubMed in August 2010 resulted in over 31,000 responses demonstrating a high level of interest in the concept and in 2006 The Journal of the CardioMetabolic Syndrome appeared. The reason for such interest is not surprising as Ford et al. (201, 202) have estimated that using the revised National Cholesterol Education Program (NCEP)/Adult Treatment Panel (ATP) III criteria (182, 240, 241) showed that between 32% and 34% all US adults (31–34% of men and 33–35% of women) have MS. Based on International Diabetes Federation (IDF) criteria, estimates were 39%, with 40% of men and 38% of women (201); similar classification occurred 93% of the time for the two definitions. This equates to greater than 100 million in the US based on a population estimate of 310 million. A multiethnic representative US sample of 12,363 men and women 20 years and older from the third National Health and Nutrition Examination Survey (NHANES) were evaluated for MS as defined by the ATP III diagnostic criteria (abdominal obesity, hypertriglyceridemia, low HDL, hypertension, and fasting hyperglycemia) and the disorder was found to be present in 22.8% and 22.6% of the men and women, respectively. MS was present in 4.6%, 22.4%, and 59.6% of normal-weight, overweight, and obese men, respectively, and physical inactivity was associated with an increased risk of developing the syndrome (488). When Reaven published his paper in 1988 he stated,
“[B]ased on available data, it is possible to suggest that there is a series of related variables – Syndrome X – that tend to occur in the same individual and may be of enormous importance in the genesis of coronary artery disease (CAD)” (522).
Many studies have investigated the syndrome as a possible independent risk factor for CHD. In a 2007 meta-analysis of longitudinal studies involving 172,573 individuals, Gami et al. (219) reported that the relative risk (RR) for individuals with the MS compared to those without the MS was 1.78 for cardiovascular events and death; for women the RR was 2.63. After adjusting for traditional cardiovascular risk factors those with the MS still had a RR of 1.54 for cardiovascular events and death. In 2006, results from another meta-analysis conducted by Galassi et al. (217) showed that the MS was associated with increased incidence of cardiovascular disease (CVD) (RR 1.53), CHD (RR 1.52), and stroke (RR 1.76). Individuals with the MS had increased all-cause mortality (RR 1.35) and cardiovascular mortality (RR 1.74). Again, the risks were higher in women compared to men.
In his 1988 paper, Reaven pointed out that resistance to insulin-stimulated glucose uptake was present not only in patients with T2D but also in a majority of individuals with impaired glucose tolerance (IGT) as well as those with the MS. Thus, it was speculated that individuals with the MS and insulin resistance might be at high risk for the development of T2D; this turned out to be the case. In a meta-analysis of 16 cohort studies, Ford et al. (204) reported that the RR for T2D in individuals with the MS ranged from 4.42 to 5.17 depending on the criteria used to define the MS. The authors concluded that the MS, however defined, has a stronger association with T2D than previously demonstrated for CHD.
Although the MS appears to be a well-accepted syndrome associated with increased risk for both CHD and T2D, the use of the term MS has been questioned for a variety of reasons (326, 523, 525). These include, but are not limited to: (i) it occurs only in insulin-resistant persons, which the ATP III criteria does not directly evaluate and, currently, there is no simple clinical measure for insulin resistance; (ii) many individuals may not satisfy the arbitrary cutoffs for diagnosis, that is, might be sufficiently insulin resistant and have additional CAD risk factors to be at significant increased CVD risk; and (iii) it has low clinical utility since treating the individual factors may be a less effective approach than addressing the underlying problem, which is generally lifestyle-induced insulin resistance in genetically susceptible individuals. In fact, published data support this contention (398).
In 2005, a joint statement from the American Diabetes Association and the European Association for the Study of Diabetes questioned the existence of the MS (326). The groups undertook a review of the literature and concluded:
“While there is no question that certain CVD risk factors are prone to cluster, we found that the MS has been imprecisely defined, there is a lack of certainty regarding its pathogenesis, and there is considerable doubt regarding its value as a CVD risk marker. Our analysis indicates that too much critically important information is missing to warrant its designation as a ’syndrome.”’
The fact that the MS is imprecisely defined stems from the different definitions adopted by different organizations to identify individuals with the syndrome. Due to the fact that insulin resistance is rarely measured clinically, other criteria have been adopted to identify individuals who might be insulin resistant and possess the MS. To date at least six different definitions from five different agencies have been proposed to define adults with the MS, with the criteria being dramatically varied among agencies. The problem is more pronounced in the pediatric arena where, according to Morrison et al. (453), as many as 40 different definitions have been used to identify youth with the MS. This is important as, not surprisingly, pediatric MS predicts adult MS, although in this cohort, body mass index (BMI) risk estimates were similar (415).
In 1998, a consultation group from the World Health Organization (WHO) published the first clinical criterion to define adults with MS (18). These criteria were proposed in part as simple tools to help health professionals identify individuals likely to have a clustering of metabolic abnormalities. Following Reaven’s suggestion, this group emphasized the importance of insulin resistance and suggested several clinical measures that could be used to assess insulin resistance, that is, IGT, impaired fasting glucose (IFG), T2D mellitus, or impaired glucose disposal demonstrated with the insulin clamp test. In addition to a measure of insulin resistance to qualify for the MS, individuals must possess two of the following risk factors: obesity, hypertension, high TG, reduced HDL-C, or macroalbuminuria.
In 1999, the European Group for the Study of Insulin Resistance (EGIR) proposed a slight modification from what had been proposed by the WHO (41). This group used the term insulin resistance syndrome as opposed to the MS. In addition, they suggested also requiring evidence of insulin resistance, as measured by plasma insulin above the upper quartile for the population plus two other factors for the diagnosis, that is, abdominal obesity, hypertension, elevated TG, reduced HDL-C, or elevated plasma glucose. Interestingly, this group excluded T2D as a criterion.
In 2001, the NCEP/ATP III introduced alternative clinical criteria to Reaven’s initially proposed definition to diagnose the syndrome and did not require any measure of insulin resistance, but did require three of the following five criteria, that is, elevated fasting glucose or a diagnosis of T2D, abdominal obesity, elevated blood pressure, elevated TG, or reduced HDL-C (1). Like the EGIR, the ATP III emphasized the importance of abdominal obesity and noted that some ethnic groups show signs of insulin resistance at lower levels of waist circumference than used as criteria for diagnosis of the syndrome.
In 2003, the American Association of Clinical Endocrinologists (AACE) also used the term insulin resistance syndrome and provided their criteria for the diagnosis including IGT or IFG with no specific number of other factors required but the left the decision to be based upon the judgment of the clinician. The major additional criteria to be considered included elevated TG, elevated blood pressure, reduced HDL-C and obesity (BMI). Other factors that could be used in the judgment included family history of atherosclerotic vascular disease or T2D, polycystic ovary syndrome, and hyperglycemia. The presence of T2D was excluded.
In 2005, the IDF (303) provided their criteria to define the MS. Although some of the members of the IDF writing group were also on the WHO consultation group, they replaced the requirement for a direct measure of insulin resistance with emphasis on abdominal obesity as it correlates well with insulin resistance. When abdominal obesity is present, two additional factors listed in the ATP III criteria were sufficient to define the syndrome. The definition of abdominal obesity involved waist circumference that was adjusted for different ethnic groups. For people of European origin (Europeans and Americans) thresholds were set at 94 cm or more for men and 80 cm or more for women. For Asian populations, excluding Japanese, the thresholds were set at 90 cm or more for men and 80 cm or more for women; while the thresholds for Japanese were set at 85 cm or more for men and 90 cm or more for women. The other difference from the ATP III criteria was a lower IFG value (100 mg/dL). This same value was also adopted by the ATP III group in 2005 (ATP III-R) (241). In addition, the lower fasting glucose, ATP III-R added the presence of drug treatment for TG, reduced HDL-C, hypertension, or elevated glucose as additional criteria. The precise cut values for the various criteria used by the different agencies have been outlined in the article describing ATP III-R criteria by Grundy et al. (241).
Thus, one would have to agree with the statement from the two diabetes groups, “ . . . the MS has been imprecisely defined . . .” (326). This lack of precision in defining the syndrome has led to different results in different studies depending on the criteria used to define the syndrome. Katzmarzyk et al. (339) conducted a longitudinal study of 20,789 US men aged 20 to 83 years who were followed for 11.4 years. They identified men with the MS using three different methods, the original ATP III, ATP III-R, and IDF. It was found that at baseline the percentage of men with the MS was 19.7, 27, and 30, according to the different criteria. The RR for cardiovascular mortality for men with the MS was 1.79, 1.67, and 1.67 according to ATP III, ATP III-R, and IDF, respectively; however, there were only 213 cardiovascular deaths total. In a similar study, Benetos et al. (59) followed 84,730 French men and women 40 years or more of age for 4.7 years. They also used three different methods to identify the MS, ATP III, IDF, and ATP III-R. The percent of individuals identified as having the MS were 9.6, 21.6, and 16.5. The RR for cardiovascular mortality was 2.05, 1.77, or 1.64 for the ATP III, ATP III-R, and IDF, respectively; again, the total number of cardiovascular deaths was small (104) over the 4.7 years. The results from these two studies on large populations clearly show that the different criteria used by the different agencies to define individuals with the MS varies dramatically, by as much as 50%. Regardless of the criteria used to identify the MS, both studies showed that the presence of the MS by any of three different definitions increased the risk for cardiovascular mortality but also with significant variability in risk.
Although the data clearly show that the presence of the MS increases the risk for CVD and cardiovascular mortality, some have questioned whether there is a need to identify such patients, especially since: (i) MS may not predict cardiovascular events better than the sum of its components and (ii) none of the definitions include traditional, well-established cardiovascular risk factors, such as smoking, total or LDL cholesterol and age factors that are standard clinical measures, along with blood pressure and HDL-C that are incorporated into the Framingham Risk Score (FRS). Wannamethee et al. (677) reported a prospective study of 5128 British men with no initial history of CHD or T2D, followed for 20 years. They compared the FRS with the MS identified by ATP III-R criteria as predictors for CHD, stroke, or T2D. Using ATP III-R criteria for MS the RR for CHD was 1.64, for stroke was 1.61 and for T2D was 3.57. When they compared the MS with the FRS, they found that the FRS was superior to the MS in predicting CHD and stroke but found it to be inferior in predicting T2D. de Zeeuw and Bakker (146) conducted a similar study of 8217 Dutch men and women followed for a median 6.5 years. The results showed that the FRS was superior to the MS in predicting cardiovascular mortality/morbidity. The FRS appears to be superior in identifying CHD risk not only because it contains the well-established CHD risk factors not included in the MS, but also because it is based on continuous data, as opposed to the discrete cutoff points used to identify the MS. Even Reaven (524) criticized the ATP III criteria, stating that the cut points are arbitrary and not based on sound scientific data, while insulin sensitivity is continuous in normal populations with at least a sixfold variation between the most sensitive and the most insulin-resistant individuals. Thus, he states that there is no simple, objective way to classify an individual as being insulin resistant, which he claimed was the basis for his syndrome X.
Despite the criticisms, the MS has been strongly defended, especially since it is a powerful predictor for T2D [80% of those with T2D have MS (309)] and a major risk factor for CHD (233, 309). For example, in a cohort of 3323 middle-aged adults, the MS RR was 2.88 for CVD, 2.54 for CHD, and 6.92 for T2D in men and 2.25, 1.54, and 6.90 in women, respectively (695). Sattar et al. (571) noted that men with four or five features of the syndrome have been estimated to have a 3.7-fold increase in risk for CAD and a 24-fold increased risk for T2D compared with men with none, while Klein et al. (358) reported 2.5% and 1.1% incidence of CVD and T2D, respectively, in those with one component of the MS; meanwhile 15% and 18% developed these diseases in those with four or more components of the syndrome.
MS is also a predictor of mortality. For example Lakka et al. (377) reported that middle-aged men with the MS exhibited a 2.9- to 4.2-fold risk of CHD death over an 11-year follow-up compared to healthy men and after adjustment for conventional risk factors. In addition, all-cause mortality was increased 2.3-fold in those in the highest quartile for MS factors. In the Botnia study of 4483 subjects from Finland and Sweden, Isomaa et al. (309) estimated the risk for cardiovascular mortality over a 7-year follow-up was increased markedly in those with MS (12.0% vs. 2.2%). A meta-analysis noted increased risks of CVD [odds ratio (OR): 2.40] and all-cause (OR: 1.58) mortality in subjects with MS (454).
Grundy (239) stated that the intended definition of the MS was not a tool to estimate absolute risk, but rather a tool to be used by clinicians to improve obesity counseling. In an attempt to clear up some of the controversy and unify the clinical definitions of the MS, a meeting was convened with representatives from the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. In 2009, a “joint interim statement” was published in Circulation (17), attempting to establish criteria to identify patients with the MS as shown in Table 1. It was agreed that there should not be an obligatory component, although there was agreement regarding the importance of central obesity, and thus waist measurement would continue to be a useful preliminary screening tool. Three abnormal findings out of five would qualify a person for MS. A single set of cut points would be used for all components except waist circumference, for which further work is required, and, at present, would be based upon population/country-specific definitions. The statement reiterated that patients with MS have two and five times the risk of developing CVD and T2D, respectively, over the next 5 to 10 years, as compared to individuals without MS.
Table 1.
Established Criteria Proposed for Clinical Diagnosis of Metabolic Syndrome
| Clinical measure | WHO (1998) | IDF (2005) | Joint IDR/NHLBI/AHA |
|---|---|---|---|
| Insulin resistance | IGT, IFT, T2DM, or lowered insulin sensitivity* Plus any two of the following |
None | None But any three of the following five features |
| Body metric | Men: waist-to-hip ratio >0.90 Women: waist-to-hip ratio >0.85 and/or BMI >30kg/m2 |
Increased WC (population specific) plus any two of the following | Population- and country-specific definitions |
| Lipid | TG 150 mg/dL and/or HDL-C <35 mg/dL in men or <39 mg/dL in women | TG > 150 mg/dL or on TG Rx | ≥150 mg/dL (1.7 mmol/L) |
|
|
|
||
| HDL-C <40 mg/dL in men or <50 mg/dL in women or on HDL-C Rx | <40 mg/dL (1.0 mmol/L) in males; <50 mg/dL (1.3 mmol/L) in females | ||
| Blood pressure | ≥140/90 mmHg | ≥130 mmHg systolic or 85 mmHg diastolic or on hypertension Rx | Systolic ≥130 and/or diastolic ≥85 mmHg |
| Glucose | IGT, IFG, or T2DM | ≥100 mg/dL (includes diabetes) | ≥100 mg/dL |
| Other | Microalbuminuria |
MS criteria as defined by WHO, IDF, and the joint IDR/NHLBI/AHA are compared. T2DM indicates type 2 diabetes mellitus; WC, waist circumferences; BMI, body mass index; and TG, triglycerides. All other abbreviations are in text.
Insulin sensitivity measured under hyperinsulinemic euglycemic conditions, glucose uptake below lowest quartile for background population under investigation.
As the debate continues more and more data are published relating insulin resistance and/or the MS to other common health problems found in the industrialized nations. In 2006, the Harvard School of Nutrition hosted a conference, Metabolic Syndrome and the Onset of Cancer, where several papers were presented showing that hyperinsulinemia was related to breast, prostate, and colon cancers. The proceedings were published in September 2007 in a supplement to the American Journal of Clinical Nutrition. Barnard (48) suggested that the MS was linked to prostate cancer risk as a result of hyperinsulinemia acting on the liver to increase production of insulin-like growth factor-I (IGF-I), a factor known to stimulate tumor growth and block apoptosis. Kalmijn et al. (327) reported that men diagnosed with the MS in their early 50s were more likely to develop dementia, especially vascular-related dementia, later in life. In a recent review, Galluzzo et al. (218) pointed out that several recent studies recommend that women with polycystic ovary syndrome should be evaluated for MS and that lifestyle modification should be the first-line therapy. Kawamoto et al. (343) tested over 3000 men and women and reported that those with the MS had a risk ratio of 1.53 for chronic kidney disease. Tsochatzis et al. (649) reported that insulin resistance was associated with chronic liver disease, especially hepatitis C and nonalcoholic fatty liver disease (449), and MS predicts hepatic steatosis (252). Additionally, accumulating evidence supports that sleep apnea is a manifestation associated with MS (665). D’Aiuto et al. (139) analyzed data from 13,994 men and women from the Third National Health and Nutrition Examination Survey (NHANES) and found that individuals with severe periodontitis were 2.31 times more likely to have the MS compared to individuals without periodontitis. D’Aiuto et al. (139) even suggested that the chronic low-grade inflammation characteristic of periodontitis might contribute to the development of the MS. The mechanisms linking the MS to CHD and T2D are well understood; however, the links to these other health problems are not well understood and require further study.
It is obvious that much more research is needed before we understand how all of these factors are related from a mechanistic point of view. It is also likely that the true underlying factors are inappropropriate diet and physical inactivity, characteristic of industrialized nations. In an analysis of the 1999–2004 NHANES data, Wildman et al. (693), showed support for the value of physical activity in preventing the MS. They found that 23.5% of normal weight (BMI < 25) were metabolically abnormal while 51.3% and 31.7% of overweight or obese (BMI > 30) were metabolically healthy. Low physical activity was an independent correlate of clustering of cardiometabolic factors in the normal-weight individuals while high physical activity correlated with zero or only one cardiometabolic abnormality in the overweight or obese individuals. The importance of exercise, and to some extent diet, will be discussed in the later sections.
Insulin Action
Section “Overview” will focus on an overview of the molecular aspects of insulin resistance. Insulin resistance (i.e., low insulin sensitivity) has been suggested as the major underpinning link between physical inactivity and MS. Many tissues, including skeletal muscle, liver, and adipose tissue may exhibit insulin resistance. Given the clinical benefit of treating those with insulin resistance, techniques have been developed to assess insulin sensitivity in vivo. The euglycemic-hyperinsulinemic glucose clamp (EHC) involves injecting a fixed dose of insulin to increase insulin to postprandial or to supraphysiological levels with normal glucose concentrations being maintained by infusing glucose. The glucose infusion rate (often referred to as M-value) reflects insulin sensitivity and is generally considered an inverse measure of insulin resistance; glucose disposal is often also determined. The FSIGT can estimate insulin sensitivity and acute insulin response using the Bergman minimal model (65), yielding a hyperbolic relationship between insulin secretion and insulin sensitivity (9). The product of these two indices is referred to as the disposition index (DI), a marker of β-cell function. As depicted in Figure 1, exercise training/physically activity status modify insulin sensitivity and insulin secretion in accordance with this relationship, and failure of insulin secretion to compensate for a fall in insulin sensitivity leads to elevated fasting glucose and prediabetes (impaired glucose tolerance), and depending on genetic predisposition a continued progressive decline in both insulin secretion and insulin sensitivity to T2D. Generally, use of the EHC and FSIGT are the gold standard methods for estimation of insulin sensitivity and/or β-cell function. However, these methods are expensive, not simple to perform and generally not applicable in standard clinical practice. The oral glucose tolerance test (OGTT) is less expensive and its simplicity allows for more widespread use. Other techniques used include insulin suppression testing, insulin tolerance testing (ITT) and continuous glucose monitoring systems (CGMSs).
Figure 1.

Graph depicting the hyperbolic relation between insulin secretion and insulin sensitivity. Insulin secretion rises as insulin sensitivity falls when an individual goes from a state of exercise training/being physically active (point A) to detraining/sedentary (point B) and vice versa, that is, bidirectionality of the two arrows from B to A when undergoing exercise training/increasing physical activity levels. A failure of insulin secretion to compensate for a fall in insulin sensitivity is noted when both insulin secretion and insulin sensitivity decline from points B to C, leading to elevated fasting glucose and prediabetes (impaired glucose tolerance). A progressive decline in both insulin secretion and insulin sensitivity to point D indicates type 2 diabetes. Adapted from reference (9) with permission.
Overview
Insulin is a polypeptide hormone that was first discovered in 1921 by Frederick G. Banting and Charles H. Best while working in the laboratory of J.R. Macleod at the University of Toronto. The hormone was later purified in 1923 by James B. Collip and is now used in clinical practice to treat insulin deficiency diseases, including type 1 and T2D. Insulin is secreted from the β-cells of the pancreatic islets of Langerhans in response to glucose and amino acids consumed during a meal. Insulin is a central regulatory hormone in the maintenance of glucose homeostasis and is also involved in anabolic processes including tissue growth and development. In a healthy person, glucose is controlled within very narrow limits in the blood. This is achieved by the regulation of glucose production by the liver, and to a lesser extent the kidney, as well as uptake by peripheral tissues, primarily skeletal muscle, liver, and adipose tissue. In addition to the control of blood glucose, insulin also exerts strong control over lipid metabolism by stimulating lipid synthesis in liver and fat cells and by attenuating lipolysis, that is, TG breakdown to fatty acids.
Glucose is utilized by cells to produce potential energy in the form of adenosine triphosphate (ATP). The entry of glucose into the cell is achieved primarily via a carrier-mediated process, which includes a family of transporters known as GLUT proteins. GLUT, proteins encoded by SLC2A family members, are membrane proteins found in most mammalian cells and contain 12 membrane-spanning helices with both the amino and carboxy terminal regions exposed on the cytoplasmic side of the plasma membrane. To date, over 14 GLUT family members have been identified (317). Each transporter isoform performs a specific role in hexose metabolism as dictated by expression patterns within tissues, protein transport kinetics, substrate specificity and the physiological conditions controlling gene expression. The GLUT family is divided into three subclasses based upon sequence similarities; however for the purposes of this review, we will focus on the well-characterized class I glucose transporters, GLUT1–GLUT4, as these are primarily expressed in glucoregulatory tissues. Insulin-stimulated transport of glucose into cells is achieved by insulin binding to its cell surface receptor and the initiation of a cascade of signaling events culminating in the redistribution of GLUT4 (the insulin responsive glucose transporter) to the plasma membrane. Glucose is then transported across the plasma membrane where it is immediately phosphorylated and either stored as glycogen or metabolized to produce ATP. In the subsequent sections we will provide an overview of the insulin signal transduction pathway and glucose transport system as well as discuss the mechanisms contributing to impaired insulin action, insulin resistance. We will explore both myocyte-related mechanisms contributing to skeletal muscle insulin resistance as well as describe insulin resistance producing factors secreted from adipose tissue and liver. We will close this section discussing the impact of muscle-secreted factors on metabolism and propose that myokines may in part mediate aspects of exercise-induced effects. Much of the focus of this section will be centered on muscle insulin action as exercise-induced improvements in insulin sensitivity appear related to gains in muscle rather than hepatic insulin action (356, 696).
Insulin Signal Transduction
Insulin and the insulin receptor
Insulin is a peptide hormone, consisting of 51 amino acids with a molecular weight of 5808 Da, secreted by the pancreas as either the full length proprotein or as the fully biologically active form in which the c-peptide is cleaved. Because insulin release into the portal circulation is susceptible to first pass degradation by the liver, c-peptide escapes this fate and is therefore a more accurate marker of insulin secretion. Insulin binds to its receptor (IR) in target tissues including skeletal muscle, liver, and adipose tissue. The IR gene is located on chromosome 19 and is comprised of 22 exons and 21 introns, spanning 150 kb (580). IR is synthesized as a preproreceptor. Following cleavage of a 30-aa signal peptide, the proreceptor undergoes glycosylation, folding, and dimerization. The final IR product consists of a heterotetrameric complex of two α-subunits and two β-subunits linked by disulphide bonds (Fig. 2). In glucoregulatory tissues, including adipose and muscle, the IR is thought to be more highly localized to caveolae located in the plasma membrane (247).
Figure 2.

Schematic of the insulin receptor and critical sites of tyrosine phosphorylation. CR, cysteine-rich region; JM, juxtamembrane; KD, kinase domain; CT, C-terminal domain; Y: tyrosine residue.
The basal form of the insulin receptor has very low kinase activity, as the activation loop, which traverses the N-and C-terminal lobes in the unliganded state, blocks ATP, and substrate binding (295). Insulin, following binding to the extracellular α-subunits, yields a conformational change in the receptor and transmits a signal across the plasma membrane, which activates the intrinsic tyrosine kinase domain of the intracellular β-subunit. This results in a series of intermolecular autophosphorylation reactions on tyrosine residues that are now known to serve distinct functional roles (138, 391).
Comprehensive studies using selective mutations in the IR as well as computational models from crystal structure analyses have yielded specific details regarding these molecular events (296, 727). Specifically, insulin binding causes the phosphorylation of three key tyrosine residues (Y1158, Y1162, and Y1163), allowing for movement in the A-loop and exposure of the ATP and substrate binding sites (178, 296, 551). Additionally, auto-phosphorylation of tyrosine residues 965 and 972 in the juxtamembrane region, 1158, 1162, and 1163 in the regulatory region (also known as the activation loop of the kinase domain), and 1328 and 1334 in the C-terminus of the cytoplasmic domain of the IR are essential for full kinase activity (687). Moreover it was shown that pTyr 960 is critical for appropriate IR substrate recognition (392), and pTyr972 serves as a binding site for the phospotyrosine binding domains (PTB) of IRS-1, Shc, and STAT5 (110, 246, 321, 573) (Fig. 2). Considered an important feature of hormone signaling, the autophosphorylated IR is rapidly internalized following ligand binding. Endocytosis of the IR leads to proteolytic degradation of the ligand receptor complex, thus terminating ligand action. Recent work by Fagerhom et al. (184) shows that this process is caveolae-mediated and involves the tyrosine phosphorylation of caveolin-1.
Proximal insulin signaling
The IR, upon phosphorylation, recruits various substrates and scaffolding proteins to exert downstream effects. These include the four well-described insulin receptor substrate (IRS) proteins, or IRS1–4, as well as Gab1, SIRPs, Cbl, Shc, and APS (Fig. 3) (500, 632). Upon phosphorylation, these substrates serve as docking or scaffolding platforms for distinct cellular kinases or effectors that mediate the divergent biological actions of insulin. In addition, each of these substrates may be compartmentalized to distinct cellular locations also owning to the specificity to which interactions with other proteins or lipids occur and to which unique downstream effects are achieved. For example, IRS and Shc are recruited to the juxtamembrane region in IR containing a critical arginine-proline-any amino acid-tyrosine (NPXY)-binding motif (342, 468), while APS binds directly to the activation loop.
Figure 3.
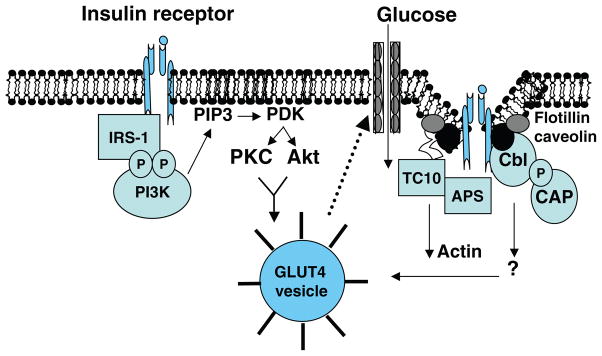
Schematic of insulin signal transduction through canonical IRS1/PI3K pathway and through abbreviations: Cbl/CAP/TC10 pathway associated with lipid rafts in the plasma membrane. Akt or PKB, protein kinase B; APS, adapter protein with a PH and SH2 domain; CAP, c-Cbl-associated protein; Cbl, protooncogene; GLUT4, insulin responsive glucose transporter highly expressed in myocytes and adipocytes; IRS, insulin receptor substrate; PDK1, phosphoinositide-dependent kinase 1; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; PKC, protein kinase C; TC10, small Ras-related GTPase, member of the Rho family.
Insulin receptor substrate-1, 2
IRSs are the major substrates of the insulin receptor and the IGF)-I receptor tyrosine kinases. Of the several IRS proteins described, IRS-1, 2 are shown to have biological relevance in peripheral tissues for glucose transport. Briefly, tyrosine phosphorylation of IRSs creates recognition sites for additional effector molecules containing Src homology 2 (SH2) domains, including the adaptor proteins Grb2 and Nck, the SHP2 protein phosphatases and the 85-kDa, p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) (686). The pleckstrin homology (PH) domain of the substrate protein is obligatory to elicit the full signal response, as insulin-stimulated tyrosine phosphorylation is significantly reduced in by PH deletion (460). Both IRS-1 and IRS-2 contain multiple YXXM motifs that are phosphorylated by the activated IR (687). In vitro, IRS-1 and IRS-2 display similar capacities to bind p85 of PI3Ks. With respect to insulin action and substrate metabolism, critical roles for IRS-1 and IRS-2 are well established (627, 697). Furthermore, human genetic inactivating mutations in either Isr1 or Irs2 are associated with insulin resistance and T2D (47, 427). While the IRS family shares a high degree of homology, studies in genetically engineered rodents delineate specific roles for these protein isoforms (637, 697). Genetic ablation of either Irs1 or Irs2 in rodents or cells leads to dire physiological consequences, including impaired insulin signal transduction and growth retardation (25, 627).
IRS-1
IRS-1 was first identified by Sun et al. in 1991 and was found to be expressed in a wide variety of tissues (619, 620). Irs-1 null mice are growth restricted and display marked skeletal muscle insulin resistance, but the development of frank T2D in this model is prevented by pancreatic Irs2-induced hyper-insulinemic compensation (25, 627). Furthermore, short-term inhibition of Irs-1 by anti-sense oligonucleotide administration in rats recapitulated much of the insulin resistance phenotype observed in Irs-1 knockout mice (26). In the context of findings for Irs-1 deletion relating to insulin action, recent evidence obtained from Irs-1 transgenic mice suggests that increased expression of IRS-1 is not associated with improved insulin sensitivity (455). In fact, transgenic mice displayed glucose intolerance, increased visceral adipose mass, and increased levels of circulating inflammatory cytokines (455). Interestingly, while Irs-1 mRNA was significantly elevated in all tissues, IRS-1 protein levels were preferentially elevated by 50% and 200% in skeletal muscle and adipose tissue, respectively. The IGT seen in the transgenic mice is explained by reduced tyrosine phosphorylation of IR and YXXM motif of IRS-1 in liver, which may be caused by indirect action given that IRS-1 protein levels were unchanged in liver (455). Taken together these data indicate a relatively narrow window in which IRS-1 levels must be maintained to preserve glucose homeostasis and healthy tissue morphology.
IRS-2
IRS-2 is expressed in all of the primary glucoregulatory tissues including pancreas, liver, adipose, and skeletal muscle. Mice with an Irs2 homozygous null mutation display metabolic defects in liver, muscle, adipose, and pancreas but develop frank diabetes as a result of defects in signal transduction in pancreatic β-cells (511, 697). Furthermore, the importance of IRS-2 in regulating β-cells mass and in the prevention of type 1 diabetes was recently shown by Norquay et al. (467) in nonobese diabetic (NOD) mice overexpressing IRS-2 in β-cells. Remarkably, glucose tolerance was markedly improved and the risk of diabetes decreased 50% in NOD Irs2 Tg mice due to a tenfold greater β-cell mass. In contrast to pancreas, IRS-2 is not necessary for insulin or exercise-stimulated glucose transport in skeletal muscle (268). In liver, insulin-stimulated regulation of sterol regulatory element-binding protein (SREBP)-1c and fatty acid homeostasis is previously thought to depend almost exclusively on the IRS-2 arm for activation of PI3K, PDK1, and atypical protein kinase C isoforms (aPKCs) (187, 561, 602). Again, recent work from the laboratory of Morris White using Lox Cre approach to generate mice with hepatic specific deletions of IRS-1 and or IRS-2 calls into question the established role of IRS-2 in the liver. Findings suggest that IRS-1, not IRS-2, plays a dominant role in regulating hepatic nutrient homeostasis (243). Whether these findings in rodents can extend to isoform specific roles in regulating insulin action in human tissues requires further investigation.
Insulin Receptor Downstream Effectors
Phosphoinositide 3,4,5 (PI3)-kinases
It is well accepted that class IA PI3K is required for insulin-dependent GLUT4-mediated glucose transport in muscle and adipose. In mammals, this class of kinase comprises a p110 catalytic subunit (p110 α, β, or δ) bound to one of five “p85” regulatory subunits (p85α, p85β, p55α, p55γ, or p50α). In the relative absence of insulin, p85 is bound tightly and stabilizes basal p110 protein, inhibiting its catalytic lipid kinase activity. In contrast, during insulin stimulation, the p85–p110 complex is recruited to phosphotyrosine residues in activated receptor or adaptor molecules containing SH2 domains. Recruitment of the p-85/p110 heterodimer to a location proximal to the plasma membrane brings p110 into close proximity with its lipid substrates, thus reducing the inhibitory actions of p85 on p110. Class IA wortmannin sensitive PI3K generates phosphatidylinositol 3,4,5 triphosphate, from PIP3 PtdIns (4,5)P2 at the cell surface and this is thought to interact with specific lipid binding domains present in Ser/Thr protein kinases, including PDK1 and protein kinase B (PKB/Akt). Subsequently, these kinases then act upon GTPases and other scaffolding proteins to promote further propagation of the insulin signal. Early studies using wortmannin to inhibit PI3K showed the insulin-mediated glucose transport into cells is nearly abolished; therefore, PI3K is considered a central and essential signaling regulator for a variety of biological cell responses, notably growth, proliferation, and metabolism (222).
Mammals express seven PI species, two of which are described above and the function of five of which remains poorly understood. While PtdIns(3,4,5)P3 is shown to rise transiently in response to acute insulin treatment and is thought to be the primary PI species regulating metabolic processes, overexpression of PtdIns(3,4,5)P3 alone is not sufficient to fully stimulate transport-mediated uptake of glucose into cells (590). Furthermore, a role for class II wortmannin-insensitive PI3K C2α and the other five PI species has recently been proposed for fine tuning the overall metabolic response of a cell to insulin (185, 590). The importance of this adjunct-signaling pathway in glucose disposal and insulin sensitivity requires further investigation.
Akt
Protein kinase B, also known as Akt, is a Ser/Thr kinase involved in a large and diverse set of cellular processes including glycolysis, glycogen synthesis, lipogenesis, suppression of gluconeogenesis, and cell survival. One of Akt’s many important functions is mediating the metabolic actions of insulin to stimulate glucose transport (688). Akt is a PI 3-K effector molecule and its isoform expression dictates cell specific actions in vivo. RNA silencing studies conducted by the Czech laboratory (728) and gene deletion strategies in mice conducted by the Birnbaum laboratory (38) show that Akt2/PKBβ is critical for insulin-mediated glucose disposal. While it is well accepted and evident that it serves as an essential role for Akt2 in glucose transport, studies where Akt inhibition did not cause impaired glucose uptake have been reported. While Akt is thought to be a central effector driving glucose transport, other signaling cascades are thought to participate in parallel including the aPKCλ/ζ (187, 188, 363), p-38 mitogen-activated protein kinase (622), and the CAP/c-Cbl/TC10 cascade (57, 500).
Atypical protein kinase C isoforms, PKCλ, ζ
aPKCs are also downstream effectors of PI 3-K; however these signaling molecules may serve different functions in regulating substrate metabolism in the various glucoregulatory tissues (187). In muscle IRS-1 knockout has a marked effect on aPKCs whereas IRS-1 knockout in liver has little effect on aPKC activation (561). While a role for aPKCs in regulating glucose uptake and metabolism is shown in muscles and in the liver, aPKCs are thought to exert insulin-mediated effects preferentially on hepatic lipid synthesis via regulation of SREBP-1c (SREBP-1c) (422). While previously considered an independent and parallel effector signaling system, recent evidence supports that aPKCs and the TC10 pathways may interact at the plasma membrane (111, 331). Atypical PKCs belong to the AGC subfamily of protein kinases [reviewed by Pearce et al. (491)] and, thus, while phosphorylated by PDK1 downstream of PI3K, can be recruited to lipid rafts in a TC10-dependent manner by Par3 (partitioning-defective) and Par6 proteins (331). Thus, atypical PKCs may represent a point of convergence between PI3K signaling and the TC10 pathways (111). Furthermore, evidence from the Klip laboratory shows that PKCε translocates to the plasma membrane of C2C12 myotubes in culture in response to contraction and that GLUT4 translocation proceeds in a PKCε-dependent manner (464). Whether PKCε is critical for glucose transport into muscle during physical exercise remains to be demonstrated.
AS160
The Rab-GTPase activating protein known as AS160 (or TBC1D4) is a 160 kDa putative substrate of Akt (330) and is widely expressed in a variety of tissues including brain, testes, kidney, pancreas, liver, heart quadriceps, and white and brown adipocytes. AS160 undergoes phosphorylation in response to insulin and contains multiple Akt phosphorylation sites: Ser318, Ser341, Ser570, Ser588, Thr642, and Ser751 (330, 565). In addition to insulin, AS160 is phosphorylated in response to PDGF, activation of nPKCs and adenosine monophosphate kinase (AMPK), and following skeletal muscle contraction (216, 639). AS160 is thought to regulate GLUT4 trafficking as well as serve as a convergence point for insulin and contraction-mediated GLUT4 translocation to the plasma membrane (334, 366). Briefly, GLUT4 vesicles migrate and recycle along the cellular framework of cytoskeletal elements and are acted upon by molecular chaperone and GTP-bound Rab proteins. Insulin stimulation causes a rapid phosphorylation of AS160 and its dissociation from GLUT4 vesicles. Removal of AS160 from the vesicle causes an accelerated rate of GLUT4 exocytosis, leading to greater accumulation at the plasma membrane culminating in elevated cellular glucose uptake (723). Phosphorylation incapable AS160 abolished insulin, PDGF, K+ depolarization, and AICAR-induced GLUT4 translocation (639). Furthermore, Karlsson et al. (334) have shown that insulin-stimulated phosporylation of AS160 is significantly impaired in type 2 diabetic muscle. More recently, TBC1D1, an AS160 paralog was identified by immunoprecipitation and mass spectral analyses (635), and its expression is several-fold higher in skeletal muscle versus other insulin responsive, glucoregulatory tissues. Similar to TBC1D4 (AS160), TBC1D1 is phsophorylated in response to insulin, exercise, and AMPK activation by AICAR; however, many novel AMPK binding sites have been identified and activation studies reveal AMPK as a more robust regulator of this signaling molecule (635, 643). Furthermore, genome wide association across multiple strains of mice identified TBC1D1 as an important obesity and diabetes candidate gene (109), similar linkage was established in human subjects as well (616). Mice harboring a truncated TBC1D1 protein lacking the Rab-GTPase-activating protein domain conferred a lean phenotype despite consumption of a high-fat diet (109). These engineered mice showed impaired glucose metabolism and thus relied more heavily on fatty acids as a primary fuel source as reflected by the reduced respiratory quotient and increased rates of whole body fatty acid oxidation, a finding recapitulated in C2C12 myotubes.
Insulin signaling via PI3-kinase independent pathway: CAP, c-Cbl, and TC10
Given the evidence that PI 3-K activation alone is not fully sufficient to achieve insulin-mediated glucose transport, activation of one or more class I PI3K-independent pathways is thought to be requisite (564, 681). This alternative signaling hypothesis is also supported by the fact that glucose transport is stimulated by exercise and hypoxia, independent of any detectable alteration in PI3K. TC10, identified by Chiang et al. (115), along with the canonical PI3K pathway are thought to be required to achieve the full effects of insulin to stimulate GLUT4 translocation. TC10 is a Rho-like GTPase that is highly expressed in adipocytes and skeletal muscle (115). Activation of PI3K-C2α (wortmannin insensitive) is also proposed to occur via TC-10 (111,185). TC-10 dependent actions were found to rely on localization to caveolin-enriched lipid raft microdomains (564, 681).
Within these lipid domains resides the Cbl protooncogene and its adaptor proteins, CAP (Cbl-associated protein) and APS (Cbl-binding protein). Insulin acting through its receptor stimulates the phosphorylation of APS and then Cbl on tyrosine residues (403, 564). Phosphorylated Cbl binds to CAP and migrates to the caveolin-enriched lipid rafts where the CAP complex is anchored by the lipid raft-associated protein flotillin (403, 564). Subsequently, the Crk/C3G (a guanyl nucleotide exchange factor) complex is recruited to this microdomain, leading to the activation of TC10. In that much of this work was performed in adipocytes, findings from JeBailey et al. (314) suggest that TC10-dependent signaling may function differently within other cell types. Additional studies are required to parse out the tissue specific function of the CAP/c-Cbl/TC10 pathway in the regulation of glucose disposal in vivo.
GLUT4 expression
Adipose tissue and skeletal muscle are primary sites of postprandial glucose uptake, and GLUT4, the primary insulin responsive glucose transporter, is highly expressed in these tissues. Under fasting conditions, when circulating insulin levels are low, GLUT4 is minimally present at the plasma membrane and is instead sequestered to intracellular membrane compartments (680). Consumption of a meal stimulates insulin secretion from the pancreatic β-cells and activates a cascade of events as mentioned above that culminate in the movement of the GLUT4 vesicle to the plasma membrane thus increasing transport of glucose from the blood into the cell to participate in metabolism or storage as glycogen. The mechanisms and cellular machinery involved in GLUT4 packaging and trafficking are reviewed in detail elsewhere (89, 167, 332, 348, 383, 402, 443, 444, 501, 680). Suffice to say, an essential role for GLUT4 in insulin-mediated glucose transport is well established.
GLUT4 null mice show many severe developmental defects and a shortened life span (84, 336). Mice heterozygous for the null mutation were insulin resistant and predisposed to diabetes (336, 609). Ablation of GLUT4 specifically in skeletal muscle led to glucose intolerance and severe impairments in insulin-mediated glucose disposal into muscle in mice as young as 8 weeks of age (730). Ablation of GLUT4, specifically in adipose tissue, led to secondary phenotypes of insulin resistance in skeletal muscle and liver, suggesting that an impairment in glucose transport in adipose causes the release of a secretory factor that impairs insulin action in other glucoregulatory tissues (5). Clearly, expression of GLUT4 in skeletal muscle and adipose tissue is essential for the maintenance of glucose homeostasis; however, defects in GLUT4 expression cannot explain the insulin resistance associated with obesity and T2D in rodents or humans (45, 83, 220, 221). Despite this, genetic overexpression or exercise-induced elevation in GLUT4 expression can ameliorate insulin resistance observed in diabetic and obese rodents and humans (84, 255, 531).
Mechanisms of Insulin Resistance
Resistance to the biological effects of insulin is a hallmark feature of the MS and an important contributing factor in the pathogenesis of T2D. In the early stages of insulin resistance, the pancreas compensates by increasing the secretion of insulin into the bloodstream in an attempt to overcome defects in peripheral insulin action. In response to this increased demand for insulin production, the β-cells hypertrophy. Under fasting conditions, basal compensation is sufficient to maintain blood glucose in the normal range. Following a meal though, when glucose is rapidly absorbed from the gut, a relative lack of insulin due to inadequate compensation is detected as the glucose excursion over time is exaggerated. This inability to take up and dispose of glucose appropriately following a meal or glucose challenge is known as glucose intolerance.
It is important to note that genetic mutations or defects in the participants of the insulin-signaling cascade only in rare occasions underlie the insulin resistance and T2D. It is now well supported that lipid oversupply and alterations in substrate metabolism due to inactivity are central underpinnings of chronic tissue inflammation and contribute to the manifestation of peripheral insulin resistance. Tissue accumulation of bioactive lipid species in peripheral tissues activate proinflammatory signaling pathways and novel PKCs; and as reviewed in references (576, 651), which are shown to impair insulin signal transduction by altering key phosphorylation events and key protein-protein interactions. Postreceptor defects are thought to account for much, if not all, of the impairment in muscle insulin action observed in T2D. Many agree that impaired insulin action at the level of IRS-1 occurs as a result of stress kinase activation [e.g., c-Jun N-terminal kinase (JNK) and nuclear factor-κB (IκB) kinase (IKK)β] and impaired phosphorylation of IRS-1 (10, 11, 555). Reduced IRS-1 phosphorylation on critical tyrosine residues and prevents binding with p85 of PI3K and downstream signal transduction. Furthermore, alteration in phosphorylation status, specifically phosphorylation of serine residues, is shown to target IRS-1 for proteasomal degradation and this is a plausible explanation for reduced IRS-1 protein levels in glucoregulatory tissues harvested from obese and/or diabetic rodent (13, 495, 510, 674). Recent work from Shulman, Copps et al. calls into question this paradigm, as in vivo evidence in mice suggests that phosphorylation of IRS-1 at Serine 307 (human Ser312) may promote insulin sensitivity (130). Given conflicting findings to previous studies from the same investigators, further studies will be necessary to test whether selective phosphorylation of IRS-1 on specific serine/threonine residues can modulate insulin action in glucoregulatory tissues in human subjects.
Downstream of IRS-1, PI3K exists in a heterodimer composed of a p110 catalytic subunit and a p85 regulatory subunit as described above. Transcription of the Pik3r1 gene leads to expression of three splice variants p85α, p50α, and p55α (213) and when under normal conditions are in excess compared to the expression of the p110 catalytic subunit. All three variants can bind p110. Interestingly, all three variants are elevated in total in skeletal muscle samples from obese and type 2 diabetic subjects; this increase in expression is associated with reduced insulin resistance and diminished insulin-stimulated PI3K activity (43). Findings in mice with a heterozygous deletion of p85 splice variants as well as in mice with a homozygous deletion of p85α or p85β support the notion that while p85 is critical in recruiting the catalytic subunit of PI3K to IRS proteins, excess levels of monomeric p85 play a role in the inhibition of insulin signaling (425,655).
Lipid phosphatases—SHIP and PTEN
PI3-kinase activity is attenuated by dephosphorylation via 3′ and 5′ lipid phosphatases. SH2 domain containing inositol 5′phosphatase 2 (SHIP2) and skeletal muscle and kidney-enriched inositol phosphatase (SKIP) hydrolyze PI(3,4,5)P3 to PI(3,4)P2, while the phosphatase and tensin homolog deleted on chromosome ten (PTEN) hydrolyzes PI(3,4,5)P3 to PI(4,54)P2. SHIP2, as opposed to SHIP1, is broadly expressed and abundant in glucoregulatory tissues. SHIP2 is phosphorylated in response to insulin and IGF1 stimulation, which leads to its translocation to sites near PI3K. In humans, polymorphisms in the SHIP2 genes are associated with the MS and T2D. These findings are supported in rodents, as SHIP2 expression levels are elevated in skeletal muscle and adipose tissue from obese and type 2 diabetic mice (279). Consistent with these findings, transgenic overexpression of SHIP2 led to IGT and insulin resistance in mice fed a normal chow diet (325) while targeted disruption of SHIP2 improved insulin sensitivity and protected mice from high-fat diet-induced obesity. PTEN was originally identified as a candidate tumor suppressor and was later found to share homology with protein tyrosine phosphatases (397). Overexpression of PTEN and SKIP are also shown to inhibit insulin action in cultured cells, although homozygous deletion of PTEN results in embryonic lethality due to tumor formation. Tissue selective deletion of PTEN in liver, skeletal muscle, fat, and pancreas appears to offer protection against insulin resistance and reductions in β-cell mass in the face of high-fat feeding and streptozotocin treatment, respectively (373,614,615,691). Interestingly, glucose tolerance and insulin sensitivity are reported in human subjects who possess germline mutations in the PTEN gene. Several population-screening studies have failed to identify a relationship between PTEN polymorphisms and T2D susceptibility; however, three variants were identified in Japanese diabetic patients (307), but the differences between the ethnic groups studied to date have yet to be explained. Great care should be taken when considering the role of PTEN as a target for therapeutic intervention as mutations in this gene are associated with tumorgenesis and neurological defects and neurodegenerative diseases. Clearly, PTEN is a critical regulator of many signaling systems throughout the body. Lipid phosphatases acting as therapeutic targets to combat insulin resistance and complications associated with T2D have been reviewed in detail previously (386, 568).
Downstream of PI3K, defective activation of aPKCs has also been observed in muscle from type 2 diabetic rats, monkeys, and humans [as reviewed in reference (187)]. This defect in aPKC signaling is at least in part due to impaired upstream signaling at IRS-1 and PI3K. Furthermore, insulin-stimulated AS160 phosphorylation is reduced in patients with T2D, although the GAP activity of AS160 appears to be specific for Rabs 2A, 8A, and 14 (334).
Protein tyrosine phosphatase 1B (PTP1B)
Protein tyrosine phosphatase is a negative regulator of insulin signal transduction as it dephosphorylates phosphotyrosine residues of the IR and IRS-1. In general, PTPases are redox sensitive enzymes and all share a common catalytic motif (21). Insulin induced ROS-mediated oxidation of critical AA residues in the catalytic domain of the enzyme leads to inactivation, and thus enhanced signal transduction downstream of the IR (225, 417). Indeed, low-level ROS production during insulin stimulation is critical for certain aspects of insulin signal transduction (404, 416).
In insulin-resistant states including obesity-associated insulin resistance, PTP1B expression and activity are elevated in muscle and adipose tissue from humans and rodents (13,701). Furthermore, polymorphisms in the PTPN1 gene confer increased phosphatase express in muscle; this is associated with insulin resistance and T2D (62, 159). Overexpression of PTP1B in mouse muscle or myocytes led to impaired insulin signal transduction of reduced glucose uptake and glycogen deposition (173, 720). Conversely, high-fat fed mice with genetic deletion of PTP1B (176, 357) and diabetic or obese animals treated with PTP1B antisense oligonucleotide (242,729) showed improved insulin sensitivity. In addition, mice lacking PTP1B are also protected from TNFα-induced insulin resistance (462). Thiazolidinedione (TZD)-induced insulin sensitization as well as exercise and caloric restriction interventions that improve whole body insulin sensitivity are associated with reduced PTP1B in skeletal muscle (14, 702). PTP1B as a therapeutic target to ameliorate insulin resistance associated with T2D has received greater attention and is reviewed in reference (362).
Inflammation and insulin signaling
Nuclear factor (NF)-κB and activating protein (AP)-1 are two central proinflammatory pathways activated in glucoregulatory tissues during overnutrition and in type 2 diabetic patients (Fig. ). Studies in rodents with inactivating mutations in the upstream kinases associated with these signaling pathways, IKKβ and JNK, have shown remarkable efficacy in preventing diet-induced insulin resistance, restraining obesity, and ameliorating T2D (28, 269). Interestingly, for over a century now it has been observed that aspirin (acetylsalicylic acid) exerts glucose lowering effects and can ameliorate certain complications associated with T2D, and work by Yuan and colleagues (717) has identified NF-κβ as the pharmacologic target of this antidiabetic agent. Recently, a favorable safety profile and a remarkable efficacy in reducing glycemia, insulin resistance, and diabetic complications, such as CVD, have been observed in clinical trials where salsalate, a prodrug form of salicylate, was administered to type 2 diabetic patients (198, 224).
In addition to the effects of these stress kinases, namely, IKKβ and JNK, to activate transcriptional inflammation programs leading to increased expression of cytokines and chemokines (e.g., TNFα, IL-6, IL-1, and MCP-1), both are thought to directly alter tyrosine kinase activity of proximal insulin signaling (10,11,269). It is currently thought that these stress kinases are activated by the intracellular accumulation of proinflammatory lipid intermediates including diacylglycerol (DAG) and ceramide although activation via cell surface toll-like receptors (TLRs) has also been implicated.
Toll-like receptors and cellular inflammation
TLRs are transmembrane protein receptors that are expressed in a variety of cell types and are critical for innate immune responses. TLRs are now viewed as an important molecular link between lipid oversupply and activation of proinflammatory signaling (207). While eleven members of the TLR family have been identified in humans and 13 in mice (508), of interest, TLR2 and TLR4 are expressed in glucoregulatory tissues including adipocytes, hepatocytes, and myocytes (382, 399). Furthermore in skeletal muscle cells and adipocytes, in vitro and in vivo evidence show that lipid oversupply causes TLR2/TLR4 upregulation, and this is associated with activation of stress-linked kinases (including p38, JNK, and PKC), as well as NF-κB nuclear translocation and subsequent transcription of downstream targets (Fig. 4) (583). Moreover, Tlr4-deficient rodents are protected against the obesigenic effects of a high-fat diet and, in addition, exhibit reduced cellular NF-κB activity including diminished circulating levels of MCP-1 (144). However, recent work shows that when accumulation of intracellular lipid intermediates including ceramide is prevented pharmacologically or genetically, long chain fatty acid-induced inflammatory signaling is also prevented; however, critically, TLR2/4 signaling by their specific ligands including lipoteichoic acid and lipopolysaccharide is maintained. Whether TLR2/4 are important and/or essential in the induction of the tissue inflammatory response in obese humans remains unknown. Clearly, critical work must be undertaken to identify the central mechanism(s) linking cellular uptake of saturated fatty acids with the activation of inflammatory signaling.
Figure 4.

Schematic illustrating mechanisms promoting inflammation that is now recognized as an important underpinning contributing in the pathogenesis of insulin resistance via impairment of insulin signal transduction. Abbreviations: AP-1, adaptor protein 1; IKK, I kappa B kinase; IKKkinase; IκB-α, inhibitor of kappa B; IL, interleukin; IRAK, interleukin receptor-associated kinase; JAK, janus kinase; JNK, c-Jun N-terminal kinase. Originally identified kinase family that binds and phosphorylates c-Jun on Ser-63 and Ser-73 within its transcriptional activation domain. MAPK2, mitogen-activated protein kinase 2; MAPK3, mitogen-activated protein kinase 3; NF-κB, nuclear factor κ B (nuclear factor kappa-light-chain-enhancer of activated B cells); RIP, receptor-interacting serine/threonine-protein kinase; ROS, reactive oxygen species; STAT, signal transducer and activator of transcription; TLR, toll-like receptor; TRADD, tumor necrosis factor receptor type 1-associated DEATH domain protein (adaptor protein); TRAF, TNF receptor associated factors.
Fatty acid-binding proteins as lipid sensors
In 1981, Abumrad et al. (7) showed that cellular uptake of long chain fatty acids is saturable and involves a plasma membrane transport system. In 1991, cloning of the cDNA encoding human skeletal muscle fatty acid-binding protein (FABP), its peptide sequences and chromosomal localization was achieved by Peeters et al. (496). Two years later Abumrad et al. (6) cloned a rat adipocyte membrane protein homologous with human CD36 so named FAT/CD36, and in 1994, Schaffer et al. (574) cloned and characterized a novel adipocyte LCFA transport protein, FATP (Slc27a1). All of these are highly expressed in muscle and regulate LCFA uptake.
Fatty acid-binding proteins are abundantly expressed cytosolic (c) or membrane bound (pm) and coordinate cellular lipid metabolism, binding with high affinity, long-chain fatty acids and eicosanoids (127). FABPs are involved in FA import, storage, and export as well as cholesterol and phospholipids metabolism (258). Additionally, it is thought that FABPs are an adaptive lipid-sensing system, as their levels are altered during elevated fatty acid exposure, chronic endurance exercise (350, 653), genetic obesity, and pathology-associated nutrient changes (406, 641, 652, 654). Interestingly, recent work suggests that in addition to regulating the transport of fatty acids into the cell, FABPs may serve as coregulators of transcription factors known to modulate lipid metabolism and inflammation via a nucleocytoplasmic shuttling mechanism (266, 322, 699). The most actively studied interaction is between FABPs and the family of peroxisome proliferator-activated receptors (PPARs) including α, δ, and γ. Thus, it was hypothesized that selective modulation of FABPs may serve as a potential therapeutic mechanism to treat lipid- and inflammatory-associated diseases. Specifically, adipocyte and macrophage FABPs play a powerful role in regulating tissue inflammation and whole body glucose metabolism. Small molecule inhibitors of aP2 (FABP4) in macrophages blunt FABP activity and reduce cellular inflammation in a time- and dose-dependent fashion. aP2 inhibition is also shown to reduce lesion area in the proximal aorta of atherosclerosis prone Apoe−/− mice fed a Western diet (215). Similar findings were observed in mice with a genetic deletion of aP2 (FABP4) and mal1 (FABP5) in adipocytes and macrophages (411). Collectively, these studies provide substantial evidence that aP2 is an important regulator of cellular fatty acid handling, inflammation, and whole body glucose tolerance and insulin sensitivity. This work implicates lipid chaperones, FABPs, as potential clinically relevant therapeutic targets in the treatment of T2D and CVD; however, studies in human subjects are still required. It is also important to note that these binding proteins are upregulated with physical exercise (350,653) and fasting (652), from which a coordinated increase in fatty acid oxidation, repression of inflammation, and improved insulin sensitivity are achieved.
Protein fatty acid transporter FAT/CD36 expression is also elevated in response exercise training and the 88 kDa protein translocates rapidly to the plasma membrane during muscle contraction. Increased expression and activity of this transport protein contributes in large part to the fivefold to 15-fold increase in fatty acid uptake by contracting skeletal muscle (73,91,544). The protein is shown to colocalize with caveolin-3 and most highly abundant in type 1 oxidative muscle fibers (666). Interestingly, similar to GLUT4, FAT/CD36 is skeletal muscle is maintained under basal conditions in an intracellular pool and translocates to the plasma membrane primarily in response to contraction or insulin stimulation (74, 407). Over the past decade, it has been disputed whether FAT/CD and FATP-1 may be involved in deciding the metabolic fate of LCFA, specifically partitioning FAs to oxidation. Bezaire and colleagues put forth the notion that fatty acid transporters FAT/CD and FATP1 are involved in the transfer of FAs into the mitochondria possibly by direct binding to the mitochondrial membrane (66, 96). This work is recently disputed by Jeppesen et al. (315). Regardless of whether these proteins bind directly to mitochondria, at least in the case of FATP, skeletal muscle-specific overexpression in mice promoted increased skeletal muscle fatty acid uptake and oxidation specifically, but did not predispose animals to diet-induced insulin resistance (274). In aggregate, despite over two decades of intense research since their identification, it would seem that our understanding of the involvement of fatty acid binding proteins in regulating substrate metabolism, tissue inflammation, and insulin action requires further investigation.
Fatty acid handling
High-fat feeding or elevation of circulating fatty acid levels achieved by TG and heparin infusion are well known to cause skeletal muscle insulin resistance; however, the precise signaling events that underlie fatty acid-induced insulin resistance remain incompletely defined. It is currently held that excessive fatty acid uptake into the myocyte and reduced fat oxidation lead to the accumulation of proinflammatory lipid metabolites, such as fatty acyl CoAs, DAG, and ceramide, which, as stated above, activate serine/threonine “stress” kinases (Fig. 4). These kinases include JNK, IKKβ, and PKCθ, all of which promote inflammatory signaling and antagonize insulin action. Reduced expression of the rate limiting enzyme for LCFA entry into the mitochondria, carnitine palmitoyltransferase-1 (CPT1), and impaired mitochondrial function (decreased mitochondrial number, lower levels of oxidative enzymes, and lower ATP synthetic rates) are found in insulin-resistant skeletal muscle and coincide with muscle lipid accumulation (58, 351, 503, 541, 694). Furthermore, genes of oxidative metabolism were shown to be coordinately reduced in muscle from human patients with insulin resistance and T2D (450, 490). It is important to note that in human subjects these are merely observational and coincident changes in multiple signaling pathways; causal relationships between impairments in oxidative metabolism and mitochondrial function with insulin resistance is not established (253), and indeed there is evidence that in the condition of primary congenital insulin resistance, mitochondrial dysfunction follows (597). Additional longitudinal studies are necessary to delineate the pathogenesis and molecular underpinnings of this often-heterogeneous mix of metabolic disorders that characterize MS.
Despite this controversy and lack of mechanistic insight into the human pathology, several studies have shown that TG accumulation in skeletal muscle correlates most highly with insulin resistance, even when compared to other factors including BMI, percent body fat, and waist-to-hip ratio (313, 371, 499). TG accumulation is regulated directly and indirectly by several important enzymes located in the cytoplasm and mitochondria. For example, CPT1 is a key metabolic regulator of fatty acid oxidation, located in the outer mitochondrial membrane and in close proximity with acetyl-CoA carboxylase (ACC), a cytosolic enzyme that catalyzes the carboxylation of cytosolic acetyl CoA to form malonyl CoA, a potent inhibitor of CPT1. Malonyl-CoA levels are controlled by the relative activities of ACC and malonyl-CoA decarboxylase, the principal enzyme involved in malonyl CoA degradation. When malonyl CoA levels rise, mitochondrial fatty acid import, and oxidation are suppressed, thus favoring fatty acid storage in the form of TG. ACC inhibition, CPT1 activation, and enhanced fatty acid oxidation are previously shown to associate with insulin sensitivity and protection against diet-induced insulin resistance (118, 161). Importantly, in skeletal muscle from obese individuals, the activity of CPT1 is reduced (351) and may account for at least some of the decrement in skeletal muscle fatty acid oxidation observed in obesity and insulin resistance. This notion is consistent with recent work showing that CPT1 overexpression in myotubes (579) and murine skeletal muscle (87), or enhanced β-oxidation in mice harboring a null ACC2 mutation (118) is protective against diet-induced insulin resistance. Given that endurance exercise in rodents and humans is associated with elevated CPT1 activity levels and greater fatty acid oxidative capacity, therapies targeted at improving fatty acid flux through the tricarboxylic pathway to ameliorate insulin resistance are warranted. It is also important to note that while elevated TG levels correlate well with insulin resistance in type 2 diabetic and sedentary individuals, TG levels are also elevated in insulin sensitive highly trained athletes (556). The notion of the lipid paradox is well supported and suggests that other factors independent of TG, such as accumulation of more bioactive lipid species, including DAG or ceramide, and peroxidation of lipid bilayers promote inflammation and are the true culprits of insulin resistance (165, 226, 556). Stored muscle triacylglycerol is thought to be a biologically inert pool of stored fat in certain instances activation of TG synthesis may be advantageous in reducing the inflammatory consequences of nutrient excess. Identification of the precise causal factors underlying proinflammatory signaling and insulin resistance associated with increased muscle lipid storage in sedentary individuals compared with those engaging in daily exercise, is of biological and clinical interest.
Accumulation of bioactive lipid intermediates and insulin resistance
Indeed, there are mechanistic links between the development of insulin resistance and the accumulation of DAG and ceramide in muscle (8, 271, 272, 310), although, as put forward by Goodpaster and colleagues previously for triacyclyglycerol, a paradox for DAG in muscle of endurance-trained athletes is now also suggested (19). Dissecting the athlete’s paradox further, in sedentary individuals, a major contributor to increased lipid deposition is an impaired ability to oxidize fat as a fuel source (108, 593, 594). Elevated intracellular fatty acids are shown to induce enzymes that promote tissue sphin-golipid synthesis. Despite being a relatively small component of the total lipid pool, sphingolipids, such as ceramide and glucosylceramide, are considered the most pathogenic (618) and are elevated almost twofold in insulin-resistant obese compared with lean, sedentary or exercise-trained subjects (19). Four serial reactions promote the synthesis of ceramide from fatty acids and serine. Two of these enzymes, serine palmitoyl-transferase (SPT)-1 and dihydroceramide desaturase (Des-1), have received attention regarding their therapeutic potential to reverse insulin resistance and ameliorate complications associated with T2D. SPT-1 catalyzes the first reaction that condenses serine with palmitoyl-coenzyme A (CoA) to produce 3-ketosphinganine. This first reaction is the rate-limiting step in the synthesis of ceramide. SPT-1 is highly selective for saturated fatty acyl-CoA and the rate of this reaction is influenced largely by the availability of fatty acid substrate. Experimental elevation of fatty acids in rodents increases ceramide accumulation in skeletal muscle and liver and is associated with insulin resistance. However, coinfusion of an SPT-1 inhibitor, such as myriocin or cycloserine, prevents both lipid-induced ceramide accumulation and impaired insulin action (272). Similarly, myriosin administered to Zucker diabetic fatty rats reduces tissue ceramide levels, improves glycemia and circulating TG, and ameliorates glucose and insulin intolerance (271, 272). Des-1 oxidizes inactive dihydroceramide into active ceramide. In line with findings for myriosin-treated rodents, tissues harvested from Des-1+/− mice accumulate less ceramide and these animals are refractory to insulin resistance (271). Overall, these findings in rodents warrant further investigation in humans so as to discern whether SPT-1 and Des-1 are viable therapeutic targets to reverse insulin resistance and restrain T2D progression. Additionally, exhaustive exercise diminishes total content and saturation of ceramide and sphingomyelin-FA as well as activity of sphingomyelinase in oxidative muscles from rats (162) and vastus lateralis from humans (265).
Collectively, these findings in rodents warrant further investigation in humans to discern whether SPT-1 and Des-1 are viable therapeutic targets to reverse insulin resistance and restrain T2D progression by diminution of cellular bioactive lipids, including ceramides. Furthermore, it will be important to unravel mechanistic underpinnings of the athlete’s paradox to determine the site of uncoupling related to lipid accumulation for proinflammatory signaling. It is likely that mitochondrial content, lipid droplet localization, the precise molecular species of the lipid and the relative abundance and activity of potent transcriptional regulators of key pathways in metabolism, insulin action, and inflammation converge to explain the discrepancies in human populations and experimental mouse models.
Protective chaperones - heat shock protein response
Heat shock proteins (HSPs) are shown in murine models to block inflammation and protect against obesity-induced insulin resistance (120). HSPs are adaptive proteins that protect against cellular stress including alterations in temperature, pH, misfolded proteins, and inflammation (465). In humans, exercise training is also shown to cause increased tissue HSP levels; this adaptation is thought to be associated with disease prevention (85, 705). Thus, it is hypothesized that loss of heat shock response may underlie susceptibility to chronic disease while activation of the heat shock response may have broad therapeutic benefit in the treatment of such diseases including CVD and T2D. This notion is supported by work from Chung et al. (120), showing that upregulation of the inducible HSP, HSP72, either by heat stress, pharmacological or genetic means leads to protection against diet- and obesity-induced inflammation, glucose intolerance and insulin resistance. HSP72-mediated protection was associated with suppressed inflammatory signaling as well as improved oxidative metabolism (100); however, the precise mechanisms underlying the cellular and tissue-specific therapeutic actions of HSP72 require further elucidation. Indeed, follow-up studies by Gupte et al. (244) in heat-treated rats confirmed findings by Chung et al., showing that induction of Hsp72 protected against high fat diet-induced glucose intolerance and insulin resistance. Furthermore, reduction in activity of citrate synthase and mitochondrial cytochrome oxidase induced by high-fat feeding was prevented by one bout of thermal stress (244). Similarly, Geiger and colleagues also noted that one bout of thermal stress improves skeletal muscle insulin action in aged Fischer 344 rats, and this was associated with reduction in proinflammatory signaling (245). In addition muscle,
Given that muscle HSP72 expression is diminished in obese and type 2 diabetic patients (85, 120), studies are currently underway to investigate whether a novel investigational compound, which causes induction of HSP72, leads to improved insulin sensitivity and reversal of complications associated with T2D.
In addition to pharmaceutical intervention, endurance exercise also elevates Hsp72 mRNA and protein levels in skeletal muscle of humans and rodents (190, 191, 601) and the degree of Hsp72 induction is thought mediated by a variety of factors including muscle glycogen content (191), tissue hypoxia (636), the severity of exercise thermal stress, and or the degree of protein oxidation (601). In addition, activation of HSP72 by thermal stress conditioning is also shown to improve antioxidant capacity and this was associated with reduced muscle injury in rats following downhill running (589). The precise stimuli and mechanisms required for the induction of HSP72 during exercise requires further investigation as well as the molecular underpinnings mediating the therapeutic benefit of HSP72 elevation.
Endoplasmic reticulum stress
Evidence implicating inflammation, specifically endoplasmic reticulum (ER) stress in the etiology of β-cell apoptosis as well as liver and adipose tissue dysfunction is now well supported (281). It is clear that the capacity of the ER to adapt to stress is paramount for the maintenance of cellular health. The ER is the organelle responsible for protein folding, maturation, quality control, and trafficking. This organelle becomes “stressed” when newly synthesized unfolded proteins accumulate excessively in the lumen and, as a consequence, the unfolded protein response (UPR) is initiated to resolve the cellular stress. Importantly, the branches of the canonical UPR intersect with two main inflammatory pathways including NF-κB and JNK-AP1. In addition to the accumulation of unfolded nascent proteins, ER stress can also be induced by imbalances in calcium, glucose and energy deprivation, hypoxia, pathogen, toxins and certain lipids (281). Thus, cells specifically involved in handling and secreting large quantities of proteins, lipid, and lipid mediators are highly susceptible to ER stress and the cellular consequences of the UPR.
In the pancreas, lipotoxicity directly affects ER stress-mediated β-cell death (93). ER stress initiates a cascade of signaling events culminating in the attenuation of de novo protein synthesis and transcriptional activation of genes encoding ER chaperones to further assist in protein refolding or removal by the ubiquitin-proteosome pathway. An impaired or defective UPR leads to apoptosis. Markers of ER stress include PKR-like ER kinase (PERK), activating transcription factor (ATF), and inositol requiring (IRE)1. During cellular stress PERK becomes phosphorylated, leading to subsequent phosphorylation of eukaryotic initiation factor (EIF)2α, causing induction of ATF4. Additionally, ATF can also activate apoptotic pathways including C/EBP homologous protein (CHOP), JNK, and caspases. A strong link between ER stress signaling and β-cell function is evidenced by ER stress gene expression increased in islets from humans with T2D as well as db/ db mice (294, 385). Within the lumen of the ER, protein chaperones, such as BiP or GPR78, GPR94, calnexinm, and calrecticulin assist in the execution of proper protein folding and the elimination of misfolded or unfolded proteins. Phenylbutyrate (PBA) or BiP overexpression in INS-1 cells causes inactivation of IRE1, reduced ER stress, and prevention of palmitate-induced cell death (93). This work suggests that selective targeting of the UPR response in β-cells could prevent cellular apoptosis, preserve β-cell mass, and prevent T2D; however, evidence for this in humans is lacking.
In liver and adipose tissue samples from genetically obese or high-fat fed mice, markers of ER stress (increased PERK, EIF2α, and c-Jun phosphorylation) were elevated when compared to lean and normal chow fed controls (235, 486). This inflammatory stress response leads to suppression of insulin receptor signaling through hyperactivation of JNK and subsequent serine phosphorylation of IRS-1. X-box-binding protein (XBP)-1 is a bZIP protein that is spliced during ER stress and becomes a key transcriptional regulator of an array of genes that are important for ER stress resolution including the induction of molecular protein chaperones. In cells, the degree of ER stress induced by the chemical compound and JNK activator tunicamycin was directly impacted by XBP-1 expression; that is, cells overexpressing XBP-1 were refractory to ER stress. In vivo studies in rodents support cell-based studies as mice deficient (heterozygous null) in XBP-1 develop glucose intolerance and insulin resistance that is associated with tissue inflammation and impaired insulin signaling (486).
In follow-up studies by the same research group, 4-phenyl butyric acid and taurine-conjugated ursodeoxycholic acid were shown to alleviate ER stress in cells and rodents. Chemical or pharmaceutical chaperones, such as 4-phenyl butyric acid (PBA), trimethylamine N-oxide dihydrate (TMAO), and dimethyl sulfoxide, are a group of low molecular weight compounds known to stabilize protein conformation, improve ER folding capacity, and facilitate the trafficking of mutant proteins. Similarly, endogenous bile acids and derivatives including ursodeoxycholic acid and its taurine-conjugated derivative (TUDCA) are also shown to modulate ER function. Specifically, treatment of obese and diabetic mice with PBA and TUDCA resulted in normalization of hyperglycemia, restoration of systemic insulin sensitivity, resolution of fatty liver disease, and enhancement of insulin action in liver, muscle, and adipose tissue (487), suggesting that chemical chaperones enhance the adaptive capacity of the ER and exhibit potent antidiabetic effects in rodents.
In humans, weight loss following gastric bypass (GBP) was associated with reduced ER stress in adipose and liver and a marked improvement in hepatic, skeletal muscle, and adipose tissue insulin sensitivity (236). Markers of ER stress in adipose tissue significantly decreased with weight loss including expression of Grp78 and spliced sXBP-1, as were phosphorylated EIF2α and JNK1. Liver sections from a subset of subjects showed intense staining for Grp78 and phosphorylated EIF2α before surgery, which was reduced in post-GBP sections. Similarly, 8 weeks of daily endurance exercise reduced proinflammatory signaling and as well as PERK and EIF2α phosphorylation in adipose and liver from high-fat-fed rats (141). Interestingly, recent work from the Spiegelman laboratory shows that the UPR is initiated in skeletal muscle following an acute bout of treadmill exercise and is involved in mediating muscle adaptations when exercise is performed repetitively (700).
Collectively, these findings demonstrate that chronic ER stress is a central feature of peripheral insulin resistance and T2D and that pharmacologic manipulation of this pathway coupled with weight loss may offer a novel therapeutic strategy for treating these common chronic diseases. Whether endurance exercise provides a therapeutic benefit in ameliorating chronic ER stress associated with obesity and T2D requires further study. The role that muscle-specific ER stress plays in tissue remodeling and metabolic function is an emerging area of investigation also requiring greater delineation and mechanistic insight.
Adipose tissue as an endocrine organ
It is known for over a decade now that adipose tissue dysfunction is a central underpinning link to obesity in the pathogenesis of the MS and T2D (Fig. 5). Over the past decade adipose tissue has been redefined as a dynamic metabolic, endocrine organ secreting various cytokines, chemokines and adipokines in a paracrine, autocrine, and endocrine fashion. Much of this work is reviewed in references (71, 229, 515, 521, 669, 732). Adipose tissue is no longer simply considered a passive energy storage depot, but instead is now recognized as the largest endocrine organ in the body secreting more than a hundred factors including fatty acids, cytokines, chemokines, prostaglandins, and steroids. These factors can exert local paracrine effects or are released into the circulation yielding systemic effects on brain, liver, and skeletal muscle. These adipose-secreted factors regulate such processes such as glucose metabolism, appetite, inflammatory signaling, immune function, angiogenesis, blood pressure, and reproductive function. Given that adipose tissue comprises a heterogeneous mix of cell types, including macrophages and other immune cells, endothelial cells, vascular smooth muscle cells, fibroblasts/preadipocytes, and mature adipocytes, the interplay among these cell types and specific roles of each of these cells in adipose tissue development, substrate metabolism, and production of secretory factors are extremely complex and still not well understood.
Figure 5.

Schematic of adipose tissue-secreted factors that act on muscle and liver to promote insulin resistance.
Dysfunction within adipocytes specifically is an important contributor to the pathogenesis of obesity and T2D. Recent evidence suggests that enlarged adipocytes, relative impairment in tissue blood flow, cellular hypoxia, local inflammation, and adipose tissue infiltration of proinflammatory immune cells are interrelated processes thought to modulate adipocytes function including adipokine production and secretion; these important factors are reviewed in more detail elsewhere (229). Enlargement of adipocytes, frequently seen in obesity and consumption of a diet rich in saturated fatty acids, is associated with increased expression of proinflammatory adipocytokines whereas small adipocytes or therapies used to promote adipogenesis are associated with insulin sensitization (420, 445). While adipocyte size correlates well with insulin action and a favorable adipokine secretion profile, whether adipocytes size is a primary and central factor in determining adipose health remains to be determined, so herein we have focused on describing the effects of adipose-secreted demonstrating a regulatory role in modulating insulin sensitivity in vivo in both humans and rodents.
Leptin
The identification of leptin (726), a 16-kDa cytokine-like peptide, and its receptor (114, 389, 634) initiated the burgeoning field of study into the role of adipose tissue as an endocrine organ. Leptin gained increasing attention in the late 1990s and this work fashioned leptin into a central regulator of feeding and energy homeostasis (209). Mice with mutations in the leptin gene or its receptor are remarkably obese (114, 208, 389). Likewise, human leptin mutations recapitulate an obese phenotype. A primary effect of leptin is exerted in the brain where the receptor is highly expressed in the hypothalamus. Leptin is shown to repress orexigenic pathways including neuropeptide Y and agouti-related peptide and activate anorexigenic pathways, pro-opiomelanocortin, and cocaine and amphetamine-regulated transcript (CART) (177, 179). The leptin receptor belongs to the IL-6 receptor family of class I cytokine receptors and exerts its central and peripheral effects on metabolism via the Janus kinase (JAK)-signal transducers and activators of transcription (STAT) and PI3K (512). Leptin administration by intracerebral catheter directly into the brain decreases food intake and increases energy expenditure, and prolonged exposure leads to a reduction in total body weight (12).
Despite the known anorexic actions of leptin, administration of recombinant leptin as an obesity therapeutic proved futile given central leptin resistance that occurs with increasing adiposity. This is consistent with the many observations that leptin levels are markedly elevated in obese humans and rodents and correlate well with adiposity. Several regulators of leptin signaling have been proposed including SH2 containing SHP2 and protein tyrosine phosphatase 1B (PTP1B) and SOCS proteins. Early work by the Flier laboratory showed that leptin administration causes a rapid induction of hypothalamic Socs3 mRNA in mice and mediates feedback inhibition of the leptin receptor (67,68). Hypothalamic deletion of Socs3 (451) or whole body Socs3 haploinsufficiency (292) confers enhanced central leptin sensitivity and protection against diet-induced obesity. Furthermore, work by Levin et al. (396) showed that leptin-induced STAT3-phosphorylation, a surrogate marker for leptin receptor activation, was markedly reduced in the hypothalamic arcuate, ventromedial, and dorsomedial nuclei of high-fat fed rats even prior to obesity, while impaired leptin transport across the blood brain barrier was only observed after animals became obese. Collectively, these findings suggest that leptin resistance occurs early in the pathogenesis of obesity and that impaired receptor function is mediated by reduced receptor activation and expression mediated by Socs3 feedback inhibition.
In addition to the effects of leptin on the brain in the regulation of feeding, leptin also exerts direct effects in the periphery to improve insulin action independent of weight loss. Similar to the leptin resistance that develops in the brain, high-fat feeding can reduce leptin mediated effects in skeletal muscle (605, 607) and liver (672). Leptin resistance in muscle during high-fat feeding is thought to occur as a result of reduced leptin receptor expression (452) and elevated Socs3 mRNA (606, 608). A similar increase in Socs3 mRNA is also observed in skeletal muscle from obese subjects (606). Furthermore, Socs3 overexpression in human myotubes can prevent leptin-induced activation of AMPK (606).
Interestingly, swimming exercise improves hypothalamic leptin sensitivity and reduce food intake. Findings show that this exercise-induced effect is mediated by IL-6 (548, 549). While the dual roles of IL-6 in insulin action remain to be clarified, the collaborative work of Pedersen and Febbraio, clearly show that skeletal muscle IL-6 mRNA is rapidly induced at the onset of exercise and is secreted in abundance into the circulation. Furthermore, homozygous IL-6 deletion promotes hepatosteatosis and systemic insulin resistance (423). In addition to the effects of IL-6 on peripheral tissue metabolism, it is likely that IL-6 secreted from muscle during contraction is requisite for exercise-mediated effects in the brain given that IL-6 neutralization prevents exercise-mediated improvements in hypothalamic leptin signaling (199). Indeed, Steinberg et al. (608) also showed that endurance exercise can protect against high-fat diet-induced leptin resistance. Additional, studies are required to dissect apart the leptin-induced effects on metabolism mediated by the brain versus peripheral tissues in response to endurance exercise.
Adiponectin
Adiponectin represents the most abundant protein secreted by adipose tissue (212) and adipose transcript as well as circulating levels of this protein are reduced in humans and in rodent models of obesity and T2D, as reviewed in references (27, 293, 323, 497, 645). A diabetes-susceptibility locus to human chromosome 3q27, where the adiponectin gene is located, and a quantitative-trait locus strongly linked to the MS in individuals of European and Asian descent have previously been identified (129, 256, 664). In rodents, administration of recombinant adiponectin or genetic overexpression lead to improved insulin sensitivity and enhanced fatty acid oxidation in liver and skeletal muscle (63, 212); while in contrast, genetic deletion is associated with glucose intolerance and insulin resistance (372, 412).
Adiponectin circulates in plasma as a low-molecular weight trimer, a mid-molecular weight hexamer, and a high-molecular weight 12- to 18-mer, and all forms are shown to exert differing biological function (670). The high molecular weight of adiponectin is thought to provide the greatest biological activity of all of the forms; however, additional work to substantiate this notion is required. However, Waki et al. (670) showed that impaired multimerization of human adiponectin is associated with T2D. Two distinct receptors, AdipoR1, which is expressed ubiquitously, and AdipoR2, which is expressed most abundantly in the liver, mediate the biological actions of adiponectin (707). Adiponectin binds and stimulates interaction of the N-terminal cytoplasmic domain of its receptor with and intracellular adaptor protein to activate intracellular pathways (418) including p38 MAPK, AMPK, and PPARα.
Accordant with reductions in circulating adiponectin levels observed in mouse models of insulin resistance and obesity, expression of both receptors was also shown to be diminished (706, 708). Experimental disruption of AdipoR1 or R2 causes blunted adiponectin-induced AMPK and PPARα responses, respectively, both leading to increased hepatic glucose production and hepatic insulin resistance (708). Conversely, adenoviral restoration of AdipoR1 or R2 in liver of diabetic mice improves adiponectin action leading to a partial restoration of insulin sensitivity (708). Interestingly, polymorphisms in both adiponectin receptors are associated with insulin resistance and T2D (135, 324, 429). An important aspect of adiponectin action includes anti-inflammatory effects to inhibit NF-κB and toll-like receptor signaling and these effects on immune cells and endothelium coupled with improved metabolism in peripheral tissues is thought to provide protection against atherosclerosis. In fact, when stratified for levels of serum adiponectin, the risk of myocardial infarction was dramatically reduced in men in the highest adiponectin quintile (504).
Thus taken together, improvement in adiponectin secretion or receptor function may serve as a therapeutic strategy to ameliorate the complications associated with insulin resistance and T2D. Consistent with this notion, it is thought that elevations in adiponectin and adiponectin receptors cause in large part TZD-induced insulin sensitization in type 2 diabetic subjects. Furthermore, recent evidence from reference (346) showed that 7 days of aerobic training (AT) increased circulating HMW adiponectin by 21%, and this alteration was associated with improved basal fat oxidation, glucose tolerance, and insulin sensitivity in middle-age obese individuals. The exercise-induced mechanisms and time course underlying enhanced adiponectin production by adipose tissue and whether corresponding changes in receptor function also occur requires further characterization.
RBP4
In 2005, DNA arrays performed in adipose tissue from mice with an adipose specific deletion or overexpression of GLUT4 to identify adipose secreted factors that may be associated with insulin resistance; retinol binding protein (RBP)4 was identified (710). RBP4 is a circulating transport protein specific for retinol, vitamin A. RBP4 is elevated in tissue and in the circulation of insulin-resistant humans and rodents (234, 710). Furthermore, mice injected with recombinant RBP4 became insulin resistant while mice with a heterozygous or homozygous deletion of RBP4 were more insulin sensitive and protected from diet-induced insulin resistance. TZD and fenretinide, a synthetic retinoid, both lower circulating RBP4 levels in rodents, and this is associated with improved insulin action in rodent models of obesity and insulin resistance (710). There still remains some controversy with RBP4 and its relationship with obesity and its in vivo effects on the pathogenesis of insulin resistance in humans.
Lipocalin
Lipocalin 2 (Lcn2), also known as neutrophil gelatinase-associated lipocalin, 24p3, and siderocalin, is a member of a large family of secreted proteins, including RBP4 that are associated with insulin resistance (106, 673, 709, 725). Lcn2 is an iron transport protein and its expression is induced in 3T3L1 adipocytes by TNFα and dexamethasone (709). In addition to being expressed in adipocytes, Lcn2 is also expressed in neutrophils, liver, kidney, and macrophages (673). Importantly, Lcn2 expression is elevated in visceral fat from obese human subjects (106), as well as adipose and serum from multiple rodent models of obesity and insulin resistance (709, 725). In addition, the circulating Lcn2 concentration is positively correlated with human adiposity, hypertriglyceridemia, CRP, and hyperglycemia but negatively correlated with HDL (673). Retroviral delivery of short hairpin RNA into 3T3L1 adipocytes yielding reduced Lcn2 expression was associated with improved insulin action while exogenous Lcn2 promoted insulin resistance in cultured hepatocytes (709). In addition, thiazolidindione-induced insulin sensitization in rodent models of obesity also causes reduced adipose tissue Lcn2 expression. Therefore, taken together, it is thought that lipocalin 2 is an adipokine that may be involved in potentiating obesity-induced insulin resistance.
PEDF
The serine protease inhibitor pigment epithelium-derived factor (PEDF) is predominantly released from adipocytes and recently was shown to play a causal role in metabolic dysfunction and insulin resistance. PEDF is elevated in obese, insulin-resistant mice, and reduced upon insulin sensitization. Lean mice injected with recombinant PEDF exhibit insulin resistance during hyperinsulinemic-euglycemic clamps, while neutralizing PEDF in obese mice enhances insulin sensitivity (136). PEDF is also shown to alter whole body fatty acid metabolism by increasing adipose tissue lipolysis and decreasing fatty acid oxidation in skeletal muscle, resulting in ectopic lipid deposition in muscle and liver. Together, these results support a causal role for PEDF in obesity-induced metabolic dysfunction and insulin resistance (136, 186).
Resistin
Resistin, first described by Steppan et al. (610), is a cytokine expressed exclusively in adipocytes in mice but is expressed predominantly in macrophages in human subjects (489). Resistin was identified as a TZD-downregulated gene in mouse adipocytes (610) and TZD therapy is also shown to reduce macrophage expression and levels of circulating resistin in humans (489). Furthermore, circulating resistin levels are elevated with obesity (516); experimental resistin elevation in rodents using acute administration (516), adenoviral-mediated delivery (570), and transgenic overexpression (519) is shown to induce insulin resistance. Consistent with these observations, loss-of-function mutations, achieved by antibody neutralization (610), genetic deletion (44), and anti-sense oligonucleotide administration (459) leads to improved insulin sensitivity and glucose homeostasis.
The receptor for resistin remains unknown and the details regarding resistin action to induce insulin resistance in glucoregulatory tissues is not completely understood; however, downstream of its putative receptor, resistin appears to inhibit hepatic and skeletal muscle AMPK (44, 459, 570). AMPK is known as the master energy regulator and controls substrate production and utilization in liver and muscle, respectively. In addition, findings in adipocytes suggest that resistin is also capable of activating Socs3 that was previously shown to cause impaired insulin action (612). While a clear relationship between circulating resistin levels and obesity/T2D in humans remains ill defined, differences in assay type and the existence of multiple higher molecular weight oligomers may contribute to the discrepant observations. Resistin levels do, however, correlate well with other inflammatory factors, including CRP peptide and the presence of atherosclerosis (318,472,529,588,611). In humans, this observation is particularly relevant given that resistin is predominantly produced by macrophages, a cell type central in the pathogenesis of arterial lesion development. Lastly, there is genetic support that resistin may play a role in T2D susceptibility given that a single nucleotide polymorphism in the promoter region is linked with obesity and insulin resistance in several populations in the United States, Japan, and Europe (117, 121, 479, 598).
Visfatin, omentin, chemerin, adipsin, ASP
Visfatin is a 52 kDA protein that is highly expressed in visceral but not adipose tissue from obese type 2 diabetic rodents (214). In humans, visfatin is correlated with visceral adipose mass and is upregulated during adipogenic differentiation; thus, plasma levels track well in some studies with human obesity. Interestingly, visfatin binds to the insulin receptor with the same affinity as insulin and promotes adipogenesis, an observation consistent with increased visfatin secretion rates from adipocytes following treatment with the PPARγ agonist rosiglitazone (250). Many unanswered questions remain as to the mechanistic role that visfatin may play in the pathogenesis of obesity and insulin resistance.
Omentin is a 38 kDa protein primarily expressed in omental fat and is thought to be secreted primarily from stromal vascular cells, not adipocytes, within the tissue compartment (711). Little is known about the physiological role of this protein, however, some studies suggest it is regulated by glucose and insulin and is associated with obesity cardiovascular syndromes (630).
Chemerin, also known as tazarotene-induced gene 2 or retinoic acid receptor responder 2 is an 18-kDa novel adipokine expressed predominantly in mature 3T3L1 adipocytes and is elevated in adipose tissue collected from obese animals. Chemerin is secreted as an inactive proprotein and is converted to its biologically active form following C-terminal proteolytic cleavage. While two studies published in 2007 (230, 546) suggest that chemerin modulates adipogenesis and that receptors for the adipokine are present in immune cells, little is known regarding the functional role of this adipokine or its true relationship to disease pathology. Recent work by Ernst et al. (181) show that chemerin and its receptors chemokine-like receptor 1, C-C motif receptor-like 2, and G protein-coupled receptor 1 (341) are altered in white adipose, liver, and skeletal muscle of obese type 2 diabetic mice. Administration of chemerin exacerbates glucose intolerance in these animals, thus, suggesting a role for chemerin in glucose homeostasis (181). Two independent studies by Bozaoglu et al. (79,80) found that circulating chemerin levels associate with obesity and the MS in Caucasian and Mexican-American populations. Furthermore, experimental hyperinsu-linemia caused a rapid induction of chemerin expression in adipose tissue explants and led to increased chemerin cellular protein level and secretion of chemerin into the conditioned media. Insulin-induced adipose tissue chemerin production was markedly reduced by the addition of metformin, a clinically utilized antidiabetic drug, to the media (631). In addition, a recent report by Ress et al. (533) showed that weight loss achieved by bariatic surgery resulted in significantly reduced circulating chemerin. These findings are consistent with at least a permissive if not a regulatory role for chemerin in glucose metabolism.
In vitro findings from Sell et al. (582) suggest the latter as chemerin release from in vitro differentiated human adipocytes and adipose tissue explants were elevated in the obese versus lean subjects. Additionally, chemerin release is correlated with BMI, waist-to-hip ratio, and adipocyte volume. Higher chemerin release also associated with insulin resistance. Ex vivo chemerin treatment induced insulin resistance in human skeletal muscle cells at the level of IRS 1, Akt and glycogen synthase (GS) kinase 3 phosphorylation, and glucose uptake (582). Chemerin also activated p38 mitogen-activated protein kinase, NF-κB, and extracellular signal-regulated kinase (ERK)-1/2. Given these recent findings and the known chemoattractant properties of chemerin, it is thought that a reduction in circulating chemerin may diminish the infiltration of proinflammatory immune cells in adipose tissue leading to restrained adipose tissue growth, thus, exerting both direct and indirect effects on peripheral tissues to regulate insulin action.
Adipsin is also known as adipocyte trypsin, factor D, or complement factor D (547). The protein encoded by the CFD gene is a member of the trypsin family of peptidases and a protein component of the alternative complement pathway. Recent evidence shows that this serine protease is expressed at high levels in adipose tissue and is secreted by adipose tissue explants. Furthermore, tissue expression levels and circulating concentration levels in the blood are elevated in obese subjects. In addition, adipose tissue also releases a protein derived from the interactions of adipsin with complement C3 and factor B known as acylation-stimulated protein (ASP). ASP is produced in a two-step process in which the aforementioned three proteins of the alternative complement system in which the enzyme adipsin causes cleavage of the parent protein C3 to C3a, which is followed by desargination of the carboxyl terminus to generate C3adesArg or ASP (124). Adipocytes are one of the few cell types that contain all three complement factors necessary for the generation of ASP. ASP is thought to represent the most biologically active component of this system and circulating levels are thought to be modulated by insulin, cytokines, and the fatty acid components of chylomicrons as reflected by dietary fat consumption. ASP is shown to exert potent effects to stimulate TG synthesis and inhibit lipolysis in adipocytes. ASP administration to obese or diabetic rodents enhanced TG clearance by as much as twofold; similar but less robust findings were also observed for lean control mice (457, 562). In part, TG synthesis is promoted by ASP-stimulated increases in diglycerol acyltransferase activity and glucose uptake into adipocytes, and these effects may be mediated by activation of specific adipocyte protein kinases, for example PKC (39). Mice with a homozygous null mutation for C3, and therefore ASP, showed altered postprandial TG clearance (456). When fed a normal chow or high-fat diet, KO mice were leaner with reduced adipose tissue mass compared with wild-type littermates despite an increased caloric consumption by KO animals. In addition, KO mice had reduced circulating leptin, insulin, and glucose levels suggesting that ASP deletion may exert indirect effects to improve glucose homeostasis and protect against the detrimental effects of high-fat feeding (456). Similar to other adipose secreted factor, levels of adipsin and C3 are reduced following diet-induced weight loss in obese subjects (458,509). The direct cause underlying this reduction remains to be delineated. Thus, the rodent data supports that ASP may play an important regulatory role in energy balance; however, additional work in humans to better understand the mechanistic role of ASP in substrate handling and immune function are required.
Adipose-Secreted Proinflammatory Cytokines and Liver-Secreted Phase Reactant Proteins
TNFα
In the 1970s, TNFα was first described as an endotoxin-induced serum factor able to produce necrosis of tumors (104). Later in the 1980s, TNFα was found to be secreted by cultured macrophages treated with endotoxin and when TNFα was administered to rodents’ tissue inflammation and cachexia were induced (642). The human gene, TNFA, was cloned in 1984 (475) and the protein was shown to be expressed as a 26-kDa plasma membrane-bound monomer. Proteolytic cleavage by TNFα converting enzyme produces a 17 kDa soluble trimer of the precursor protein. Two TNFα receptors have been described, TNFR1 (55 kDa isoform) and TNFR2 (75 kDa isoform), and the subsequent downstream signal is propagated by protein complexes with cytoplasmic adaptor proteins. In 1993, Hotamisligil and colleagues showed increased expression of TNFα mRNA in adipose tissue from four different rodent models of obesity and T2D and found a strong correlation between tissue expression levels and insulin resistance (285). TNFα protein was also elevated locally and systemically. Furthermore, antibody neutralization of TNFα in obese fa/fa rats caused a significant increase in the peripheral uptake of glucose in response to insulin (284). In agreement with neutralization studies, a targeted null mutation in the gene encoding TNFα and the two receptors led to significantly improved insulin sensitivity in both diet-induced and leptin-deficient ob/ob mice (656).
Follow-up studies in obese individuals recapitulated findings in rodents and showed that TNFα mRNA is elevated 2.5-fold in adipose tissue from obese subjects relative to the lean controls (282). Similar increases in TNFα protein concentrations in plasma and in conditioned medium from explanted adipose tissue were also observed; however, it is important to note that circulating levels of this protein are quite low. A strong positive correlation was observed between TNFα mRNA expression levels in fat tissue and the level of hyperinsulinemia, an indirect index of insulin resistance. Dietary treatment in obese subjects resulted in improved insulin sensitivity and this was associated with a decrease in TNFα expression in adipose tissue. Taken together, these seminal studies conducted by Hotamisligil show that the abnormal regulation of this cytokine is involved in the pathogenesis of obesity-related insulin resistance (280, 282, 285, 656).
To understand the molecular action of TNFα on adipocytes several groups have performed incubation studies using the standard adipocyte cell line, 3T3L1. TNFα incubation has repeatedly been shown to induce lipolysis, as well as inhibit insulin-stimulated glucose transport. Inhibition of insulin action is thought to occur via alteration in phosphorylation status of key molecules in the insulin-signaling cascade (283, 284), which is purported to activate serine-threonine stress kinases as well as tyrosine phosphatases including PTP1B (462). In addition, TNFα is shown to antagonize central transcription factors that are involved in regulating substrate metabolism, insulin action, and adipogenesis (724). Furthermore, TNFα promotes the release of fatty acids from adipocytes and stimulates the production of other inflammatory cytokines, chemokines, and adipokines, for example, IL-6, PAI1, and leptin, all factors with purported insulin resistance producing actions (354). These adipose-secreted factors are elevated in circulation in vivo in mice treated with TNFα as well as in humans infused intravenously with the cytokine (660). Studies in TNFR mutant cells demonstrate that TNFα-induced lipolysis, as well as inhibition of insulin-stimulated glucose transport is predominantly mediated by TNFR1 and that the presence of TNFR2 is not necessary for these functions (585). Despite the extensive body of work that has been conducted to understand the biological role and involvement of TNFα and its receptors in the etiology of obesity and T2D, targeted therapies against this cytokine to ameliorate complications associated with these chronic diseases have yielded poor efficacy (470) [as reviewed by reference (447)].
IL-6
The precise role that IL-6 plays in the pathogenesis of obesity and insulin resistance remains controversial despite over a decade of intense research. Circulating levels of IL-6 are increased in obese subjects and are reduced with insulin sensitizing therapies (56, 376). Similar to leptin, IL-6 at rest is secreted primarily from adipose tissue and, thus, a reduction is adipose tissue mass leads to a reduction in circulating IL-6 concentration. Furthermore, variants in the IL-6 gene are associated with adiposity in human subjects (23, 513). However, whether the correlation between IL-6 and obesity/insulin resistance syndrome reflects a causal relationship has yet to be confirmed. In vitro findings provided by several laboratories indicates that IL-6 induces insulin resistance in cultured hepatocytes and hepatoma cells (584), impairs insulin action, and promotes lipolysis in adipocytes (376, 554, 646). Furthermore, IL-6 neutralization in vivo leads to improved insulin action in obese ob/ob leptin deficient and high-fat-fed mice (360). While these findings support a negative role for IL-6, recent work by Sadagurski et al. from the laboratory of White, show that mice with transgenic overexpression of IL-6 are more insulin sensitive and display increased energy expenditure compared to wild-type animals despite comparable fat pad mass and food consumption (560). Transgenic mice fed a high-fat diet were also partially protected against insulin resistance and adipose tissue weight gain. When the IL-6 transgene was expressed in leptin deficient ob/ob mice, these animals showed improved glycemia and a reduction in fed and fasting insulin levels. A role for I-L6 in energy expenditure and improved glucose uptake in rodents and humans is previously supported (101, 647).
Taken together the notion that IL-6 can exert both pro- and anti-inflammatory actions is gaining scientific support. Work by Rose-John and colleagues may resolve some of the confusion related to IL-6 function in vivo in that the pro- and anti-inflammatory actions of IL-6 may be ascribed to differences in signaling from the soluble and membrane bound IL-6 receptors. The activities of IL-6 via the soluble IL-6R, referred to as IL-6 “transsignaling” is largely responsible for proinflammatory IL-6 signaling since all cells express the gp130 receptor (gp130R), but many cells lack the membrane bound IL-6R. A disintegrin and metalloproteinases (ADAMs) are important enzymes involved in “ectodomain shedding” of a diverse group of molecules including growth factors, cytokines, receptors and adhesion molecules, and altered expression of specific ADAMs are associated with disease pathophysiology. Specifically, the membrane bound IL-6R is shed via ADAM10 and ADAM17. As a consequence of receptor shedding, the IL-6/IL6R complex can travel to sites of inflammation or cellular dysfunction and transduce a signal via the gp130R (550). Overexpression of a soluble form of gp130 (sgp130) either by administering a dose of sgp130Fc protein exogenously or by generating a transgenic mouse overexpressing this protein (515) in both cases blocked IL-6 transsignaling and inflammation to a similar extent as described for IL-6 null mice. These data support the notion that the sIL-6R is responsible for most of the tissue inflammation brought about by IL-6 signaling. Thus, in pathological cases where IL-6 signaling is heightened, it is proposed that blocking IL-6 transsignaling, by downregulation of sheddase activity using specific small molecule inhibitors, or by squelching/mopping up the soluble receptor, may serve as an effective way to repress inflammation and restrain disease progression. This notion is supported by studies conducted in human patients harboring a single nucleotide polymorphism (Gly148Arg) for the IL-6 signal transducer (IL-6ST)/gp130 gene, which is associated with decreased IL-6 responsiveness and a significantly reduced odds ratio for heart disease (61). Clinical trials are currently underway to investigate the impact of reduced inflammatory signaling via ADAM inhibition on cancer progression. Whether this mechanism may also be exploited for treatment or prevention of T2D requires further investigation.
IL1β and IL-1Ra
IL-1 is linked with various autoimmune diseases and now metabolic dysregulation. Eleven ligands and ten receptors have now been identified in the IL1 family. IL-1β is mainly produced by immune cells including blood monocytes, tissue macrophages, dendritic cells, B lymphocytes, NK cells; however there is some evidence for production by β-cells within pancreatic islets as well (264, 413). IL-1β is implicated in β-cell destruction that is purportedly mediated by ER stress and activation of apoptotic pathways. Furthermore, endogenous production or exogenous administration of the IL-1 receptor antagonist (Ra) or anti-IL1β monoclonal antibody (XOMA 052) protect against diet-induced insulin resistance and β-cell death (174, 375, 485, 572). IL-1β is induced and IL-1Ra is repressed in human islets cultured in high glucose or treated with leptin suggesting a role for adipose secreted factors in the pathogenesis of islet destruction (414). In human subjects, a strong association between variation in the IL-1 gene family and indices of glucose homeostasis and T2D prevalence have been documented (408) and circulating IL-1β levels are elevated in type 2 diabetic patients and subjects with the MS, however, show a direct causal role for IL-1β and IL-1Ra in insulin resistance and β-cell destruction have yet to be confirmed.
PAI-1
Plasminogen activator inhibitor (PAI)-1 is a 47-kDa serine protease inhibitor protein and the principal negative regulator of tissue plaminogen activator and urokinase, activators of plasminogen and hence fibrinolysis. PAI-1 is mainly produced by the endothelium but is also produced and secreted by adipose tissue and macrophages (298). Thus, circulating levels of PAI-1 are elevated with obesity and the MS (367). Therefore, the risk of thrombosis is elevated in humans with increased circulating levels of PAI-1. During inflammatory conditions fibrin is deposited at the site of injury and PAI-1 is thought to exert and important profibrotic role. ANGII as well as cytokines and growth factors are shown to promote PAI-1 synthesis, thus adipose secreted factors appear to work in concert to promote atherosclerotic lesion development and thus constitute an important nexus by which obesity elevates CVD risk.
Weight loss induced by bariatric surgery performed on morbidly obese subjects significantly diminished tissue factor, PAI-1 and prothrombin fragment 1.2 levels and the thrombotic lag phase was significantly extended which correlated well with improved insulin sensitivity (35). This relationship is supported in rodents in which PAI-1 null mice are protected against high-fat diet-induced accretion of fat and insulin resistance (409). Additional studies are needed to better understand the role of PAI-1 in the pathogenesis of obesity and insulin resistance.
Angiotensin
Angiotensin (ANG) is produced by adipose and transcript levels are elevated in adipose tissue from obese subjects (662). ANGII is a potent vasoconstrictor and may underlie the increased incidence of obesity-associated hypertension. This notion is supported in rodent models where hypertension and increased fat mass were observed in animals with an adipose selective overexpression of ANGII (419). In addition, the same group also showed that adipose selective ablation of ANGII is protective against diet-induced obesity and in part this observation was explained by increased locomotor activity (419). Given that ANGII is shown to promote cellular oxidative stress and stimulate NF-κB leading to the increased secretion of PAI-1, leptin, IL-6, and IL-8 in human primary adipocytes suggests that ANGII-induced inflammation may underlie the association between ANGII expression and increased adiposity. Given that treatment with ANG converting enzyme inhibitors (ACEIs) reverses inflammation, increases circulating adiponectin levels, and protects against high-fat diet (HFD)-induced adipose tissue weight gain supports a role for the renin-ANG pathway in the etiology of obesity and the MS (145, 193, 393, 394). Further studies in the clinic must be performed to determine whether pharmaceutical targeting of this system is efficacious and practical in ameliorating complications associated with T2D.
CRP
CRP is a member of the pentraxin family of acute phase proteins produced by liver in response to secreted factors from macrophages and adipocytes, including IL-6. CRP is thought to link chronic inflammation with atherosclerosis. Its putative role is to bind dead or dying cells to activate the complement system via c1q thus promoting cellular removal by macrophage phagocytosis. CRP is the best characterized and well-standardized marker of systemic inflammation, and several studies now confirm that CRP levels are elevated in patients with the MS. Furthermore, as many as six major prospective clinical trials support the hypothesis that CRP contributes to increased CVD risk. In the insulin resistance and atherosclerosis study, CRP was highly associated with central adiposity and insulin resistance, and additionally with blood pressure, LDL cholesterol, and TGs. Administration of recombinant CRP causes impaired insulin action and endothelial nitric oxide synthase (eNOS) production in endothelial cells (704). Syk and JNK are thought to mediate CRP-induced insulin resistance as inhibitors of these kinases (Y27632 and SP600125, respectively) restored in part insulin action and eNOS production (704). Similar findings of CRP-induced insulin resistance by a JNK-mediated mechanism to inhibit IRS-1 were observed for L6 muscle cells (140). In monocytic immune cells, CRP is shown to promote inflammation by induced expression of tissue factor, cytokines (IL-6 and TNFα), ROS, CCR2, and matrix metalloproteinases (157, 158, 263, 596). While clinical findings strongly support a relationship between CRP levels and various features of the MS, experimental studies to clarify the specific sites and molecular modes of CRP action in vivo are still needed. Additionally, CRP is inversely related to fitness level (31, 32, 587), and circulating levels are reduced in healthy and T2D patients following lifestyle intervention, including chronic aerobic exercise (15, 142); as reviewed in reference (278). Additionally, some evidence indicates that resistance training (RT) may also induce a reduction in circulating CRP (163, 262, 477). However, other evidence suggests exercise training does not alter CRP levels (131, 359, 623, 731).
Adipose tissue-secreted chemokines
Chemokines are a family of small (8–10 kD) cytokines that are involved primarily in promoting the migration, chemotaxis, of immune cells to sites of infection, cellular damage, inflammation, or altered metabolism; however, nonchemotactic homeostatic functions have also been described. In addition to the small size, many in the chemokine family members also possess conserved amino acid sequence homology including four well-characterized cysteine residues that are important for creating the characteristic chemokine three-dimensional structure. Chemokine proteins are typically produced as propeptides, in which a signal peptide of approximately 20 AA is cleaved upon mature peptide release. Members of the chemokine family are divided into four groups defined by the spacing of the first two cysteine residues: CC chemokines, CXC chemokines, C chemokines, and CX3C chemokines. Chemokines bind to cell surface G-protein coupled receptor containing on average seven transmembrane domains. As many as 20 chemokine receptors have been characterized and these are classified into four groups based upon the type of chemokine they bind. With respect to the cross talk between adipose tissue and the immune system in the regulation of whole body insulin action, the CC chemokines including monocyte chemoattractant protein (MCP)-1 (CCL2), MIP1α (CCL3), and regulated-upon-activated-normal-T-cells expressed and secreted (RANTES or CCL5) have been studied the most extensively.
CC chemokine MCP1
MCP-1 is expressed in a number of cell types including skeletal muscle, endothelial cells, smooth muscle cells, and adipocytes. MCP-1 exerts its action through CC motif receptor (CCR) 2 that is also expressed in a variety of tissues including skeletal muscle, adipocytes, and macrophages. MCP-1 was first described as a monocyte and endothelial secretory product involved in the pathogenesis of atherosclerosis (581). In addition, MCP-1 is insulin and TNFα responsive as expression levels and rates of secretion from adipocytes were markedly increased with hyperinsulinemia or TNFα treatment (567). Furthermore, MCP1 is overexpressed in adipose from obese rodents and insulin action as well as expression levels of several adipogenic genes (LPL, adipsin, GLUT4, aP2, β3-adrenergic receptor, and PPARγ) are reduced in cells treated with MCP-1 (567). Secretion of MCP-1 from adipocytes is thought to prompt the diapedesis or movement of monocytes from the vascular compartment into adipose tissue. Once differentiated, these tissue resident macrophages/professional phagocytes exert a host of functions to regulate tissue remodeling, inflammation, and substrate metabolism. The phenotypic role of tissue resident macrophages has received increasing attention in the past decade and despite the increased exploration, many unresolved issues remain.
Given the increased MCP-1 expression and secretion from adipose taken from obese, diabetic, and high-fat-fed mice, Kanda et al. (329) generated animals with an adipocyte selective overexpression of MCP-1. These mice exhibited whole body insulin resistance, had increased macrophage infiltration in adipose tissue, and hepatic TG accumulation (329). Consistent with this observation, MCP-1 homozygous null mice were protected from high-fat-diet-induced insulin resistance, hepatic steatosis, and adipose tissue macrophage infiltration. A similar amelioration of these measures was observed in db/db with a dominant negative MCP-1 mutation. Similar adipocyte-specific MCP-1 overexpression studies conducted by Kamei et al. (328) yielded identical findings.
Collectively, this work is further supported by several investigations in which the elimination or inhibition of CCR2 was protective against insulin resistance and complications associated with T2D (628,629,682). Until recently, it was unknown as to whether MCP-1 could induce insulin resistance in vivo independent of alterations in adipose tissue inflammation or obesity. Tamura et al. (628) recently showed that acute administration of purified MCP-1 to circulating levels comparable to those observed in obese rodents, was sufficient to cause insulin resistance measure by glucose clamp; this was independent of adipose tissue inflammation.
Studies in visceral adipose tissue from lean and obese human subjects show that macrophage specific markers and CC chemokines in monocyte chemotaxis as well as their receptors are elevated in obese visceral fat compared with lean controls. Furthermore, adipose tissue expression of these chemokines and receptors correlated positively with increased systemic inflammation and insulin resistance (297).
CX chemokine CXCL5
CXCL5 or epithelial neutrophil-activating peptide (ENA-78) belongs to a family of chemokines that is mainly shown to recruit and activate neutrophils to sites of inflammation through interaction with the CXCR2 receptor (671). Although this chemokine has been implicated in lung cancer, pulmonary diseases and arthritis, recent work by Aquilante et al., show that CXCL5 is highly expressed in adipose tissue from obese subjects and that circulating levels of the chemokine are elevated almost twofold over levels observed in lean subjects (24, 112). Adipose tissue explants harvested from obese and diabetic rodents support observations of increased circulating levels of CXCL5 in obese humans. Fractionation analyses performed in both rodent and human white adipose tissue shows that the stromal vascular fraction, specifically tissue resident macrophages are the primary source of CXCL5. Furthermore, this group also shows that the CXCR2 receptor is most highly expressed in skeletal muscle compared with adipose tissue and liver, suggesting that CXCL5 acting through its putative receptor, CXCR2, may exert its deleterious effects on glucose homeostasis by inhibiting insulin action in skeletal muscle. In support of this notion, Chavey et al. (112) showed that treatment of muscle ex vivo with recombinant CXCL5 markedly inhibited insulin-stimulated glucose uptake and signaling. Similar observations were made in mouse embryonic fibroblasts. Follow-up mechanistic analyses suggest that CXCL5 activates the JAK/STAT pathway and SOCS2, which were shown previously to inhibit proximal insulin signal transduction. Use of CXCL5 neutralizing antibodies and inhibitors against Jak2(AG490) and the CXCL5 receptor CXCR2 support this mechanism of action.
Interestingly, an 800 kcal/d caloric deficit that led to an average 7% reduction in body weight in obese women was correlated with a significant decrease in serum CXCL5 concentration (112). A more pronounced reduction in CXCL5 was observed following treatment with the insulin sensitizing compound rosiglitazone (112). Taken together, CXCL5 is an important adipose tissue secreted chemokine that is elevated with obesity and exerts effects directly on skeletal muscle to cause insulin resistance via JAK/STAT inhibition of proximal insulin signaling. A direct role of exercise to suppress adipose chemokine secretion independent of adipose tissue weight loss warrants further attention.
Immune Function and Regulation of Insulin Action and Adiposity
Chronic low-grade inflammation is a central underpinning in the development of obesity and insulin resistance and cells of both the innate and adaptive immune system are shown to play an important role in the pathogenesis of metabolic disease (Fig. 5). In 2003, two seminal papers were published showing an accumulation of F4/80-positive immune cells in adipose tissue of obese mice and animals fed a diet rich in fat (683, 703) (Fig. 6A). These F4/80+ adipose tissue macrophages obtained from obese animals were found to secrete proinflammatory, insulin resistance-producing factors including TNFα, IL6, IL1β, serum amyloid A3 (SAA3), and CCL2 (i.e., MCP-1) to name a few (684) (Fig. 6B). These two publications sparked a fervent burst of investigation into the role of the immune system in the regulation of glucose home-ostasis and adiposity. Subsequent studies have demonstrated that lymphocytes including regulatory T cells, effector T cells, and B cells are recruited during various stages in the expansion of adipose tissue as well (168, 463, 473). Many propose that regulatory T cells, TH2-polarized T cells as well as alternatively activated anti-inflammatory macrophages are most abundant in adipose tissue from lean rodents, whereas diet-induced adiposity is associated with the orchestrated trafficking of B cells, TH1-polarized T cells, and effector T cells with later stages of adipose tissue expansion showing the accumulation of classically activated proinflammatory macrophages and natural killer cells (168, 169, 473).
Figure 6.

The role of macrophages in the development of obesity. (A) Increased abundance of F4/80 positive macrophages in visceral perigonadal adipose tissue obtained from obese mice. (a) Lean female, (b) Ay/+ female, (c) Lep o/ob female, (d) lean male, (e) diet-induced obesity, and (f) Lep ob/ob male mice. F4/80 positive cells are small, dispersed and rarely aggregated in adipose tissue from lean animals (a and d), but found frequently in clusters “crowning” adipocytes in adipose tissue from high-fat fed and genetically obese mice (c, e, and f). Reprinted, with permission, from reference (683). (B) Schematic overview of the effects of adipose macrophage infiltration on the development of tissue inflammation and insulin resistance. Classically activated macrophages in adipose tissue from obese humans and rodents are shown to secrete insulin resistance producing proinflammatory cytokines and chemokines. Reprinted, with permission, from reference (684).
Despite the burgeoning research in this field, the metabolic factors regulating immune responses in glucoregulatory tissues including adipose, liver, and skeletal muscle remain poorly defined. To date, much of the work in this field implicates macrophages as major effectors in the pathogenesis obesity-insulin resistance phenotypes. Early studies clearly show that ablation of genes involved in inflammatory signaling within macrophages is protective against diet-induced obesity and insulin resistance (28, 94, 559), while conversely, hematopoietic/myeloid-specific ablation of transcription factors involved in inflammation repression promotes diet-induced obesity and whole body insulin resistance (267, 469) as well as accelerated atherosclerotic lesion development (538). In later part of the past decade, a theme emerged in which immune cells were rigidly ascribed as either phenotypically anti-inflammatory, alternatively activated “M2” or proinflammatory, classically activated, “M1.” Although the appealingly simplistic binary classification of M1 versus M2 macrophage phenotype has been readily accepted by the obesity/diabetes research community, the diversity and complexity of immune cell type and function favors a spectral classification taking into account the required functionality of the immune cell and the specificity of chemotactic signal and environmental milieu unique to the recruiting tissue (125, 232).
Altering the chemotactic signal from a given tissue is shown to exert either health benefit or pathogenic repercussions. For instance, there is some suggestion that deletion of MCP1 or its receptor, CCR2, confers protection against adipose tissue macrophage accumulation, insulin resistance, and atherosclerosis during high fat feeding (329, 471, 682) whereas adipocyte-specific overexpression of MCP1 promotes macrophage infiltration, inflammation, insulin resistance and atherosclerosis (328).
Work over last past decade clearly shows a critical role for the immune system in the regulation of glucose homeostasis and the pathogenesis of adiposity and atherosclerotic lesion development. In addition, more recent evidence shows that diet/caloric restriction and/or exercise are potent interventions to promote adipose tissue weight loss and alteration of immune cell phenotype (30,344,364). Thus, the key for future studies will be to further delineate the tissue-selective aspects of immune cell recruitment and function, and determine how each cell type may be exploited to ameliorate disease complications without compromising innate immunity.
Chronic exercise exerts potent anti-inflammatory effects (40, 335, 502) and these effects are likely mediated by direct effects on the immune system and a reduction in visceral fat including diminished release of proinflammatory cytokines and chemokines from adipocytes [as reviewed by reference (223)]. In part, endurance exercise reduces toll-receptor expression on monocytes and macrophages (200), and experimental reduction in Tlr expression is associated with reduced induction of proinflammatory signaling and diet-induced obesity (144, 559, 650). Furthermore, treadmill exercise reduces adipose tissue macrophage infiltration and promotes an M2 anti-inflammatory immune cell phenotype (344). During exercise, skeletal muscle is thought to produce and secrete a host of anti-inflammatory cytokines that are thought to produce and secrete a host of anti-inflammatory cytokines that are shown to experimentally alter immune cell function and phenotype. Muscle-secreted factors are discussed below.
Muscle as an Endocrine Organ—Myokines and Insulin Action
Mounting evidence suggests that skeletal muscle is an endocrine organ capable of secreting a variety of factors that act on peripheral tissues to alter metabolic function. Myokines are skeletal muscle specific-secreted factors able to exert humoral effects in vivo and may underlie the health benefits associated with daily physical exercise. Muscle-derived factors including IL-6 (100, 101, 492, 493) IL-15 (514), BNDF (365, 424, 520), FGF21 (29, 64, 132, 164, 311, 349), and Follistatin-like 1 (482, 484), and SPARC (466) are a few putative myokines currently under investigation. IL-6, one of the first myokines identified, is shown to increase in expression over 100-fold during prolonged exercise and stimulate the appearance of other circulating anti-inflammatory cytokines such as IL-1Ra and IL-10, and inhibit the production of the proinflammatory cytokine TNFα (494, 603). In addition, IL-6 enhances lipid turnover, stimulating lipolysis as well as fat oxidation (16, 101). Moreover, contrary to conventional wisdom IL-6 infusion stimulates glucose disposal in humans and rodents (101, 719), while in contrast, homozygous deletion of IL-6 promoted systemic insulin resistance in mice (423). Novel secreted proteins of the human and mouse muscle proteome are being vigorously studied and indeed several of these are now associated with improvements in immune cell function (442) and restraint of cancer cell proliferation (270). Whether these observations in culture translate directly to the reduction in cancer risk associated with exercise requires further investigation. Considering that skeletal muscle comprises approximately 40% of total body mass and is the primary tissue contributing to insulin-mediated glucose uptake and fatty acid oxidation, identification of signals emanating from muscle to regulate whole body energy balance and metabolism will likely prove to be of therapeutic benefit.
Exercise and the Metabolic Syndrome
Epidemiology/observational evidence
As previously discussed, MS is clearly a significant public health problem in the US with both fitness and physical activity playing vital roles in preventing and treating MS. Numerous studies have clearly demonstrated that the pathogenesis of the MS is largely attributable to a lack of fitness and physical activity. Some of these studies are described below. Cross-sectionally, several studies have indicated greater prevalence of MS in subjects with lower fitness. Lakka et al. (378) noted associations of leisure-time physical activity and cardiorespiratory fitness (CRF) with MS in a population-based sample of 1069 middle-aged men without T2D, CVD, or cancer. Men who engaged in moderate-intensity leisure-time physical activity 1.0 h/wk or less were 60% more likely to have MS than those engaging in 3.0 h/wk or more. In addition, men with a VO2max equal to 29.1 mL/kg/min or less were approximately seven times more likely to exhibit MS than those with a VO2max is equal to 35.5 mL/kg/min or more. Across CRF quintiles, as measured by maximal treadmill exercise test, prevalence of MS in a population of 7104 women was lower as fitness increased, with prevalence ranging from 19.0% in the least fit quintile to 2.3% in the most fit quintile (189) (Fig. 7). Additionally, the prevalence of MS in the different age groups for women who achieved a maximal metabolic equivalent (MET) level of 11 or higher was one-third to one-fourth that of women who achieved lower maximal MET levels (189). An additional study in middle-aged men noted age and BMI-adjusted OR for MS of 0.55 and 0.26 for moderate and high fitness, respectively (103).
Figure 7.

Prevalence of metabolic syndrome according to cardiorespiratory fitness quintiles in more than 7000 women enrolled in the Aerobics Center Longitudinal Study from 1979 to 2000. Number of subjects in each quintile is I: 796; II: 1173; III: 1241; IV: 1754; and V: 2140. Adapted from reference (189) with permission.
In a prospective population-based study, LaMonte et al. (379) noted that in 9007 middle-aged men and 1491 women, multivariate OR for incident MS in the low, middle, and upper third of fitness were 1.0, 0.74, and 0.47 for men and 1.0, 0.80, and 0.37 for women, respectively. Similar patterns of significant inverse associations between fitness and MS incidence were seen when men were stratified according to categories of BMI, age, and number of baseline metabolic risk factors. Because lifestyle factors potentially exhibit similar patterns and/or exercise may impact food choices, Finley et al. (197) noted that adjustment for macronutrient intake and other potential confounding variables did not alter the association between CRF and prevalent MS. It is also important to note that in most cases controlling for obesity does not impact the link between fitness and MS.
Muscle strength and lean body mass (LBM) are also related to MS. Jurca et al. (320) demonstrated in men from the Aerobics Center Longitudinal Study (ACLS) that greater muscle strength is associated with lower prevalence of abnormal MS components. Each of the five MS components were inversely associated with muscular strength, as determined by one-repetition maximum (1-RM) bench press and leg press when adjusted for age and smoking status. The OR for MS was 0.33 in the quartile with the highest versus the quartile with the lowest muscle strength. Interestingly, the effects of muscle strength and CRF were independent of each other and largely of BMI (Fig. 8A). The effects of muscle strength were modestly attenuated when controlling for CRF, likely due to the overlap between muscular and aerobic fitness. This suggests that both aerobic fitness and muscular strength have preventive effects on MS risk. In this analysis for CRF, the OR for MS with the highest fitness was 0.08 compared to the lowest. This group followed up their work to demonstrate that muscle strength was inversely associated with incident MS in men, independent of age (319) (Fig. 8B). In addition, in a cohort of 1019 adults, muscle strength was inversely associated with MS risk score; although, adjustment for aerobic fitness attenuated this inverse association (692). Atlantis et al. (33) also noted increased prevalence of MS (by both the ATP III and IDF criteria) in subjects with low peak handgrip strength.
Figure 8.

(A) Age and smoking adjusted prevalence of metabolic syndrome in men according to level of muscular strength and cardiorespiratory fitness. Q1 represents the lowest and Q4 the highest muscular strength quartile. (B) Incidence of MS across muscular strength categories by age groups. Incidence rates per 1000 man-years are shown labeled with bars. The number of subjects for each age group is 20–39: 1239; 40–49: 1249; and 50+: 745. Q1 represents the lowest and Q4 the highest muscular strength quartile. The linear trend p values for the age groups 20–39, 40–49, and 50+ are less < 0.001, 0.01, and 0.05, respectively. Adapted from references (319, 320) with permission.
Obviously, investigation of mortality outcomes related to MS and fitness is important. Along these lines, in the ACLS, Katzmarzyk et al. (337) investigated the effects of CRF and mortality in 19,223 men 20 to 83 years old, separated into “healthy” men and those with MS. During 196,466 man-years of follow-up RR of all-cause and CVD mortality were 1.29 and 1.89, respectively, for men with the MS compared with healthy men. However, after accounting for CRF, the associations were no longer significant (0.98 for all-cause and 1.23 for CVD) (Fig. 9). The RRs for all-cause mortality comparing unfit and fit men were similar in men deemed healthy and in those with MS (2.18 and 2.01 vs. fit groups, respectively). Overall mortality risk was 5.18% in the healthy unfit group compared with 1.95% in the healthy fit group and the number needed to treat (NNT) to prevent one death was 31. In those with MS, overall mortality risk was 5.15% in the unfit MS group compared with 2.69% in the fit MS group, with an NNT of 40.6. More importantly, the results indicated that unfit healthy men exhibited higher all-cause and CVD death rates per 10,000 man-years of follow-up than fit men with MS (Fig. 10A and B). Additionally, a significant dose-response relationship between CRF and mortality was observed in men with MS. These data indicate that CRF may be as powerful of a predictor of mortality as all the MS characteristics combined, indicating that CRF should be included as a feature of MS (257). This group went on to demonstrate in 19,173 men (338) that at baseline 19.5% of the men had MS, and the ORs of MS at baseline were 4.7 in overweight and 30.6 in obese men compared with normal weight men. In an average 10-year follow-up, the risks of all-cause mortality were 1.11 in normal weight, 1.09 in overweight, and 1.55 in obese men with MS compared with normal weight healthy men (Fig. 11A). Corresponding risks for CVD mortality were 2.06 in normal weight, 1.80 in overweight, and 2.83 in obese men with MS compared with normal weight healthy men (Fig. 11B). However, after inclusion of CRF in the statistical model, the risks associated with obesity and MS were no longer significant. This data suggests that fitness is likely a better predictor of CVD mortality than obesity or MS. Given the aforementioned increased risk of cardiovascular and all-cause mortality with MS (454), it is likely that a significant portion of this risk is explained by low fitness. These data are compelling evidence that exercise provides protection against mortality risk in subjects with MS.
Figure 9.

Impact of fitness on relative risk for all-cause and cardiovascular disease (CVD) mortality associated with metabolic syndrome before and after the inclusion of cardiorespiratory fitness (CRF) as a covariate in more than 19,000 men 20–83 years of age from Aerobics Center Longitudinal Study. Error bars represent 95% confidence intervals and demonstrate that after inclusion of CRF as a covariate, all-cause and CVD mortality were no longer statistically significant. Adapted from reference (337) with permission.
Figure 10.

All-cause (A) and cardiovascular disease (B) mortality death rates per 10,000 mean-years of follow-up in “healthy” and subjects with metabolic syndrome (MS), adjusted for age and year of examination more than 19,000 men 20–83 years of age from Aerobics Center Longitudinal Study. The theoretical contributions of fitness and MS are depicted by brackets. Adapted from reference (337) with permission.
Figure 11.

Relative risk (RR) of (A) all-cause and (B) cardiovascular disease (CVD) mortality in more than 19,000 men from Aerobics Center Longitudinal Study, adjusted for age, examination year, smoking, alcohol consumption, possible existence of CVD, and parental history of premature CVD. Second and forth bars within a body mass index category refer to cardiorespiratory fitness (CRF)-adjusted RR’s. Data, with permission, from reference (338).
The odds of having risk factors for MS (elevated systolic blood pressure, serum TG, fasting glucose, and central adiposity) was reported as 3.0 and 10.1 for least-fit men compared to moderately fit and most-fit men, respectively (685). Furthermore, in the Coronary Artery Risk Development in Young Adults (CARDIA) study, low fitness predicted risk of MS as powerfully as conventional factors (102). Additionally, in a study of 5159 men ages 40–59, physical activity level was associated with MS factors, as well as risk of CAD and T2D (676). CRF has been consistently associated with many components of MS, including insulin resistance, HDL cholesterol, TG levels, and blood pressure (685).
While fitness quantitation is the optimal measure when assessing risk, physical activity questionnaire data can be helpful when assessing large datasets. Nevertheless, physical activity energy expenditure is inherently difficult to measure precisely, and previous epidemiological studies have primarily relied on self-reported physical activity when examining associations with MS. Several studies have demonstrated that the pathogenesis of MS is largely attributable to activity levels. Cross-sectionally, Churilla and Fitzhugh (123) used NHANES 1999–2004 data from 5620 adults and the newer AHA/NHLBI definition, again, similar to ATP III except for the fasting glucose cutpoint of more than 100 mg/dL, estimated MS prevalence among US adults at 21.9% and 36.3% for the WHO and AHA/NLHBI criteria, respectively. The highest quintile of physical activity predicted an OR of 0.54 in men and 0.50 in women, compared to the lowest (123). In the Whitehall II study comprising 5153 Caucasian Europeans, the ORs for having MS among participants engaging in vigorous (METs ≥5) and moderate (3 ≤ METs <5) activity were 0.52 and 0.78, respectively (532). The OR for MS in the high leisure-time physical activity group (intense activity more than two times per week, at least 30 min each time) was 0.33 among 4228 60-year-old Swedish men and women (251). In the Second Manifestations of ARTerial disease (SMART) study examining 1097 patients, the prevalence of MS was lower in physically active patients (>15 MET/h per week) compared to those who were physically inactive (OR: 0.50) (82). Using self-reported physical activity data obtained from 2164 participants, aged 18 to 92 years old living in Portugal, both higher total physical activity during transportation, work, and household (OR: 0.63 in women; OR: 0.55 in men) and higher leisure-time physical activity levels (OR: 0.86 in women; OR: 0.59 in men) were significantly associated with a lower prevalence of MS (566). Ford et al. (203) noted in 1626 men and women from NHANES 1999–2000 that participants who did not engage in any moderate or vigorous physical activity during leisure-time had approximately twice the odds of having MS (OR: 1.90) as those who reported engaging in 150 min/wk or more of such activity. Adjustment for age, sex, race or ethnicity, educational status, smoking status, and alcohol use attenuated the OR (1.46). The adjusted odds ratio for having MS in the high leisure-time physical activity group was 0.33 using the low leisure-time PA group as reference. However, no such inverse association was noted for work-related PA. In the aforementioned study by Carroll et al. (103), findings for CRF were also found for physical activity index (OR for moderate: 0.28 and high fitness 0.12). Interestingly, although CRF is generally considered more precise in the determination of the relationships between exercise training status and disease risk, Franks et al. (206) noted the association between physical activity and MS was much greater than for CRF. Although CRF modified the relationship between physical activity energy expenditure and MS, it suggested a strong inverse association between physical activity and MS, especially in unfit individuals.
These relationships were extended to African-American and native American women, as the OR for MS was 0.18 for women in the highest category of physical activity compared with the lowest, while the OR was 0.07 comparing the duration of groups with the shortest versus longest maximal treadmill time (306). In urban Mexican adults (428), where prevalence of MS was 24.4% (25.3% in men and 21.8% in women), MS risk was reduced among men (OR: 0.72) and women (OR: 0.78) who reported 30 min/d or more of leisure-time activity, and among women who reported an amount of 3 h/d or more of workplace activity (OR 0.75). Furthermore, similar relationships were noted in Asian men for both CRF (390) and physical activity (116) in men and women. These results indicate that the fitness/MS relationship holds across ethnicity. Furthermore, DuBose et al. (166) noted that aerobic fitness modified the BMI-MS relationship, with higher fitness decreasing MS score in normal weight, overweight, and obese children.
Longitudinal studies have also confirmed the development of MS is related to physical activity levels. Over a 4-year follow-up period, the Kuopio Ischemic Heart Disease Risk Factor Study reported an inverse relationship between leisure-time physical activity (defined as >3 h/wk of structured or lifestyle physical activity of ≥4.5 METs) and the development of MS in middle-aged men (374) (Fig. 12). In the Medical Research Council Ely Study, over 5.6 years of follow-up, the physical activity energy expenditure measure predicted a progression toward MS that was independent of aerobic fitness, obesity, and other confounding factors in middle-aged healthy Caucasians (175). In the Oslo study, leisure-time physical activity was a significant predictor of incident MS (OR: 0.65) over 28 years of follow-up in men (275).
Figure 12.
Adjusted cases of metabolic syndrome (MS) based on minutes per week of leisure time physical activity in fit and unfit men after an average 4-year follow-up (374). Copyright 2002 American Diabetes Association. From Diabetes Care®, Vol. 25, 2002; 1612–1618. Reprinted by permission of the American Diabetes Association.
In addition to the link between MS factors and fitness or physical activity, other risk factors in subjects with MS are also linked with fitness. For instance, greater white blood cell count is noted in subjects with MS factors and low CRF (517). In another example, Pitsavos et al. (505) noted that after controlling for various potential confounders, physically active individuals with MS had lower levels of CRP, white blood cells, serum amyloid A, TNFα, and IL-6 when compared to sedentary subjects. Additional data indicates that in subjects with MS, higher CRF is associated with lower CRP concentration (31).
Intervention Effects on Metabolic Syndrome
Early on it was suggested that physical activity and exercise prevented or controlled MS (51, 180), and numerous studies have investigated the effects of exercise on MS. Many studies should be acknowledged as they have investigated MS components, such as insulin resistance/hyperglycemia, dyslipidemia, hypertension, and obesity, and/or have assessed markers of metabolic health other than those that makeup the definition of MS. Many of these studies were not designed to test effects of physical activity on MS per se, given its more recent characterization. Relatively few studies, have investigated the effects of exercise on MS per se, which we discuss here, while the individual components of MS are discussed in Chapter by Booth et al. (76).
The Studies of a Targeted Risk Reduction Intervention through Defined Exercise (STRRIDE) study noted that a low amount of moderate intensity exercise (~19 km/wk of walking) and a high amount of vigorous intensity (~32 km/wk of jogging), but not a low amount of vigorous intensity (~19 km/wk of jogging), improved MS compared to controls, with only the high-vigorous reporting a decrease in BMI (316). Katzmarzyk et al. (340) investigated the efficacy of exercise training in treating MS, as defined according to the NCEP criteria in 621 Black and White participants (males and females, age 17–65 years) from the HERITAGE Family Study. The presence of MS and component risk factors were determined before and after 20 weeks of supervised aerobic exercise training (3 d/wk at 55% of VO2max for 30 min and progressing up to 75% VO2max for 50 min). The prevalence of the MS using ATP III criteria was 16.9% in this sample (105/621) of apparently healthy participants. Of the 105 participants with MS at baseline, 30.5% were no longer classified as having MS after training.
Kemmler et al. (347) using 4 d/wk group exercise sessions combining aerobic (20 min, 70–85% HRmax) and RT (2 sets, 12–15 repetitions) in older women with MS noted that although several components of MS decreased significantly in the women, a nonsignificant decrease (P = 0.15) in the number of MS factors by IDF criteria was noted. Stewart et al. (613) investigated the effect of a combined strength and endurance exercise protocol (3 times per week, seven exercises, two sets of 10–15 repetitions at 50% 1-RM; 45 min of endurance exercise at 60–90% HRmax) on MS prevalence in older subjects (55–75 years old). There was no significant difference in the number of subjects without MS in the intervention versus control groups, although the number of subject MS factors decreased by 0.65 factors in the exercise group and 0.30 in the control group (P = 0.06).
No studies on RT alone have investigated effects on MS per se. However, among 137 subjects who participated in RT in the Finnish Diabetes Trial, MS components of hyperglycemia, hypertriglyceridemia, and low HDL were favorable in individuals who were in the upper third of participation rate (median 51 times per year) compared with individuals in the lowest third (median 8.5 times per year) (305).
Regarding interval training, a recent study compared moderate-intensity AT (70% of HRmax) to interval training (90% HRmax) three times per week, and noted decreases in number of MS criteria (5.9-4.0 for interval training vs. 5.7-5.0 for AT), independent of differences in body weight changes, which were similar. Interval training was superior to AT in enhancing endothelial function, insulin signaling in fat and skeletal muscle, skeletal muscle biogenesis, and in reducing blood glucose, and lipogenesis in adipose tissue, which likely contributed to mechanisms underlying the beneficial effects of interval training on MS (640).
Overall, aside from the STRRIDE and HERITAGE Family Study, relatively few studies have investigated the effects of exercise training on MS per se and in those that have, decreases in MS components are noted; however, the effects on MS per se need to be investigated further.
Although not the focus of this review, numerous other intervention studies have suggested that combined interventions with diet and exercise/activity help in the treatment and prevention of MS. Combined studies tend to show better effects, with some comparisons made between isolated and combined interventions, and likely the mechanisms for improvements with diet and exercise being, at least in part, independent. Anderssen et al. (22) evaluated the effect of a supervised endurance program (3 d/wk, 60 min at 60–80% HRmax) with or without caloric restriction on IDF-defined MS prevalence in middle-aged men recruited from the larger Oslo Diet and Exercise Study. In the exercise group, the MS prevalence was reduced 24% (1% drop in body weight) and the combined caloric restriction and exercise group noted a 67% prevalence reduction (7% drop in body weight), although the MS reduction in exercise group was not significantly different from the control group (12%). Although exercise was not investigated alone, Okura et al. (474) noted that exercise plus a low-calorie diet reversed MS in 36 of 38 subjects compared with 15 of 21 subjects with a low-calorie diet alone who completed a 14-week intervention (eight subjects did not complete the intervention).
In the Diabetes Prevention Program, lifestyle intervention resulted in 3-year cumulative diabetes incidence of 51%, 45%, and 34% in placebo, metformin, and lifestyle groups associated with a 41% reduction in the lifestyle group and a 17% reduction in the metformin group (478). In the Finnish Diabetes Prevention Study, after adjustments for changes in dietary intakes of total and saturated fat, fiber, and energy, and change in BMI, increased moderate-to-vigorous leisure-time physical activity was associated with a greater likelihood for diabetes resolution (29.7 vs. 19.1% in the upper vs. lower third of change) and a lesser likelihood for development (23.5 vs. 44.7%) of MS (305). Additionally, the prevalence of MS decreased (OR: 0.62) in the intervention group compared to the control group throughout the intervention (304).
Lifestyle intervention significantly reduced MS compared with standard physician information (OR = 0.28), with a 31% absolute risk reduction, corresponding to 3.2 patients needing to be treated to prevent 1 case after the 12-month intervention (72). Camhi et al. (95) noted that a 1-year combined diet and exercise intervention improved MS score compared to control, with the authors suggesting changes in body fat explaining the intervention effects. Roberts et al. (542) noted reversal of MS in 9 of 15 subjects with an intensive 21-day diet and exercise intervention. The latter study incorporated truly intensive programs that require follow-up randomized-controlled studies to determine if interventions of this magnitude can be translated to a large segment in the population who might benefit from rapid reductions and maintenance of reduced risk for MS-related complications. Similar findings have demonstrated that combined interventions are also effective in children in some (113, 448, 530) but not all studies (690). In a nonrandomized 14-day intervention that incorporated an ad libitum high-fiber, plant-based diet along with approximately 2 h/d of exercise and game play, Chen et al. (113) noted reversal of MS in seven of seven children. In a 1-year multidisciplinary intervention including aerobic exercise and nutrition therapy, prevalence of MS was 27% prior and 8% after the intervention (98). It is interesting to note that in these studies, despite reversal of MS, the subjects remained obese. In addition, Thomas et al. (638) noted that in subjects who underwent a caloric restriction and aerobic exercise intervention to improve MS factors (and elicit 10% weight loss), in subjects that continued to exercise during a subsequent regain phase, improvements in some MS and other related risk factors were maintained compared with subjects that did not exercise during the regain phase.
It is important to note that other factors, the more salient being diet as well as smoking, sleep, alcohol intake, and stress all may contribute to MS; and there are potential interactions between several of these lifestyle factors (566, 678), as alluded to above. In many subjects, these other factors are not considered but are likely related to patterns of lifestyle in individuals. In addition, treating individual risk factors with lifestyle change might be as beneficial as treating MS as a separate disease.
Exercise and Insulin Sensitivity
As discussed above, insulin resistance has been suggested as the major underpinning link between physical inactivity and MS. It is well known that trained subjects and those with high levels of physical activity exhibit high levels of insulin sensitivity/insulin action. Additionally, changes in exercise through initiation or cessation of training programs and increases or decreases in physical activity modify insulin action. We will touch on exercise training per se, rather than acute exercise and briefly discuss the effects of aerobic, resistance, and interval training as well as increased and decreased physical activity levels. Below is a discussion of selected studies in these areas. The majority of studies discussed will focus on studies using the EHC, FSIGT, or OGTT. We will only discuss training studies with the caveat that the timing of the insulin sensitivity measure after the last bout may impact the effects noted, as considered below.
Aerobic Training
It is well known that AT results in increases in insulin action in skeletal muscle of healthy individuals. The effects of AT on insulin sensitivity have been noted both cross-sectionally in exercise-trained compared to untrained individuals and for untrained subjects after periods of training. One of the earliest demonstrations of increased insulin action was a cross-sectional study of trained and untrained men, which noted lower insulin and glucose during an OGTT in the trained subjects (70). Several other studies early on noted that aerobically/endurance-trained subjects exhibit improved insulin responses to intravenous glucose (388,405), even differing in highly trained versus trained (387), and higher insulin sensitivity using the EHC in younger (353,569) or older (273) trained compared to untrained subjects, including whole body and nonoxidative glucose disposal (172).
Many studies have subsequently demonstrated that aerobic/endurance training elicits increases in insulin sensitivity, in general, from 25% to 50% in a variety of age and population groups, including normal healthy men (153, 289) and women (183), overweight and obese (149, 227, 591), young (712), middle-aged (291), and older subjects (160), in addition to first degree relatives of T2D patients (55, 483, 498), subjects with T2D (152), and even adolescents using GCMS (658).
Differences in aerobic exercise intensity have also been investigated. For example, Seals et al. (578) noted in older subjects that oral glucose tolerance was not significantly changed after training, although the area under the curve (AUC) for insulin was 8% lower after 6 months of lower intensity training (walking 30 min, 3–4 d/wk, ~40% of HRreserve), and decreased an additional 23% after an additional 6 months of higher intensity training (jogging 30–45 min, 3–4 days/wk, ~75% of HRreserve), although the study design was sequential as opposed to crossover. More recently, in a 9-month study of older women, DiPietro et al. (160) noted that only high-intensity training (~80% Vo2max) elicited an increase in insulin sensitivity (21%) compared with moderate-intensity (~65% Vo2max) training (16%), independent of changes in body composition or Vo2max with the same training volume. Second, in a 12-wk program in overweight elderly women, despite similar changes in fitness and no changes in body fat or BMI, high-intensity training (75% of Vo2max) led to a 28% increase in glucose disposal via EHC when compared to no change with moderate intensity at 50% Vo2max, mediated by increase in nonoxidative glucose disposal (128). On the other hand, in the aforementioned STRRIDE study, Houmard et al. (290) suggested that duration should be a consideration, as low volume/moderate intensity and high volume/high intensity exhibited a greater response than low-volume high-intensity AT in sedentary, overweight subjects. In addition, Braun et al. (81), using an insulin suppression test, noted similar changes in insulin sensitivity in women with T2D using different intensities of 50% and 75% of Vo2max.
Also of note are the numerous studies that have found increases in insulin sensitivity independent of changes in body weight and/or body composition. For example, Duncan et al. (170) noted increases in insulin sensitivity of 40% or more in middle-aged, previously sedentary adults after a moderate-intensity walking intervention, independent of any changes in BMI or waist circumference. These findings have also been noted in younger subjects with a training program for 1 year independent of any BMI changes (481), in older subjects without changes in LBM or fat mass (299) and subjects with T2D exhibited a 44% improvement during an ITT without a change in body weight. Furthermore, a decrease AUC for insulin during an OGTT was shown in children, with no changes in body weight, LBM, or visceral fat (461).
Because of the timing of the posttraining test of insulin sensitivity, it has been difficult to dissect the acute versus chronic effects, and thus one aspect to consider is the timing of the measure of insulin sensitivity after training to establish the acute versus chronic effects of exercise on insulin sensitivity. This conundrum is complex given that there are likely effects of a single bout (388) and accumulated bouts that are difficult to separate, as training results in a diminished insulin response during an OGTT more so than does a single exercise bout (261). This also has implications for what constitutes “inactivity” as discussed below. Hence, there is a fine line between the “training” effect per se and the effects that can be attributed to the last exercise bout. For example, Mikines et al. (435) noted that trained subjects exhibited higher insulin sensitivity compared to untrained subjects, and although less pronounced, one bout in untrained subjects did lead to increased insulin sensitivity of glucose uptake, an effect that lasted 48 h, but not 5 days (434). While waiting 4 to 5 days will allow for studying the training effects per se, it might actually be at a time when the training effects have waned and therefore underestimate the effect. To complicate matters, it is also evident that the time frame for this effect has been highly variable across studies, with some showing affects waning after as little as 38 to 60 h postfinal bout (92,431,480) and others, such as LeBlanc et al. (388), demonstrating that when trained subjects did not exercise for 3 days, intravenous glucose induced greater elevations in insulin, however, still remaining smaller than chronically sedentary subjects. Interestingly, 1 week of vigorous aerobic-exercise training produced increases in both insulin sensitivity and responsiveness of glucose disposal, as well as enhanced suppression of hepatic glucose production in overweight patients with T2D, without weight loss (posttesting was performed 18–20 h after last bout) (356). This finding was also noted by Mikus et al. (438), who used CGMS to document improved glycemic control with 1 week of training, independent of changes in fitness or adiposity.
Mechanisms
As previously discussed, many studies have documented that AT improved whole body insulin sensitivity. Skeletal muscle, the most important target tissue for insulin action, has received the most attention for mechanistic studies with emphasis on insulin signaling pathway studied from biopsy samples. While the effects of AT-induced increases in glucose disposal have been attributed primarily to increases in insulin signaling protein and/or activities, the data are not consistent. Studies comparing endurance-trained subjects versus untrained subjects have indicated differences in glycogen storage in response to insulin (172), insulin signaling proteins, such as increased GS, GLUT4, GLUT4 vesicle-associated protein (insulin-regulated aminopeptidase), and PI3-K activation, but decreased IRS-1, IRS-2, insulin receptor, and no difference in AKT (172, 286, 355, 716).
Using 31P NMR spectroscopy, Perseghin et al. (498) noted that despite lower glucose transport phosphorylation in offspring of T2D patients compared with normal subjects, both exhibited increases in whole body insulin sensitivity after AT by 40% or more and whole body nonoxidative glucose metabolism by 60% to 70%, mediated by an increase in glucose transport-phosphorylation (leading to greater glycogen synthesis). The training-induced improvement in glucose disposal has been attributed, at least in part, to increased insulin-stimulated glucose storage and GS fractional velocity, which correlated with insulin-stimulated glucose storage (119). Dela et al. (152) also noted increases in nonoxidative glucose disposal associated with increased GS mRNA and Frosig et al. (211) noted increased GS activity with short-term training. Ferrara et al. (195) noted similar effects on GS fractional activity in response to insulin, with no change in total activity. Dela et al. (150) noted maximum insulin binding and that basal- and insulin-stimulated receptor kinase activity did not change; however, GLUT4 protein concentration increased and the training-induced increase in GLUT4 (26%) matched the increase in maximum insulin-stimulated leg glucose uptake (25%) in the same subjects (153), and individual values of the two variables were correlated (r=0.84). In subjects with IGT, independent of changes in body composition, glucose disposal at high insulin concentration during an EHC increased approximately 60% and GLUT4 and glycogen increased after training (299). Several additional studies have noted increases in GLUT4 content. For example, 60% in middle-aged subjects (291), approximately 80% in nondiabetic men (289), 38% and 22% in overweight nondiabetic and diabetic men (119), 40% in healthy men and 23% in men with T2D (154) (mRNA also increased). Short et al. (592) noted increases in GLUT4 mRNA and protein content, independent of age in subjects ranging from age 21 to 87. Furthermore, short-term training increased GLUT4 protein similarly in both younger and older subjects (133).
Frosig et al. (211) noted AT-induced increases in insulin-stimulated glucose uptake of 60% and increases in muscle Akt1/2, AS160, GLUT4, HK2 and insulin-responsive aminopeptidase protein content and activities Akt1, GS and AS160 phosphorylation. However, IRS-1-associated PI3K activity was reduced. Short-term training studies reported no change in mRNA expression of IR, IRS-1, IRS-2 and the p85α-subunit of PI3K (668), but there were noted increases after short-term training in insulin receptor autophosphorylation (714) and PI3K activity (288, 355) in concert with increased insulin sensitivity, and PI3K activity in chronic-trained young men (288, 355). On the other hand, Christ-Roberts et al. (119) noted along with increased insulin sensitivity increased GS fractional activity, AKT protein expression but not IRS-1-associated PI3K activity after AT in both insulin-resistant, nondiabetic and type 2 diabetic subjects. Furthermore, Tanner et al. (633) also noted that short-term training did not increase PI3K activity with increased insulin sensitivity. Other pathways have also been investigated, such as those related to mitochondrial biogenesis (PPAR, PCG1α, etc), inflammation and stress kinases (JNK, IKKβ, etc), and AMPK, but it is unclear if modulation of these pathways directly caused the increase in insulin sensitivity with AT. For example, Frøsig et al. (210) also noted increases in AMPK α1, β2, and γ1. Therefore, it is apparent that there are not generalized improvements in insulin signaling after AT.
One issue to acknowledge in these studies is that the timing of the biopsy after the last bout of exercise may contribute to differences noted (675), given that biopsies taken within the first day may in part reflect acute effects of the last bout and the time course and effects of training may differ for different types of proteins.
Other factors such as fiber type changes (289), increased oxidative capacity/mitochondria or blood flow (152, 172), altered muscle lipid metabolism (86, 88), and endocrine/paracrine mediators and reduced visceral obesity (592) may also contribute. For example, Ebeling et al. (172) noted that skeletal muscle GS fractional activity was highly correlated (r = 0.88) with blood flow, suggesting they may be causally related.
Interval Training
An additional modality of training that has received renewed interest is high-intensity sprint interval training (SIT), also referred to as high-intensity interval training (HIT or HIIT). Although it has long been known to induce muscular adaptations, for example, increase in peak power output, VO2max, glycolytic (hexokinase, phosphofructokinase) and aerobic [citrate synthase, succinate dehydrogenase) enzyme activities (410) and as reviewed by (552)], since 2005 SIT has been the subject of greater investigation (90). However, in 1994, Tremblay et al. (644) noted that 20 weeks of SIT led to greater reductions in skinfold thickness as an indicator of body composition, compared with AT. Interestingly, these improvements were despite the SIT having <50% of the total energy cost compared to AT, demonstrating that changes in fat mass are not a function of exercise energy expenditure. Of note, recent studies have demonstrated increased insulin sensitivity with SIT. For example, Babraj et al. (36) noted that six sessions of four to six 30-s Wingate cycling sprints in healthy, young recreationally active men led to 12% and 37% decreases in AUC of glucose and insulin, respectively during an OGTT. Whyte et al. (689) performed a similar study design, and noted decreased AUC for insulin, but not glucose, 24 h, but not 72 h after the last training session. Again, using a similar 2-wk paradigm, SIT increased glucose infusion rate by 21% in young men and women 72 h after the last training session during EHC (539). More recently, eight patients with T2D performed six sessions of HIT (10 × 60-s cycling bouts at ~90% HRmax) over 2 wk, and average 24-h blood glucose concentration was reduced after training, as was the three meal sum 3-h postprandial glucose AUC (400) (as determined by CGMS). Additionally, Metcalfe et al. (430) used a 3×/wk for 6-week protocol that consisted of 10-min-exercise sessions which included progressing to 2 to 20 s all out sprints and noted a 28% increase in insulin sensitivity in males, but not females, a finding that requires further study.
As for potential mechanisms, SIT was found to increase IR phosphorylation more than moderate AT (640). Little et al. (401) used a more practical six sessions of 8 to 12 60-s cycle sprints at 100% of peak power output achieved during a VO2max test and noted 119% increases in GLUT-4. Also from the Gibala laboratory, Hood et al. (277), using a similar protocol in middle-aged subjects, noted a 2.5-fold increase in GLUT-4 content and Little et al. (400) noted a similar increase in GLUT-4 in older subjects with T2D. It has also been suggested that glycogen depletion during SIT may induce the improvements in insulin sensitivity (36, 689), since its depletion correlated with several aspects of enhanced insulin signaling.
Resistance Training
Yki-Jarvinen and Koivisto (713) demonstrated cross-sectionally that weight lifters exhibited similar glucose disposal to long-distance runners. Szczypaczewska et al. (624) also noted that bodybuilders exhibit greater glucose tolerance and insulin action compared to controls. These findings led to research demonstrating that RT also enhances insulin sensitivity and improves glucose tolerance in a wide range of study groups, including younger (506, 586) and older individuals (721), postmenopausal women (558), those with hypertension (536) and T2D (308).
Insulin sensitivity has been demonstrated to improve with RT in several studies using the EHC (Table 2). For example, in healthy, middle-aged men, glucose infusion increased 24% during EHC after RT (440). In subjects with T2D (308) improvements in the glucose infusion rates have been noted, generally attributable to increases in nonoxidative glucose metabolism. Reynolds et al. (536) noted improved glucose disposal during EHC in older hypertensive subjects. Ryan et al. (558) noted an increase in a small cohort of post-menopausal women. Interestingly, the training program intensities and durations in these studies were highly variable, suggesting that multiple RT paradigms are capable of improving insulin sensitivity. In addition, Reynolds et al. (537) used the EHC and noted no significant changes in fractional glucose extraction or glucose clearance with RT. On the other hand, two studies (143, 557) found non-significant increases in insulin sensitivity. In the first (557), the increase was ~10% and did not achieve significance (P < 0.06). In the latter (143), the weekly training was 60 minutes in duration, suggesting that a potential threshold of overload may exist.
Table 2.
Studies Incorporating Use of FSIGT or EHC Methodologies to Determine Effect of Resistance Training on Insulin Sensitivity
| Study | Subject population | Design | Testing | Major findings | Hour after last bout |
|---|---|---|---|---|---|
| Zachwieja et al. (721) | 64–75 years M | 4×/wk, 16 weeks 4×4–10, 75–90% 1 RM |
FSIGT | 33% ⇑ in insulin sensitivity | 168 |
| Ibanez et al. (302) | 66±3 years M, T2D | 2×/wk, 16 weeks 50–70% 1 RM, then 70–80% 1 RM, 10–15 repetitions |
FSIGT | 25% ⇑ in insulin sensitivity, ⇓ fasting glucose | 24 |
| Shaibi et al. (586) | 15±0.5 years M Hispanic | 2×/wk, 16weeks Progressive increase in workload |
FSIGT | 45% ⇑ in insulin sensitivity | 48–72 |
| Van Der Heijden et al. (659) | 15±0.5 years M/F Hispanic | 2×/wk, 12weeks Progressive increase in workload |
FSIGT (plus tracer) | ⇑ peripheral in 8 of 12 subjects, but ⇓ in 4 of 12; hepatic insulin sensitivity ⇑ 24% | 72 |
| Miller et al. (440) | 58±1 years M | 1 set 90% 1 RM for three repetitions ⇓ load up to 15 repetitions 3×/wk, 16 weeks |
EHC OGTT |
24% ⇑ glucose infusion w/EHC ⇓ insulin during OGTT, fasting insulin ⇔ glucose during OGTT |
22–24 |
| Ryan et al. (558) | 58±2 years F | 3×/wk, ~16 weeks 1–2 sets 15 repetitions |
EHC | 16% ⇓ in insulin response | 24 |
| Ishii et al. (308) | 47±9 (SD) years, T2D | 5×/wk, 4–6 weeks 2×10–20 repetitions 40–50% of 1 RM |
EHC | 48% ⇑ in glucose disposal | 48 |
| Poehlman et al. (506) | 28±3 years F | 3×/wk, 6 months 80% 1 RM |
EHC | 9% ⇑ in glucose infusion | 96±24 |
| Holten et al. (276) | 61±2 years, healthy and T2D | 3×/wk, 6 weeks, 1 legged training, 8–12 repetitions | EHC | ~10% ⇑ in leg glucose clearance | 16 |
| Ryan et al. (557) | 69±1 years M/F | 3×/wk, 6 months 1–2 sets, 8–15 repetitions |
EHC | Nonsignificant ⇑ in SI | 24–36 |
| Andersen et al. (20) | 26±1 years M | RT cessation for 90 d, RT was 3×/wk, 3 months Periodized workload |
EHC | 11% ⇓ in insulin sensitivity | – |
| Reynolds et al. (536) | 67±2 years M/F | 3×/wk, 16 weeks 2×10–12 repetitions |
EHC | 15% ⇑ in glucose disposal ⇔ insulin response, fasting insulin |
24 |
| Klimcakova et al. (359) | 50±2 M | 3×/wk, 3 months 1×12–15 repetitions |
EHC | 24% ⇑ glucose disposal | 48–72 |
| Davidson et al. (143) | 60±4 | 3×/wk, 1 set up to 15 reps (20 min/session) | EHC | nonsignificant ⇑ in SI | 36–48 |
Several of these studies demonstrated an effect of RT without altering aerobic capacity or body weight/composition. In older men, Zachwieja et al. (721) noted a 33% increase in insulin sensitivity and an increase in glucose rate of disappearance, using a stable glucose isotope FSIGT, 7 days after the last bout of training, without a change in body weight, secondary to reciprocal increases and decreases in LBM and fat mass. In obese, middle-aged men, Klimcakova et al. (359) noted a 24% increase in glucose disposal rate, without any change in body weight, fat mass or VO2max. Poehlman et al. (506) also noted a modest increase, which occurred independent of changes in total body, subcutaneous or visceral fat. In the study by Ryan et al. (558), body weight, fat mass, percent body fat, LBM, and VO2max did not change with RT intervention. The addition of a calorically restricted diet augmented the response and induced weight loss, suggesting the possibility of additive effects with multiple lifestyle interventions. Increases in insulin sensitivity during an FSIGT have also been noted in children, despite no change in weight and an increase in LBM compared to a control group (586). Van Der Heijden et al. (659) noted highly variable responses in youth in peripheral insulin sensitivity, but a 24% increase in liver insulin sensitivity. Additionally, improvement in adipose tissue insulin sensitivity has been noted (507).
Similar findings have been noted in subjects with T2D. Ishii et al. (308) noted that RT increased glucose disposal by 48% in lean subjects with T2D without a change in VO2max, weight loss or body composition. In older men with T2D, a 2 d/wk RT program led to a 45% increase in insulin sensitivity and a 10% decrease in abdominal fat, without a change in body weight (302) and an estimated increase in energy intake of 15%. In addition, Misra et al. (446) noted improved insulin sensitivity by an ITT in South Indians with T2D.
Many RT studies have also used an OGTT as an index of insulin action, and some (599, 600), but not all (134, 301, 440,441), have noted improvements in glucose tolerance after RT. Plasma insulin concentrations have been demonstrated to decrease during an OGTT (134, 440, 441, 599, 600). Using the OGTT method, RT improves insulin action including subjects with IGT or T2D (137, 171, 276, 600). Alternatively, glucose and insulin AUCs were not altered by 6 weeks of RT in women with T2D (192). Another aspect to consider is while the EHC estimates glucose disposal and the FSIGT insulin secretion and β-cell function, the OGTT has classically been used to estimate glucose tolerance via AUCs or the Matsuda index (421) of insulin sensitivity. However, given the advantages of the technique, Abdul-Ghani et al. (3) validated a muscle insulin sensitivity index (ISI) and hepatic insulin resistance index (IRI) with EHC data and oral DI (4, 534), an estimate of β-cell function, from FSIGT testing. Roberts et al. (unpublished observations) recently demonstrated that 12 weeks of RT improved muscle ISI and oral DI, without a change in hepatic IRI, suggesting that RT affects insulin sensitivity in skeletal muscle, but not liver.
Overall, a systemic review of 20 studies found that supervised resistance exercise training improved glycemic control and insulin sensitivity in a wide variety of study groups (231). However, without supervision, RT compliance and glycemic control are generally less, suggesting either the need for supervision or alternative incentives to maximize training-induced benefits.
Aerobic Versus Resistance Training
A handful of studies have directly compared the effects of AT and RT on insulin action. Smutok et al. (599) noted that both modalities led to decreased AUC for both glucose and insulin and that no significant difference between the interventions were noted, nor were there any changes in body weight, although body fat percentage dropped slightly in the AT group. This group also studied the effects of AT or RT in subjects with T2D or IGT, noting similar results (600). Cauza et al. (107) reported greater benefits of RT as opposed to endurance training, on glycemic control (mean blood glucose decreased 15%) in patients with T2D using CGMS. Also RT and AT were compared in older obese men 3 days/wk for 6 months and noted similar 20–25% increases with EHC in both groups, despite a decrease in body weight of 2% in the aerobic group and an increase in body weight of 2% in the RT group (195). In subjects with T2D, both AT and RT for 4 months resulted in increases in glucose disposal rates during EHC (37). Cuff et al. (137) did not compare the effects of AT versus RT; however, they did investigate the effects of added RT to an AT intervention in postmenopausal women with T2D and noted greater glucose infusion rate during the combined intervention, in the absence of an effect of AT alone. In addition, the effects of combined training versus each modality in isolation noted that combined was superior to RT, but not to AT (143). It should be noted that the total exercise time was 60 min/wk in the RT group and 150 min/wk in the aerobic and combined groups, and suggests when making comparisons of different training modalities, attempting to match the training overload is critical. However, given differences in energy expenditure that exist with different RT prescriptions, there is inherent difficulty in trying to match the overload of the different training modalities. Because of this conundrum, from a feasibility standpoint, one option is to match the duration of the training. Thus, we propose it is important to report the exercise time and to attempt to match the relative intensity when comparing these different training modalities.
Mechanisms
Although the mechanism for the increase in insulin sensitivity with aerobic exercise training has been often investigated, the increase with resistance exercise training is far less studied. Some insight has been provided by cross-sectional and comparison studies. Takala et al. (626) noted no effect of RT on insulin-stimulated glucose uptake per kilogram of muscle mass, and the positive effect of RT was attributed to the larger muscle mass. Yki-Jarvinen el al. (713) noted that both trained weight lifters and long-distance runners exhibited higher glucose disposal compared to controls during EHC; however, when calculating per LBM, only the runners exhibited higher values, suggesting that, at least in part, the mechanism for increased glucose disposal in resistance-trained subjects is due to increased skeletal muscle mass. This is in agreement with AT versus RT (506), which noted increases in glucose disposal rate during an EHC with both modalities of training and when these rates were express per fat-free mass (FFM), the improved insulin sensitivity persisted in the AT group, but not in the RT group. In other aforementioned studies, the EHC clamp increases were expressed relative to FFM (276, 308, 440), suggesting effects independent of LBM changes. Miller et al. (441) noted a significant correlation between insulin AUC and increase in LBM (r = 0.89).
These data suggest that the mechanism for the improvement with RT and AT, at least in part, may be distinct. Dela and Kjaer (151) suggested that RT improves insulin action by unknown mechanisms in addition to increased muscle mass. Despite numerous studies investigating the effects of RT on insulin sensitivity, few studies have attempted to investigate the mechanism(s) responsible for improved insulin sensitivity. Miller et al. (440) noted a 40% increase in nonoxidative glucose disposal during insulin infusion, suggesting increased glycogen synthesis. Holten et al. (276) noted, using the single-leg RT model, increased glucose clearance more than what could be explained by increases in LBM alone. In addition, these authors reported increased protein content of GLUT4 (T2D subjects only), insulin receptor, protein kinase B-α/β, GS, as well as GS total activity; however, no training effect was observed for protein content of IRS-1 or the p85 subunit of PI3K. On the other hand, RT did not alter muscle GS total activity, glycogen content, or levels of PI3K in overweight/obese older men (195). Roberts et al. (unpublished observations) recently demonstrated that 12 weeks of RT increased GLUT4 and HKII in obese young men. Alternatively, GLUT4 was not altered in older Hispanic adults with T2D, although in the latter, sodium-dependent glucose cotransporter system (hSGLT3) transcript levels in the vastus lateralis muscle was positively correlated with glucose uptake (105). In subjects undergoing bed rest, GLUT-4 decreased in vastus lateralis after bed rest but increased in subjects undergoing RT during bed rest (625). Given the dearth of evidence in humans on the molecular mechanism, it is interesting to note that Krisan et al. (368) noted, using a rodent model of RT, increased GLUT4 content and IRS-1 associated PI-3K, Akt, and atypical PKC-ζ/λ activities. Some further insight has been provided by Andersen et al. (20), who noted that the decrease in leg glucose uptake, expressed relative to leg muscle mass, was associated with decreased glycogen content and increase myosin heavy chain IIx; however, GLUT4 mRNA, enzymatic changes, or capillary density did not change with detraining. Additionally, regarding the possibility that AMPK might be involved, RT resulted in similar changes to various AMPK subunit isoforms in subjects with T2D and healthy controls (698).
To try to gain some insight into the potential contribution of adipose tissue to altered insulin sensitivity in muscle, Klimcakova et al. (359) found no effects of RT on the mRNA levels of adiponectin, leptin, Il-1β, IL-6, and TNFα from subcutaneous adipose tissue. Overall, it is evident that there is a lack of clarity with respect to what are the molecular mechanisms responsible for the improvement in insulin sensitivity with RT, and this remains an important area for future research (Fig. 13).
Figure 13.

Schematic diagram of future directions to determine mechanisms by which aerobic training (AT) and resistance training (RT) increase insulin sensitivity. Although there is preliminary evidence, more research is needed to clearly identify the mechanisms that are involved, as denoted by the question marks linking AT and RT to enhance insulin signaling.
Physical Activity
Although exercise training is typically imposed to assess the impact of physical activity on insulin sensitivity, physical activity levels per se are also related to insulin action. For example, Kriska et al. (369) noted that higher physical activity was associated with lower insulin concentration in more than 5000 Pimas and Mauritians of difference body compositions, suggesting influences of activity independent of body composition. In the IRAS study, insulin sensitivity determined by FSIGT was approximately 80% higher for those who participated in vigorous activity five times per week or more compared with those who rarely or never participated (426). More recently, objectively measured sedentary time, light-intensity physical activity, and moderate- to vigorous-intensity activity were all related to 2-h glucose after an OGTT (260). In addition, in a cohort of 801 men and women, the European Relationship between insulin sensitivity and cardiovascular risk study (42) demonstrated that total physical activity, activity intensity and sedentary time as recorded objectively by accelerometry were all associated with insulin sensitivity as determined by EHC. In addition, the dual-peak effects of accelerometry-measured physical activity and percent sedentary time suggest the potential for independent effects of activity and inactivity (Fig. 14). Interestingly, the most sedentary group exhibited a significant relation between insulin sensitivity and total activity; however, the effects in other quartiles of sedentariness, these trends were not as strong, nor were there trends between insulin sensitivity and sedentary time after accounting for physical activity. These data suggest that the interactions between sedentary behavior, physical activity, and structured exercise are very complex and relationships between sedentary behavior and physical activity are not simply inversely associated. Furthermore, the interactions between exercise training and habitual activity (i.e., from ambulatory patterns, occupation, etc.) are generally unknown.
Figure 14.

Insulin sensitivity and physical activity measured by accelerometer by quartiles of average number of counts/min and quartiles of percent time sedentary in the insulin sensitivity and cardiovascular risk study. M/I is the unit measurement of insulin sensitivity (μmol.min−1kgFFM−1nmol/L−1) (42). Copyright 2008 American Diabetes Association. From Diabetes Care®, Vol. 57, 2008; 2613–2618. Reprinted by permission of the American Diabetes Association.
Decreased Physical Activity
Physical inactivity via either modification of habitual physical activity or from a trained state to training cessation also has effects on insulin action. For the former, several studies have suggested that becoming physically inactive may decrease insulin sensitivity. The original paradigm for this was bed rest, which demonstrated significant reductions in insulin action within 7 days (617) (432, 433). More recently, however, studies have indicated that simply decreasing ambulatory physical activity leads to blunted insulin action. For example, physical inactivity in a free-living environment decreases glucose tolerance during an OGTT after 1 week of daily step reduction from 6203 to 1394 and decreases glucose infusion rate during an EHC after 2 weeks of reducing daily step number from 10,501 to 1344 in healthy, young men (476). Mikus et al. (439) noted that 3 days of reduced activity (step reduction of 12,956 to 4319 steps/d) in healthy, young adult volunteers led to increased postprandial glucose responses as measured by CGMS, despite compensatory increases in insulin response to an OGTT. Peripheral uptake of 2-deoxyglucose was also reduced without change in hepatic insulin resistance after 2 weeks of reduced stepping (370). More recently, Knudsen et al. (361) noted that an average daily step reduction from 10,278 to 1,521 led to significant reductions in OGTT estimated Matsuda index, which preceded significant changes in fat mass.
In particular, previous studies have noted that training induced increases in insulin sensitivity could overcome effects of aging, leading to the concept that exercise and activity per se, as opposed to aging per se are largely the true underlying cause of insulin resistance commonly observed in older individuals (see inactivity section) (126, 577) (Fig. 15). In addition, as mentioned above, there is a fine line between the “training” effects of exercise training-induced increases in insulin sensitivity and when “detraining” or “inactivity” ensues. Nevertheless, it is evident that training effects are rapidly lost in response to training cessation. For example, one of the first studies in this area noted that AT effects were lost within 10 days of detraining in well-trained subjects, independent of any changes in CRF, body weight or body fat. Plasma glucose and insulin responses increased to levels similar to that commonly found in untrained subjects after 10 days of training cessation, and one bout of exercise significantly improved the response (Fig. 16) (261).
Figure 15.

Effects of training and age on area under the curve for (A) glucose and (B) insulin during an oral glucose tolerance test. Adapted, with permission, from reference (577).
Figure 16.

Effects of chronic training (dashed line), inactivity (solid line) for 10 days and one single bout (dotted line) of aerobic exercise on (A) blood glucose and (B) insulin during an oral glucose tolerance test in well-trained subjects. Adapted, with permission, from reference (261).
Studies using the EHC procedure (at submaximal insulin concentrations) demonstrated that elevated insulin-stimulated glucose disposal rates, and therefore the improved insulin sensitivity, in trained individuals disappear after as little as 2 (480), 3 (and sustained at 7 days) (92), or 10 days (352) of training cessation. Interestingly, 5 days of detraining decreased insulin action to levels comparable to untrained subjects (434, 436, 437), and this was reported to be associated with decreased GS activity (436). In addition, Dela et al. (153), using the well-known single-leg training model, noted that the cycling-induced increases in insulin sensitivity returned to near pretraining levels after 6 days of detraining. In a follow-up study by this group, the effect was lost after 6 days in T2D patients, but lasted longer in nondiabetic controls (152). Vukovich et al. (667) also noted decreased glucose disposal after 6 days of detraining, as well as a decrease in GLUT4 content. However, Houmard et al. (287) noted that both AT and RT training cessation led to decreased insulin sensitivity to oral glucose, but GLUT4 remained unaltered. Andersen et al. (20) also noted cessation of RT training for 3 months after 3 months of training led to a drop in whole body glucose uptake of 11% and leg glucose uptake 33%, which occurred in concert with increased IIx fiber percentage, but no difference in GLUT4 or GS mRNA. The authors attributed this decrease to reduced nonoxidative glucose disposal.
On the other hand, Rodgers et al. (545) noted after 10 days of inactivity, glucose tolerance was still not at untrained levels, so there appears to remain a training component with longer term training (average 13 years). Evidently, the effects are highly variable depending on the degree of training of subjects, aspects of the training program performed, and maybe due to genetic contribution. Similarly, one way to avoid the effects of detraining is to continue to exercise, albeit at a reduced training load. For example, Houmard et al. (291) noted maintained insulin sensitivity and elevated GLUT-4 when subjects decreased training frequency by 50% for 2 weeks; however, training cessation led to a return of values to sedentary levels after 2 weeks.
Summary
MS, although only more recently defined and investigated, exhibits a prevalence of nearly 25% of the US adult population, and epitomizes the integrative nature of modern chronic disease, given its endocrine, metabolic, and cardiovascular underpinnings. Most notable is the relation between cardiovascular fitness and MS, given data that the mortality risk in unfit versus fit individuals with the MS is similar to the same comparison in healthy men. In addition, many studies not discussed, although not designed to investigate the effect of exercise on MS per se, have shown amelioration of risk factors comprising the MS, including insulin resistance, hypertension, dyslipidemia, inflammation, and endothelial dysfunction. Various modalities of training, including AT, RT, and SIT, as well as activity/inactivity can alter insulin action in a short period of time.
Perspectives
Drug mimetics for exercise
Drug therapies and exercise training/physical activity have been utilized to ameliorate MS and its associated risk factors. In conjunction with this, there has been controversy surrounding the ability of exercise mimetics to target different phenotypes with hopes to reduce, among other diseases, MS-related disease outcomes (75, 99, 122, 228, 259, 540, 679). Although attempts have been made to engineer drugs to improve insulin sensitivity and ameliorate MS phenotypes, due to the myriad of beneficial effects that exercise and physical activity provide, virtually the entire population will benefit from increased physical activity levels. Although for select individuals physically unable to perform physical activity, drug mimetics for exercise might have credence for selected phenotypes; no drug therapies will be able to mimic the array of adaptations that occur with regular physical activity in the context of MS and its related causes and complications. Even if a polypill for many of these adaptations was possible, it is likely that for many as discussed below, the pill would not even be taken.
Comparison with drug therapy
Studies have demonstrated that there are numerous barriers that invariably lead to discontinuance of exercise training programs and regular physical activity leading to MS and ultimately T2D and CVD. It is clear that translation of the efficacy studies clearly demonstrating benefits of a wide variety of exercise and activity modalities is vital. Case in point are studies suggesting that supervised training programs [commonly implemented in randomized controlled trials (RCTs)] yield superior results to unsupervised programs. Alternatively, it is assumed that for many, since exercise (and other lifestyle therapies, such as diet) is difficult to sustain, it will be easier for individuals to sustain health improvements through drug therapy. However, cohort studies of patients prescribed medications suggest variable but disappointingly high rates of discontinuation of therapy and poor adherence to drug regimens. As an example, lipid-lowering drugs are some of the most commonly prescribed drugs in many countries. Estimates have suggested that approximately 60% of patients discontinue their medication over 12 months, with half of the discontinuations occurring within 3 months. Main reasons for discontinuation included 32% unconvinced about need for treatment and 32% indicating poor efficacy (595). In accordance with this, approximately 60% of older patients discontinued statins for primary prevention within 1 year, and 75% after 2 years (312). Avorn and colleagues (34) noted that patients 65 years of age or more failed to fill prescriptions for lipid-lowering drugs for about 40% of the study year and that approximately 45% were adherent to statin medications after 6 months (60). Alternatively, age and standard of care may impact this effect as much greater discontinuation in younger Danish patients occurred within the first year (~60%) compared with older patients (~40%) (384). Whether other drug therapies for chronic disease risk factors exhibit similar patterns of discontinuation is largely unknown. This demonstrates that similar to RCTs, exercise in which shortcomings include ability to translate the intervention to the public at large, although underappreciated, clinical trials using lipid-lowering drugs likely succumb to the same shortcomings in the ability to translate the therapy to the population at large, albeit likely for different reasons. Even a therapy as simple as taking a pill, as opposed to changing physical activity, is also subject to high degrees of noncompliance. Ways to manage this conundrum should be investigated in the future.
Weight loss
As discussed above many of the changes in phenotypic outcomes, for example, insulin sensitivity and MS factors (HDL, TG, blood glucose, and blood pressure) can occur with aerobic, interval or resistance exercise training, independent of weight loss, and with or without accompanying changes in body composition and fat distribution. Nevertheless, at present there is a continued focus on weight loss and a misconception that weight loss is required for benefits. However, it is likely that the exercise-induced effects related to MS have mechanisms that, at least in part, are distinct from body weight loss per se. We need to understand the mechanisms for improvement of MS factors, as likely in some, fat loss may occur and contribute to the benefits noted; however, in others weight loss will not occur. Future studies should be carried out to investigate this question further.
Individualized programs
One aspect alluded to but not discussed in detail are individualized effects of training on insulin sensitivity and MS factors. It is apparent that in those with insulin resistance or MS, not all will respond similarly. Several studies, including the HERITAGE Family Study (77) and STRRIDE [Figure G2.4 in reference (2)] studies have noted this highly individualized effect. Case in point in the HERITAGE study, Boulé et al. (78) noted that although a majority of subjects exhibited an improvement in insulin sensitivity with aerobic training, the individual response was highly variable, with some subjects being unresponsive; additionally, they noted the change in insulin secretion was dependent on the baseline glucose tolerance. Therefore, as we learn more about the interaction between genetic variation and exercise, we can identify those that may respond to exercise interventions and better individualize training programs to optimize risk factor modification.
Future directions
Overall, future trials are needed to (i) establish the effects of long-term training programs on prevention and reversal of MS; (ii) compare long-term effects of exercise versus drug therapy sustainability, as well as the potential synergistic or antagonizing effects of common drug therapies (statins, metformin, etc.) on training-induced effects; (iii) explore further the mechanisms by which different training modalities increase insulin sensitivity (Fig. 13) and prevent MS; (iv) further investigate the potential of exercise to promote metabolic health independent of significant weight loss; and (v) provide insight into the factors and clues that will assist with optimization of individualized training programs.
Acknowledgments
The authors wish to thank Jodi J. Tam for help with figures and editorial comments.
Note: Due to space limitations, the authors have not discussed all articles for each aspect/section.
Footnotes
References
- 1.Executive summary of The Third Report of The National Cholesterol Education Program (NCEP). . Expert panel on detection, evaluation and treatment of high blood cholesterol in adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 2.Physical Activity Guideline Advisory Committee Report. Washington, DC: U.S. Department of Health and Human Services; 2008. [Google Scholar]
- 3.Abdul-Ghani MA, Matsuda M, Balas B, DeFronzo RA. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care. 2007;30:89–94. doi: 10.2337/dc06-1519. [DOI] [PubMed] [Google Scholar]
- 4.Abdul-Ghani MA, Williams K, DeFronzo RA, Stern M. What is the best predictor of future type 2 diabetes? Diabetes Care. 2007;30:1544–1548. doi: 10.2337/dc06-1331. [DOI] [PubMed] [Google Scholar]
- 5.Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- 6.Abumrad NA, el-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. Homology with human CD36. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- 7.Abumrad NA, Perkins RC, Park JH, Park CR. Mechanism of long chain fatty acid permeation in the isolated adipocyte. J Biol Chem. 1981;256:9183–9191. [PubMed] [Google Scholar]
- 8.Adams JM, II, Pratipanawatr T, Berria R, Wang E, DeFronzo RA, Sullards MC, Mandarino LJ. Ceramide content is increased in skeletal muscle from obese insulin-resistant humans. Diabetes. 2004;53:25–31. doi: 10.2337/diabetes.53.1.25. [DOI] [PubMed] [Google Scholar]
- 9.Ader M, Bergman RN. Insulin sensitivity in the intact organism. Baillieres Clin Endocrinol Metab. 1987;1:879–910. doi: 10.1016/s0950-351x(87)80010-1. [DOI] [PubMed] [Google Scholar]
- 10.Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- 11.Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem. 2002;277:1531–1537. doi: 10.1074/jbc.M101521200. [DOI] [PubMed] [Google Scholar]
- 12.Ahima RS, Kelly J, Elmquist JK, Flier JS. Distinct physiologic and neuronal responses to decreased leptin and mild hyperleptinemia. Endocrinology. 1999;140:4923–4931. doi: 10.1210/endo.140.11.7105. [DOI] [PubMed] [Google Scholar]
- 13.Ahmad F, Azevedo JL, Cortright R, Dohm GL, Goldstein BJ. Alterations in skeletal muscle protein-tyrosine phosphatase activity and expression in insulin-resistant human obesity and diabetes. J Clin Invest. 1997;100:449–458. doi: 10.1172/JCI119552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahmad F, Considine RV, Bauer TL, Ohannesian JP, Marco CC, Goldstein BJ. Improved sensitivity to insulin in obese subjects following weight loss is accompanied by reduced protein-tyrosine phosphatases in adipose tissue. Metabolism. 1997;46:1140–1145. doi: 10.1016/s0026-0495(97)90206-7. [DOI] [PubMed] [Google Scholar]
- 15.Ahmadi N, Eshaghian S, Huizenga R, Sosnin K, Ebrahimi R, Siegel R. Effects of intense exercise and moderate caloric restriction on cardiovascular risk factors and inflammation. Am J Med. 2011;124:978–982. doi: 10.1016/j.amjmed.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 16.Al-Khalili L, Bouzakri K, Glund S, Lonnqvist F, Koistinen HA, Krook A. Signaling specificity of interleukin-6 action on glucose and lipid metabolism in skeletal muscle. Mol Endocrinol. 2006;20:3364–3375. doi: 10.1210/me.2005-0490. [DOI] [PubMed] [Google Scholar]
- 17.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC., Jr Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and international association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 18.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 19.Amati F, Dubé JJ, Alvarez-Carnero E, Edreira MM, Chomentowski P, Coen PM, Switzer GE, Bickel PE, Stefanovic-Racic M, Toledo FGS, Goodpaster BH. Skeletal muscle triglycerides, diacylglycerols, and ceramides in insulin resistance. Diabetes. 2011;60:2588–2597. doi: 10.2337/db10-1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andersen JL, Schjerling P, Andersen LL, Dela F. Resistance training and insulin action in humans: Effects of detraining. J Physiol (Lond) 2003;551:1049–1058. doi: 10.1113/jphysiol.2003.043554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol Cell Biol. 2001;21:7117–7136. doi: 10.1128/MCB.21.21.7117-7136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderssen SA, Carroll S, Urdal P, Holme I. Combined diet and exercise intervention reverses the metabolic syndrome in middle-aged males: Results from the Oslo Diet and Exercise Study. Scand J Med Sci Sports. 2007;17:687–695. doi: 10.1111/j.1600-0838.2006.00631.x. [DOI] [PubMed] [Google Scholar]
- 23.Andersson N, Strandberg L, Nilsson S, Adamovic S, Karlsson MK, Ljunggren O, Mellstrom D, Lane NE, Zmuda JM, Nielsen C, Orwoll E, Lorentzon M, Ohlsson C, Jansson JO. A variant near the interleukin-6 gene is associated with fat mass in Caucasian men. Int J Obes (Lond) 2010;34:1011–1019. doi: 10.1038/ijo.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aquilante CL, Kosmiski LA, Knutsen SD, Zineh I. Relationship between plasma resistin concentrations, inflammatory chemokines, and components of the metabolic syndrome in adults. Metabolism. 2008;57:494–501. doi: 10.1016/j.metabol.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 25.Araki E, Lipes MA, Patti ME, Bruning JC, Haag BL, II, Johnson RS, Kahn CR. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature. 1994;372:186–190. doi: 10.1038/372186a0. [DOI] [PubMed] [Google Scholar]
- 26.Araujo EP, De Souza CT, Gasparetti AL, Ueno M, Boschero AC, Saad MJA, Velloso LA. Short-term in vivo inhibition of insulin receptor substrate-1 expression leads to insulin resistance, hyperinsulinemia, and increased adiposity. Endocrinology. 2005;146:1428–1437. doi: 10.1210/en.2004-0778. [DOI] [PubMed] [Google Scholar]
- 27.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J-i, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 28.Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 29.Arner P, Pettersson A, Mitchell PJ, Dunbar JD, Kharitonenkov A, Ryden M. FGF21 attenuates lipolysis in human adipocytes - a possible link to improved insulin sensitivity. FEBS Lett. 2008;582:1725–1730. doi: 10.1016/j.febslet.2008.04.038. [DOI] [PubMed] [Google Scholar]
- 30.Aron-Wisnewsky J, Tordjman J, Poitou C, Darakhshan F, Hugol D, Basdevant A, Aissat A, Guerre-Millo M, Clement K. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;94:4619–4623. doi: 10.1210/jc.2009-0925. [DOI] [PubMed] [Google Scholar]
- 31.Aronson D, Sella R, Sheikh-Ahmad M, Kerner A, Avizohar O, Rispler S, Bartha P, Markiewicz W, Levy Y, Brook GJ. The association between cardiorespiratory fitness and C-reactive protein in subjects with the metabolic syndrome. J Am Coll Cardiol. 2004;44:2003–2007. doi: 10.1016/j.jacc.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 32.Aronson D, Sheikh-Ahmad M, Avizohar O, Kerner A, Sella R, Bartha P, Markiewicz W, Levy Y, Brook GJ. C-Reactive protein is inversely related to physical fitness in middle-aged subjects. Atherosclerosis. 2004;176:173–179. doi: 10.1016/j.atherosclerosis.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 33.Atlantis E, Martin SA, Haren MT, Taylor AW, Wittert GA. Inverse associations between muscle mass, strength, and the metabolic syndrome. Metabolism. 2009;58:1013–1022. doi: 10.1016/j.metabol.2009.02.027. [DOI] [PubMed] [Google Scholar]
- 34.Avorn J, Monette J, Lacour A, Bohn RL, Monane M, Mogun H, LeLorier J. Persistence of use of lipid-lowering medications: A cross-national study. JAMA. 1998;279:1458–1462. doi: 10.1001/jama.279.18.1458. [DOI] [PubMed] [Google Scholar]
- 35.Ay L, Kopp H-P, Brix J-M, Ay C, Quehenberger P, Schernthaner G-H, Pabinger I, Schernthaner G. Thrombin generation in morbid obesity: Significant reduction after weight loss. J Thromb Haemost. 2010;8:759–765. doi: 10.1111/j.1538-7836.2010.03766.x. [DOI] [PubMed] [Google Scholar]
- 36.Babraj J, Vollaard N, Keast C, Guppy F, Cottrell G, Timmons J. Extremely short duration high intensity interval training substantially improves insulin action in young healthy males. BMC Endocrine Disorders. 2009;9:3. doi: 10.1186/1472-6823-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bacchi E, Negri C, Zanolin ME, Milanese C, Faccioli N, Trombetta M, Zoppini G, Cevese A, Bonadonna RC, Schena F, Bonora E, Lanza M, Moghetti P. Metabolic Effects of Aerobic Training and Resistance Training in Type 2 Diabetic Subjects. Diabetes Care. 2012;35:676–682. doi: 10.2337/dc11-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bae SS, Cho H, Mu J, Birnbaum MJ. Isoform-specific regulation of insulin-dependent glucose uptake by Akt/protein kinase B. J Biol Chem. 2003;278:49530–49536. doi: 10.1074/jbc.M306782200. [DOI] [PubMed] [Google Scholar]
- 39.Baldo A, Sniderman A, St Luce S, Zhang X, Cianflone K. Signal transduction pathway of acylation stimulating protein: Involvement of protein kinase C. J Lipid Res. 1995;36:1415–1426. [PubMed] [Google Scholar]
- 40.Balducci S, Zanuso S, Nicolucci A, De Feo P, Cavallo S, Cardelli P, Fallucca S, Alessi E, Fallucca F, Pugliese G. Effect of an intensive exercise intervention strategy on modifiable cardiovascular risk factors in subjects with type 2 diabetes mellitus: A randomized controlled trial: The Italian Diabetes and Exercise Study (IDES) Arch Intern Med. 2010;170:1794–1803. doi: 10.1001/archinternmed.2010.380. [DOI] [PubMed] [Google Scholar]
- 41.Balkau B, Charles MA. Comment on the provisional report from the WHO consultation. European Group for the Study of Insulin Resistance (EGIR) Diabet Med. 1999;16:442–443. doi: 10.1046/j.1464-5491.1999.00059.x. [DOI] [PubMed] [Google Scholar]
- 42.Balkau B, Mhamdi L, Oppert JM, Nolan J, Golay A, Porcellati F, Laakso M, Ferrannini E. Physical activity and insulin sensitivity: The RISC study. Diabetes. 2008;57:2613–2618. doi: 10.2337/db07-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased p85/55/50 expression and decreased phosphotidylinositol 3-kinase activity in insulin-resistant human skeletal muscle. Diabetes. 2005;54:2351–2359. doi: 10.2337/diabetes.54.8.2351. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–1198. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- 45.Banks EA, Brozinick JT, Jr, Yaspelkis BB, III, Kang HY, Ivy JL. Muscle glucose transport, GLUT-4 content, and degree of exercise training in obese Zucker rats. Am J Physiol. 1992;263:E1010–E1015. doi: 10.1152/ajpendo.1992.263.5.E1015. [DOI] [PubMed] [Google Scholar]
- 46.Barakat HA, Carpenter JW, McLendon VD, Khazanie P, Leggett N, Heath J, Marks R. Influence of obesity, impaired glucose tolerance, and NIDDM on LDL structure and composition. Possible link between hyperinsulinemia and atherosclerosis. Diabetes. 1990;39:1527–1533. doi: 10.2337/diab.39.12.1527. [DOI] [PubMed] [Google Scholar]
- 47.Barbieri M, Rizzo MR, Papa M, Boccardi V, Esposito A, White MF, Paolisso G. The IRS2 Gly1057Asp variant is associated with human longevity. J Gerontol A Biol Sci Med Sci. 2009;65:282–286. doi: 10.1093/gerona/glp154. [DOI] [PubMed] [Google Scholar]
- 48.Barnard RJ. Prostate cancer prevention by nutritional means to alleviate metabolic syndrome. Am J Clin Nutr. 2007;86:s889–s893. doi: 10.1093/ajcn/86.3.889S. [DOI] [PubMed] [Google Scholar]
- 49.Barnard RJ, Faria DJ, Menges JE, Martin DA. Effects of a high-fat, sucrose diet on serum insulin and related atherosclerotic risk factors in rats. Atherosclerosis. 1993;100:229–236. doi: 10.1016/0021-9150(93)90209-d. [DOI] [PubMed] [Google Scholar]
- 50.Barnard RJ, Roberts CK, Varon SM, Berger JJ. Diet-induced insulin resistance precedes other aspects of the metabolic syndrome. J Appl Physiol. 1998;84:1311–1315. doi: 10.1152/jappl.1998.84.4.1311. [DOI] [PubMed] [Google Scholar]
- 51.Barnard RJ, Wen SJ. Exercise and diet in the prevention and control of the metabolic syndrome. Sports Med. 1994;18:218–228. doi: 10.2165/00007256-199418040-00002. [DOI] [PubMed] [Google Scholar]
- 52.Barnard RJ, Youngren JF, Martin DA. Diet, not aging, causes skeletal muscle insulin resistance. Gerontology. 1995;41:205–211. doi: 10.1159/000213683. [DOI] [PubMed] [Google Scholar]
- 53.Barnard RJ, Youngren JF, Scheck SH. Reversibility of diet-induced skeletal muscle insulin resistance. Diabetes Research. 1997;32:213–221. [Google Scholar]
- 54.Baron AD. Hemodynamic actions of insulin. Am J Physiol. 1994;267:E187–E202. doi: 10.1152/ajpendo.1994.267.2.E187. [DOI] [PubMed] [Google Scholar]
- 55.Barwell N, Malkova D, Moran C, Cleland S, Packard C, Zammit V, Gill J. Exercise training has greater effects on insulin sensitivity in daughters of patients with type 2 diabetes than in women with no family history of diabetes. Diabetologia. 2008;51:1912–1919. doi: 10.1007/s00125-008-1097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J Clin Endocrinol Metab. 2000;85:3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- 57.Baumann CA, Ribon V, Kanzaki M, Thurmond DC, Mora S, Shigematsu S, Bickel PE, Pessin JE, Saltiel AR. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 58.Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, Shulman GI. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56:1376–1381. doi: 10.2337/db06-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benetos A, Thomas F, Pannier B, Bean K, Jego B, Guize L. All-cause and cardiovascular mortality using the different definitions of metabolic syndrome. Am J Cardiol. 2008;102:188–191. doi: 10.1016/j.amjcard.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 60.Benner JS, Glynn RJ, Mogun H, Neumann PJ, Weinstein MC, Avorn J. Long-term persistence in use of statin therapy in elderly patients. JAMA. 2002;288:455–461. doi: 10.1001/jama.288.4.455. [DOI] [PubMed] [Google Scholar]
- 61.Benrick A, Jirholt P, Wernstedt I, Gustafsson M, Scheller J, Eriksson AL, Boren J, Hedner T, Ohlsson C, Hard T, Rose-John S, Jansson JO. A non-conservative polymorphism in the IL-6 signal transducer (IL6ST)/gp130 is associated with myocardial infarction in a hypertensive population. Regul Pept. 2008;146:189–196. doi: 10.1016/j.regpep.2007.09.031. [DOI] [PubMed] [Google Scholar]
- 62.Bento JL, Palmer ND, Mychaleckyj JC, Lange LA, Langefeld CD, Rich SS, Freedman BI, Bowden DW. Association of protein tyrosine phosphatase 1B gene polymorphisms with type 2 diabetes. Diabetes. 2004;53:3007–3012. doi: 10.2337/diabetes.53.11.3007. [DOI] [PubMed] [Google Scholar]
- 63.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 64.Berglund ED, Li CY, Bina HA, Lynes SE, Michael MD, Shanafelt AB, Kharitonenkov A, Wasserman DH. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology. 2009;150:4084–4093. doi: 10.1210/en.2009-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergman RN. Lilly lecture 1989. Toward physiological understanding of glucose tolerance. Minimal-model approach. Diabetes. 1989;38:1512–1527. doi: 10.2337/diab.38.12.1512. [DOI] [PubMed] [Google Scholar]
- 66.Bezaire V, Bruce CR, Heigenhauser GJ, Tandon NN, Glatz JF, Luiken JJ, Bonen A, Spriet LL. Identification of fatty acid translocase on human skeletal muscle mitochondrial membranes: Essential role in fatty acid oxidation. Am J Physiol Endocrinol Metab. 2006;290:E509–E515. doi: 10.1152/ajpendo.00312.2005. [DOI] [PubMed] [Google Scholar]
- 67.Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- 68.Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG., Jr SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem. 2000;275:40649–40657. doi: 10.1074/jbc.M007577200. [DOI] [PubMed] [Google Scholar]
- 69.Bjorntorp P. “Portal” adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis. 1990;10:493–496. [PubMed] [Google Scholar]
- 70.Björntorp P, Fahlén M, Grimby G, Gustafson A, Holm J, Renström P, Scherstén T. Carbohydrate and lipid metabolism in middle-aged, physically well-trained men. Metabolism. 1972;21:1037–1044. doi: 10.1016/0026-0495(72)90034-0. [DOI] [PubMed] [Google Scholar]
- 71.Blüher M. Adipose tissue dysfunction in obesity. Exp Clin Endocrinol Diabetes. 2009;117:241–250. doi: 10.1055/s-0029-1192044. [DOI] [PubMed] [Google Scholar]
- 72.Bo S, Ciccone G, Baldi C, Benini L, Dusio F, Forastiere G, Lucia C, Nuti C, Durazzo M, Cassader M, Gentile L, Pagano G. Effectiveness of a lifestyle intervention on metabolic syndrome. A randomized controlled trial. J Gen Intern Med. 2007;22:1695–1703. doi: 10.1007/s11606-007-0399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bonen A, Dyck DJ, Ibrahimi A, Abumrad NA. Muscle contractile activity increases fatty acid metabolism and transport and FAT/CD36. Am J PhysiolAm J Physiol. 1999;276:E642–E649. doi: 10.1152/ajpendo.1999.276.4.E642. [DOI] [PubMed] [Google Scholar]
- 74.Bonen A, Luiken JJ, Arumugam Y, Glatz JF, Tandon NN. Acute regulation of fatty acid uptake involves the cellular redistribution of fatty acid translocase. J Biol Chem. 2000;275:14501–14508. doi: 10.1074/jbc.275.19.14501. [DOI] [PubMed] [Google Scholar]
- 75.Booth FW, Laye MJ. Lack of adequate appreciation of physical exercise’s complexities can pre-empt appropriate design and interpretation in scientific discovery. J Physiol. 2009;587:5527–5539. doi: 10.1113/jphysiol.2009.179507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Booth FW, Roberts CK, Laye MJ. Comprehensive Physiology. John Wiley & Sons, Inc; 2011. Lack of exercise is a major cause of chronic diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bouchard C, An P, Rice T, Skinner JS, Wilmore JH, Gagnon J, Perusse L, Leon AS, Rao DC. Familial aggregation of VO2 max response to exercise training: Results from the HERITAGE Family Study. J Appl Physiol. 1999;87:1003–1008. doi: 10.1152/jappl.1999.87.3.1003. [DOI] [PubMed] [Google Scholar]
- 78.Boule NG, Weisnagel SJ, Lakka TA, Tremblay A, Bergman RN, Rankinen T, Leon AS, Skinner JS, Wilmore JH, Rao DC, Bouchard C. Effects of exercise training on glucose homeostasis: The heritage family study. Diabetes Care. 2005;28:108–114. doi: 10.2337/diacare.28.1.108. [DOI] [PubMed] [Google Scholar]
- 79.Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, Walder K, Segal D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology. 2007;148:4687–4694. doi: 10.1210/en.2007-0175. [DOI] [PubMed] [Google Scholar]
- 80.Bozaoglu K, Segal D, Shields KA, Cummings N, Curran JE, Comuzzie AG, Mahaney MC, Rainwater DL, VandeBerg JL, MacCluer JW, Collier G, Blangero J, Walder K, Jowett JB. Chemerin is associated with metabolic syndrome phenotypes in a Mexican-American population. J Clin Endocrinol Metab. 2009;94:3085–3088. doi: 10.1210/jc.2008-1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Braun B, Zimmermann MB, Kretchmer N. Effects of exercise intensity on insulin sensitivity in women with non-insulin-dependent diabetes mellitus. J Appl Physiol. 1995;78:300–306. doi: 10.1152/jappl.1995.78.1.300. [DOI] [PubMed] [Google Scholar]
- 82.Brouwer BG, Visseren FLJ, van der Graaf Y. The effect of leisure-time physical activity on the presence of metabolic syndrome in patients with manifest arterial disease. The SMART study. Am Heart J. 2007;154:1146–1152. doi: 10.1016/j.ahj.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 83.Brozinick JT, Jr, Etgen GJ, Jr, Yaspelkis BB, III, Ivy JL. Glucose uptake and GLUT-4 protein distribution in skeletal muscle of the obese Zucker rat. Am J Physiol. 1994;267:R236–R243. doi: 10.1152/ajpregu.1994.267.1.R236. [DOI] [PubMed] [Google Scholar]
- 84.Brozinick JT, Jr, McCoid SC, Reynolds TH, Nardone NA, Hargrove DM, Stevenson RW, Cushman SW, Gibbs EM. GLUT4 overexpression in db/db mice dose-dependently ameliorates diabetes but is not a lifelong cure. Diabetes. 2001;50:593–600. doi: 10.2337/diabetes.50.3.593. [DOI] [PubMed] [Google Scholar]
- 85.Bruce CR, Carey AL, Hawley JA, Febbraio MA. Intramuscular heat shock protein 72 and heme oxygenase-1 mRNA are reduced in patients with type 2 diabetes. Diabetes. 2003;52:2338–2345. doi: 10.2337/diabetes.52.9.2338. [DOI] [PubMed] [Google Scholar]
- 86.Bruce CR, Hawley JA. Improvements in insulin resistance with aerobic exercise training: A lipocentric approach. Med Sci Sports Exerc. 2004;36:1196–1201. [PubMed] [Google Scholar]
- 87.Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, Cooney GJ, Febbraio MA, Kraegen EW. Overexpression of carnitine palmitoyltransferase-1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high-fat diet–induced insulin resistance. Diabetes. 2009;58:550–558. doi: 10.2337/db08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bruce CR, Thrush AB, Mertz VA, Bezaire V, Chabowski A, Heigen-hauser GJ, Dyck DJ. Endurance training in obese humans improves glucose tolerance and mitochondrial fatty acid oxidation and alters muscle lipid content. Am J Physiol Endocrinol Metab. 2006;291:E99–E107. doi: 10.1152/ajpendo.00587.2005. [DOI] [PubMed] [Google Scholar]
- 89.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 90.Burgomaster KA, Hughes SC, Heigenhauser GJF, Bradwell SN, Gibala MJ. Six sessions of sprint interval training increases muscle oxidative potential and cycle endurance capacity in humans. J Appl Physiol. 2005;98:1985–1990. doi: 10.1152/japplphysiol.01095.2004. [DOI] [PubMed] [Google Scholar]
- 91.Burguera B, Proctor D, Dietz N, Guo Z, Joyner M, Jensen MD. Leg free fatty acid kinetics during exercise in men and women. Am J Physiol Endocrinol Metab. 2000;278:E113–E117. doi: 10.1152/ajpendo.2000.278.1.E113. [DOI] [PubMed] [Google Scholar]
- 92.Burstein R, Polychronakos C, Toews CJ, MacDougall JD, Guyda HJ, Posner BI. Acute reversal of the enhanced insulin action in trained athletes. Association with insulin receptor changes. Diabetes. 1985;34:756–760. doi: 10.2337/diab.34.8.756. [DOI] [PubMed] [Google Scholar]
- 93.Busch AK, Gurisik E, Cordery DV, Sudlow M, Denyer GS, Lay-butt DR, Hughes WE, Biden TJ. Increased fatty acid desaturation and enhanced expression of stearoyl coenzyme A desaturase protects pancreatic beta-cells from lipoapoptosis. Diabetes. 2005;54:2917–2924. doi: 10.2337/diabetes.54.10.2917. [DOI] [PubMed] [Google Scholar]
- 94.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Camhi SM, Stefanick ML, Katzmarzyk PT, Young DR. Metabolic syndrome and changes in body fat from a low-fat diet and/or exercise randomized controlled trial. Obesity. 2009;18:548–554. doi: 10.1038/oby.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Campbell SE, Tandon NN, Woldegiorgis G, Luiken JJ, Glatz JF, Bonen A. A novel function for fatty acid translocase (FAT)/CD36: Involvement in long chain fatty acid transfer into the mitochondria. J Biol Chem. 2004;279:36235–36241. doi: 10.1074/jbc.M400566200. [DOI] [PubMed] [Google Scholar]
- 97.Camus JP. Gout, diabetes, hyperlipemia: A metabolic trisyndrome. Rev Rhum Mal Osteoartic. 1966;33:10–14. [PubMed] [Google Scholar]
- 98.Caranti DA, de Mello MT, Prado WL, Tock L, Siqueira KO, de Piano A, Lofrano MC, Cristofalo DMJ, Lederman H, Tufik S, Dâmaso AR. Short- and long-term beneficial effects of a multidisciplinary therapy for the control of metabolic syndrome in obese adolescents. Metabolism. 2007;56:1293–1300. doi: 10.1016/j.metabol.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 99.Carey A, Kingwell B. Novel pharmacological approaches to combat obesity and insulin resistance: Targeting skeletal muscle with ‘exercise mimetics’. Diabetologia. 2009;52:2015–2026. doi: 10.1007/s00125-009-1420-x. [DOI] [PubMed] [Google Scholar]
- 100.Carey A, Petersen E, Bruce C, Southgate R, Pilegaard H, Hawley J, Pedersen B, Febbraio M. Discordant gene expression in skeletal muscle and adipose tissue of patients with type 2 diabetes: Effect of interleukin-6 infusion. Diabetologia. 2006;49:1000–1007. doi: 10.1007/s00125-006-0178-7. [DOI] [PubMed] [Google Scholar]
- 101.Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK, Febbraio MA. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2006;55:2688–2697. doi: 10.2337/db05-1404. [DOI] [PubMed] [Google Scholar]
- 102.Carnethon MR, Gidding SS, Nehgme R, Sidney S, Jacobs DR, Jr, Liu K. Cardiorespiratory fitness in young adulthood and the development of cardiovascular disease risk factors. JAMA. 2003;290:3092–3100. doi: 10.1001/jama.290.23.3092. [DOI] [PubMed] [Google Scholar]
- 103.Carroll S, Cooke CB, Butterly RJ. Metabolic clustering, physical activity and fitness in nonsmoking, middle-aged men. Med Sci Sports Exerc. 2000;32:2079–2086. doi: 10.1097/00005768-200012000-00018. [DOI] [PubMed] [Google Scholar]
- 104.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Castaneda F, Layne JE, Castaneda C. Skeletal muscle sodium glucose co-transporters in older adults with type 2 diabetes undergoing resistance training. Int J Med Sci. 2006;3:84–91. doi: 10.7150/ijms.3.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotel-lar F, Gil M, Cienfuegos J, Salvador J, Frühbeck G. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med. 2009;87:803–813. doi: 10.1007/s00109-009-0486-8. [DOI] [PubMed] [Google Scholar]
- 107.Cauza E, Hanusch-Enserer U, Strasser B, Kostner K, Dunky A, Haber P. Strength and endurance training lead to different post exercise glucose profiles in diabetic participants using a continuous subcutaneous glucose monitoring system. Eur J Clin Invest. 2005;35:745–751. doi: 10.1111/j.1365-2362.2005.01573.x. [DOI] [PubMed] [Google Scholar]
- 108.Cha BS, Ciaraldi TP, Park KS, Carter L, Mudaliar SR, Henry RR. Impaired fatty acid metabolism in type 2 diabetic skeletal muscle cells is reversed by PPARgamma agonists. Am J Physiol Endocrinol Metab. 2005;289:E151–E159. doi: 10.1152/ajpendo.00141.2004. [DOI] [PubMed] [Google Scholar]
- 109.Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, Bernhardt U, Dreja T, Vogel H, Schmolz K, Kluge R, Zierath JR, Hultschig C, Hoeben RC, Schurmann A, Joost HG, Al-Hasani H. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet. 2008;40:1354–1359. doi: 10.1038/ng.244. [DOI] [PubMed] [Google Scholar]
- 110.Chaika OV, Chaika N, Volle DJ, Hayashi H, Ebina Y, Wang L-M, Pierce JH, Lewis RE. Mutation of tyrosine 960 within the insulin receptor juxtamembrane domain impairs glucose transport but does not inhibit ligand-mediated phosphorylation of insulin receptor substrate-2 in 3T3-L1 adipocytes. J Biol Chem. 1999;274:12075–12080. doi: 10.1074/jbc.274.17.12075. [DOI] [PubMed] [Google Scholar]
- 111.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chavey C, Lazennec G, Lagarrigue S, Clapé C, Iankova I, Teyssier J, Annicotte J-S, Schmidt J, Mataki C, Yamamoto H, Sanches R, Guma A, Stich V, Vitkova M, Jardin-Watelet B, Renard E, Strieter R, Tuthill A, Hotamisligil GS, Vidal-Puig A, Zorzano A, Langin D, Fajas L. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metabolism. 2009;9:339–349. doi: 10.1016/j.cmet.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen AK, Roberts CK, Barnard RJ. Effect of a short-term diet and exercise intervention on metabolic syndrome in overweight children. Metabolism. 2006;55:871–878. doi: 10.1016/j.metabol.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 114.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, Ellis SJ, Lakey ND, Culpepper J, Moore KJ, Breitbart RE, Duyk GM, Tepper RI, Morgenstern JP. Evidence that the diabetes gene encodes the leptin receptor: Identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. doi: 10.1016/s0092-8674(00)81294-5. [DOI] [PubMed] [Google Scholar]
- 115.Chiang S-H, Baumann CA, Kanzaki M, Thurmond DC, Watson RT, Neudauer CL, Macara IG, Pessin JE, Saltiel AR. Insulin-stimulated GLUT4 translocation requires the CAP-dependent activation of TC10. Nature. 2001;410:944–948. doi: 10.1038/35073608. [DOI] [PubMed] [Google Scholar]
- 116.Cho ER, Shin A, Kim J, Jee SH, Sung J. Leisure-time physical activity is associated with a reduced risk for metabolic syndrome. Ann Epidemiol. 2009;19:784–792. doi: 10.1016/j.annepidem.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 117.Cho YM, Youn BS, Chung SS, Kim KW, Lee HK, Yu KY, Park HJ, Shin HD, Park KS. Common genetic polymorphisms in the promoter of resistin gene are major determinants of plasma resistin concentrations in humans. Diabetologia. 2004;47:559–565. doi: 10.1007/s00125-003-1319-x. [DOI] [PubMed] [Google Scholar]
- 118.Choi CS, Savage DB, Abu-Elheiga L, Liu Z-X, Kim S, Kulkarni A, Distefano A, Hwang Y-J, Reznick RM, Codella R, Zhang D, Cline GW, Wakil SJ, Shulman GI. Continuous fat oxidation in acetyl–CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc Nat Acad Sci U S A. 2007;104:16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Christ-Roberts CY, Pratipanawatr T, Pratipanawatr W, Berria R, Belfort R, Kashyap S, Mandarino LJ. Exercise training increases glycogen synthase activity and GLUT4 expression but not insulin signaling in overweight nondiabetic and type 2 diabetic subjects. Metabolism. 2004;53:1233–1242. doi: 10.1016/j.metabol.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 120.Chung J, Nguyen A-K, Henstridge DC, Holmes AG, Chan MHS, Mesa JL, Lancaster GI, Southgate RJ, Bruce CR, Duffy SJ, Horvath I, Mestril R, Watt MJ, Hooper PL, Kingwell BA, Vigh L, Hevener A, Febbraio MA. HSP72 protects against obesity-induced insulin resistance. Proc Nat Acad Sci U S A. 2008;105:1739–1744. doi: 10.1073/pnas.0705799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chung SS, Choi HH, Kim KW, Cho YM, Lee HK, Park KS. Regulation of human resistin gene expression in cell systems: An important role of stimulatory protein 1 interaction with a common promoter polymorphic site. Diabetologia. 2005;48:1150–1158. doi: 10.1007/s00125-005-1762-y. [DOI] [PubMed] [Google Scholar]
- 122.Church TS, Blair SN. When will we treat physical activity as a legitimate medical therapy . . . even though it does not come in a pill? Br J Sports Med. 2009;43:80–81. doi: 10.1136/bjsm.2008.053850. [DOI] [PubMed] [Google Scholar]
- 123.Churilla JR, Fitzhugh EC. Relationship between leisure-time physical activity and metabolic syndrome using varying definitions: 1999–2004 NHANES. Diab Vasc Dis Res. 2009;6:100–109. doi: 10.1177/1479164109336040. [DOI] [PubMed] [Google Scholar]
- 124.Cianflone K, Xia Z, Chen LY. Critical review of acylation-stimulating protein physiology in humans and rodents. Biochim Biophys Acta. 2003;1609:127–143. doi: 10.1016/s0005-2736(02)00686-7. [DOI] [PubMed] [Google Scholar]
- 125.Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular T(regs): A unique population of adipose-tissue-resident Foxp3+CD4+ T cells that impacts organismal metabolism. Semin Immunol. 2011;23:431–437. doi: 10.1016/j.smim.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 126.Clevenger CM, Parker Jones P, Tanaka H, Seals DR, DeSouza CA. Decline in insulin action with age in endurance-trained humans. J Appl Physiol. 2002;93:2105–2111. doi: 10.1152/japplphysiol.00315.2002. [DOI] [PubMed] [Google Scholar]
- 127.Coe NR, Bernlohr DA. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim Biophys Acta. 1998;1391:287–306. doi: 10.1016/s0005-2760(97)00205-1. [DOI] [PubMed] [Google Scholar]
- 128.Coker RH, Hays NP, Williams RH, Brown AD, Freeling SA, Kortebein PM, Sullivan DH, Starling RD, Evans WJ. Exercise-induced changes in insulin action and glycogen metabolism in elderly adults. Med Sci Sports Exerc. 2006;38:433–438. doi: 10.1249/01.mss.0000191417.48710.11. [DOI] [PubMed] [Google Scholar]
- 129.Comuzzie AG, Funahashi T, Sonnenberg G, Martin LJ, Jacob HJ, Black AEK, Maas D, Takahashi M, Kihara S, Tanaka S, Matsuzawa Y, Blangero J, Cohen D, Kissebah A. The genetic basis of plasma variation in adiponectin, a global endophenotype for obesity and the metabolic syndrome. J Clin Endocrinol Metab. 2001;86:4321–4325. doi: 10.1210/jcem.86.9.7878. [DOI] [PubMed] [Google Scholar]
- 130.Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cortez-Cooper MY, DeVan AE, Anton MM, Farrar RP, Beckwith KA, Todd JS, Tanaka H. Effects of high intensity resistance training on arterial stiffness and wave reflection in women. Am J Hypertens. 2005;18:930–934. doi: 10.1016/j.amjhyper.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 132.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, Moller DE, Kharitonenkov A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 133.Cox JH, Cortright RN, Dohm GL, Houmard JA. Effect of aging on response to exercise training in humans: Skeletal muscle GLUT-4 and insulin sensitivity. J Appl Physiol. 1999;86:2019–2025. doi: 10.1152/jappl.1999.86.6.2019. [DOI] [PubMed] [Google Scholar]
- 134.Craig BW, Everhart J, Brown R. The influence of high-resistance training on glucose tolerance in young and elderly subjects. Mech Ageing Dev. 1989;49:147–157. doi: 10.1016/0047-6374(89)90098-5. [DOI] [PubMed] [Google Scholar]
- 135.Crimmins NA, Martin LJ. Polymorphisms in adiponectin receptor genes ADIPOR1 and ADIPOR2 and insulin resistance. Obesity Reviews. 2007;8:419–423. doi: 10.1111/j.1467-789X.2007.00348.x. [DOI] [PubMed] [Google Scholar]
- 136.Crowe S, Wu LE, Economou C, Turpin SM, Matzaris M, Hoehn KL, Hevener AL, James DE, Duh EJ, Watt MJ. Pigment epithelium-derived factor contributes to insulin resistance in obesity. Cell Metab. 2009;10:40–47. doi: 10.1016/j.cmet.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 137.Cuff DJ, Meneilly GS, Martin A, Ignaszewski A, Tildesley HD, Frohlich JJ. Effective exercise modality to reduce insulin resistance in women with type 2 diabetes. Diabetes Care. 2003;26:2977–2982. doi: 10.2337/diacare.26.11.2977. [DOI] [PubMed] [Google Scholar]
- 138.Czech MP. The nature and regulation of the insulin receptor: Structure and function. Annu Rev Physiol. 1985;47:357–381. doi: 10.1146/annurev.ph.47.030185.002041. [DOI] [PubMed] [Google Scholar]
- 139.D’Aiuto FSW, Netuveli G, Donos N, Hingorani AD, Deanfield J, Tsakos G. Association of the metabolic syndrome with severe periodontitis in a large U.S. population-based syrvey. J Clin Endocrinol Metab. 2008;93:3989–3994. doi: 10.1210/jc.2007-2522. [DOI] [PubMed] [Google Scholar]
- 140.D’Alessandris C, Lauro R, Presta I, Sesti G. C-reactive protein induces phosphorylation of insulin receptor substrate-1 on Ser307 and Ser612 in L6 myocytes, thereby impairing the insulin signalling pathway that promotes glucose transport. Diabetologia. 2007;50:840–849. doi: 10.1007/s00125-006-0522-y. [DOI] [PubMed] [Google Scholar]
- 141.da Luz G, Frederico MJ, da Silva S, Vitto MF, Cesconetto PA, de Pinho RA, Pauli JR, Silva AS, Cintra DE, Ropelle ER, De Souza CT. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur J Appl Physiol. 2011;111:2015–2023. doi: 10.1007/s00421-010-1802-2. [DOI] [PubMed] [Google Scholar]
- 142.Daray LA, Henagan TM, Zanovec M, Earnest CP, Johnson LG, Winchester J, Tuuri G, Stewart LK. Endurance and resistance training lowers C-reactive protein in young, healthy females. Appl Physiol Nutr Metab. 2011;36:660–670. doi: 10.1139/h11-077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Davidson LE, Hudson R, Kilpatrick K, Kuk JL, McMillan K, Janiszewski PM, Lee S, Lam M, Ross R. Effects of exercise modality on insulin resistance and functional limitation in older adults: A randomized controlled trial. Arch Intern Med. 2009;169:122–131. doi: 10.1001/archinternmed.2008.558. [DOI] [PubMed] [Google Scholar]
- 144.Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity (Silver Spring) 2008;16:1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- 145.de Kloet AD, Krause EG, Kim D-H, Sakai RR, Seeley RJ, Woods SC. The effect of angiotensin-converting enzyme inhibition using captopril on energy balance and glucose homeostasis. Endocrinology. 2009;150:4114–4123. doi: 10.1210/en.2009-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.de Zeeuw D, Bakker SJ. Does the metabolic syndrome add to the diagnosis and treatment of cardiovascular disease? Nat Clin Pract Cardiovasc Med. 2008;5 (Suppl 1):S10–S14. doi: 10.1038/ncpcardio1271. [DOI] [PubMed] [Google Scholar]
- 147.DeFronzo RA. Lilly lecture 1987. The triumvirate: Beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. doi: 10.2337/diab.37.6.667. [DOI] [PubMed] [Google Scholar]
- 148.Defronzo RA, Ferrannini E. Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14:173–194. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 149.DeFronzo RA, Sherwin RS, Kraemer N. Effect of physical training on insulin action in obesity. Diabetes. 1987;36:1379–1385. doi: 10.2337/diab.36.12.1379. [DOI] [PubMed] [Google Scholar]
- 150.Dela F, Handberg A, Mikines KJ, Vinten J, Galbo H. GLUT 4 and insulin receptor binding and kinase activity in trained human muscle. J Physiol. 1993;469:615–624. doi: 10.1113/jphysiol.1993.sp019833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dela F, Kjaer M. Resistance training, insulin sensitivity and muscle function in the elderly. Essays Biochem. 2006;42:75–88. doi: 10.1042/bse0420075. [DOI] [PubMed] [Google Scholar]
- 152.Dela F, Larsen JJ, Mikines KJ, Ploug T, Petersen LN, Galbo H. Insulin-stimulated muscle glucose clearance in patients with NIDDM. Effects of one-legged physical training. Diabetes. 1995;44:1010–1020. doi: 10.2337/diab.44.9.1010. [DOI] [PubMed] [Google Scholar]
- 153.Dela F, Mikines KJ, von Linstow M, Secher NH, Galbo H. Effect of training on insulin-mediated glucose uptake in human muscle. Am J Physiol. 1992;263:E1134–E1143. doi: 10.1152/ajpendo.2006.263.6.E1134. [DOI] [PubMed] [Google Scholar]
- 154.Dela F, Ploug T, Handberg A, Petersen LN, Larsen JJ, Mikines KJ, Galbo H. Physical training increases muscle GLUT4 protein and mRNA in patients with NIDDM. Diabetes. 1994;43:862–865. doi: 10.2337/diab.43.7.862. [DOI] [PubMed] [Google Scholar]
- 155.Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006;444:881–887. doi: 10.1038/nature05488. [DOI] [PubMed] [Google Scholar]
- 156.Despres JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, Rodes-Cabau J, Bertrand OF, Poirier P. Abdominal obesity and the metabolic syndrome: Contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–1049. doi: 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
- 157.Devaraj S, Dasu MR, Singh U, Rao LV, Jialal I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. 2009;203:67–74. doi: 10.1016/j.atherosclerosis.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Devaraj S, Singh U, Jialal I. Human C-reactive protein and the metabolic syndrome. Curr Opin Lipidol. 2009;20:182–189. doi: 10.1097/MOL.0b013e32832ac03e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Di Paola R, Frittitta L, Miscio G, Bozzali M, Baratta R, Centra M, Spampinato D, Santagati MG, Ercolino T, Cisternino C, Soccio T, Mastroianno S, Tassi V, Almgren P, Pizzuti A, Vigneri R, Trischitta V. A variation in 3′ UTR of hPTP1B increases specific gene expression and associates with insulin resistance. Am J Hum Genet. 2002;70:806–812. doi: 10.1086/339270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.DiPietro L, Dziura J, Yeckel CW, Neufer PD. Exercise and improved insulin sensitivity in older women: Evidence of the enduring benefits of higher intensity training. J Appl Physiol. 2006;100:142–149. doi: 10.1152/japplphysiol.00474.2005. [DOI] [PubMed] [Google Scholar]
- 161.Dobbins RL, Szczepaniak LS, Bentley B, Esser V, Myhill J, McGarry JD. Prolonged inhibition of muscle carnitine palmitoyltransferase-1 promotes intramyocellular lipid accumulation and insulin resistance in rats. Diabetes. 2001;50:123–130. doi: 10.2337/diabetes.50.1.123. [DOI] [PubMed] [Google Scholar]
- 162.Dobrzyn A, Gorski J. Ceramides and sphingomyelins in skeletal muscles of the rat: Content and composition. Effect of prolonged exercise. Am J Physiol Endocrinol Metab. 2002;282:E277–E285. doi: 10.1152/ajpendo.00151.2001. [DOI] [PubMed] [Google Scholar]
- 163.Donges CE, Duffield R, Drinkwater EJ. Effects of resistance or aerobic exercise training on interleukin-6, C-reactive protein, and body composition. Med Sci Sports Exerc. 2010;42:304–313. doi: 10.1249/MSS.0b013e3181b117ca. [DOI] [PubMed] [Google Scholar]
- 164.Dostalova I, Haluzikova D, Haluzik M. Fibroblast growth factor 21: A novel metabolic regulator with potential therapeutic properties in obesity/type 2 diabetes mellitus. Physiol Res. 2009;58:1–7. doi: 10.33549/physiolres.931610. [DOI] [PubMed] [Google Scholar]
- 165.Dube JJ, Amati F, Stefanovic-Racic M, Toledo FGS, Sauers SE, Goodpaster BH. Exercise-induced alterations in intramyocellular lipids and insulin resistance: The athlete’s paradox revisited. Am J Physiol Endocrinol Metab. 2008;294:E882–E888. doi: 10.1152/ajpendo.00769.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.DuBose KD, Eisenmann JC, Donnelly JE. Aerobic fitness attenuates the metabolic syndrome score in normal-weight, at-risk-for-overweight, and overweight children. Pediatrics. 2007;120:e1262–e1268. doi: 10.1542/peds.2007-0443. [DOI] [PubMed] [Google Scholar]
- 167.Dudek RW, Dohm GL, Holman GD, Cushman SW, Wilson CM. Glucose transporter localization in rat skeletal muscle. Autoradiographic study using ATB-(2–3H]BMPA photolabel. FEBS Lett. 1994;339:205–208. doi: 10.1016/0014-5793(94)80416-8. [DOI] [PubMed] [Google Scholar]
- 168.Duffaut C, Galitzky J, Lafontan M, Bouloumie A. Unexpected trafficking of immune cells within the adipose tissue during the onset of obesity. Biochem Biophys Res Commun. 2009;384:482–485. doi: 10.1016/j.bbrc.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 169.Duffaut C, Zakaroff-Girard A, Bourlier V, Decaunes P, Maumus M, Chiotasso P, Sengenes C, Lafontan M, Galitzky J, Bouloumie A. Interplay between human adipocytes and T lymphocytes in obesity: CCL20 as an adipochemokine and T lymphocytes as lipogenic modulators. Arterioscler Thromb Vasc Biol. 2009;29:1608–1614. doi: 10.1161/ATVBAHA.109.192583. [DOI] [PubMed] [Google Scholar]
- 170.Duncan GE, Perri MG, Theriaque DW, Hutson AD, Eckel RH, Stacpoole PW. Exercise training, without weight loss, increases insulin sensitivity and postheparin plasma lipase activity in previously sedentary adults. Diabetes Care. 2003;26:557–562. doi: 10.2337/diacare.26.3.557. [DOI] [PubMed] [Google Scholar]
- 171.Dunstan DW, Daly RM, Owen N, Jolley D, De Courten M, Shaw J, Zimmet P. High-intensity resistance training improves glycemic control in older patients with type 2 diabetes. Diabetes Care. 2002;25:1729–1736. doi: 10.2337/diacare.25.10.1729. [DOI] [PubMed] [Google Scholar]
- 172.Ebeling P, Bourey R, Koranyi L, Tuominen JA, Groop LC, Henriksson J, Mueckler M, Sovijarvi A, Koivisto VA. Mechanism of enhanced insulin sensitivity in athletes. Increased blood flow, muscle glucose transport protein (GLUT-4) concentration, and glycogen synthase activity. J Clin Invest. 1993;92:1623–1631. doi: 10.1172/JCI116747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Egawa K, Maegawa H, Shimizu S, Morino K, Nishio Y, Bryer-Ash M, Cheung AT, Kolls JK, Kikkawa R, Kashiwagi A. Protein-tyrosine phosphatase-1B negatively regulates insulin signaling in L6 myocytes and fao hepatoma cells. J Biol Chem. 2001;276:10207–10211. doi: 10.1074/jbc.M009489200. [DOI] [PubMed] [Google Scholar]
- 174.Ehses JA, Lacraz G, Giroix M-H, Schmidlin F, Coulaud J, Kassis N, Irminger J-C, Kergoat M, Portha B, Homo-Delarche F, Donath MY. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Nat Acad Sci U S A. 2009;106:13998–14003. doi: 10.1073/pnas.0810087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ekelund U, Brage, Franks PW, Hennings S, Emms S, Wareham NJ. Physical activity energy expenditure predicts progression toward the metabolic syndrome independently of aerobic fitness in middle-aged healthy Caucasians. Diabetes Care. 2005;28:1195–1200. doi: 10.2337/diacare.28.5.1195. [DOI] [PubMed] [Google Scholar]
- 176.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan C-C, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283:1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 177.Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- 178.Ellis L, Morgan DO, Koshland DE, Jr, Clauser E, Moe GR, Bollag G, Roth RA, Rutter WJ. Linking functional domains of the human insulin receptor with the bacterial aspartate receptor. Proc Natl Acad Sci U S A. 1986;83:8137–8141. doi: 10.1073/pnas.83.21.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: Hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 180.Eriksson J, Taimela S, Koivisto VA. Exercise and the metabolic syndrome. Diabetologia. 1997;40:125–135. doi: 10.1007/s001250050653. [DOI] [PubMed] [Google Scholar]
- 181.Ernst MC, Issa M, Goralski KB, Sinal CJ. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology. 2010;151:1998–2007. doi: 10.1210/en.2009-1098. [DOI] [PubMed] [Google Scholar]
- 182.Ervin RB. National Health Statistics Report. Hyattsville, MD: 2009. Prevalence of Metabolic Syndrome Among Adults 20 Years of Age and Over, by Sex, Age, Race and Ethnicity, and Body Mass Index: United States, 2003–2006. [PubMed] [Google Scholar]
- 183.Evans EM, Van Pelt RE, Binder EF, Williams DB, Ehsani AA, Kohrt WM. Effects of HRT and exercise training on insulin action, glucose tolerance, and body composition in older women. J Appl Physiol. 2001;90:2033–2040. doi: 10.1152/jappl.2001.90.6.2033. [DOI] [PubMed] [Google Scholar]
- 184.Fagerholm S, Ortegren U, Karlsson M, Ruishalme I, Stralfors P. Rapid insulin-dependent endocytosis of the insulin receptor by caveolae in primary adipocytes. PLoS ONE. 2009;4:e5985. doi: 10.1371/journal.pone.0005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Falasca M, Hughes WE, Dominguez V, Sala G, Fostira F, Fang MQ, Cazzolli R, Shepherd PR, James DE, Maffucci T. The role of phosphoinositide 3-kinase C2α in insulin signaling. J Biol Chem. 2007;282:28226–28236. doi: 10.1074/jbc.M704357200. [DOI] [PubMed] [Google Scholar]
- 186.Famulla S, Lamers D, Hartwig S, Passlack W, Horrighs A, Cramer A, Lehr S, Sell H, Eckel J. Pigment epithelium-derived factor is one of the most abundant proteins secreted by human adipocytes and induces insulin resistance and inflammatory signaling in muscle and fat cells. Int J Obes (Lond) 2010;35:762–772. doi: 10.1038/ijo.2010.212. [DOI] [PubMed] [Google Scholar]
- 187.Farese RV, Sajan MP, Standaert ML. Atypical protein kinase C in insulin action and insulin resistance. Biochem Soc Trans. 2005;33:350–353. doi: 10.1042/BST0330350. [DOI] [PubMed] [Google Scholar]
- 188.Farese RV, Sajan MP, Yang H, Li P, Mastorides S, Gower WR, Jr, Nimal S, Choi CS, Kim S, Shulman GI, Kahn CR, Braun U, Leitges M. Muscle-specific knockout of PKC-lambda impairs glucose transport and induces metabolic and diabetic syndromes. J Clin Invest. 2007;117:2289–2301. doi: 10.1172/JCI31408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Farrell SW, Cheng YJ, Blair SN. Prevalence of the metabolic syndrome across cardiorespiratory fitness levels in women. Obes Res. 2004;12:824–830. doi: 10.1038/oby.2004.99. [DOI] [PubMed] [Google Scholar]
- 190.Febbraio MA, Koukoulas I. HSP72 gene expression progressively increases in human skeletal muscle during prolonged, exhaustive exercise. J Appl Physiol. 2000;89:1055–1060. doi: 10.1152/jappl.2000.89.3.1055. [DOI] [PubMed] [Google Scholar]
- 191.Febbraio MA, Steensberg A, Walsh R, Koukoulas I, van Hall G, Saltin B, Pedersen BK. Reduced glycogen availability is associated with an elevation in HSP72 in contracting human skeletal muscle. J Physiol. 2002;538:911–917. doi: 10.1113/jphysiol.2001.013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fenicchia LM, Kanaley JA, Azevedo JL, Jr, Miller CS, Weinstock RS, Carhart RL, Ploutz-Snyder LL. Influence of resistance exercise training on glucose control in women with type 2 diabetes. Metabolism. 2004;53:284–289. doi: 10.1016/j.metabol.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 193.Ferder L, Inserra F, Martinez-Maldonado M. Inflammation and the metabolic syndrome: Role of angiotensin II and oxidative stress. Curr Hypertens Rep. 2006;8:191–198. doi: 10.1007/s11906-006-0050-7. [DOI] [PubMed] [Google Scholar]
- 194.Ferrannini E, Haffner SM, Mitchell BD, Stern MP. Hyperinsulinaemia: The key feature of a cardiovascular and metabolic syndrome. Diabetologia. 1991;34:416–422. doi: 10.1007/BF00403180. [DOI] [PubMed] [Google Scholar]
- 195.Ferrara CM, Goldberg AP, Ortmeyer HK, Ryan AS. Effects of aerobic and resistive exercise training on glucose disposal and skeletal muscle metabolism in older men. J Gerontol A Biol Sci Med Sci. 2006;61:480–487. doi: 10.1093/gerona/61.5.480. [DOI] [PubMed] [Google Scholar]
- 196.Festa A, D’Agostino R, Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM. Chronic subclinical inflammation as part of the insulin resistance syndrome: The Insulin Resistance Atherosclerosis Study (IRAS) Circulation. 2000;102:42–47. doi: 10.1161/01.cir.102.1.42. [DOI] [PubMed] [Google Scholar]
- 197.Finley CE, LaMonte MJ, Waslien CI, Barlow CE, Blair SN, Nichaman MZ. Cardiorespiratory fitness, macronutrient intake, and the metabolic syndrome: The Aerobics Center Longitudinal Study. J Am Diet Assoc. 2006;106:673–679. doi: 10.1016/j.jada.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 198.Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. 2008;31:289–294. doi: 10.2337/dc07-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Flores MB, Fernandes MF, Ropelle ER, Faria MC, Ueno M, Velloso LA, Saad MJ, Carvalheira JB. Exercise improves insulin and leptin sensitivity in hypothalamus of Wistar rats. Diabetes. 2006;55:2554–2561. doi: 10.2337/db05-1622. [DOI] [PubMed] [Google Scholar]
- 200.Flynn MG, McFarlin BK. Toll-like receptor 4: Link to the anti-inflammatory effects of exercise? Exerc Sport Sci Rev. 2006;34:176–181. doi: 10.1249/01.jes.0000240027.22749.14. [DOI] [PubMed] [Google Scholar]
- 201.Ford ES. Prevalence of the metabolic syndrome defined by the International Diabetes Federation among adults in the U.S. Diabetes Care. 2005;28:2745–2749. doi: 10.2337/diacare.28.11.2745. [DOI] [PubMed] [Google Scholar]
- 202.Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among U.S. Adults. Diabetes Care. 2004;27:2444–2449. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 203.Ford ES, Kohl HW, Mokdad AH, Ajani UA. Sedentary behavior, physical activity, and the metabolic syndrome among U.S. adults[ast][ast] Obesity. 2005;13:608–614. doi: 10.1038/oby.2005.65. [DOI] [PubMed] [Google Scholar]
- 204.Ford ES, Li C, Sattar N. Metabolic syndrome and incident diabetes: Current state of the evidence. Diabetes Care. 2008;31:1898–1904. doi: 10.2337/dc08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Foster DW. Insulin resistance–a secret killer? N Engl J Med. 1989;320:733–734. doi: 10.1056/NEJM198903163201111. [DOI] [PubMed] [Google Scholar]
- 206.Franks PW, Ekelund U, Brage, Wong M-Y, Wareham NJ. Does the association of habitual physical activity with the metabolic syndrome differ by level of cardiorespiratory fitness? Diabetes Care. 2004;27:1187–1193. doi: 10.2337/diacare.27.5.1187. [DOI] [PubMed] [Google Scholar]
- 207.Fresno M, Alvarez R, Cuesta N. Toll-like receptors, inflammation, metabolism and obesity. Arch Physiol Biochem. 2011;117:151–164. doi: 10.3109/13813455.2011.562514. [DOI] [PubMed] [Google Scholar]
- 208.Friedman JE, Dohm GL, Leggett-Frazier N, Elton CW, Tapscott EB, Pories WP, Caro JF. Restoration of insulin responsiveness in skeletal muscle of morbidly obese patients after weight loss. Effect on muscle glucose transport and glucose transporter GLUT4. J Clin Invest. 1992;89:701–705. doi: 10.1172/JCI115638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 210.Frøsig C, Jørgensen SB, Hardie DG, Richter EA, Wojtaszewski JFP. 5′-AMP-activated protein kinase activity and protein expression are regulated by endurance training in human skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286:E411–E417. doi: 10.1152/ajpendo.00317.2003. [DOI] [PubMed] [Google Scholar]
- 211.Frosig C, Rose AJ, Treebak JT, Kiens B, Richter EA, Wojtaszewski JF. Effects of endurance exercise training on insulin signaling in human skeletal muscle: Interactions at the level of phosphatidylinositol 3-kinase, Akt, and AS160. Diabetes. 2007;56:2093–2102. doi: 10.2337/db06-1698. [DOI] [PubMed] [Google Scholar]
- 212.Fruebis J, Tsao T-S, Javorschi S, Ebbets-Reed D, Erickson MRS, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Nat Acad Sci U S A. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fruman DA, Cantley LC, Carpenter CL. Structural organization and alternative splicing of the murine phosphoinositide 3-kinase p85 alpha gene. Genomics. 1996;37:113–121. doi: 10.1006/geno.1996.0527. [DOI] [PubMed] [Google Scholar]
- 214.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, Watanabe E, Takagi T, Akiyoshi M, Ohtsubo T, Kihara S, Yamashita S, Makishima M, Funahashi T, Yamanaka S, Hiramatsu R, Matsuzawa Y, Shimomura I. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 215.Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, Sulsky R, Robl JA, Parker RA, Hotamisligil GS. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature. 2007;447:959–965. doi: 10.1038/nature05844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gaidhu MP, Perry RL, Noor F, Ceddia RB. Disruption of AMPKalpha1 signaling prevents AICAR-induced inhibition of AS160/TBC1D4 phosphorylation and glucose uptake in primary rat adipocytes. Mol Endocrinol. 2010;24:1434–1440. doi: 10.1210/me.2009-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Galassi A, Reynolds K, He J. Metabolic syndrome and risk of cardiovascular disease: A meta-analysis. Am J Med. 2006;119:812–819. doi: 10.1016/j.amjmed.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 218.Galluzzo A, Amato MC, Giordano C. Insulin resistance and polycystic ovary syndrome. Nutr Metab Cardiovasc Dis. 2008;18:511–518. doi: 10.1016/j.numecd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 219.Gami AS, Witt BJ, Howard DE, Erwin PJ, Gami LA, Somers VK, Montori VM. Metabolic syndrome and risk of incident cardiovascular events and death: A systematic review and meta-analysis of longitudinal studies. J Am Coll Cardiol. 2007;49:403–414. doi: 10.1016/j.jacc.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 220.Garvey WT, Maianu L, Hancock JA, Golichowski AM, Baron A. Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 1992;41:465–475. doi: 10.2337/diab.41.4.465. [DOI] [PubMed] [Google Scholar]
- 221.Garvey WT, Maianu L, Zhu JH, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386. doi: 10.1172/JCI1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Geering B, Cutillas PR, Vanhaesebroeck B. Regulation of class IA PI3Ks: Is there a role for monomeric PI3K subunits? Biochem Soc Trans. 2007;035:199–203. doi: 10.1042/BST0350199. [DOI] [PubMed] [Google Scholar]
- 223.Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: Mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. 2011;11:607–615. doi: 10.1038/nri3041. [DOI] [PubMed] [Google Scholar]
- 224.Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci. 2008;1:36–43. doi: 10.1111/j.1752-8062.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Goldstein BJ, Mahadev K, Wu X, Zhu L, Motoshima H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid Redox Signal. 2005;7:1021–1031. doi: 10.1089/ars.2005.7.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Goodpaster BH, He J, Watkins S, Kelley DE. Skeletal muscle lipid content and insulin resistance: Evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab. 2001;86:5755–5761. doi: 10.1210/jcem.86.12.8075. [DOI] [PubMed] [Google Scholar]
- 227.Goodpaster BH, Katsiaras A, Kelley DE. Enhanced fat oxidation through physical activity is associated with improvements in insulin sensitivity in obesity. Diabetes. 2003;52:2191–2197. doi: 10.2337/diabetes.52.9.2191. [DOI] [PubMed] [Google Scholar]
- 228.Goodyear LJ. The exercise pill - too good to be true? N Engl J Med. 2008;359:1842–1844. doi: 10.1056/NEJMcibr0806723. [DOI] [PubMed] [Google Scholar]
- 229.Goossens GH. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiol Behav. 2008;94:206–218. doi: 10.1016/j.physbeh.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 230.Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, Muruganandan S, Sinal CJ. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem. 2007;282:28175–28188. doi: 10.1074/jbc.M700793200. [DOI] [PubMed] [Google Scholar]
- 231.Gordon BA, Benson AC, Bird SR, Fraser SF. Resistance training improves metabolic health in type 2 diabetes: A systematic review. Diabetes Res Clin Pract. 2009;83:157–175. doi: 10.1016/j.diabres.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 232.Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 233.Gotto AM, Jr, Blackburn GL, Dailey GE, III, Garber AJ, Grundy SM, Sobel BE, Weir MR. The metabolic syndrome: A call to action. Coron Artery Dis. 2006;17:77–80. doi: 10.1097/00019501-200602000-00013. [DOI] [PubMed] [Google Scholar]
- 234.Graham TE, Yang Q, Bluher M, Hammarstedt A, Ciaraldi TP, Henry RR, Wason CJ, Oberbach A, Jansson P-A, Smith U, Kahn BB. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N Engl J Med. 2006;354:2552–2563. doi: 10.1056/NEJMoa054862. [DOI] [PubMed] [Google Scholar]
- 235.Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte biology. Adipocyte stress: The endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48:1905–1914. doi: 10.1194/jlr.R700007-JLR200. [DOI] [PubMed] [Google Scholar]
- 236.Gregor MF, Yang L, Fabbrini E, Mohammed BS, Eagon JC, Hotamisligil GS, Klein S. Endoplasmic reticulum stress is reduced in tissues of obese subjects after weight loss. Diabetes. 2009;58:693–700. doi: 10.2337/db08-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Grimditch GK, Barnard RJ, Hendricks L, Weitzman D. Peripheral insulin sensitivity as modified by diet and exercise training. Am J Clin Nutr. 1988;48:38–43. doi: 10.1093/ajcn/48.1.38. [DOI] [PubMed] [Google Scholar]
- 238.Grimditch GK, Barnard RJ, Sternlicht E, Whitson RH, Kaplan SA. Effect of diet on insulin binding and glucose transport in rat sarcolemmal vesicles. Am J Physiol. 1987;252:E420–E425. doi: 10.1152/ajpendo.1987.252.3.E420. [DOI] [PubMed] [Google Scholar]
- 239.Grundy SM. Point: The metabolic syndrome still lives. Clin Chem. 2005;51:1352–1354. doi: 10.1373/clinchem.2005.050989. [DOI] [PubMed] [Google Scholar]
- 240.Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C for the Conference Participants. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 241.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Jr, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation. 2005;112:2735–2752. doi: 10.1161/CIRCULATIONAHA.105.169404. [DOI] [PubMed] [Google Scholar]
- 242.Gum RJ, Gaede LL, Koterski SL, Heindel M, Clampit JE, Zinker BA, Trevillyan JM, Ulrich RG, Jirousek MR, Rondinone CM. Reduction of protein tyrosine phosphatase 1B increases insulin-dependent signaling in ob/ob mice. Diabetes. 2003;52:21–28. doi: 10.2337/diabetes.52.1.21. [DOI] [PubMed] [Google Scholar]
- 243.Guo S, Copps KD, Dong X, Park S, Cheng Z, Pocai A, Rossetti L, Sajan M, Farese RV, White MF. The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis. Mol Cell Biol. 2009;29:5070–5083. doi: 10.1128/MCB.00138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes. 2009;58:567–578. doi: 10.2337/db08-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gupte AA, Bomhoff GL, Touchberry CD, Geiger PC. Acute heat treatment improves insulin-stimulated glucose uptake in aged skeletal muscle. J Appl Physiol. 2011;110:451–457. doi: 10.1152/japplphysiol.00849.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Gustafson TA, He W, Craparo A, Schaub CD, O’Neill TJ. Phosphotyrosine-dependent interaction of SHC and insulin receptor substrate 1 with the NPEY motif of the insulin receptor via a novel non-SH2 domain. Mol Cell Biol. 1995;15:2500–2508. doi: 10.1128/mcb.15.5.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Gustavsson J, Parpal S, Karlsson M, Ramsing C, Thorn H, Borg M, Lindroth M, Peterson KH, Magnusson KE, Stralfors P. Localization of the insulin receptor in caveolae of adipocyte plasma membrane. FASEB J. 1999;13:1961–1971. [PubMed] [Google Scholar]
- 248.Haffner SM, Katz MS, Dunn JF. Increased upper body and overall adiposity is associated with decreased sex hormone binding globulin in postmenopausal women. Int J Obes (Lond) 1991;15:471–478. [PubMed] [Google Scholar]
- 249.Haffner SM, Valdez RA, Hazuda HP, Mitchell BD, Morales PA, Stern MP. Prospective analysis of the insulin-resistance syndrome (syndrome X) Diabetes. 1992;41:715–722. doi: 10.2337/diab.41.6.715. [DOI] [PubMed] [Google Scholar]
- 250.Haider DG, Mittermayer F, Schaller G, Artwohl M, Baumgartner-Parzer SM, Prager G, Roden M, Wolzt M. Free fatty acids normalize a rosiglitazone-induced visfatin release. Am J Physiol Endocrinol Metab. 2006;291:E885–E890. doi: 10.1152/ajpendo.00109.2006. [DOI] [PubMed] [Google Scholar]
- 251.Halldin M, Rosell M, de Faire U, Hellénius ML. The metabolic syndrome: Prevalence and association to leisure-time and work-related physical activity in 60-year-old men and women. Nutr Metab Cardiovasc Dis. 2007;17:349–357. doi: 10.1016/j.numecd.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 252.Hamaguchi M, Kojima T, Takeda N, Nakagawa T, Taniguchi H, Fujii K, Omatsu T, Nakajima T, Sarui H, Shimazaki M, Kato T, Okuda J, Ida K. The metabolic syndrome as a predictor of nonalcoholic fatty liver disease. Ann Intern Med. 2005;143:722–728. doi: 10.7326/0003-4819-143-10-200511150-00009. [DOI] [PubMed] [Google Scholar]
- 253.Han DH, Hancock CR, Jung SR, Higashida K, Kim SH, Holloszy JO. Deficiency of the mitochondrial electron transport chain in muscle does not cause insulin resistance. PLoS ONE. 2011;6:e19739. doi: 10.1371/journal.pone.0019739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hansel B, Giral P, Nobecourt E, Chantepie S, Bruckert E, Chapman MJ, Kontush A. Metabolic syndrome is associated with elevated oxidative stress and dysfunctional dense high-density lipoprotein particles displaying impaired antioxidative activity. J Clin Endocrinol Metab. 2004;89:4963–4971. doi: 10.1210/jc.2004-0305. [DOI] [PubMed] [Google Scholar]
- 255.Hansen PA, Gulve EA, Marshall BA, Gao J, Pessin J, Holloszy JO, Mueckler M. Skeletal muscle glucose transport and metabolism are enhanced in transgenic mice overexpressing the Glut4 glucose transporter. J Biol Chem. 1995;270:1679–1684. doi: 10.1074/jbc.270.5.1679. [DOI] [PubMed] [Google Scholar]
- 256.Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, Kadowaki H, Hagura R, Akanuma Y, Yazaki Y, Nagai R, Taniyama M, Matsubara K, Yoda M, Nakano Y, Kimura S, Tomita M, Ito C, Froguel P, Kadowaki T. Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes. 2002;51:536–540. doi: 10.2337/diabetes.51.2.536. [DOI] [PubMed] [Google Scholar]
- 257.Hassinen M, Lakka TA, Savonen K, Litmanen H, Kiviaho L, Laaksonen DE, Komulainen P, Rauramaa R. Cardiorespiratory fitness as a feature of metabolic syndrome in older men and women. Diabetes Care. 2008;31:1242–1247. doi: 10.2337/dc07-2298. [DOI] [PubMed] [Google Scholar]
- 258.Haunerland NH, Spener F. Fatty acid-binding proteins–insights from genetic manipulations. Prog Lipid Res. 2004;43:328–349. doi: 10.1016/j.plipres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 259.Hawley JA, Holloszy JO. Exercise: It’s the real thing! Nutr Rev. 2009;67:172–178. doi: 10.1111/j.1753-4887.2009.00185.x. [DOI] [PubMed] [Google Scholar]
- 260.Healy GN, Dunstan DW, Salmon J, Cerin E, Shaw JE, Zimmet PZ, Owen N. Objectively measured light-intensity physical activity is independently associated with 2-h plasma glucose. Diabetes Care. 2007;30:1384–1389. doi: 10.2337/dc07-0114. [DOI] [PubMed] [Google Scholar]
- 261.Heath GW, Gavin JR, III, Hinderliter JM, Hagberg JM, Bloomfield SA, Holloszy JO. Effects of exercise and lack of exercise on glucose tolerance and insulin sensitivity. J Appl Physiol. 1983;55:512–517. doi: 10.1152/jappl.1983.55.2.512. [DOI] [PubMed] [Google Scholar]
- 262.Heffernan KS, Jae SY, Vieira VJ, Iwamoto GA, Wilund KR, Woods JA, Fernhall B. C-reactive protein and cardiac vagal activity following resistance exercise training in young African-American and white men. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1098–R1105. doi: 10.1152/ajpregu.90936.2008. [DOI] [PubMed] [Google Scholar]
- 263.Hein TW, Singh U, Vasquez-Vivar J, Devaraj S, Kuo L, Jialal I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis. 2009;206:61–68. doi: 10.1016/j.atherosclerosis.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Heitmeier MR, Arnush M, Scarim AL, Corbett JA. Pancreatic beta-cell damage mediated by beta-cell production of interleukin-1. A novel mechanism for virus-induced diabetes. J Biol Chem. 2001;276:11151–11158. doi: 10.1074/jbc.M009159200. [DOI] [PubMed] [Google Scholar]
- 265.Helge JW, Dobrzyn A, Saltin B, Gorski J. Exercise and training effects on ceramide metabolism in human skeletal muscle. Exp Physiol. 2004;89:119–127. doi: 10.1113/expphysiol.2003.002605. [DOI] [PubMed] [Google Scholar]
- 266.Helledie T, Antonius M, Sorensen RV, Hertzel AV, Bernlohr DA, Kolvraa S, Kristiansen K, Mandrup S. Lipid-binding proteins modulate ligand-dependent trans-activation by peroxisome proliferator-activated receptors and localize to the nucleus as well as the cytoplasm. J Lipid Res. 2000;41:1740–1751. [PubMed] [Google Scholar]
- 267.Hevener AL, Olefsky JM, Reichart D, Nguyen MT, Bandyopadyhay G, Leung HY, Watt MJ, Benner C, Febbraio MA, Nguyen AK, Folian B, Subramaniam S, Gonzalez FJ, Glass CK, Ricote M. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J Clin Invest. 2007;117:1658–1669. doi: 10.1172/JCI31561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Higaki Y, Wojtaszewski JF, Hirshman MF, Withers DJ, Towery H, White MF, Goodyear LJ. Insulin receptor substrate-2 is not necessary for insulin- and exercise-stimulated glucose transport in skeletal muscle. J Biol Chem. 1999;274:20791–20795. doi: 10.1074/jbc.274.30.20791. [DOI] [PubMed] [Google Scholar]
- 269.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 270.Hojman P, Dethlefsen C, Brandt C, Hansen J, Pedersen L, Pedersen BK. Exercise-induced muscle-derived cytokines inhibit mammary cancer cell growth. Am J Physiol Endocrinol Metab. 2011;301:E504–E510. doi: 10.1152/ajpendo.00520.2010. [DOI] [PubMed] [Google Scholar]
- 271.Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, Narra K, Hoehn KL, Knotts TA, Siesky A, Nelson DH, Karathanasis SK, Fontenot GK, Birnbaum MJ, Summers SA. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–179. doi: 10.1016/j.cmet.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 272.Holland WL, Summers SA. Sphingolipids, insulin resistance, and metabolic disease: New insights from in vivo manipulation of sphingolipid metabolism. Endocr Rev. 2008;29:381–402. doi: 10.1210/er.2007-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Hollenbeck CB, Haskell W, Rosenthal M, Reaven GM. Effect of habitual physical activity on regulation of insulin-stimulated glucose disposal in older males. J Am Geriatr Soc. 1985;33:273–277. doi: 10.1111/j.1532-5415.1985.tb07116.x. [DOI] [PubMed] [Google Scholar]
- 274.Holloway GP, Chou CJ, Lally J, Stellingwerff T, Maher AC, Gavrilova O, Haluzik M, Alkhateeb H, Reitman ML, Bonen A. Increasing skeletal muscle fatty acid transport protein 1 (FATP1) targets fatty acids to oxidation and does not predispose mice to diet-induced insulin resistance. Diabetologia. 2011;54:1457–1467. doi: 10.1007/s00125-011-2114-8. [DOI] [PubMed] [Google Scholar]
- 275.Holme I, Tonstad S, Sogaard A, Larsen P, Haheim L. Leisure time physical activity in middle age predicts the metabolic syndrome in old age: Results of a 28-year follow-up of men in the Oslo study. BMC Public Health. 2007;7:154. doi: 10.1186/1471-2458-7-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Holten MK, Zacho M, Gaster M, Juel C, Wojtaszewski JFP, Dela F. Strength training increases insulin-mediated glucose uptake, GLUT4 content, and insulin signaling in skeletal muscle in patients with type 2 diabetes. Diabetes. 2004;53:294–305. doi: 10.2337/diabetes.53.2.294. [DOI] [PubMed] [Google Scholar]
- 277.Hood MS, Little JP, Tarnopolsky MA, Myslik F, Gibala MJ. Low-volume interval training improves muscle oxidative capacity in sedentary adults. Med Sci Sports Exerc. 2011;43:1849–1856. doi: 10.1249/MSS.0b013e3182199834. [DOI] [PubMed] [Google Scholar]
- 278.Hopps E, Canino B, Caimi G. Effects of exercise on inflammation markers in type 2 diabetic subjects. Acta diabetologica. 2011;48:183–189. doi: 10.1007/s00592-011-0278-9. [DOI] [PubMed] [Google Scholar]
- 279.Hori H, Sasaoka T, Ishihara H, Wada T, Murakami S, Ishiki M, Kobayashi M. Association of SH2-containing inositol phosphatase 2 with the insulin resistance of diabetic db/db mice. Diabetes. 2002;51:2387–2394. doi: 10.2337/diabetes.51.8.2387. [DOI] [PubMed] [Google Scholar]
- 280.Hotamisligil GS. The role of TNF-alpha and TNF receptors in obesity and insulin resistance. J Internal Medicine. 1999;245:621–625. doi: 10.1046/j.1365-2796.1999.00490.x. [DOI] [PubMed] [Google Scholar]
- 281.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. 1994;94:1543–1549. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271:665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 285.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 286.Houmard JA, Egan PC, Neufer PD, Friedman JE, Wheeler WS, Israel RG, Dohm GL. Elevated skeletal muscle glucose transporter levels in exercise-trained middle-aged men. J Physiol Endocrinol Metab. 1991;261:E437–E443. doi: 10.1152/ajpendo.1991.261.4.E437. [DOI] [PubMed] [Google Scholar]
- 287.Houmard JA, Hortobagyi T, Neufer PD, Johns RA, Fraser DD, Israel RG, Dohm GL. Training cessation does not alter GLUT-4 protein levels in human skeletal muscle. J Appl Physiol. 1993;74:776–781. doi: 10.1152/jappl.1993.74.2.776. [DOI] [PubMed] [Google Scholar]
- 288.Houmard JA, Shaw CD, Hickey MS, Tanner CJ. Effect of short-term exercise training on insulin-stimulated PI 3- kinase activity in human skeletal muscle. Am J Physiol. 1999;277:E1055–E1060. doi: 10.1152/ajpendo.1999.277.6.E1055. [DOI] [PubMed] [Google Scholar]
- 289.Houmard JA, Shinebarger MH, Dolan PL, Leggett-Frazier N, Bruner RK, McCammon MR, Israel RG, Dohm GL. Exercise training increases GLUT-4 protein concentration in previously sedentary middle-aged men. Am J Physiol. 1993;264:E896–E901. doi: 10.1152/ajpendo.1993.264.6.E896. [DOI] [PubMed] [Google Scholar]
- 290.Houmard JA, Tanner CJ, Slentz CA, Duscha BD, McCartney JS, Kraus WE. Effect of the volume and intensity of exercise training on insulin sensitivity. J Appl Physiol. 2004;96:101–106. doi: 10.1152/japplphysiol.00707.2003. [DOI] [PubMed] [Google Scholar]
- 291.Houmard JA, Tyndall GL, Midyette JB, Hickey MS, Dolan PL, Gavigan KE, Weidner ML, Dohm GL. Effect of reduced training and training cessation on insulin action and muscle GLUT-4. J Appl Physiol. 1996;81:1162–1168. doi: 10.1152/jappl.1996.81.3.1162. [DOI] [PubMed] [Google Scholar]
- 292.Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- 293.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 294.Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC. High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes. 2007;56:2016–2027. doi: 10.2337/db07-0197. [DOI] [PubMed] [Google Scholar]
- 295.Hubbard S, Wei L, Ellis l, Hendrickson W. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature. 1994;372:746–754. doi: 10.1038/372746a0. [DOI] [PubMed] [Google Scholar]
- 296.Hubbard SR. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 1997;16:5572–5581. doi: 10.1093/emboj/16.18.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Huber J, Kiefer FW, Zeyda M, Ludvik B, Silberhumer GR, Prager G, Zlabinger GJ, Stulnig TM. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J Clin Endocrinol Metab. 2008;93:3215–3221. doi: 10.1210/jc.2007-2630. [DOI] [PubMed] [Google Scholar]
- 298.Huber K. Plasminogen activator inhibitor type-1 (part two): Role for failure of thrombolytic therapy. PAI-1 resistance as a potential benefit for new fibrinolytic agents. J Thromb Thrombolysis. 2001;11:195–202. doi: 10.1023/a:1011952602122. [DOI] [PubMed] [Google Scholar]
- 299.Hughes VA, Fiatarone MA, Fielding RA, Kahn BB, Ferrara CM, Shepherd P, Fisher EC, Wolfe RR, Elahi D, Evans WJ. Exercise increases muscle GLUT-4 levels and insulin action in subjects with impaired glucose tolerance. Am J Physiol. 1993;264:E855–E862. doi: 10.1152/ajpendo.1993.264.6.E855. [DOI] [PubMed] [Google Scholar]
- 300.Hung J, McQuillan BM, Thompson PL, Beilby JP. Circulating adiponectin levels associate with inflammatory markers, insulin resistance and metabolic syndrome independent of obesity. Int J Obes (Lond) 2008;32:772–779. doi: 10.1038/sj.ijo.0803793. [DOI] [PubMed] [Google Scholar]
- 301.Hurley BF, Hagberg JM, Goldberg AP, Seals DR, Ehsani AA, Brennan RE, Holloszy JO. Resistive training can reduce coronary risk factors without altering VO2max or percent body fat. Med Sci Sports Exerc. 1988;20:150–154. doi: 10.1249/00005768-198820020-00008. [DOI] [PubMed] [Google Scholar]
- 302.Ibanez J, Izquierdo M, Arguelles I, Forga L, Larrion JL, Garcia-Unciti M, Idoate F, Gorostiaga EM. Twice-weekly progressive resistance training decreases abdominal fat and improves insulin sensitivity in older men with type 2 diabetes. Diabetes Care. 2005;28:662–667. doi: 10.2337/diacare.28.3.662. [DOI] [PubMed] [Google Scholar]
- 303.IDF. Worldwide definition of the metabolic syndrome. 2005 http://www.idf.org.
- 304.Ilanne-Parikka P, Eriksson JG, Lindström J, Peltonen M, Aunola S, Hämäläinen H, Keinänen-Kiukaanniemi S, Laakso M, Valle TT, Lahtela J, Uusitupa M, Tuomilehto J. Effect of lifestyle intervention on the occurrence of metabolic syndrome and its components in the Finnish diabetes prevention study. Diabetes Care. 2008;31:805–807. doi: 10.2337/dc07-1117. [DOI] [PubMed] [Google Scholar]
- 305.Ilanne-Parikka P, Laaksonen DE, Eriksson JG, Lakka TA, Lindstr J, Peltonen M, Aunola S, Keinänen-Kiukaanniemi S, Uusitupa M, Tuomilehto J. Leisure-time physical activity and the metabolic syndrome in the Finnish diabetes prevention study. Diabetes Care. 2010;33:1610–1617. doi: 10.2337/dc09-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Irwin ML, Ainsworth BE, Mayer-Davis EJ, Addy CL, Pate RR, Durstine JL. Physical activity and the metabolic syndrome in a tri-ethnic sample of women. Obes Res. 2002;10:1030–1037. doi: 10.1038/oby.2002.140. [DOI] [PubMed] [Google Scholar]
- 307.Ishihara H, Sasaoka T, Kagawa S, Murakami S, Fukui K, Kawagishi Y, Yamazaki K, Sato A, Iwata M, Urakaze M, Ishiki M, Wada T, Yaguchi S, Tsuneki H, Kimura I, Kobayashi M. Association of the polymorphisms in the 5′-untranslated region of PTEN gene with type 2 diabetes in a Japanese population. FEBS Lett. 2003;554:450–454. doi: 10.1016/s0014-5793(03)01225-0. [DOI] [PubMed] [Google Scholar]
- 308.Ishii T, Yamakita T, Sato T, Tanaka S, Fujii S. Resistance training improves insulin sensitivity in NIDDM subjects without altering maximal oxygen uptake. Diabetes Care. 1998;21:1353–1355. doi: 10.2337/diacare.21.8.1353. [DOI] [PubMed] [Google Scholar]
- 309.Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, Taskinen MR, Groop L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 310.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 311.Izumiya Y, Bina HA, Ouchi N, Akasaki Y, Kharitonenkov A, Walsh K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008;582:3805–3810. doi: 10.1016/j.febslet.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA. 2002;288:462–467. doi: 10.1001/jama.288.4.462. [DOI] [PubMed] [Google Scholar]
- 313.Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen C-D, Haring H-U. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of tpe 2 diabetic subjects. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 314.JeBailey L, Rudich A, Huang X, Ciano-Oliveira CD, Kapus A, Klip A. Skeletal muscle cells and adipocytes differ in their reliance on TC10 and Rac for insulin-induced actin remodeling. Mol Endocrinol. 2004;18:359–372. doi: 10.1210/me.2003-0294. [DOI] [PubMed] [Google Scholar]
- 315.Jeppesen J, Mogensen M, Prats C, Sahlin K, Madsen K, Kiens B. FAT/CD36 is localized in sarcolemma and in vesicle-like structures in subsarcolemma regions but not in mitochondria. J Lipid Res. 2010;51:1504–1512. doi: 10.1194/jlr.M003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Johnson JL, Slentz CA, Houmard JA, Samsa GP, Duscha BD, Aiken LB, McCartney JS, Tanner CJ, Kraus WE. Exercise training amount and intensity effects on metabolic syndrome (from Studies of a Targeted Risk Reduction Intervention through Defined Exercise) Am J Cardiol. 2007;100:1759–1766. doi: 10.1016/j.amjcard.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Joost HG, Thorens B. The extended GLUT-family of sugar/polyol transport facilitators: Nomenclature, sequence characteristics, and potential function of its novel members (review) Mol Membr Biol. 2001;18:247–256. doi: 10.1080/09687680110090456. [DOI] [PubMed] [Google Scholar]
- 318.Jung HS, Park KH, Cho YM, Chung SS, Cho HJ, Cho SY, Kim SJ, Kim SY, Lee HK, Park KS. Resistin is secreted from macrophages in atheromas and promotes atherosclerosis. Cardiovasc Res. 2006;69:76–85. doi: 10.1016/j.cardiores.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 319.Jurca R, Lamonte MJ, Barlow CE, Kampert JB, Church TS, Blair SN. Association of muscular strength with incidence of metabolic syndrome in men. Med Sci Sports Exerc. 2005;37:1849–1855. doi: 10.1249/01.mss.0000175865.17614.74. [DOI] [PubMed] [Google Scholar]
- 320.Jurca R, Lamonte MJ, Church TS, Earnest CP, Fitzgerald SJ, Barlow CE, Jordan AN, Kampert JB, Blair SN. Associations of muscle strength and fitness with metabolic syndrome in men. Med Sci Sports Exerc. 2004;36:1301–1307. doi: 10.1249/01.mss.0000135780.88930.a9. [DOI] [PubMed] [Google Scholar]
- 321.Kaburagi Y, Yamamoto-Honda R, Tobe K, Ueki K, Yachi M, Akanuma Y, Stephens R, Kaplan D, Yazaki Y, Kadowaki T. The role of the NPXY motif in the insulin receptor in tyrosine phosphorylation of insulin receptor substrate-1 and Shc. Endocrinology. 1995;136:3437–3443. doi: 10.1210/endo.136.8.7543044. [DOI] [PubMed] [Google Scholar]
- 322.Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG. Fatty acid binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem. 2011;287:3415–3424. doi: 10.1074/jbc.M111.304907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev. 2005;26:439–451. doi: 10.1210/er.2005-0005. [DOI] [PubMed] [Google Scholar]
- 324.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Kagawa S, Soeda Y, Ishihara H, Oya T, Sasahara M, Yaguchi S, Oshita R, Wada T, Tsuneki H, Sasaoka T. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology. 2008;149:642–650. doi: 10.1210/en.2007-0820. [DOI] [PubMed] [Google Scholar]
- 326.Kahn R, Buse J, Ferrannini E, Stern M. The metabolic syndrome: Time for a critical appraisal: Joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2005;28:2289–2304. doi: 10.2337/diacare.28.9.2289. [DOI] [PubMed] [Google Scholar]
- 327.Kalmijn S, Foley D, White L, Burchfiel CM, Curb JD, Petrovitch H, Ross GW, Havlik RJ, Launer LJ. Metabolic cardiovascular syndrome and risk of dementia in japanese- american elderly men: The honolulu-asia aging study. Arterioscler Thromb Vasc Biol. 2000;20:2255–2260. doi: 10.1161/01.atv.20.10.2255. [DOI] [PubMed] [Google Scholar]
- 328.Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, Yamauchi T, Ueki K, Oishi Y, Nishimura S, Manabe I, Hashimoto H, Ohnishi Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Nagai R, Kadowaki T. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem. 2006;281:26602–26614. doi: 10.1074/jbc.M601284200. [DOI] [PubMed] [Google Scholar]
- 329.Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, Kasuga M. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, Lienhard GE. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem. 2002;277:22115–22118. doi: 10.1074/jbc.C200198200. [DOI] [PubMed] [Google Scholar]
- 331.Kanzaki M, Mora S, Hwang JB, Saltiel AR, Pessin JE. Atypical protein kinase C (PKCzeta/lambda) is a convergent downstream target of the insulin-stimulated phosphatidylinositol 3-kinase and TC10 signaling pathways. J Cell Biol. 2004;164:279–290. doi: 10.1083/jcb.200306152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Kanzaki M, Watson RT, Khan AH, Pessin JE. Insulin stimulates actin comet tails on intracellular GLUT4-containing compartments in differentiated 3T3L1 adipocytes. J Biol Chem. 2001;276:49331–49336. doi: 10.1074/jbc.M109657200. [DOI] [PubMed] [Google Scholar]
- 333.Kaplan NM. The deadly quartet. Upper-body obesity, glucose intolerance, hypertriglyceridemia, and hypertension. Arch Intern Med. 1989;149:1514–1520. doi: 10.1001/archinte.149.7.1514. [DOI] [PubMed] [Google Scholar]
- 334.Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54:1692–1697. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- 335.Kasapis C, Thompson PD. The effects of physical activity on serum C-reactive protein and inflammatory markers: A systematic review. J Am Coll Cardiol. 2005;45:1563–1569. doi: 10.1016/j.jacc.2004.12.077. [DOI] [PubMed] [Google Scholar]
- 336.Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature. 1995;377:151–155. doi: 10.1038/377151a0. [DOI] [PubMed] [Google Scholar]
- 337.Katzmarzyk PT, Church TS, Blair SN. Cardiorespiratory fitness attenuates the effects of the metabolic syndrome on all-cause and cardiovascular disease mortality in men. Arch Intern Med. 2004;164:1092–1097. doi: 10.1001/archinte.164.10.1092. [DOI] [PubMed] [Google Scholar]
- 338.Katzmarzyk PT, Church TS, Janssen I, Ross R, Blair SN. Metabolic syndrome, obesity, and mortality. Diabetes Care. 2005;28:391–397. doi: 10.2337/diacare.28.2.391. [DOI] [PubMed] [Google Scholar]
- 339.Katzmarzyk PT, Janssen I, Ross R, Church TS, Blair SN. The importance of waist circumference in the definition of metabolic syndrome: Prospective analyses of mortality in men. Diabetes Care. 2006;29:404–409. doi: 10.2337/diacare.29.02.06.dc05-1636. [DOI] [PubMed] [Google Scholar]
- 340.Katzmarzyk PT, Leon AS, Wilmore JH, Skinner JS, Rao DC, Rankinen T, Bouchard C. Targeting the metabolic syndrome with exercise: Evidence from the HERITAGE Family Study. Med Sci Sports Exerc. 2003;35:1703–1709. doi: 10.1249/01.MSS.0000089337.73244.9B. [DOI] [PubMed] [Google Scholar]
- 341.Kaur J, Adya R, Tan BK, Chen J, Randeva HS. Identification of chemerin receptor (ChemR23) in human endothelial cells: Chemerin-induced endothelial angiogenesis. Biochem Biophys Res Commun. 2010;391:1762–1768. doi: 10.1016/j.bbrc.2009.12.150. [DOI] [PubMed] [Google Scholar]
- 342.Kavanaugh WM, Williams LT. An alternative to SH2 domains for binding tyrosine-phosphorylated proteins. Science. 1994;266:1862–1865. doi: 10.1126/science.7527937. [DOI] [PubMed] [Google Scholar]
- 343.Kawamoto R, Kohara K, Tabara Y, Miki T. An association between metabolic syndrome and the estimated glomerular filtration rate. Intern Med. 2008;47:1399–1406. doi: 10.2169/internalmedicine.47.1202. [DOI] [PubMed] [Google Scholar]
- 344.Kawanishi N, Yano H, Yokogawa Y, Suzuki K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc Immunol Rev. 2010;16:105–118. [PubMed] [Google Scholar]
- 345.Keaney JF, Jr, Larson MG, Vasan RS, Wilson PWF, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in The Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 346.Kelly KR, Blaszczak A, Haus JM, Patrick-Melin A, Fealy CE, Solomon TPJ, Kalinski MI, Kirwan JP. A 7-d exercise program increases high–molecular weight adiponectin in obese adults. Med Sci Sports Exerc. 2012;44:69–74. doi: 10.1249/MSS.0b013e318228bf85. [DOI] [PubMed] [Google Scholar]
- 347.Kemmler W, Von Stengel S, Engelke K, Kalender WA. Exercise decreases the risk of metabolic syndrome in elderly females. Med Sci Sports Exerc. 2009;41:297–305. doi: 10.1249/MSS.0b013e31818844b7. [DOI] [PubMed] [Google Scholar]
- 348.Khan AH, Capilla E, Hou JC, Watson RT, Smith JR, Pessin JE. Entry of newly synthesized GLUT4 into the insulin-responsive storage compartment is dependent upon both the amino terminus and the large cytoplasmic loop. J Biol Chem. 2004;279:37505–37511. doi: 10.1074/jbc.M405694200. [DOI] [PubMed] [Google Scholar]
- 349.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Kiens B, Kristiansen S, Jensen P, Richter EA, Turcotte LP. Membrane associated fatty acid binding protein (FABPpm) in human skeletal muscle is increased by endurance training. Biochem Biophys Res Commun. 1997;231:463–465. doi: 10.1006/bbrc.1997.6118. [DOI] [PubMed] [Google Scholar]
- 351.Kim JY, Hickner RC, Cortright RL, Dohm GL, Houmard JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. 2000;279:E1039–E1044. doi: 10.1152/ajpendo.2000.279.5.E1039. [DOI] [PubMed] [Google Scholar]
- 352.King DS, Dalsky GP, Clutter WE, Young DA, Staten MA, Cryer PE, Holloszy JO. Effects of exercise and lack of exercise on insulin sensitivity and responsiveness. J Appl Physiol. 1988;64:1942–1946. doi: 10.1152/jappl.1988.64.5.1942. [DOI] [PubMed] [Google Scholar]
- 353.King DS, Dalsky GP, Staten MA, Clutter WE, Van Houten DR, Holloszy JO. Insulin action and secretion in endurance-trained and untrained humans. J Appl Physiol. 1987;63:2247–2252. doi: 10.1152/jappl.1987.63.6.2247. [DOI] [PubMed] [Google Scholar]
- 354.Kirchgessner TG, Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Tumor necrosis factor-alpha contributes to obesity-related hyper-leptinemia by regulating leptin release from adipocytes. J Clin Invest. 1997;100:2777–2782. doi: 10.1172/JCI119824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Kirwan JP, del Aguila LF, Hernandez JM, Williamson DL, O’Gorman DJ, Lewis R, Krishnan RK. Regular exercise enhances insulin activation of IRS-1-associated PI3- kinase in human skeletal muscle. J Appl Physiol. 2000;88:797–803. doi: 10.1152/jappl.2000.88.2.797. [DOI] [PubMed] [Google Scholar]
- 356.Kirwan JP, Solomon TP, Wojta DM, Staten MA, Holloszy JO. Effects of 7 days of exercise training on insulin sensitivity and responsiveness in type 2 diabetes mellitus. Am J Physiol Endocrinol Metab. 2009;297:E151–E156. doi: 10.1152/ajpendo.00210.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim Y-B, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20:5479–5489. doi: 10.1128/mcb.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Klein BE, Klein R, Lee KE. Components of the metabolic syndrome and risk of cardiovascular disease and diabetes in beaver dam. Diabetes Care. 2002;25:1790–1794. doi: 10.2337/diacare.25.10.1790. [DOI] [PubMed] [Google Scholar]
- 359.Klimcakova E, Polak J, Moro C, Hejnova J, Majercik M, Viguerie N, Berlan M, Langin D, Stich V. Dynamic strength training improves insulin sensitivity without altering plasma levels and gene expression of adipokines in subcutaneous adipose tissue in obese men. J Clin Endocrinol Metab. 2006;91:5107–5112. doi: 10.1210/jc.2006-0382. [DOI] [PubMed] [Google Scholar]
- 360.Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi: 10.2337/diabetes.52.11.2784. [DOI] [PubMed] [Google Scholar]
- 361.Knudsen SH, Hansen LS, Pedersen M, Dejgaard T, Hansen J, Hall GV, Thomsen C, Solomon TPJ, Pedersen BK, Krogh-Madsen R. Changes in insulin sensitivity precede changes in body composition during 14 days of step reduction combined with overfeeding in healthy young men. J Appl Physiol. 2012;113:7–15. doi: 10.1152/japplphysiol.00189.2011. [DOI] [PubMed] [Google Scholar]
- 362.Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab. 2007;21:621–640. doi: 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 363.Kotani K, Ogawa W, Matsumoto M, Kitamura T, Sakaue H, Hino Y, Miyake K, Sano W, Akimoto K, Ohno S, Kasuga M. Requirement of atypical protein kinase clambda for insulin stimulation of glucose uptake but not for Akt activation in 3T3-L1 adipocytes. Mol Cell Biol. 1998;18:6971–6982. doi: 10.1128/mcb.18.12.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Kovacikova M, Sengenes C, Kovacova Z, Siklova-Vitkova M, Klimcakova E, Polak J, Rossmeislova L, Bajzova M, Hejnova J, Hnevkovska Z, Bouloumie A, Langin D, Stich V. Dietary intervention-induced weight loss decreases macrophage content in adipose tissue of obese women. Int J Obes (Lond) 2010;35:91–98. doi: 10.1038/ijo.2010.112. [DOI] [PubMed] [Google Scholar]
- 365.Krabbe K, Nielsen A, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, Fischer C, Lindegaard B, Petersen A, Taudorf S, Secher N, Pilegaard H, Bruunsgaard H, Pedersen B. Brain-derived neu-rotrophic factor (BDNF) and type 2 diabetes. Diabetologia. 2007;50:431–438. doi: 10.1007/s00125-006-0537-4. [DOI] [PubMed] [Google Scholar]
- 366.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem. 2006;281:31478–31485. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- 367.Kressel G, Trunz B, Bub A, Hülsmann O, Wolters M, Lichtinghagen R, Stichtenoth DO, Hahn A. Systemic and vascular markers of inflammation in relation to metabolic syndrome and insulin resistance in adults with elevated atherosclerosis risk. Atherosclerosis. 2009;202:263–271. doi: 10.1016/j.atherosclerosis.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 368.Krisan AD, Collins DE, Crain AM, Kwong CC, Singh MK, Bernard JR, Yaspelkis BB., III Resistance training enhances components of the insulin signaling cascade in normal and high-fat-fed rodent skeletal muscle. J Appl Physiol. 2004;96:1691–1700. doi: 10.1152/japplphysiol.01054.2003. [DOI] [PubMed] [Google Scholar]
- 369.Kriska AM, Pereira MA, Hanson RL, de Courten MP, Zimmet PZ, Alberti KG, Chitson P, Bennett PH, Narayan KM, Knowler WC. Association of physical activity and serum insulin concentrations in two populations at high risk for type 2 diabetes but differing by BMI. Diabetes Care. 2001;24:1175–1180. doi: 10.2337/diacare.24.7.1175. [DOI] [PubMed] [Google Scholar]
- 370.Krogh-Madsen R, Thyfault JP, Broholm C, Mortensen OH, Olsen RH, Mounier R, Plomgaard P, van Hall G, Booth FW, Pedersen BK. A 2-wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J Appl Physiol. 2010;108:1034–1040. doi: 10.1152/japplphysiol.00977.2009. [DOI] [PubMed] [Google Scholar]
- 371.Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: A 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 372.Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, Yano W, Froguel P, Nagai R, Kimura S, Kadowaki T, Noda T. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866. doi: 10.1074/jbc.C200251200. [DOI] [PubMed] [Google Scholar]
- 373.Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Insulin hypersensitivity and resistance to streptozotocin-induced diabetes in mice lacking PTEN in adipose tissue. Mol Cell Biol. 2005;25:2498–2510. doi: 10.1128/MCB.25.6.2498-2510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Laaksonen DE, Lakka HM, Salonen JT, Niskanen LK, Rauramaa R, Lakka TA. Low levels of leisure-time physical activity and cardiorespiratory fitness predict development of the metabolic syndrome. Diabetes Care. 2002;25:1612–1618. doi: 10.2337/diacare.25.9.1612. [DOI] [PubMed] [Google Scholar]
- 375.Lacraz G, Giroix M-H, Kassis N, Coulaud J, Galinier A, Noll C, Cornut M, Schmidlin F, Paul J-L, Janel N, Irminger J-C, Kergoat M, Portha B, Donath MY, Ehses JA, Homo-Delarche F. Islet endothelial activation and oxidative stress gene expression is reduced by IL-1Ra treatment in the type 2 diabetic GK rat. PLoS One. 2009;4:e6963. doi: 10.1371/journal.pone.0006963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: Prevention by rosiglitazone. Biochem Biophys Res Commun. 2003;311:372–379. doi: 10.1016/j.bbrc.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 377.Lakka HM, Laaksonen DE, Lakka TA, Niskanen LK, Kumpusalo E, Tuomilehto J, Salonen JT. The metabolic syndrome and total and cardiovascular disease mortality in middle-aged men. JAMA. 2002;288:2709–2716. doi: 10.1001/jama.288.21.2709. [DOI] [PubMed] [Google Scholar]
- 378.Lakka TA, Laaksonen DE, Lakka HM, Mannikko N, Niskanen LK, Rauramaa R, Salonen JT. Sedentary lifestyle, poor cardiorespiratory fitness, and the metabolic syndrome. Med Sci Sports Exerc. 2003;35:1279–1286. doi: 10.1249/01.MSS.0000079076.74931.9A. [DOI] [PubMed] [Google Scholar]
- 379.LaMonte MJ, Barlow CE, Jurca R, Kampert JB, Church TS, Blair SN. Cardiorespiratory fitness is inversely associated with the incidence of metabolic syndrome: A prospective study of men and women. Circulation. 2005;112:505–512. doi: 10.1161/CIRCULATIONAHA.104.503805. [DOI] [PubMed] [Google Scholar]
- 380.Landin K, Tengborn L, Smith U. Elevated fibrinogen and plasminogen activator inhibitor (PAI-1) In hypertension are related to metabolic risk factors for cardiovascular disease. J Intern Med. 1990;227:273–278. doi: 10.1111/j.1365-2796.1990.tb00157.x. [DOI] [PubMed] [Google Scholar]
- 381.Landsberg L. Body fat distribution and cardiovascular risk: A tale of 2 sites. Arch Intern Med. 2008;168:1607–1608. doi: 10.1001/archinte.168.15.1607. [DOI] [PubMed] [Google Scholar]
- 382.Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA. Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock. 2003;19:538–546. doi: 10.1097/01.shk.0000055237.25446.80. [DOI] [PubMed] [Google Scholar]
- 383.Larance M, Ramm G, James DE. The GLUT4 code. Mol Endocrinol. 2008;22:226–233. doi: 10.1210/me.2007-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Larsen J, Andersen M, Kragstrup J, Gram LF. High persistence of statin use in a Danish population: Compliance study 1993–1998. Br J Clin Pharmacol. 2002;53:375–378. doi: 10.1046/j.1365-2125.2002.01563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 386.Lazar DF, Saltiel AR. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov. 2006;5:333–342. doi: 10.1038/nrd2007. [DOI] [PubMed] [Google Scholar]
- 387.LeBlanc J, Nadeau A, Boulay M, Rousseau-Migneron S. Effects of physical training and adiposity on glucose metabolism and 125I-insulin binding. J Appl Physiol. 1979;46:235–239. doi: 10.1152/jappl.1979.46.2.235. [DOI] [PubMed] [Google Scholar]
- 388.LeBlanc J, Nadeau A, Richard D, Tremblay A. Studies on the sparing effect of exercise on insulin requirements in human subjects. Metabolism. 1981;30:1119–1124. doi: 10.1016/0026-0495(81)90057-3. [DOI] [PubMed] [Google Scholar]
- 389.Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 390.Lee J, Kim S-U, Kang H-S. Low cardio/respiratory fitness as an independent predictor of metabolic syndrome in Korean young men. Eur J Appl Physiol. 2010;108:633–639. doi: 10.1007/s00421-009-1251-y. [DOI] [PubMed] [Google Scholar]
- 391.Lee J, O’Hare T, Pilch PF, Shoelson SE. Insulin receptor autophosphorylation occurs asymmetrically. J Biol Chem. 1993;268:4092–4098. [PubMed] [Google Scholar]
- 392.Lee J, Pilch PF. The insulin receptor: Structure, function, and signaling. Am J Physiol. 1994;266:C319–C334. doi: 10.1152/ajpcell.1994.266.2.C319. [DOI] [PubMed] [Google Scholar]
- 393.Lee JM, Kim JH, Son HS, Hong EG, Yu JM, Han KA, Min KW, Chang SA. Valsartan increases circulating adiponectin levels without changing HOMA-IR in patients with type 2 diabetes mellitus and hypertension. J Int Med Res. 2010;38:234–241. doi: 10.1177/147323001003800128. [DOI] [PubMed] [Google Scholar]
- 394.Leiter LA, Lewanczuk RZ. Of the renin-angiotensin system and reactive oxygen species Type 2 diabetes and angiotensin II inhibition. Am J Hypertens. 2005;18:121–128. doi: 10.1016/j.amjhyper.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 395.Lemieux I, Pascot A, Prud’homme D, Almeras N, Bogaty P, Nadeau A, Bergeron J, Despres JP. Elevated C-reactive protein: Another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21:961–967. doi: 10.1161/01.atv.21.6.961. [DOI] [PubMed] [Google Scholar]
- 396.Levin BE, Dunn-Meynell AA, Banks WA. Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol. 2004;286:R143–R150. doi: 10.1152/ajpregu.00393.2003. [DOI] [PubMed] [Google Scholar]
- 397.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 398.Liao Y, Kwon S, Shaughnessy S, Wallace P, Hutto A, Jenkins AJ, Klein RL, Garvey WT. Critical evaluation of adult treatment panel III criteria in identifying insulin resistance with dyslipidemia. Diabetes Care. 2004;27:978–983. doi: 10.2337/diacare.27.4.978. [DOI] [PubMed] [Google Scholar]
- 399.Lin Y, Lee H, Berg AH, Lisanti MP, Shapiro L, Scherer PE. The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem. 2000;275:24255–24263. doi: 10.1074/jbc.M002137200. [DOI] [PubMed] [Google Scholar]
- 400.Little JP, Gillen JB, Percival ME, Safdar A, Tarnopolsky MA, Punthakee Z, Jung ME, Gibala MJ. Low-volume high-intensity interval training reduces hyperglycemia and increases muscle mitochondrial capacity in patients with type 2 diabetes. J Appl Physiol. 2011;111:1554–1560. doi: 10.1152/japplphysiol.00921.2011. [DOI] [PubMed] [Google Scholar]
- 401.Little JP, Safdar A, Wilkin GP, Tarnopolsky MA, Gibala MJ. A practical model of low-volume high-intensity interval training induces mitochondrial biogenesis in human skeletal muscle: Potential mechanisms. J Physiol. 2010;588:1011–1022. doi: 10.1113/jphysiol.2009.181743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Liu G, Hou JC, Watson RT, Pessin JE. Initial entry of IRAP into the insulin-responsive storage compartment occurs prior to basal or insulin-stimulated plasma membrane recycling. Am J Physiol Endocrinol Metab. 2005;289:E746–E752. doi: 10.1152/ajpendo.00175.2005. [DOI] [PubMed] [Google Scholar]
- 403.Liu J, Kimura A, Baumann CA, Saltiel AR. APS facilitates c-Cbl tyrosine phosphorylation and GLUT4 translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell Biol. 2002;22:3599–3609. doi: 10.1128/MCB.22.11.3599-3609.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Loh K, Deng H, Fukushima A, Cai X, Boivin B, Galic S, Bruce C, Shields BJ, Skiba B, Ooms LM, Stepto N, Wu B, Mitchell CA, Tonks NK, Watt MJ, Febbraio MA, Crack PJ, Andrikopoulos S, Tiganis T. Reactive oxygen species enhance insulin sensitivity. Cell metabolism. 2009;10:260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Lohmann D, Liebold F, Heilmann W, Senger H, Pohl A. Diminished insulin response in highly trained athletes. Metabolism. 1978;27:521–524. doi: 10.1016/0026-0495(78)90017-3. [DOI] [PubMed] [Google Scholar]
- 406.Luiken JJ, Arumugam Y, Dyck DJ, Bell RC, Pelsers MM, Turcotte LP, Tandon NN, Glatz JF, Bonen A. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem. 2001;276:40567–40573. doi: 10.1074/jbc.M100052200. [DOI] [PubMed] [Google Scholar]
- 407.Luiken JJ, Dyck DJ, Han XX, Tandon NN, Arumugam Y, Glatz JF, Bonen A. Insulin induces the translocation of the fatty acid transporter FAT/CD36 to the plasma membrane. Am J Physiol Endocrinol Metab. 2002;282:E491–E495. doi: 10.1152/ajpendo.00419.2001. [DOI] [PubMed] [Google Scholar]
- 408.Luotola K, Paakkonen R, Alanne M, Lanki T, Moilanen L, Surakka I, Pietila A, Kahonen M, Nieminen MS, Kesaniemi YA, Peters A, Jula A, Perola M, Salomaa V for the Health AIRGENE Study Groups. Association of variation in the interleukin-1 gene family with diabetes and glucose homeostasis. J Clin Endocrinol Metab. 2009;94:4575–4583. doi: 10.1210/jc.2009-0666. [DOI] [PubMed] [Google Scholar]
- 409.Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, Brown NJ, Swift LL, McGuinness OP, Wasserman DH, Vaughan DE, Fogo AB. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. 2004;53:336–346. doi: 10.2337/diabetes.53.2.336. [DOI] [PubMed] [Google Scholar]
- 410.MacDougall JD, Hicks AL, MacDonald JR, McKelvie RS, Green HJ, Smith KM. Muscle performance and enzymatic adaptations to sprint interval training. J Appl Physiol. 1998;84:2138–2142. doi: 10.1152/jappl.1998.84.6.2138. [DOI] [PubMed] [Google Scholar]
- 411.Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, Minokoshi Y, Kahn BB, Parker RA, Hotamisligil GS. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab. 2005;1:107–119. doi: 10.1016/j.cmet.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 412.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, Komuro R, Ouchi N, Kihara S, Tochino Y, Okutomi K, Horie M, Takeda S, Aoyama T, Funahashi T, Matsuzawa Y. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 413.Maedler K, Dharmadhikari G, Schumann DM, Størling J. Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin Biol Ther. 2009;9:1177–1188. doi: 10.1517/14712590903136688. [DOI] [PubMed] [Google Scholar]
- 414.Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, Dayer J-M, Reinecke M, Halban PA, Donath MY. Leptin modulates β cell expression of IL-1 receptor antagonist and release of IL-1β in human islets. Proc Nat Acad Sci U S A. 2004;101:8138–8143. doi: 10.1073/pnas.0305683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Magnussen CG, Koskinen J, Chen W, Thomson R, Schmidt MD, Srinivasan SR, Kivimaki M, Mattsson N, Kahonen M, Laitinen T, Taittonen L, Ronnemaa T, Viikari JSA, Berenson GS, Juonala M, Raitakari OT. Pediatric metabolic syndrome predicts adulthood metabolic syndrome, subclinical atherosclerosis, and type 2 diabetes mellitus but is no better than body mass index alone: The Bogalusa Heart Study and the Cardiovascular Risk in Young Finns Study. Circulation. 2010;122:1604–1611. doi: 10.1161/CIRCULATIONAHA.110.940809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JT, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. J Biol Chem. 2001;276:48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- 417.Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276:21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 418.Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim R-Y, Liu F, Dong LQ. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- 419.Massiera F, Bloch-Faure M, Ceiler D, Murakami K, Fukamizu A, Gasc JM, Quignard-Boulange A, Negrel R, Ailhaud G, Seydoux J, Meneton P, Teboul M. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J. 2001;15:2727–2729. doi: 10.1096/fj.01-0457fje. [DOI] [PubMed] [Google Scholar]
- 420.Matsubara Y, Kano K, Kondo D, Mugishima H, Matsumoto T. Differences in adipocytokines and fatty acid composition between two adipocyte fractions of small and large cells in high-fat diet-induced obese mice. Ann Nutr Metabol. 2009;54:258–267. doi: 10.1159/000229506. [DOI] [PubMed] [Google Scholar]
- 421.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: Comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- 422.Matsumoto M, Ogawa W, Akimoto K, Inoue H, Miyake K, Furukawa K, Hayashi Y, Iguchi H, Matsuki Y, Hiramatsu R, Shimano H, Yamada N, Ohno S, Kasuga M, Noda T. PKClambda in liver mediates insulin-induced SREBP-1c expression and determines both hepatic lipid content and overall insulin sensitivity. J Clin Invest. 2003;112:935–944. doi: 10.1172/JCI18816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Matthews VB, Allen TL, Risis S, Chan MH, Henstridge DC, Watson N, Zaffino LA, Babb JR, Boon J, Meikle PJ, Jowett JB, Watt MJ, Jansson JO, Bruce CR, Febbraio MA. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53:2431–2441. doi: 10.1007/s00125-010-1865-y. [DOI] [PubMed] [Google Scholar]
- 424.Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH, Penkowa M, Hojman P, Zankari A, Watt MJ, Bruunsgaard H, Pedersen BK, Febbraio MA. Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia. 2009;52:1409–1418. doi: 10.1007/s00125-009-1364-1. [DOI] [PubMed] [Google Scholar]
- 425.Mauvais-Jarvis F, Ueki K, Fruman DA, Hirshman MF, Sakamoto K, Goodyear LJ, Iannacone M, Accili D, Cantley LC, Kahn CR. Reduced expression of the murine p85alpha subunit of phosphoinositide 3-kinase improves insulin signaling and ameliorates diabetes. J Clin Invest. 2002;109:141–149. doi: 10.1172/JCI13305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Mayer-Davis EJ, D’Agostino R, Jr, Karter AJ, Haffner SM, Rewers MJ, Saad M, Bergman RN. Intensity and amount of physical activity in relation to insulin sensitivity: The Insulin Resistance Atherosclerosis Study. JAMA. 1998;279:669–674. doi: 10.1001/jama.279.9.669. [DOI] [PubMed] [Google Scholar]
- 427.McGettrick AJ, Feener EP, Kahn CR. Human insulin receptor substrate- 1 (IRS-1) polymorphism G972R causes IRS-1 to associate with the insulin receptor and inhibit receptor autophosphorylation. J Biol Chem. 2005;280:6441–6446. doi: 10.1074/jbc.M412300200. [DOI] [PubMed] [Google Scholar]
- 428.Mendez-Hernandez P, Flores Y, Siani C, Lamure M, Dosamantes-Carrasco LD, Halley-Castillo E, Huitron G, Talavera J, Gallegos-Carrillo K, Salmeron J. Physical activity and risk of metabolic syndrome in an urban Mexican cohort. BMC Public Health. 2009;9:276. doi: 10.1186/1471-2458-9-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Menzaghi C, Trischitta V, Doria A. Genetic influences of adiponectin on insulin resistance, type 2 diabetes, and cardiovascular disease. Diabetes. 2007;56:1198–1209. doi: 10.2337/db06-0506. [DOI] [PubMed] [Google Scholar]
- 430.Metcalfe RS, Babraj JA, Fawkner SG, Vollaard NB. Towards the minimal amount of exercise for improving metabolic health: Beneficial effects of reduced-exertion high-intensity interval training. Eur J Appl Physiol. 2011 doi: 10.1007/s00421-011-2254-z. [DOI] [PubMed] [Google Scholar]
- 431.Mikines KJ. The influence of physical activity and inactivity on insulin action and secretion in man. Acta Physiol Scand Suppl. 1992;609:1–43. [PubMed] [Google Scholar]
- 432.Mikines KJ, Dela F, Tronier B, Galbo H. Effect of 7 days of bed rest on dose-response relation between plasma glucose and insulin secretion. Am J Physiol. 1989;257:E43–E48. doi: 10.1152/ajpendo.1989.257.1.E43. [DOI] [PubMed] [Google Scholar]
- 433.Mikines KJ, Richter EA, Dela F, Galbo H. Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J Appl Physiol. 1991;70:1245–1254. doi: 10.1152/jappl.1991.70.3.1245. [DOI] [PubMed] [Google Scholar]
- 434.Mikines KJ, Sonne B, Farrell PA, Tronier B, Galbo H. Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am J Physiol. 1988;254:E248–E259. doi: 10.1152/ajpendo.1988.254.3.E248. [DOI] [PubMed] [Google Scholar]
- 435.Mikines KJ, Sonne B, Farrell PA, Tronier B, Galbo H. Effect of training on the dose-response relationship for insulin action in men. J Appl Physiol. 1989;66:695–703. doi: 10.1152/jappl.1989.66.2.695. [DOI] [PubMed] [Google Scholar]
- 436.Mikines KJ, Sonne B, Tronier B, Galbo H. Effects of acute exercise and detraining on insulin action in trained men. J Appl Physiol. 1989a;66:704–711. doi: 10.1152/jappl.1989.66.2.704. [DOI] [PubMed] [Google Scholar]
- 437.Mikines KJ, Sonne B, Tronier B, Galbo H. Effects of training and detraining on dose-response relationship between glucose and insulin secretion. Am J Physiol. 1989b;256:E588–E596. doi: 10.1152/ajpendo.1989.256.5.E588. [DOI] [PubMed] [Google Scholar]
- 438.Mikus CR, Oberlin DJ, Libla J, Boyle LJ, Thyfault JP. Glycaemic control is improved by 7 days of aerobic exercise training in patients with type 2 diabetes. Diabetologia. 2012;55:1417–1423. doi: 10.1007/s00125-012-2490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Mikus CR, Oberlin DJ, Libla JL, Taylor AM, Booth FW, Thyfault JP. Lowering physical activity impairs glycemic control in healthy volunteers. Med Sci Sports Exerc. 2012;44:225–231. doi: 10.1249/MSS.0b013e31822ac0c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Miller JP, Pratley RE, Goldberg AP, Gordon P, Rubin M, Treuth MS, Ryan AS, Hurley BF. Strength training increases insulin action in healthy 50- to 65-yr-old men. J Appl Physiol. 1994;77:1122–1127. doi: 10.1152/jappl.1994.77.3.1122. [DOI] [PubMed] [Google Scholar]
- 441.Miller WJ, Sherman WM, Ivy JL. Effect of strength training on glucose tolerance and post-glucose insulin response. Med Sci Sports Exerc. 1984;16:539–543. [PubMed] [Google Scholar]
- 442.Mills PJ, Hong S, Redwine L, Carter SM, Chiu A, Ziegler MG, Dimsdale JE, Maisel AS. Physical fitness attenuates leukocyte-endothelial adhesion in response to acute exercise. J Appl Physiol. 2006;101:785–788. doi: 10.1152/japplphysiol.00135.2006. [DOI] [PubMed] [Google Scholar]
- 443.Min J, Okada S, Kanzaki M, Elmendorf JS, Coker KJ, Ceresa BP, Syu LJ, Noda Y, Saltiel AR, Pessin JE. Synip: A novel insulin-regulated syn-taxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol Cell. 1999;3:751–760. doi: 10.1016/s1097-2765(01)80007-1. [DOI] [PubMed] [Google Scholar]
- 444.Min L, Leung YM, Tomas A, Watson RT, Gaisano HY, Halban PA, Pessin JE, Hou JC. Dynamin is functionally coupled to insulin granule exocytosis. J Biol Chem. 2007;282:33530–33536. doi: 10.1074/jbc.M703402200. [DOI] [PubMed] [Google Scholar]
- 445.Minoura H, Takeshita S, Kimura C, Hirosumi J, Takakura S, Kawamura I, Seki J, Manda T, Mutoh S. Mechanism by which a novel non-thiazolidinedione peroxisome proliferator-activated receptor gamma agonist, FK614, ameliorates insulin resistance in Zucker fatty rats. Diabetes Obes Metab. 2007;9:369–378. doi: 10.1111/j.1463-1326.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 446.Misra A, Alappan NK, Vikram NK, Goel K, Gupta N, Mittal K, Bhatt S, Luthra K. Effect of supervised progressive resistance-exercise training protocol on insulin sensitivity, glycemia, lipids, and body composition in Asian Indians with type 2 diabetes. Diabetes Care. 2008;31:1282–1287. doi: 10.2337/dc07-2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Moller DE. Potential role of TNF-[alpha] in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab. 2000;11:212–217. doi: 10.1016/s1043-2760(00)00272-1. [DOI] [PubMed] [Google Scholar]
- 448.Monzavi R, Dreimane D, Geffner ME, Braun S, Conrad B, Klier M, Kaufman FR. Improvement in risk factors for metabolic syndrome and insulin resistance in overweight youth who are treated with lifestyle intervention. Pediatrics. 2006;117:e1111–e1118. doi: 10.1542/peds.2005-1532. [DOI] [PubMed] [Google Scholar]
- 449.Moore JB. Non-alcoholic fatty liver disease: The hepatic consequence of obesity and the metabolic syndrome. Proc Nutr Soc. 2010;69:211–220. doi: 10.1017/S0029665110000030. [DOI] [PubMed] [Google Scholar]
- 450.Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. Erralpha and Gabpa/b specify PGC-1alpha-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- 452.Morris MJ, Chen H, Watts R, Shulkes A, Cameron-Smith D. Brain neuropeptide Y and CCK and peripheral adipokine receptors: Temporal response in obesity induced by palatable diet. Int J Obes (Lond) 2008;32:249–258. doi: 10.1038/sj.ijo.0803716. [DOI] [PubMed] [Google Scholar]
- 453.Morrison JA, Ford ES, Steinberger J. The pediatric metabolic syndrome. Minerva Med. 2008;99:269–287. [PubMed] [Google Scholar]
- 454.Mottillo S, Filion KB, Genest J, Joseph L, Pilote L, Poirier P, Rinfret S, Schiffrin EL, Eisenberg MJ. The metabolic syndrome and cardiovascular risk: A systematic review and meta-analysis. J Am Coll Cardiol. 2010;56:1113–1132. doi: 10.1016/j.jacc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 455.Murata Y, Tsuruzoe K, Kawashima J, Furukawa N, Kondo T, Motoshima H, Igata M, Taketa K, Sasaki K, Kishikawa H, Kahn CR, Toyonaga T, Araki E. IRS-1 transgenic mice show increased epididymal fat mass and insulin resistance. Biochem Biophys Res Commun. 2007;364:301–307. doi: 10.1016/j.bbrc.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 456.Murray I, Havel PJ, Sniderman AD, Cianflone K. Reduced body weight, adipose tissue, and leptin levels despite increased energy intake in female mice lacking acylation-stimulating protein. Endocrinology. 2000;141:1041–1049. doi: 10.1210/endo.141.3.7364. [DOI] [PubMed] [Google Scholar]
- 457.Murray I, Sniderman AD, Cianflone K. Enhanced triglyceride clearance with intraperitoneal human acylation stimulating protein in C57BL/6 mice. Am J Physiol. 1999;277:E474–E480. doi: 10.1152/ajpendo.1999.277.3.E474. [DOI] [PubMed] [Google Scholar]
- 458.Muscari A, Bozzoli C, Puddu GM, Rovinetti C, Fiorentini GP, Roversi RA, Puddu P. Correlations between serum lipids and complement components in adults without demonstrated atherosclerotic disease. Atherosclerosis. 1990;81:111–118. doi: 10.1016/0021-9150(90)90017-d. [DOI] [PubMed] [Google Scholar]
- 459.Muse ED, Obici S, Bhanot S, Monia BP, McKay RA, Rajala MW, Scherer PE, Rossetti L. Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest. 2004;114:232–239. doi: 10.1172/JCI21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Myers MG, Jr, Grammer TC, Brooks J, Glasheen EM, Wang LM, Sun XJ, Blenis J, Pierce JH, White MF. The pleckstrin homology domain in insulin receptor substrate-1 sensitizes insulin signaling. J Biol Chem. 1995;270:11715–11718. doi: 10.1074/jbc.270.20.11715. [DOI] [PubMed] [Google Scholar]
- 461.Nassis GP, Papantakou K, Skenderi K, Triandafillopoulou M, Kavouras SA, Yannakoulia M, Chrousos GP, Sidossis LS. Aerobic exercise training improves insulin sensitivity without changes in body weight, body fat, adiponectin, and inflammatory markers in overweight and obese girls. Metabolism. 2005;54:1472–1479. doi: 10.1016/j.metabol.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 462.Nieto-Vazquez I, Fernández-Veledo S, de Alvaro C, Rondinone CM, Valverde AM, Lorenzo M. Protein–tyrosine phosphatase 1B–deficient myocytes show increased insulin sensitivity and protection against tumor necrosis factor-α–induced insulin resistance. Diabetes. 2007;56:404–413. doi: 10.2337/db06-0989. [DOI] [PubMed] [Google Scholar]
- 463.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 464.Niu W, Bilan PJ, Yu J, Gao J, Boguslavsky S, Schertzer JD, Chu G, Yao Z, Klip A. PKCepsilon regulates contraction-stimulated GLUT4 traffic in skeletal muscle cells. J Cell Physiol. 2011;226:173–180. doi: 10.1002/jcp.22320. [DOI] [PubMed] [Google Scholar]
- 465.Nollen EAA, Morimoto RI. Chaperoning signaling pathways: Molecular chaperones as stress-sensing ‘heat shock’ proteins. J Cell Sci. 2002;115:2809–2816. doi: 10.1242/jcs.115.14.2809. [DOI] [PubMed] [Google Scholar]
- 466.Norheim F, Raastad T, Thiede B, Rustan AC, Drevon CA, Haugen F. Proteomic identification of secreted proteins from human skeletal muscle cells and expression in response to strength training. Am J Physiol Endocrinol Metab. 2011;301:E1013–E1021. doi: 10.1152/ajpendo.00326.2011. [DOI] [PubMed] [Google Scholar]
- 467.Norquay LD, D’Aquino KE, Opare-Addo LM, Kuznetsova A, Haas M, Bluestone JA, White MF. Insulin receptor substrate-2 in beta-cells decreases diabetes in nonobese diabetic mice. Endocrinology. 2009;150:4531–4540. doi: 10.1210/en.2009-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.O’Neill TJ, Craparo A, Gustafson TA. Characterization of an interaction between insulin receptor substrate 1 and the insulin receptor by using the two-hybrid system. Mol Cell Biol. 1994;14:6433–6442. doi: 10.1128/mcb.14.10.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R. Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes. 1996;45:881–885. doi: 10.2337/diab.45.7.881. [DOI] [PubMed] [Google Scholar]
- 471.Ohman MK, Wright AP, Wickenheiser KJ, Luo W, Russo HM, Eitzman DT. Monocyte chemoattractant protein-1 deficiency protects against visceral fat-induced atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1151–1158. doi: 10.1161/ATVBAHA.110.205914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Ohmori R, Momiyama Y, Kato R, Taniguchi H, Ogura M, Ayaori M, Nakamura H, Ohsuzu F. Associations between serum resistin levels and insulin resistance, inflammation, and coronary artery disease. J Am Coll Cardiol. 2005;46:379–380. doi: 10.1016/j.jacc.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 473.Ohmura K, Ishimori N, Ohmura Y, Tokuhara S, Nozawa A, Horii S, Andoh Y, Fujii S, Iwabuchi K, Onoe K, Tsutsui H. Natural killer T cells are involved in adipose tissues inflammation and glucose intolerance in diet-induced obese mice. Arterioscler Thromb Vasc Biol. 2010;30:193–199. doi: 10.1161/ATVBAHA.109.198614. [DOI] [PubMed] [Google Scholar]
- 474.Okura T, Nakata Y, Ohkawara K, Numao S, Katayama Y, Matsuo T, Tanaka K. Effects of aerobic exercise on metabolic syndrome improvement in response to weight reduction[ast][ast] Obesity. 2007;15:2478–2484. doi: 10.1038/oby.2007.294. [DOI] [PubMed] [Google Scholar]
- 475.Old LJ. Tumor necrosis factor (TNF) Science. 1985;230:630–632. doi: 10.1126/science.2413547. [DOI] [PubMed] [Google Scholar]
- 476.Olsen RH, Krogh-Madsen R, Thomsen C, Booth FW, Pedersen BK. Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA. 2008;299:1261–1263. doi: 10.1001/jama.299.11.1259. [DOI] [PubMed] [Google Scholar]
- 477.Olson TP, Dengel DR, Leon AS, Schmitz KH. Changes in inflammatory biomarkers following one-year of moderate resistance training in overweight women. Int J Obes (Lond) 2007;31:996–1003. doi: 10.1038/sj.ijo.0803534. [DOI] [PubMed] [Google Scholar]
- 478.Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, Marcovina S, Fowler S for the Diabetes Prevention Program Research Group. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: The diabetes prevention program randomized trial. Ann Intern Med. 2005;142:611–619. doi: 10.7326/0003-4819-142-8-200504190-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Osawa H, Yamada K, Onuma H, Murakami A, Ochi M, Kawata H, Nishimiya T, Niiya T, Shimizu I, Nishida W, Hashiramoto M, Kanatsuka A, Fujii Y, Ohashi J, Makino H. The G/G genotype of a resistin single-nucleotide polymorphism at −420 increases type 2 diabetes mellitus susceptibility by inducing promoter activity through specific binding of Sp1/3. Am J Hum Genet. 2004;75:678–686. doi: 10.1086/424761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Oshida Y, Yamanouchi K, Hayamizu S, Nagasawa J, Ohsawa I, Sato Y. Effects of training and training cessation on insulin action. Int J Sports Med. 1991;12:484–486. doi: 10.1055/s-2007-1024718. [DOI] [PubMed] [Google Scholar]
- 481.Oshida Y, Yamanouchi K, Hayamizu S, Sato Y. Long-term mild jogging increases insulin action despite no influence on body mass index or VO2 max. J Appl Physiol. 1989;66:2206–2210. doi: 10.1152/jappl.1989.66.5.2206. [DOI] [PubMed] [Google Scholar]
- 482.Oshima Y, Ouchi N, Sato K, Izumiya Y, Pimentel DR, Walsh K. Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117:3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Ostergard T, Andersen JL, Nyholm B, Lund S, Nair KS, Saltin B, Schmitz O. Impact of exercise training on insulin sensitivity, physical fitness, and muscle oxidative capacity in first-degree relatives of type 2 diabetic patients. Am J Physiol Endocrinol Metab. 2006;290:E998–E1005. doi: 10.1152/ajpendo.00012.2005. [DOI] [PubMed] [Google Scholar]
- 484.Ouchi N, Oshima Y, Ohashi K, Higuchi A, Ikegami C, Izumiya Y, Walsh K. Follistatin-like 1, a secreted muscle protein, promotes endothelial cell function and revascularization in ischemic tissue through a nitric-oxide synthase-dependent mechanism. J Biol Chem. 2008;283:32802–32811. doi: 10.1074/jbc.M803440200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Owyang AM, Maedler K, Gross L, Yin J, Esposito L, Shu L, Jadhav J, Domsgen E, Bergemann J, Lee S, Kantak S. XOMA 052, an anti-IL-1{beta} monoclonal antibody, improves glucose control and {beta}-cell function in the diet-induced obesity mouse model. Endocrinology. 2010;151:2515–2527. doi: 10.1210/en.2009-1124. [DOI] [PubMed] [Google Scholar]
- 486.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 487.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of tType 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Park YW, Zhu S, Palaniappan L, Heshka S, Carnethon MR, Heymsfield SB. The metabolic syndrome: Prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch Intern Med. 2003;163:427–436. doi: 10.1001/archinte.163.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Patel L, Buckels AC, Kinghorn IJ, Murdock PR, Holbrook JD, Plumpton C, Macphee CH, Smith SA. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun. 2003;300:472–476. doi: 10.1016/s0006-291x(02)02841-3. [DOI] [PubMed] [Google Scholar]
- 490.Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11:9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 492.Pedersen BK, Febbraio M. Muscle-derived interleukin-6–A possible link between skeletal muscle, adipose tissue, liver, and brain. Brain Behav Immun. 2005;19:371–376. doi: 10.1016/j.bbi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 493.Pedersen BK, Febbraio MA. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol Rev. 2008;88:1379–1406. doi: 10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- 494.Pedersen BK, Steensberg A, Keller P, Keller C, Fischer C, Hiscock N, van Hall G, Plomgaard P, Febbraio MA. Muscle-derived interleukin-6: Lipolytic, anti-inflammatory and immune regulatory effects. Pflügers Arch. 2003;446:9–16. doi: 10.1007/s00424-002-0981-z. [DOI] [PubMed] [Google Scholar]
- 495.Pederson TM, Kramer DL, Rondinone CM. Serine/threonine phosphorylation of IRS-1 triggers its degradation: Possible regulation by tyrosine phosphorylation. Diabetes. 2001;50:24–31. doi: 10.2337/diabetes.50.1.24. [DOI] [PubMed] [Google Scholar]
- 496.Peeters RA, Veerkamp JH, Geurts van Kessel A, Kanda T, Ono T. Cloning of the cDNA encoding human skeletal-muscle fatty-acid-binding protein, its peptide sequence and chromosomal localization. Biochem J. 1991;276 (Pt 1):203–207. doi: 10.1042/bj2760203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Pellmé F, Smith U, Funahashi T, Matsuzawa Y, Brekke H, Wiklund O, Taskinen M-R, Jansson P-A. Circulating adiponectin levels are reduced in nonobese but insulin-resistant first-degree relatives of type 2 diabetic patients. Diabetes. 2003;52:1182–1186. doi: 10.2337/diabetes.52.5.1182. [DOI] [PubMed] [Google Scholar]
- 498.Perseghin G, Price TB, Petersen KF, Roden M, Cline GW, Gerow K, Rothman DL, Shulman GI. Increased glucose transport-phosphorylation and muscle glycogen synthesis after exercise training in insulin-resistant subjects. N Engl J Med. 1996;335:1357–1362. doi: 10.1056/NEJM199610313351804. [DOI] [PubMed] [Google Scholar]
- 499.Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G, Del Maschio A, Luzi L. Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: A 1H-13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes. 1999;48:1600–1606. doi: 10.2337/diabetes.48.8.1600. [DOI] [PubMed] [Google Scholar]
- 500.Pessin JE, Saltiel AR. Signaling pathways in insulin action: Molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Pessin JE, Thurmond DC, Elmendorf JS, Coker KJ, Okada S. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J Biol Chem. 1999;274:2593–2596. doi: 10.1074/jbc.274.5.2593. [DOI] [PubMed] [Google Scholar]
- 502.Petersen AMW, Pedersen BK. The anti-inflammatory effect of exercise. J Appl Physiol. 2005;98:1154–1162. doi: 10.1152/japplphysiol.00164.2004. [DOI] [PubMed] [Google Scholar]
- 503.Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350:664–671. doi: 10.1056/NEJMoa031314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA. 2004;291:1730–1737. doi: 10.1001/jama.291.14.1730. [DOI] [PubMed] [Google Scholar]
- 505.Pitsavos C, Panagiotakos DB, Chrysohoou C, Kavouras S, Stefanadis C. The associations between physical activity, inflammation, and coagulation markers, in people with metabolic syndrome: The ATTICA study. Eur J Cardiovasc Prev Rehabil. 2005;12:151–158. doi: 10.1097/01.hjr.0000164690.50200.43. [DOI] [PubMed] [Google Scholar]
- 506.Poehlman ET, Dvorak RV, DeNino WF, Brochu M, Ades PA. Effects of resistance training and endurance training on insulin sensitivity in nonobese, young women: A controlled randomized trial. J Clin Endocrinol Metab. 2000;85:2463–2468. doi: 10.1210/jcem.85.7.6692. [DOI] [PubMed] [Google Scholar]
- 507.Polak J, Moro C, Klimcakova E, Hejnova J, Majercik M, Viguerie N, Langin D, Lafontan M, Stich V, Berlan M. Dynamic strength training improves insulin sensitivity and functional balance between adrenergic alpha 2A and beta pathways in subcutaneous adipose tissue of obese subjects. Diabetologia. 2005;48:2631–2640. doi: 10.1007/s00125-005-0003-8. [DOI] [PubMed] [Google Scholar]
- 508.Poltorak A, Smirnova I, He X, Liu M-Y, Van Huffel C, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EKL, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B. Genetic and physical mapping of the Lps locus: Identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998;24:340–355. doi: 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- 509.Pomeroy C, Mitchell J, Eckert E, Raymond N, Crosby R, Dalmasso AP. Effect of body weight and caloric restriction on serum complement proteins, including factor D/adipsin: Studies in anorexia nervosa and obesity. Clin Exp Immunol. 1997;108:507–515. doi: 10.1046/j.1365-2249.1997.3921287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Potashnik R, Bloch-Damti A, Bashan N, Rudich A. IRS1 degradation and increased serine phosphorylation cannot predict the degree of metabolic insulin resistance induced by oxidative stress. Diabetologia. 2003;46:639–648. doi: 10.1007/s00125-003-1097-5. [DOI] [PubMed] [Google Scholar]
- 511.Previs SF, Withers DJ, Ren J-M, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem. 2000;275:38990–38994. doi: 10.1074/jbc.M006490200. [DOI] [PubMed] [Google Scholar]
- 512.Prodi E, Obici S. Minireview: The brain as a molecular target for diabetic therapy. Endocrinology. 2006;147:2664–2669. doi: 10.1210/en.2006-0143. [DOI] [PubMed] [Google Scholar]
- 513.Qi L, Zhang C, van Dam RM, Hu FB. Interleukin-6 genetic variability and adiposity: Associations in two prospective cohorts and systematic review in 26,944 individuals. J Clin Endocrinol Metab. 2007;92:3618–3625. doi: 10.1210/jc.2007-0877. [DOI] [PubMed] [Google Scholar]
- 514.Quinn LS, Anderson BG, Strait-Bodey L, Stroud AM, Argiles JM. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am J Physiol Endocrinol Metab. 2009;296:E191–E202. doi: 10.1152/ajpendo.90506.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Rabe B, Chalaris A, May U, Waetzig GH, Seegert D, Williams AS, Jones SA, Rose-John S, Scheller J. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 2008;111:1021–1028. doi: 10.1182/blood-2007-07-102137. [DOI] [PubMed] [Google Scholar]
- 516.Rajala MW, Qi Y, Patel HR, Takahashi N, Banerjee R, Pajvani UB, Sinha MK, Gingerich RL, Scherer PE, Ahima RS. Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes. 2004;53:1671–1679. doi: 10.2337/diabetes.53.7.1671. [DOI] [PubMed] [Google Scholar]
- 517.Rana JS, Nasir K, Santos RD, Roguin A, Orakzai SH, Carvalho JA, Meneghello R, Blumenthal RS. Increased level of cardiorespiratory fitness blunts the inflammatory response in metabolic syndrome. Int J Cardiol. 2006;110:224–230. doi: 10.1016/j.ijcard.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 518.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789. doi: 10.1016/s0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 519.Rangwala SM, Rich AS, Rhoades B, Shapiro JS, Obici S, Rossetti L, Lazar MA. Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes. 2004;53:1937–1941. doi: 10.2337/diabetes.53.8.1937. [DOI] [PubMed] [Google Scholar]
- 520.Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, Secher NH, Pedersen BK, Pilegaard H. Evidence for a release of BDNF from the brain during exercise. Exp Physiol. 2009;94:1062–106. doi: 10.1113/expphysiol.2009.048512. [DOI] [PubMed] [Google Scholar]
- 521.Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93:s64–s73. doi: 10.1210/jc.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 523.Reaven GM. Insulin resistance, cardiovascular disease, and the metabolic syndrome: How well do the emperor’s clothes fit? Diabetes Care. 2004;27:1011–1012. doi: 10.2337/diacare.27.4.1011. [DOI] [PubMed] [Google Scholar]
- 524.Reaven GM. The metabolic syndrome: Requiescat in pace. Clin Chem. 2005;51:931–938. doi: 10.1373/clinchem.2005.048611. [DOI] [PubMed] [Google Scholar]
- 525.Reaven GM. The metabolic syndrome: Is this diagnosis necessary? Am J Clin Nutr. 2006;83:1237–1247. doi: 10.1093/ajcn/83.6.1237. [DOI] [PubMed] [Google Scholar]
- 526.Reaven GM, Chen YD, Jeppesen J, Maheux P, Krauss RM. Insulin resistance and hyperinsulinemia in individuals with small, dense low density lipoprotein particles. J Clin Invest. 1993;92:141–146. doi: 10.1172/JCI116541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Reaven GM, Risser TE, Chen Y-DI, Reaven EP. Characterizaiton of a model of dietary-induced hypetriglyceridemia in young, nonobese rats. J Lipid Res. 1979;20:371–378. [PubMed] [Google Scholar]
- 528.Reil TD, Barnard RJ, Kashyap VS, Roberts CK, Gelabert HA. Diet-induced changes in endothelial dependent relaxation of the rat aorta. J Surg Res. 1999;85:96–100. doi: 10.1006/jsre.1999.5666. [DOI] [PubMed] [Google Scholar]
- 529.Reilly MP, Lehrke M, Wolfe ML, Rohatgi A, Lazar MA, Rader DJ. Resistin is an inflammatory marker of atherosclerosis in humans. Circulation. 2005;111:932–939. doi: 10.1161/01.CIR.0000155620.10387.43. [DOI] [PubMed] [Google Scholar]
- 530.Reinehr T, Kleber M, Toschke AM. Lifestyle intervention in obese children is associated with a decrease of the metabolic syndrome prevalence. Atherosclerosis. 2009;207:174–180. doi: 10.1016/j.atherosclerosis.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 531.Ren JM. Overexpression of Glut4 protein in muscle increases basal and insulin-stimulated whole body glucose disposal in conscious mice. J Clin Invest. 1995;95:429–432. doi: 10.1172/JCI117673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Rennie KL, McCarthy N, Yazdgerdi S, Marmot M, Brunner E. Association of the metabolic syndrome with both vigorous and moderate physical activity. Int J Epidemiol. 2003;32:600–606. doi: 10.1093/ije/dyg179. [DOI] [PubMed] [Google Scholar]
- 533.Ress C, Tschoner A, Engl J, Klaus A, Tilg H, Ebenbichler CF, Patsch JR, Kaser S. Effect of bariatric surgery on circulating chemerin levels. Eur J Clin Invest. 2010;40:277–280. doi: 10.1111/j.1365-2362.2010.02255.x. [DOI] [PubMed] [Google Scholar]
- 534.Retnakaran R, Shen S, Hanley AJ, Vuksan V, Hamilton JK, Zinman B. Hyperbolic relationship between insulin secretion and sensitivity on oral glucose tolerance test. Obesity. 2008;16:1901–1907. doi: 10.1038/oby.2008.307. [DOI] [PubMed] [Google Scholar]
- 535.Rewers M, Zaccaro D, D’Agostino R, Haffner S, Saad MF, Selby JV, Bergman R, Savage P. Insulin sensitivity, insulinemia, and coronary artery disease: The Insulin Resistance Atherosclerosis Study. Diabetes Care. 2004;27:781–787. doi: 10.2337/diacare.27.3.781. [DOI] [PubMed] [Google Scholar]
- 536.Reynolds IVTH, Supiano MA, Dengel DR. Resistance training enhances insulin-mediated glucose disposal with minimal effect on the tumor necrosis factor-alpha system in older hypertensives. Metabolism. 2004;53:397–402. doi: 10.1016/j.metabol.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 537.Reynolds THIV, Supiano MA, Dengel DR. Regional differences in glucose clearance: Effects of insulin and resistance training on arm and leg glucose clearance in older hypertensive individuals. J Appl Physiol. 2007;102:985–991. doi: 10.1152/japplphysiol.00914.2006. [DOI] [PubMed] [Google Scholar]
- 538.Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, Mohammad L, Henstridge DC, Febbraio MA, Hewitt SC, Korach KS, Bensinger SJ, Hevener AL. Myeloid-specific estrogen receptor {alpha} deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Nat Acad Sci U S A. 2011;108:16457–16462. doi: 10.1073/pnas.1104533108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Richards JC, Johnson TK, Kuzma JN, Lonac MC, Schweder MM, Voyles WF, Bell C. Short-term sprint interval training increases insulin sensitivity in healthy adults but does not affect the thermogenic response to beta-adrenergic stimulation. J Physiol. 2010;588:2961–2972. doi: 10.1113/jphysiol.2010.189886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Richter EA, Kiens B, Wojtaszewski JFP. Can exercise mimetics substitute for exercise? Cell Metabolism. 2008;8:96–98. doi: 10.1016/j.cmet.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 541.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 542.Roberts CK, Ng C, Hama S, Eliseo AJ, Barnard RJ. Effect of a short-term diet and exercise intervention on inflammatory/anti-inflammatory properties of HDL in overweight/obese men with cardiovascular risk factors. J Appl Physiol. 2006;101:1727–1732. doi: 10.1152/japplphysiol.00345.2006. [DOI] [PubMed] [Google Scholar]
- 543.Roberts CK, Vaziri ND, Liang KH, Barnard RJ. Reversibility of chronic experimental syndrome X by diet. Hypertension. 2001;37:1323–1328. doi: 10.1161/01.hyp.37.5.1323. [DOI] [PubMed] [Google Scholar]
- 544.Roepstorff C, Steffensen CH, Madsen M, Stallknecht B, Kanstrup IL, Richter EA, Kiens B. Gender differences in substrate utilization during submaximal exercise in endurance-trained subjects. Am J Physiol Endocrinol Metab. 2002;282:E435–E447. doi: 10.1152/ajpendo.00266.2001. [DOI] [PubMed] [Google Scholar]
- 545.Rogers MA, King DS, Hagberg JM, Ehsani AA, Holloszy JO. Effect of 10 days of physical inactivity on glucose tolerance in master athletes. J Appl Physiol. 1990;68:1833–1837. doi: 10.1152/jappl.1990.68.5.1833. [DOI] [PubMed] [Google Scholar]
- 546.Roh SG, Song SH, Choi KC, Katoh K, Wittamer V, Parmentier M, Sasaki S. Chemerin–a new adipokine that modulates adipogenesis via its own receptor. Biochem Biophys Res Commun. 2007;362:1013–1018. doi: 10.1016/j.bbrc.2007.08.104. [DOI] [PubMed] [Google Scholar]
- 547.Ronti T, Lupattelli G, Mannarino E. The endocrine function of adipose tissue: An update. Clin Endocrinol. 2006;64:355–365. doi: 10.1111/j.1365-2265.2006.02474.x. [DOI] [PubMed] [Google Scholar]
- 548.Ropelle ER, Fernandes MF, Flores MB, Ueno M, Rocco S, Marin R, Cintra DE, Velloso LA, Franchini KG, Saad MJ, Carvalheira JB. Central exercise action increases the AMPK and mTOR response to leptin. PLoS One. 2008;3:e3856. doi: 10.1371/journal.pone.0003856. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 549.Ropelle ER, Pauli JR, Prada PO, de Souza CT, Picardi PK, Faria MC, Cintra DE, Fernandes MF, Flores MB, Velloso LA, Saad MJ, Carvalheira JB. Reversal of diet-induced insulin resistance with a single bout of exercise in the rat: The role of PTP1B and IRS-1 serine phosphorylation. J Physiol. 2006;577:997–1007. doi: 10.1113/jphysiol.2006.120006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550.Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J Leukoc Biol. 2006;80:227–236. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 551.Rosen OM, Herrera R, Olowe Y, Petruzzelli LM, Cobb MH. Phosphorylation activates the insulin receptor tyrosine protein kinase. Proc Natl Acad Sci U S A. 1983;80:3237–3240. doi: 10.1073/pnas.80.11.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Ross A, Leveritt M. Long-term metabolic and skeletal muscle adaptations to short-sprint training: Implications for sprint training and tapering. Sports Med. 2001;31:1063–1082. doi: 10.2165/00007256-200131150-00003. [DOI] [PubMed] [Google Scholar]
- 553.Ross R, Aru J, Freeman J, Hudson R, Janssen I. Abdominal adiposity and insulin resistance in obese men. Am J Physiol Endocrinol Metab. 2002;282:E657–E663. doi: 10.1152/ajpendo.00469.2001. [DOI] [PubMed] [Google Scholar]
- 554.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem. 2003;278:45777–45784. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 555.Rui L, Aguirre V, Kim JK, Shulman GI, Lee A, Corbould A, Dunaif A, White MF. Insulin/IGF-1 and TNF-alpha stimulate phosphorylation of IRS-1 at inhibitory Ser307 via distinct pathways. J Clin Invest. 2001;107:181–189. doi: 10.1172/JCI10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Russell AP. Lipotoxicity: The obese and endurance-trained paradox. Int J Obes Relat Metab Disord. 2004;28:S66–S71. doi: 10.1038/sj.ijo.0802859. [DOI] [PubMed] [Google Scholar]
- 557.Ryan AS, Hurlbut DE, Lott ME, Ivey FM, Fleg J, Hurley BF, Goldberg AP. Insulin action after resistive training in insulin resistant older men and women. J Am Geriatr Soc. 2001;49:247–253. doi: 10.1046/j.1532-5415.2001.4930247.x. [DOI] [PubMed] [Google Scholar]
- 558.Ryan AS, Pratley RE, Goldberg AP, Elahi D. Resistive training increases insulin action in postmenopausal women. J Gerontol A Biol Sci Med Sci. 1996;51:M199–M205. doi: 10.1093/gerona/51a.5.m199. [DOI] [PubMed] [Google Scholar]
- 559.Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Sadagurski M, Norquay L, Farhang J, D’Aquino K, Copps K, White MF. Human IL6 enhances leptin action in mice. Diabetologia. 2009 doi: 10.1007/s00125-009-1580-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Sajan MP, Standaert ML, Miura A, Kahn CR, Farese RV. Tissue-specific differences in activation of atypical protein kinase C and protein kinase B in muscle, liver, and adipocytes of insulin receptor substrate-1 knockout mice. Mol Endocrinol. 2004;18:2513–2521. doi: 10.1210/me.2004-0045. [DOI] [PubMed] [Google Scholar]
- 562.Saleh J, Blevins JE, Havel PJ, Barrett JA, Gietzen DW, Cianflone K. Acylation stimulating protein (ASP) acute effects on postprandial lipemia and food intake in rodents. Int J Obes Relat Metab Disord. 2001;25:705–713. doi: 10.1038/sj.ijo.0801613. [DOI] [PubMed] [Google Scholar]
- 563.Saltevo J, Vanhala M, Kautiainen H, Kumpusalo E, Laakso M. Association of C-reactive protein, interleukin-1 receptor antagonist and adiponectin with the metabolic syndrome. Mediators Inflamm. 2007;2007:93573. doi: 10.1155/2007/93573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Saltiel AR, Pessin JE. Insulin signaling in microdomains of the plasma membrane. Traffic. 2003;4:711–716. doi: 10.1034/j.1600-0854.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 565.Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- 566.Santos AC, Ebrahim S, Barros H. Alcohol intake, smoking, sleeping hours, physical activity and the metabolic syndrome. Preventive Medicine. 2007;44:328–334. doi: 10.1016/j.ypmed.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 567.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Nat Acad Sci U S A. 2003;100:7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Sasaoka T, Wada T, Tsuneki H. Lipid phosphatases as a possible therapeutic target in cases of type 2 diabetes and obesity. Pharmacol Ther. 2006;112:799–809. doi: 10.1016/j.pharmthera.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 569.Sato Y, Iguchi A, Sakamoto N. Biochemical determination of training effects using insulin clamp technique. Horm Metab Res. 1984;16:483–486. doi: 10.1055/s-2007-1014825. [DOI] [PubMed] [Google Scholar]
- 570.Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic “hyperresistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114:224–231. doi: 10.1172/JCI20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Sattar N, Gaw A, Scherbakova O, Ford I, O’Reilly DSJ, Haffner SM, Isles C, Macfarlane PW, Packard CJ, Cobbe SM, Shepherd J. Metabolic syndrome with and without C-reactive protein as a predictor of coronary heart disease and diabetes in the West of Scotland Coronary Prevention Study. Circulation. 2003;108:414–419. doi: 10.1161/01.CIR.0000080897.52664.94. [DOI] [PubMed] [Google Scholar]
- 572.Sauter NS, Schulthess FT, Galasso R, Castellani LW, Maedler K. The antiinflammatory cytokine interleukin-1 receptor antagonist protects from high-fat diet-induced hyperglycemia. Endocrinology. 2008;149:2208–2218. doi: 10.1210/en.2007-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573.Sawka-Verhelle D, Filloux C, Tartare-Deckert S, Mothe I, Obberghen E. Identification of stat 5B as a substrate of the insulin receptor. Eur J Biochem. 1997;250:411–417. doi: 10.1111/j.1432-1033.1997.0411a.x. [DOI] [PubMed] [Google Scholar]
- 574.Schaffer JE, Lodish HF. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell. 1994;79:427–436. doi: 10.1016/0092-8674(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 575.Scheck SH, Barnard RJ, Lawani LO, Youngren IF, Martin DA, Singh R. Effects of NIDDM on the glucose transport system in human skeletal muscle. Diabetes Res. 1991;16:111–119. [PubMed] [Google Scholar]
- 576.Schmitz-Peiffer C. Protein kinase C and lipid-induced insulin resistance in skeletal muscle. Ann N Y Acad Sci. 2002;967:146–157. doi: 10.1111/j.1749-6632.2002.tb04272.x. [DOI] [PubMed] [Google Scholar]
- 577.Seals DR, Hagberg JM, Allen WK, Hurley BF, Dalsky GP, Ehsani AA, Holloszy JO. Glucose tolerance in young and older athletes and sedentary men. J Appl Physiol. 1984;56:1521–1525. doi: 10.1152/jappl.1984.56.6.1521. [DOI] [PubMed] [Google Scholar]
- 578.Seals DR, Hagberg JM, Hurley BF, Ehsani AA, Holloszy JO. Effects of endurance training on glucose tolerance and plasma lipid levels in older men and women. JAMA. 1984;252:645–649. [PubMed] [Google Scholar]
- 579.Sebastian D, Herrero L, Serra D, Asins G, Hegardt FG. CPT I over-expression protects L6E9 muscle cells from fatty acid-induced insulin resistance. Am J Physiol Endocrinol Metab. 2007;292:E677–E686. doi: 10.1152/ajpendo.00360.2006. [DOI] [PubMed] [Google Scholar]
- 580.Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A. 1989;86:114–118. doi: 10.1073/pnas.86.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Seino Y, Ikeda U, Takahashi M, Hojo Y, Irokawa M, Kasahara T, Shimada K. Expression of monocyte chemoattractant protein-1 in vascular tissue. Cytokine. 1995;7:575–579. doi: 10.1006/cyto.1995.0078. [DOI] [PubMed] [Google Scholar]
- 582.Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A, Arner P, Eckel J. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes. 2009;58:2731–2740. doi: 10.2337/db09-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Senn JJ. Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem. 2006;281:26865–26875. doi: 10.1074/jbc.M513304200. [DOI] [PubMed] [Google Scholar]
- 584.Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51:3391–3399. doi: 10.2337/diabetes.51.12.3391. [DOI] [PubMed] [Google Scholar]
- 585.Sethi JK, Xu H, Uysal KT, Wiesbrock SM, Scheja L, Hotamisligil GS. Characterisation of receptor-specific TNF[alpha] functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. FEBS Letters. 2000;469:77–82. doi: 10.1016/s0014-5793(00)01250-3. [DOI] [PubMed] [Google Scholar]
- 586.Shaibi GQ, Cruz ML, Ball GD, Weigensberg MJ, Salem GJ, Crespo NC, Goran MI. Effects of resistance training on insulin sensitivity in overweight Latino sdolescent males. Med Sci Sports Exerc. 2006;38:1208–1215. doi: 10.1249/01.mss.0000227304.88406.0f. [DOI] [PubMed] [Google Scholar]
- 587.Shanely RA, Nieman DC, Henson DA, Jin F, Knab AM, Sha W. Inflammation and oxidative stress are lower in physically fit and active adults. Scand J Med Sci Sports. 2011 doi: 10.1111/j.1600-0838.2011.01373.x. [DOI] [PubMed] [Google Scholar]
- 588.Shetty GK, Economides PA, Horton ES, Mantzoros CS, Veves A. Circulating adiponectin and resistin levels in relation to metabolic factors, inflammatory markers, and vascular reactivity in diabetic patients and subjects at risk for diabetes. Diabetes Care. 2004;27:2450–2457. doi: 10.2337/diacare.27.10.2450. [DOI] [PubMed] [Google Scholar]
- 589.Shima Y, Kitaoka K, Yoshiki Y, Maruhashi Y, Tsuyama T, Tomita K. Effect of heat shock preconditioning on ROS scavenging activity in rat skeletal muscle after downhill running. J Physiol Sci. 2008;58:341–348. doi: 10.2170/physiolsci.RP004808. [DOI] [PubMed] [Google Scholar]
- 590.Shisheva A. Phosphoinositides in insulin action on GLUT4 dynamics: Not just PtdIns(3,4,5)P3. Am J Physiol Endocrinol Metab. 2008;295:E536–E544. doi: 10.1152/ajpendo.90353.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591.Shojaee-Moradie F, Baynes K, Pentecost C, Bell J, Thomas E, Jackson N, Stolinski M, Whyte M, Lovell D, Bowes S, Gibney J, Jones R, Umpleby A. Exercise training reduces fatty acid availability and improves the insulin sensitivity of glucose metabolism. Diabetologia. 2007;50:404–413. doi: 10.1007/s00125-006-0498-7. [DOI] [PubMed] [Google Scholar]
- 592.Short KR, Vittone JL, Bigelow ML, Proctor DN, Rizza RA, Coenen-Schimke JM, Nair KS. Impact of aerobic exercise training on age-related changes in insulin sensitivity and muscle oxidative capacity. Diabetes. 2003;52:1888–1896. doi: 10.2337/diabetes.52.8.1888. [DOI] [PubMed] [Google Scholar]
- 593.Simoneau JA, Kelley DE. Altered glycolytic and oxidative capacities of skeletal muscle contribute to insulin resistance in NIDDM. J Appl Physiol. 1997;83:166–171. doi: 10.1152/jappl.1997.83.1.166. [DOI] [PubMed] [Google Scholar]
- 594.Simoneau JA, Veerkamp JH, Turcotte LP, Kelley DE. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999;13:2051–2060. doi: 10.1096/fasebj.13.14.2051. [DOI] [PubMed] [Google Scholar]
- 595.Simons LA, Levis G, Simons J. Apparent discontinuation rates in patients prescribed lipid-lowering drugs. Med J Aust. 1996;164:208–211. doi: 10.5694/j.1326-5377.1996.tb94138.x. [DOI] [PubMed] [Google Scholar]
- 596.Singh U, Devaraj S, Jialal I. C-reactive protein stimulates myeloperoxidase release from polymorphonuclear cells and monocytes: Implications for acute coronary syndromes. Clin Chem. 2009;55:361–364. doi: 10.1373/clinchem.2008.109207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Sleigh A, Raymond-Barker P, Thackray K, Porter D, Hatunic M, Vottero A, Burren C, Mitchell C, McIntyre M, Brage S, Carpenter TA, Murgatroyd PR, Brindle KM, Kemp GJ, O’Rahilly S, Semple RK, Savage DB. Mitochondrial dysfunction in patients with primary congenital insulin resistance. J Clin Invest. 2011;121:2457–2461. doi: 10.1172/JCI46405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Smith SR, Bai F, Charbonneau C, Janderova L, Argyropoulos G. A promoter genotype and oxidative stress potentially link resistin to human insulin resistance. Diabetes. 2003;52:1611–1618. doi: 10.2337/diabetes.52.7.1611. [DOI] [PubMed] [Google Scholar]
- 599.Smutok MA, Kokkinos PF, Farmer C, Dawson P, Shulman R, DeVane-Bell J, Patterson J, Charabogos C, Goldberg AP, Hurley BF. Aerobic versus strength training for risk factor intervension in middle-aged men at high risk for coronary heart disease. Metabolism. 1993;42 (2):177–184. doi: 10.1016/0026-0495(93)90032-j. [DOI] [PubMed] [Google Scholar]
- 600.Smutok MA, Reece C, Kokkinos PF, Farmer CM, Dawson PK, DeVane J, Patterson J, Goldberg AP, Hurley BF. Effects of exercise training modality on glucose tolerance in men with abnormal glucose regulation. Int J Sports Med. 1994;15:283–289. doi: 10.1055/s-2007-1021061. [DOI] [PubMed] [Google Scholar]
- 601.Staib JL, Tumer N, Powers SK. Increased temperature and protein oxidation lead to HSP72 mRNA and protein accumulation in the in vivo exercised rat heart. Exp Physiol. 2009;94:71–80. doi: 10.1113/expphysiol.2008.044685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Standaert ML, Sajan MP, Miura A, Kanoh Y, Chen HC, Farese RV, Jr, Farese RV. Insulin-induced activation of atypical protein kinase C, but not protein kinase B, is maintained in diabetic (ob/ob and Goto-Kakazaki) liver. Contrasting insulin signaling patterns in liver versus muscle define phenotypes of type 2 diabetic and high fat-induced insulin-resistant states. J Biol Chem. 2004;279:24929–24934. doi: 10.1074/jbc.M402440200. [DOI] [PubMed] [Google Scholar]
- 603.Steensberg A, Keller C, Starkie RL, Osada T, Febbraio MA, Pedersen BK. IL-6 and TNF-alpha expression in, and release from, contracting human skeletal muscle. Am J Physiol Endocrinol Metab. 2002;283:E1272–E1278. doi: 10.1152/ajpendo.00255.2002. [DOI] [PubMed] [Google Scholar]
- 604.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 605.Steinberg GR, Dyck DJ. Development of leptin resistance in rat soleus muscle in response to high-fat diets. Am J Physiol Endocrinol Metab. 2000;279:E1374–E1382. doi: 10.1152/ajpendo.2000.279.6.E1374. [DOI] [PubMed] [Google Scholar]
- 606.Steinberg GR, McAinch AJ, Chen MB, O’Brien PE, Dixon JB, Cameron-Smith D, Kemp BE. The suppressor of cytokine signaling 3 inhibits leptin activation of AMP-kinase in cultured skeletal muscle of obese humans. J Clin Endocrinol Metab. 2006;91:3592–3597. doi: 10.1210/jc.2006-0638. [DOI] [PubMed] [Google Scholar]
- 607.Steinberg GR, Parolin ML, Heigenhauser GJ, Dyck DJ. Leptin increases FA oxidation in lean but not obese human skeletal muscle: Evidence of peripheral leptin resistance. Am J Physiol Endocrinol Metab. 2002;283:E187–E192. doi: 10.1152/ajpendo.00542.2001. [DOI] [PubMed] [Google Scholar]
- 608.Steinberg GR, Smith AC, Wormald S, Malenfant P, Collier C, Dyck DJ. Endurance training partially reverses dietary-induced leptin resistance in rodent skeletal muscle. Am J Physiol Endocrinol Metab. 2004;286:E57–E63. doi: 10.1152/ajpendo.00302.2003. [DOI] [PubMed] [Google Scholar]
- 609.Stenbit AE, Tsao TS, Li J, Burcelin R, Geenen DL, Factor SM, Houseknecht K, Katz EB, Charron MJ. GLUT4 heterozygous knockout mice develop muscle insulin resistance and diabetes. Nat Med. 1997;3:1096–1101. doi: 10.1038/nm1097-1096. [DOI] [PubMed] [Google Scholar]
- 610.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 611.Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, Enders GH, Silberg DG, Wen X, Wu GD, Lazar MA. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci U S A. 2001;98:502–506. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 612.Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol. 2005;25:1569–1575. doi: 10.1128/MCB.25.4.1569-1575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Stewart KJ, Bacher AC, Turner K, Lim JG, Hees PS, Shapiro EP, Tayback M, Ouyang P. Exercise and risk factors associated with metabolic syndrome in older adults. Am J Prev Med. 2005;28:9–18. doi: 10.1016/j.amepre.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 614.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim Y-J, Sherwin R, Devaskar S, Lesche R, Magnuson MA, Wu H. Live-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity. Proc Nat Acad Sci U S A. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, Magnuson MA, Wu H. Selective deletion of pten in pancreatic {beta} cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. 2006;26:2772–2781. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Stone S, Abkevich V, Russell DL, Riley R, Timms K, Tran T, Trem D, Frank D, Jammulapati S, Neff CD, Iliev D, Gress R, He G, Frech GC, Adams TD, Skolnick MH, Lanchbury JS, Gutin A, Hunt SC, Shattuck D. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum Mol Genet. 2006;15:2709–2720. doi: 10.1093/hmg/ddl204. [DOI] [PubMed] [Google Scholar]
- 617.Stuart CA, Shangraw RE, Prince MJ, Peters EJ, Wolfe RR. Bed-rest-induced insulin resistance occurs primarily in muscle. Metabolism. 1988;37:802–806. doi: 10.1016/0026-0495(88)90018-2. [DOI] [PubMed] [Google Scholar]
- 618.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 619.Sun X, MM, MGM, EMG, JMB, CRK, MFW Expression and function of IRS-1 in insulin signal transmission. J Biol Chem. 1992;267:22662–22672. [PubMed] [Google Scholar]
- 620.Sun X, Rothenberg P, Kahn CR, Backer J, Araki E, Wilden P, Cahill D, Goldstein B, White MF. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature. 1991;352:73–77. doi: 10.1038/352073a0. [DOI] [PubMed] [Google Scholar]
- 621.Suzuki T, Hirata K, Elkind MS, Jin Z, Rundek T, Miyake Y, Boden-Albala B, Di Tullio MR, Sacco R, Homma S. Metabolic syndrome, endothelial dysfunction, and risk of cardiovascular events: The Northern Manhattan Study (NOMAS) Am Heart J. 2008;156:405–410. doi: 10.1016/j.ahj.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Sweeney G, Somwar R, Ramlal T, Volchuk A, Ueyama A, Klip A. An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J Biol Chem. 1999;274:10071–10078. doi: 10.1074/jbc.274.15.10071. [DOI] [PubMed] [Google Scholar]
- 623.Swift DL, Johannsen NM, Earnest CP, Blair SN, Church TS. Effect of exercise training modality on C-reactive protein in type-2 diabetes. Med Sci Sports Exerc. 2011 doi: 10.1249/MSS.0b013e31824526cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Szczypaczewska M, Nazar K, Kaciuba-Uscilko H. Glucose tolerance and insulin response to glucose load in body builders. Int J Sports Med. 1989;10:34–37. doi: 10.1055/s-2007-1024870. [DOI] [PubMed] [Google Scholar]
- 625.Tabata I, Suzuki Y, Fukunaga T, Yokozeki T, Akima H, Funato K. Resistance training affects GLUT-4 content in skeletal muscle of humans after 19 days of head-down bed rest. J Appl Physiol. 1999;86:909–914. doi: 10.1152/jappl.1999.86.3.909. [DOI] [PubMed] [Google Scholar]
- 626.Takala TO, Nuutila P, Knuuti J, Luotolahti M, Yki-Jarvinen H. Insulin action on heart and skeletal muscle glucose uptake in weight lifters and endurance athletes. Am J Physiol. 1999;276:E706–E711. doi: 10.1152/ajpendo.1999.276.4.E706. [DOI] [PubMed] [Google Scholar]
- 627.Tamemoto H. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. 1994;372:182–186. doi: 10.1038/372182a0. [DOI] [PubMed] [Google Scholar]
- 628.Tamura Y, Sugimoto M, Murayama T, Minami M, Nishikaze Y, Ariyasu H, Akamizu T, Kita T, Yokode M, Arai H. C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice. J Atheroscler Thromb. 2010;17:219–228. doi: 10.5551/jat.3368. [DOI] [PubMed] [Google Scholar]
- 629.Tamura Y, Sugimoto M, Murayama T, Ueda Y, Kanamori H, Ono K, Ariyasu H, Akamizu T, Kita T, Yokode M, Arai H. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol. 2008;28:2195–2201. doi: 10.1161/ATVBAHA.108.168633. [DOI] [PubMed] [Google Scholar]
- 630.Tan BK, Adya R, Farhatullah S, Lewandowski KC, O’Hare P, Lehnert H, Randeva HS. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: Ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. 2008;57:801–808. doi: 10.2337/db07-0990. [DOI] [PubMed] [Google Scholar]
- 631.Tan BK, Chen J, Farhatullah S, Adya R, Kaur J, Heutling D, Lewandowski KC, O’Hare JP, Lehnert H, Randeva HS. Insulin and metformin regulate circulating and adipose tissue chemerin. Diabetes. 2009;58:1971–1977. doi: 10.2337/db08-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: Insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 633.Tanner CJ, Koves TR, Cortright RL, Pories WJ, Kim Y-B, Kahn BB, Dohm GL, Houmard JA. Effect of short-term exercise training on insulin-stimulated PI 3-kinase activity in middle-aged men. J Physiol Endocrinol Metab. 2002;282:E147–E153. doi: 10.1152/ajpendo.2002.282.1.E147. [DOI] [PubMed] [Google Scholar]
- 634.Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 635.Taylor EB, An D, Kramer HF, Yu H, Fujii NL, Roeckl KS, Bowles N, Hirshman MF, Xie J, Feener EP, Goodyear LJ. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem. 2008;283:9787–9796. doi: 10.1074/jbc.M708839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Taylor L, Midgley AW, Chrismas B, Madden LA, Vince RV, Mc-Naughton LR. The effect of acute hypoxia on heat shock protein 72 expression and oxidative stress in vivo. Eur J Appl Physiol. 2010;109:849–855. doi: 10.1007/s00421-010-1430-x. [DOI] [PubMed] [Google Scholar]
- 637.Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab. 2006;17:72–78. doi: 10.1016/j.tem.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 638.Thomas TR, Warner SO, Dellsperger KC, Hinton PS, Whaley-Connell AT, Rector RS, Liu Y, Linden MA, Chockalingam A, Thyfault JP, Huyette DR, Wang Z, Cox RH. Exercise and the metabolic syndrome with weight regain. J Appl Physiol. 2010;109:3–10. doi: 10.1152/japplphysiol.01361.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 639.Thong FS, Bilan PJ, Klip A. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes. 2007;56:414–423. doi: 10.2337/db06-0900. [DOI] [PubMed] [Google Scholar]
- 640.Tjonna AE, Lee SJ, Rognmo O, Stolen TO, Bye A, Haram PM, Loen-nechen JP, Al-Share QY, Skogvoll E, Slordahl SA, Kemi OJ, Najjar SM, Wisloff U. Aerobic interval training versus continuous moderate exercise as a treatment for the metabolic syndrome: A pilot study. Circulation. 2008;118:346–354. doi: 10.1161/CIRCULATIONAHA.108.772822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Todd MK, Yaspelkis BB, III, Turcotte LP. Short-term leptin treatment increases fatty acids uptake and oxidation in muscle of high fat-fed rats. Metabolism. 2005;54:1218–1224. doi: 10.1016/j.metabol.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 642.Tracey KJ, Wei H, Manogue KR, Fong Y, Hesse DG, Nguyen HT, Kuo GC, Beutler B, Cotran RS, Cerami A, Lowry SF. Cachectin/tumor necrosis factor induces cachexia, anemia, and inflammation. J Exp Med. 1988;167:1211–1227. doi: 10.1084/jem.167.3.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Treebak JT, Taylor EB, Witczak CA, An D, Toyoda T, Koh HJ, Xie J, Feener EP, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C377–C385. doi: 10.1152/ajpcell.00297.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 644.Tremblay A, Simoneau JA, Bouchard C. Impact of exercise intensity on body fatness and skeletal muscle metabolism. Metabolism. 1994;43:814–818. doi: 10.1016/0026-0495(94)90259-3. [DOI] [PubMed] [Google Scholar]
- 645.Trujillo ME, Scherer PE. Adiponectin–journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Inter Med. 2005;257:167–175. doi: 10.1111/j.1365-2796.2004.01426.x. [DOI] [PubMed] [Google Scholar]
- 646.Trujillo ME, Sullivan S, Harten I, Schneider SH, Greenberg AS, Fried SK. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J Clin Endocrinol Metab. 2004;89:5577–5582. doi: 10.1210/jc.2004-0603. [DOI] [PubMed] [Google Scholar]
- 647.Tsigos C, Papanicolaou DA, Defensor R, Mitsiadis CS, Kyrou I, Chrousos GP. Dose effects of recombinant human interleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology. 1997;66:54–62. doi: 10.1159/000127219. [DOI] [PubMed] [Google Scholar]
- 648.Tsimikas S, Willerson JT, Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol. 2006;47:C19–C31. doi: 10.1016/j.jacc.2005.10.066. [DOI] [PubMed] [Google Scholar]
- 649.Tsochatzis EA, Manolakopoulos S, Papatheodoridis GV, Archimandritis AJ. Insulin resistance and metabolic syndrome in chronic liver diseases: Old entities with new implications. Scand J Gastroenterol. 2009;44:6–14. doi: 10.1080/00365520802273058. [DOI] [PubMed] [Google Scholar]
- 650.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 651.Turban S, Hajduch E. Protein kinase C isoforms: Mediators of reactive lipid metabolites in the development of insulin resistance. FEBS letters. 2011;585:269–274. doi: 10.1016/j.febslet.2010.12.022. [DOI] [PubMed] [Google Scholar]
- 652.Turcotte LP, Srivastava AK, Chiasson J-L. Fasting increases plasma membrane fatty acid-binding protein (FABPPM) in red skeletal muscle. Molecular and Cellular Biochemistry. 1997;166:153–158. doi: 10.1023/a:1006846907394. [DOI] [PubMed] [Google Scholar]
- 653.Turcotte LP, Swenberger JR, Tucker MZ, Yee AJ. Training-induced elevation in FABPPM is associated with increased palmitate use in contracting muscle. J Appl Physiol. 1999;87:285–293. doi: 10.1152/jappl.1999.87.1.285. [DOI] [PubMed] [Google Scholar]
- 654.Turcotte LP, Swenberger JR, Zavitz Tucker M, Yee AJ. Increased fatty acid uptake and altered fatty acid metabolism in insulin-resistant muscle of obese Zucker rats. Diabetes. 2001;50:1389–1396. doi: 10.2337/diabetes.50.6.1389. [DOI] [PubMed] [Google Scholar]
- 655.Ueki K, Yballe CM, Brachmann SM, Vicent D, Watt JM, Kahn CR, Cantley LC. Increased insulin sensitivity in mice lacking p85β subunit of phosphoinositide 3-kinase. Proc Nat Acad Sci U S A. 2002;99:419–424. doi: 10.1073/pnas.012581799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 656.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-[alpha] function. Nature. 1997;389:610–614. doi: 10.1038/39335. [DOI] [PubMed] [Google Scholar]
- 657.Vague J. La differentiation sexuelle-facteur determinant des formes de l’obesite’. Presse Med. 1947;30 [PubMed] [Google Scholar]
- 658.van der Heijden G-J, Toffolo G, Manesso E, Sauer PJJ, Sunehag AL. Aerobic exercise increases peripheral and hepatic insulin sensitivity in sedentary adolescents. J Clin Endocrinol Metab. 2009;94:4292–4299. doi: 10.1210/jc.2009-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 659.Van Der Heijden G-J, Wang ZJ, Chu Z, Toffolo G, Manesso E, Sauer PJJ, Sunehag AL. Strength exercise improves muscle mass and hepatic insulin sensitivity in obese youth. Med Sci Sports Exerc. 2010;42:1973–1980. doi: 10.1249/MSS.0b013e3181df16d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660.Van der Poll T, Romijn JA, Endert E, Borm JJ, Buller HR, Sauerwein HP. Tumor necrosis factor mimics the metabolic response to acute infection in healthy humans. Am J Physiol Endocrinol Metab. 1991;261:E457–E465. doi: 10.1152/ajpendo.1991.261.4.E457. [DOI] [PubMed] [Google Scholar]
- 661.Van Guilder GP, Hoetzer GL, Greiner JJ, Stauffer BL, DeSouza CA. Influence of metabolic syndrome on biomarkers of oxidative stress and inflammation in obese adults. Obesity. 2006;14:2127–2131. doi: 10.1038/oby.2006.248. [DOI] [PubMed] [Google Scholar]
- 662.Van Harmelen V, Ariapart P, Hoffstedt J, Lundkvist I, Bringman S, Arner P. Increased adipose angiotensinogen gene expression in human obesity. Obesity. 2000;8:337–341. doi: 10.1038/oby.2000.40. [DOI] [PubMed] [Google Scholar]
- 663.Van Zwieten PA. Endothelial dysfunction in hypertension. A critical evaluation. Blood Press Suppl. 1997;2:67–70. [PubMed] [Google Scholar]
- 664.Vasseur F, Helbecque N, Dina C, Lobbens S, Delannoy V, Gaget S, Boutin P, Vaxillaire M, Lepretre F, Dupont S, Hara K, Clement K, Bihain B, Kadowaki T, Froguel P. Single-nucleotide polymorphism haplotypes in the both proximal promoter and exon 3 of the APM1 gene modulate adipocyte-secreted adiponectin hormone levels and contribute to the genetic risk for type 2 diabetes in French Caucasians. Hum Mol Genet. 2002;11:2607–2614. doi: 10.1093/hmg/11.21.2607. [DOI] [PubMed] [Google Scholar]
- 665.Vgontzas AN, Bixler EO, Chrousos GP. Sleep apnea is a manifestation of the metabolic syndrome. Sleep Med Rev. 2005;9:211–224. doi: 10.1016/j.smrv.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 666.Vistisen B, Roepstorff K, Roepstorff C, Bonen A, van Deurs B, Kiens B. Sarcolemmal FAT/CD36 in human skeletal muscle colocalizes with caveolin-3 and is more abundant in type 1 than in type 2 fibers. J Lipid Res. 2004;45:603–609. doi: 10.1194/jlr.M300424-JLR200. [DOI] [PubMed] [Google Scholar]
- 667.Vukovich MD, Arciero PJ, Kohrt WM, Racette SB, Hansen PA, Holloszy JO. Changes in insulin action and GLUT-4 with 6 days of inactivity in endurance runners. J Appl Physiol. 1996;80:240–244. doi: 10.1152/jappl.1996.80.1.240. [DOI] [PubMed] [Google Scholar]
- 668.Wadley GD, Tunstall RJ, Sanigorski A, Collier GR, Hargreaves M, Cameron-Smith D. Differential effects of exercise on insulin-signaling gene expression in human skeletal muscle. J Appl Physiol. 2001;90:436–440. doi: 10.1152/jappl.2001.90.2.436. [DOI] [PubMed] [Google Scholar]
- 669.Waki H, Tontonoz P. Endocrine functions of adipose tissue. Annu Rev Pathol. 2007;2:31–56. doi: 10.1146/annurev.pathol.2.010506.091859. [DOI] [PubMed] [Google Scholar]
- 670.Waki H, Yamauchi T, Kamon J, Ito Y, Uchida S, Kita S, Hara K, Hada Y, Vasseur F, Froguel P, Kimura S, Nagai R, Kadowaki T. Impaired multimerization of human adiponectin mutants associated with diabetes. J Biol Chem. 2003;278:40352–40363. doi: 10.1074/jbc.M300365200. [DOI] [PubMed] [Google Scholar]
- 671.Walz A, Strieter RM, Schnyder S. Neutrophil-activating peptide ENA-78. Adv Exp Med Biol. 1993;351:129–137. doi: 10.1007/978-1-4615-2952-1_14. [DOI] [PubMed] [Google Scholar]
- 672.Wang J, Obici S, Morgan K, Barzilai N, Feng Z, Rossetti L. Overfeeding rapidly induces leptin and insulin resistance. Diabetes. 2001;50:2786–2791. doi: 10.2337/diabetes.50.12.2786. [DOI] [PubMed] [Google Scholar]
- 673.Wang Y, Lam KSL, Kraegen EW, Sweeney G, Zhang J, Tso AWK, Chow W-S, Wat NMS, Xu JY, Hoo RLC, Xu A. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. doi: 10.1373/clinchem.2006.075614. [DOI] [PubMed] [Google Scholar]
- 674.Wang Y, Nishina PM, Naggert JK. Degradation of IRS1 leads to impaired glucose uptake in adipose tissue of the type 2 diabetes mouse model TALLYHO/Jng. J Endocrinol. 2009;203:65–74. doi: 10.1677/JOE-09-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 675.Wang Y, Simar D, Fiatarone Singh MA. Adaptations to exercise training within skeletal muscle in adults with type 2 diabetes or impaired glucose tolerance: A systematic review. Diabetes Metab Res Rev. 2009;25:13–40. doi: 10.1002/dmrr.928. [DOI] [PubMed] [Google Scholar]
- 676.Wannamethee SG, Shaper AG, Alberti KG. Physical activity, metabolic factors, and the incidence of coronary heart disease and type 2 diabetes. Arch Intern Med. 2000;160:2108–2116. doi: 10.1001/archinte.160.14.2108. [DOI] [PubMed] [Google Scholar]
- 677.Wannamethee SG, Shaper AG, Lennon L, Morris RW. Metabolic syndrome vs Framingham Risk Score for prediction of coronary heart disease, stroke, and type 2 diabetes mellitus. Arch Intern Med. 2005;165:2644–2650. doi: 10.1001/archinte.165.22.2644. [DOI] [PubMed] [Google Scholar]
- 678.Wannamethee SG, Shaper AG, Whincup PH. Modifiable lifestyle factors and the metabolic syndrome in older men: Effects of lifestyle changes. J Am Geriatr Soc. 2006;54:1909–1914. doi: 10.1111/j.1532-5415.2006.00974.x. [DOI] [PubMed] [Google Scholar]
- 679.Warden SJ, Fuchs RK. Are “exercise pills” the answer to the growing problem of physical inactivity? Br J Sports Med. 2008;42:562–563. doi: 10.1136/bjsm.2008.053512. [DOI] [PubMed] [Google Scholar]
- 680.Watson RT, Pessin JE. GLUT4 translocation: The last 200 nanometers. Cell Signal. 2007;19:2209–2217. doi: 10.1016/j.cellsig.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 681.Watson RT, Pessin JE. Transmembrane domain length determines intracellular membrane compartment localization of syntaxins 3, 4, and 5. Am J Physiol Cell Physiol. 2001;281:C215–C223. doi: 10.1152/ajpcell.2001.281.1.C215. [DOI] [PubMed] [Google Scholar]
- 682.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 684.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 685.Whaley MH, Kampert JB, Kohl HW, III, Blair SN. Physical fitness and clustering of risk factors associated with the metabolic syndrome. Med Sci Sports Exerc. 1999;31:287–293. doi: 10.1097/00005768-199902000-00013. [DOI] [PubMed] [Google Scholar]
- 686.White MF. The IRS-signalling system: A network of docking proteins that mediate insulin action. Mol Cell Biochem. 1998;182:3–11. [PubMed] [Google Scholar]
- 687.White MF, Kahn CR. The insulin signaling system. J Biol Chem. 1994;269:1–4. [PubMed] [Google Scholar]
- 688.Whiteman EL, Cho H, Birnbaum MJ. Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab. 2002;13:444–451. doi: 10.1016/s1043-2760(02)00662-8. [DOI] [PubMed] [Google Scholar]
- 689.Whyte LJ, Gill JMR, Cathcart AJ. Effect of 2 weeks of sprint interval training on health-related outcomes in sedentary overweight/obese men. Metabolism. 2010;59:1421–1428. doi: 10.1016/j.metabol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 690.Wickham EP, Stern M, Evans RK, Bryan DL, Moskowitz WB, Clore JN, Laver JH. Prevalence of the metabolic syndrome among obese adolescents enrolled in a multidisciplinary weight management program: Clinical correlates and response to treatment. Metab Syndr Relat Disord. 2009;7:179–186. doi: 10.1089/met.2008.0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 691.Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, Crackower M, Suzuki A, Mak TW, Kahn CR, Klip A, Woo M. Muscle-specific pten deletion protects against insulin resistance and diabetes. Mol Cell Biol. 2005;25:1135–1145. doi: 10.1128/MCB.25.3.1135-1145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Wijndaele K, Duvigneaud N, Matton L, Duquet W, Thomis M, Beunen G, Lefevre J, Philippaerts RM. Muscular strength, aerobic fitness, and metabolic syndrome risk in Flemish adults. Med Sci Sports Exerc. 2007;39:233–240. doi: 10.1249/01.mss.0000247003.32589.a6. [DOI] [PubMed] [Google Scholar]
- 693.Wildman RP, Muntner P, Reynolds K, McGinn AP, Rajpathak S, Wylie-Rosett J, Sowers MR. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: Prevalence and correlates of 2 phenotypes among the US population (NHANES 1999–2004) Arch Intern Med. 2008;168:1617–1624. doi: 10.1001/archinte.168.15.1617. [DOI] [PubMed] [Google Scholar]
- 694.Wilson FH, Hariri A, Farhi A, Zhao H, Petersen KF, Toka HR, Nelson-Williams C, Raja KM, Kashgarian M, Shulman GI, Scheinman SJ, Lifton RP. A cluster of metabolic defects caused by mutation in a mitochondrial tRNA. Science. 2004;306:1190–1194. doi: 10.1126/science.1102521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 695.Wilson PWF, D’Agostino RB, Parise H, Sullivan L, Meigs JB. Metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–3072. doi: 10.1161/CIRCULATIONAHA.105.539528. [DOI] [PubMed] [Google Scholar]
- 696.Winnick JJ, Sherman WM, Habash DL, Stout MB, Failla ML, Belury MA, Schuster DP. Short-term aerobic exercise training in obese humans with type 2 diabetes mellitus improves whole-body insulin sensitivity through gains in peripheral, not hepatic insulin sensitivity. J Clin Endocrinol Metab. 2008;93:771–778. doi: 10.1210/jc.2007-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 698.Wojtaszewski JFP, Birk JB, Frosig C, Holten M, Pilegaard H, Dela F. 5′AMP activated protein kinase expression in human skeletal muscle: Effects of strength training and type 2 diabetes. J Physiol (Lond) 2005;564:563–573. doi: 10.1113/jphysiol.2005.082669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 699.Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypolipidemic drugs regulate peroxisome proliferator-activated receptors alpha - and gamma-mediated gene expression via liver fatty acid binding protein: A signaling path to the nucleus. Proc Nat Acad Sci U S A. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 700.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Bostrom P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell metabolism. 2011;13:160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 701.Wu X, Hoffstedt J, Deeb W, Singh R, Sedkova N, Zilbering A, Zhu L, Park PK, Arner P, Goldstein BJ. Depot-specific variation in protein-tyrosine phosphatase activities in human omental and subcutaneous adipose tissue: A pPotential contribution to differential insulin sensitivity. J Clin Endocrinol Metab. 2001;86:5973–5980. doi: 10.1210/jcem.86.12.8109. [DOI] [PubMed] [Google Scholar]
- 702.Wu Y, Ouyang JP, Wu K, Wang SS, Wen CY, Xia ZY. Rosiglita-zone ameliorates abnormal expression and activity of protein tyrosine phosphatase 1B in the skeletal muscle of fat-fed, streptozotocin-treated diabetic rats. British Journal of Pharmacology. 2005;146:234–243. doi: 10.1038/sj.bjp.0706306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 703.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Xu J-W, Morita I, Ikeda K, Miki T, Yamori Y. C-reactive protein suppresses insulin signaling in endothelial cells: Role of Sspleen tyrosine knase. Mol Endocrinol. 2007;21:564–573. doi: 10.1210/me.2006-0354. [DOI] [PubMed] [Google Scholar]
- 705.Yamada P, Amorin F, Moseley P, Schneider S. Heat shock protein 72 response to exercise in humans. Sports Medicine. 2008;38:715. doi: 10.2165/00007256-200838090-00002. [DOI] [PubMed] [Google Scholar]
- 706.Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond) 2008;32:S13–S18. doi: 10.1038/ijo.2008.233. [DOI] [PubMed] [Google Scholar]
- 707.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, Murakami K, Ohteki T, Uchida S, Takekawa S, Waki H, Tsuno NH, Shibata Y, Terauchi Y, Froguel P, Tobe K, Koyasu S, Taira K, Kitamura T, Shimizu T, Nagai R, Kadowaki T. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 708.Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, Okada-Iwabu M, Kawamoto S, Kubota N, Kubota T, Ito Y, Kamon J, Tsuchida A, Kumagai K, Kozono H, Hada Y, Ogata H, Tokuyama K, Tsunoda M, Ide T, Murakami K, Awazawa M, Takamoto I, Froguel P, Hara K, Tobe K, Nagai R, Ueki K, Kadowaki T. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–339. doi: 10.1038/nm1557. [DOI] [PubMed] [Google Scholar]
- 709.Yan Q-W, Yang Q, Mody N, Graham TE, Hsu C-H, Xu Z, Houstis NE, Kahn BB, Rosen ED. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56:2533–2540. doi: 10.2337/db07-0007. [DOI] [PubMed] [Google Scholar]
- 710.Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM, Kotani K, Quadro L, Kahn BB. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362. doi: 10.1038/nature03711. [DOI] [PubMed] [Google Scholar]
- 711.Yang RZ, Lee MJ, Hu H, Pray J, Wu HB, Hansen BC, Shuldiner AR, Fried SK, McLenithan JC, Gong DW. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: Possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290:E1253–E1261. doi: 10.1152/ajpendo.00572.2004. [DOI] [PubMed] [Google Scholar]
- 712.Yfanti C, Nielsen AR, Åkerström T, Nielsen S, Rose AJ, Richter EA, Lykkesfeldt J, Fischer CP, Pedersen BK. Effect of antioxidant supplementation on insulin sensitivity in response to endurance exercise training. J Physiol Endocrinol Metab. 2011;300:E761–E770. doi: 10.1152/ajpendo.00207.2010. [DOI] [PubMed] [Google Scholar]
- 713.Yki-Jarvinen H, Koivisto VA. Effects of body composition on insulin sensitivity. Diabetes. 1983;32:965–969. doi: 10.2337/diab.32.10.965. [DOI] [PubMed] [Google Scholar]
- 714.Youngren JF, Keen S, Kulp JL, Tanner CJ, Houmard JA, Goldfine ID. Enhanced muscle insulin receptor autophosphorylation with short-term aerobic exercise training. Am J Physiol Endocrinol Metab. 2001;280:E528–E533. doi: 10.1152/ajpendo.2001.280.3.E528. [DOI] [PubMed] [Google Scholar]
- 715.Youngren JF, Paik J, Barnard RJ. Impaired insulin-receptor autophosphorylation is an early defect in fat-fed, insulin-resistant rats. J Appl Physiol. 2001;91:2240–2247. doi: 10.1152/jappl.2001.91.5.2240. [DOI] [PubMed] [Google Scholar]
- 716.Yu M, Blomstrand E, Chibalin AV, Wallberg-Henriksson H, Zierath JR, Krook A. Exercise-associated differences in an array of proteins involved in signal transduction and glucose transport. J Appl Physiol. 2001;90:29–34. doi: 10.1152/jappl.2001.90.1.29. [DOI] [PubMed] [Google Scholar]
- 717.Yuan M, Konstantopoulos N, Lee J, Hansen L, Li Z-W, Karin M, Shoelson SE. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of ikkbeta. Science. 2001;293:1673–1677. doi: 10.1126/science.1061620. [DOI] [PubMed] [Google Scholar]
- 718.Yudkin JS, Stehouwer CD, Emeis JJ, Coppack SW. C-reactive protein in healthy subjects: Associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue? Arterioscler Thromb Vasc Biol. 1999;19:972–978. doi: 10.1161/01.atv.19.4.972. [DOI] [PubMed] [Google Scholar]
- 719.Yuen DY, Dwyer RM, Matthews VB, Zhang L, Drew BG, Neill B, Kingwell BA, Clark MG, Rattigan S, Febbraio MA. Interleukin-6 attenuates insulin-mediated increases in endothelial cell signaling but augments skeletal muscle insulin action via differential effects on tumor necrosis factor-alpha expression. Diabetes. 2009;58:1086–1095. doi: 10.2337/db08-0775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 720.Zabolotny JM, Haj FG, Kim Y-B, Kim H-J, Shulman GI, Kim JK, Neel BG, Kahn BB. Transgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J Biol Chem. 2004;279:24844–24851. doi: 10.1074/jbc.M310688200. [DOI] [PubMed] [Google Scholar]
- 721.Zachwieja JJ, Toffolo G, Cobelli C, Bier DM, Yarasheski KE. Resistance exercise and growth hormone administration in older men: effects on insulin sensitivity and secretion during a stable-label intravenous glucose tolerance test. Metabolism. 1996;45:254–260. doi: 10.1016/s0026-0495(96)90063-3. [DOI] [PubMed] [Google Scholar]
- 722.Zavaroni I, Sander S, Scott S, Reaven GM. Effect of fructose feeding on insulin secretion and insulin action in the rat. Metabolism. 1980;29:970–973. doi: 10.1016/0026-0495(80)90041-4. [DOI] [PubMed] [Google Scholar]
- 723.Zeigerer A, McBrayer MK, McGraw TE. Insulin stimulation of GLUT4 exocytosis, but not its inhibition of endocytosis, is dependent on Rab-GAP AS160. Mol Biol Cell. 2004;15:4406–4415. doi: 10.1091/mbc.E04-04-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 724.Zhang B, Berger J, Hu E, Szalkowski D, White-Carrington S, Spiegelman B, Moller D. Negative regulation of peroxisome proliferator-activated receptor-gamma gene expression contributes to the antiadi-pogenic effects of tumor necrosis factor-alpha. Mol Endocrinol. 1996;10:1457–1466. doi: 10.1210/mend.10.11.8923470. [DOI] [PubMed] [Google Scholar]
- 725.Zhang J, Wu Y, Zhang Y, LeRoith D, Bernlohr DA, Chen X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol. 2008;22:1416–1426. doi: 10.1210/me.2007-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 726.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 727.Zhou B, Wong CF. A computational study of the phosphorylation mechanism of the insulin receptor tyrosine kinase. Jf Phys Chem A. 2009;113:5144–5150. doi: 10.1021/jp810827w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Zhou QL, Park JG, Jiang ZY, Holik JJ, Mitra P, Semiz S, Guilherme A, Powelka AM, Tang X, Virbasius J, Czech MP. Analysis of insulin signalling by RNAi-based gene silencing. Biochem Soc Trans. 2004;32:817–821. doi: 10.1042/BST0320817. [DOI] [PubMed] [Google Scholar]
- 729.Zinker BA, Rondinone CM, Trevillyan JM, Gum RJ, Clampit JE, Waring JF, Xie N, Wilcox D, Jacobson P, Frost L, Kroeger PE, Reilly RM, Koterski S, Opgenorth TJ, Ulrich RG, Crosby S, Butler M, Murray SF, McKay RA, Bhanot S, Monia BP, Jirousek MR. PTP1B antisense oligonucleotide lowers PTP1B protein, normalizes blood glucose, and improves insulin sensitivity in diabetic mice. Proc Nat Acad Sci U S A. 2002;99:11357–11362. doi: 10.1073/pnas.142298199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 730.Zisman A, Peroni OD, Abel ED, Michael MD, Mauvais-Jarvis F, Lowell BB, Wojtaszewski JF, Hirshman MF, Virkamaki A, Goodyear LJ, Kahn CR, Kahn BB. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 731.Zoppini G, Targher G, Zamboni C, Venturi C, Cacciatori V, Moghetti P, Muggeo M. Effects of moderate-intensity exercise training on plasma biomarkers of inflammation and endothelial dysfunction in older patients with type 2 diabetes. Nutr Metab Cardiovasc Dis. 2006;16:543–549. doi: 10.1016/j.numecd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 732.Zou C, Shao J. Role of adipocytokines in obesity-associated insulin resistance. J Nutr Biochem. 2008;19:277–286. doi: 10.1016/j.jnutbio.2007.06.006. [DOI] [PubMed] [Google Scholar]