

Abstract
BACKGROUND:
Retinoid X receptor (RXR) plays a central role in the regulation of intracellular receptor signaling pathways. The activation of RXR has protective effect on H2O2-induced apoptosis of H9c2 ventricular cells in rats. But the protective effect and mechanism of activating RXR in cardiomyocytes against hypoxia/reoxygenation (H/R)-induced oxidative iniury are still unclear.
METHODS:
The model of H/R injury was established through hypoxia for 2 hours and reoxygenation for 4 hours in H9c2 cardiomyocytes of rats. 9-cis-retinoic acid (9-cis RA) was obtained as an RXR agonist, and HX531 as an RXR antagonist. Cultured cardiomyocytes were randomly divided into four groups: sham group, H/R group, H/R+9-cis RA -pretreated group (100 nmol/L 9-cis RA), and H/R+9-cis RA+HX531-pretreated group (2.5 μmol/L HX531). The cell viability was measured by MTT, apoptosis rate of cardiomyocytes by flow cytometry analysis, and mitochondrial membrane potential (ΔΨm) by JC-1 fluorescent probe, and protein expressions of Bcl-2, Bax and cleaved caspase-9 with Western blotting. All measurement data were expressed as mean±standard deviation, and analyzed using one-way ANOVA and the Dunnett test. Differences were considered significant when P was <0.05.
RESULTS:
Pretreatment with RXR agonist enhanced cell viability, reduced apoptosis ratio, and stabled ΔΨm. Dot blotting experiments showed that under H/R stress conditions, Bcl-2 protein level decreased, while Bax and cleaved caspase-9 were increased. 9-cis RA administration before H/R stress prevented these effects, but the protective effects of activating RXR on cardiomyocytes against H/R induced oxidative injury were abolished when pretreated with RXR pan-antagonist HX531.
CONCLUSION:
The activation of RXR has protective effects against H/R injury in H9c2 cardiomyocytes of rats through attenuating signaling pathway of mitochondria apoptosis.
KEY WORDS: Retinoid X receptor, Cardiomyocytes, Apoptosis, Mitochondria, Hypoxia reoxygenation
INTRODUCTION
Retinoid X receptor (RXR) is a member of the nuclear receptor superfamily, which mediates the biological effects of many hormones, vitamins, and drugs. RXR is unique among nuclear receptors because it forms heterodimers with many nuclear receptors, including retinoic acid receptor (RAR), peroxisome proliferator-activated receptor (PPAR) and liver X receptor (LXR),[1,2] which play a critical role in regulating cardiovascular functions. Therefore, ligand activation of RXR has potentially pleiotropic effects on numerous biological pathways. We have recently demonstrated that pharmacological activation of RXR exerts protective effects against H2O2-induced apoptosis in H9c2 ventricular cells of rats.[3] However, the underlying mechanism is still unclear. To determine the protective effects and mechanism of activating RXR on H9c2 cardiomyocytes against hypoxia/reoxygenation (H/R) induced oxidative injury, we established a model of H/ R injury through hypoxia for 2 hours and reoxygenation for 4 hours, by which 9-cis-retinoic acid (9-cis RA) was obtained as an RXR agonist and HX531 as an RXR antagonist.
METHODS
Drugs and chemicals
Dulbecco's modified Eagle's medium (DMEM) with high glucose, fetal bovine serum and trypsin was purchased from Gibco BRL Co (Grand Island, NY, USA). MTT [3-(4’5-dimethylthiazol-2-yl)-2’5- diphenyltetrazolium bromide] and RXR natural agonist ligand, 9-cis RA were purchased from Sigma Aldrich (St. Louis, MO, USA). RXR pan-antagonist (HX531) was kindly provided by Dr. Kagechika from Graduate School of Pharmaceutical Sciences, University of Tokyo, Japan. The polyclonal antibodies against Bcl-2, Bax, cleaved caspase-9 and β-actin were acquired from Cell Signaling Technology (Beverly, MA, USA).
Cell culture and experimental treatments
The rat ventricular myocardial cell line H9c2 was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The cells were maintained in DMEM with 10% FBS and 1% penicillin/streptomycin in a humidified incubator. Experiments were carried out using mononucleated myoblasts after cells were serum starved for 24 hours. Cultured cardiomyocytes were randomly divided into four groups: sham group (normoxic condition for 6 hours in DMSO 0.1%), H/R group (hypoxia for 2 hours and reoxygenation for 4 hours), H/ R+9-cis RA-pretreated group (100 nmol/L 9-cis RA for 1 hour), and H/R+9-cis RA+HX531-pretreated group (2.5 μmol/L HX531 for 1 hour).
Cultured cells induced by hypoxia/reoxygenation
H9c2 cardiomyocytes were cultured under normoxic condition (5% CO2, 21% O2, and 74% N2) in a humidified incubator at 37 °C. These cells grew up to 70% confluence, changed to fresh medium without FBS, and transferred to a triple gas incubator with hypoxic settings (5% CO2, 1% O2, and 94% N2) for 2 hours, followed by reoxygenation (5% CO2, 21% O2, and 74% N2) for 4 hours.[4]
Quantification of cell viability by MTT test
Cell viability was evaluated using MTT with a method described previously.[3] Briefly, MTT labeling reagent (5 mg/mL MTT) was added to the culture to a final concentration of 0.5 mg/mL, and the cells were incubated at 37 °C for 4 hours. The cell viability was evaluated by measuring the absorbance at 570 nm using a microplate reader Elx800 (BIO-TEK Instruments, Inc.).
Flow cytometric analysis of apoptosis by JC-1 assay
Apoptotic cells were assessed by flow cytometric analysis using the MitoProbeTM JC-1 Assay Kit (M34152, Molecular Probes, Inc.). JC-1 fluorescent probe was used for the quantitation of depolarized mitochondria, which was one of the earliest characteristics of apoptotic cells. Briefly, the cells were loaded with 5 μg /mL JC-1 fluorescent probe at 37 °C for 30 minutes in the dark, and flow cytometric analysis was conducted on a FACScan, using CellQuest™ Pro Software (Becton Dickinson).[5]
Measurement of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential (ΔΨm) was detected and quantified in live cells by either a laser scanning confocal microscope or flow cytometry with the JC-1 Assay Kit (Molecular Probes, Inc., Eugene, OR,USA). The cells were loaded with 5 μg/mL JC-1 at 37 °C for 30 minutes, and flow cytometric analysis was conducted by FACScan using 488 nm excitation with 530 nm and 585 nm emission filters. For confocal microscopy analysis, the cells on coverslips were loaded with 10 μg/mL of JC-1 at 37 °C for 15 minutes, and fluorescence was viewed with a Leica TCS SP2 confocal[5] microscope.
Cellular fractionation and Western blot
Proteins of cells or tissues were prepared according to the standard protocols, and protein concentrations of lysates were determined using the BCA protein assay kit. Equal quantities of proteins (30–50 μg/lane) were submitted to 10% or 15% SDS-PAGE depending on the target proteins, electrotransferred onto polyvinylidene fluoride membranes, and then incubated with primary antibodies against Bax (1:500), Bcl-2 (1:500), caspase-9 (1:500), and β-actin (1:10 000). After incubation with the corresponding second antibodies, protein bands were detected using enhanced chemiluminescence (Pierce, IL, USA), and quantitation was performed using the Quantity One 4.4.0 software (Bio-Rad, CA, USA).
Statistical analysis
The data of the study were presented as mean ±standard deviation. Statistical analyses were performed using SAS9.13 (SAS Institute Inc., Cary, NC, USA). The normality assumption was assessed by the Shapiro- Wilk test. Statistical significance of multiple treatments was determined by one-way analysis of variance (ANOVA) followed by the Dunnett test when appropriate. Probabilities of 0.05 or less were considered to be statistically significant.
RESULTS
RXR agonist protects against H/R injury-induced decreases in cell viability
The H/R injury was found to produce significant reduction in cell viability (65.7%±8.9%, F=30.11, P <0.01). When the cells were pretreated for 1 hour with RXR agonist (100 nmol/L 9-cis RA) before the H/R injury, significant increase in cell viability was observed (85.7%±11.4%, F= 10.21, P<0.01). To investigate cytoprotective effect of RXR activation, the cells were pretreated with RXR antagonist HX531 for 1 hour, treated with 9-cis RA for 1 hour and then underwent H/ R injury. HX531 totally inhibited the beneficial effect of 9-cis RA (61.5%±10.7%, F=14.9, P<0.01).
RXR agonist protects H9c2 cells from H/R injury-induced apoptosis
Next, we examined the effect of RXR agonists on H/R injury-induced apoptosis. In JC-1 assay, apoptotic cells with depolarized mitochondria appeared in the lower right (LR) quadrant of the flow cytometric scatter plot (Figure 1A). H/R injury caused an increase in the proportion of cells in the LR quadrant of the plot (31.60±3.4)% compared with untreated control H9c2 cells (5.3%±1.5%, F=194.6, P<0.01). Pretreatment with 9-cis RA significantly inhibited the percentage of H/ R injury-induced apoptosis to (13.63±0.98)% (F=92.4, P <0.01). Pretreated with RXR antagonist, HX531 significantly inhibited the anti-apoptotic effect of 9-cis RA described above (29.40%±1.2%, F=73.5, P<0.01) (Figure 1B). These data strongly suggested that the activation of RXR prevented the apoptotic death of H/R injury-induced H9c2 cells.
Figure 1.
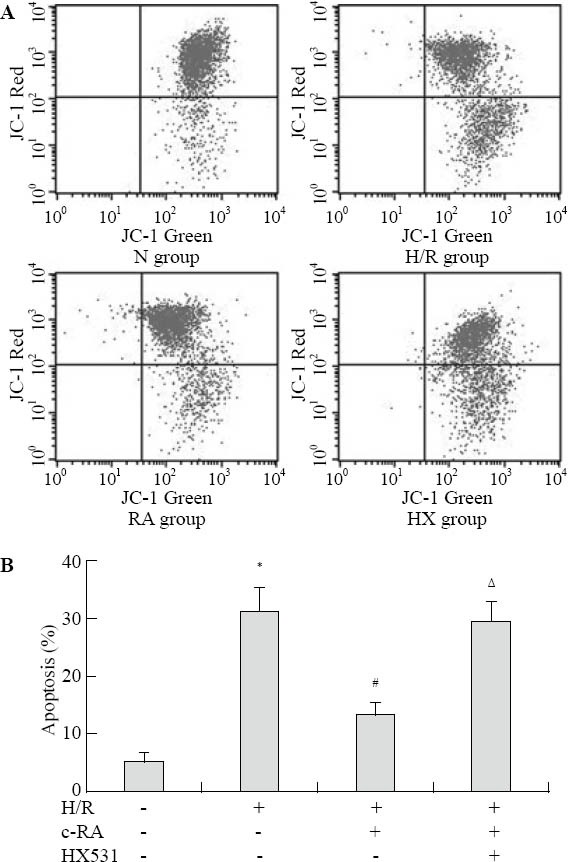
Activating RXR suppressed H/R-induced myocardial cell apoptosis by flow cytometric analysis with JC-1 assay (n=6, mean±SD). A: The percentage of apoptotic cells was determined by calculating the ratio of JC-1 green-stained cells (LR) to total cells. B: All data were expressed by the bar graph showing means±standard deviation (n=6); compared with the control group, *P<0.01; compared with the H/R group, #P<0.01; compared with the RA group, ΔP<0.01.
RXR agonist attenuates H/R injury-induced loss of ΔΨm
The ΔΨm is a critical factor in maintaining the integrity of mitochondria and subsequent regulation of apoptosis. In this experiment, we used both confocal microscope and flow cytometry to examine the changes in ΔΨm. As shown in Figure 2A, untreated control cells exhibit red mitochondrial staining, indicative of normal high membrane potentials. In contrast, H/R injury results in a decreased red mitochondrial staining and an increased green fluorescence signal, indicating a loss of ΔΨm. Pretreatment with 9-cis RA protects against the loss of ΔΨm induced by H/R injury, as evidenced by an increased fluorescent red intensity. We further quantified the change in the ΔΨm by flow cytometric analysis (Figure 2B). Exposure to H/R injury induced a decrease in the red/green fluorescence intensity ratio, compared with the control group (2.08±0.21 vs. 4.11±0.22, F=144.8, P<0.01). In agreement with the confocal image analysis, 9-cis RA prevented the H/R injury-induced dissipation of ΔΨm, as evidenced by an increase in the red/green ratio (3.41±0.17 vs. 2.08±0.21, F=57.88, P <0.01). The protective effect of 9-cis RA was blocked by HX-531 (1.67±0.28 vs. 4.11±0.22, F=92.2, P<0.01).
Figure 2.
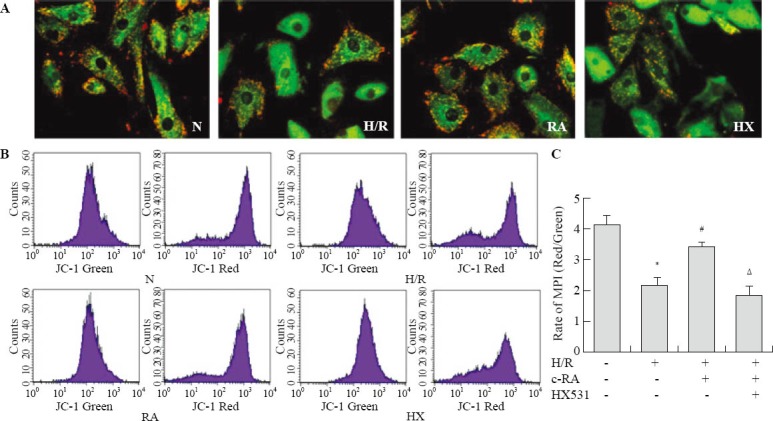
Activation of RXR stabled H/R injury-induced ΔΨm in myocardial cells. A: The cells were stained with JC-1 and visualized by a fluorescence microscope. The regions of high mitochondrial membrane polarization are indicated by red fluorescence due to J-aggregate formation by concentrated dye. The regions of reduced polarization are indicated by the green fluorescence of JC-1 monomers. B: ΔΨm was monitored using JC-1, by flow cytometry. The excitation wave length was 488 nm. The emission fluorescence was monitored at 530 nm with green fluorescence (left panel) and 585 nm with red fluorescence (right panel). C: Mitochondrial transmembrane potential was expressed as red/green fluorescence intensity ratio (n=6); compared with the control group, *P<0.01; compared with the H/R group, #P<0.01; compared with the RA group, ΔP<0.01.
RXR agonist inhibits the mitochondrial- mediated apoptotic pathway
The ratio between Bcl-2 and Bax expression has been proposed as an important marker of myocardial cell survival probability. Caspase 9 is a key protein involved in the mitochondrial apoptosis pathway. To determine whether the anti-apoptotic effect of RXR agonist was accompanied by a change in the ratio of Bcl-2 to Bax, and activation of caspase 9, western blot was conducted (Figure 3). The results showed a relative decrease in Bcl-2 and an increase in Bax and cleaved caspase 9 expression in H/R injury-induced cells, compared with the control group. 9-cis RA increased the expression of Bcl-2 and prevented H/R injury-induced increase in the expression of Bax and cleaved caspase 9. The effect of 9-cis RA was significantly attenuated by pre-incubation with HX531.
Figure 3.
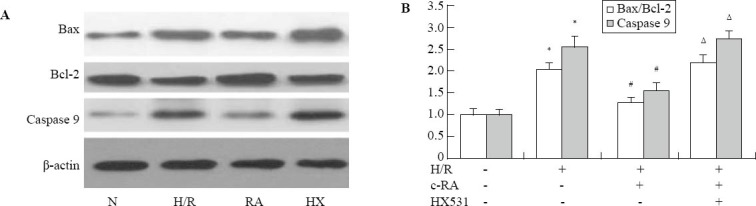
Activation of RXR inhibited the H/R-induced mitochondrial apoptosis pathway (n=3,  ). A: Western blot analysis of Bcl-2/Bax and cleaved caspase 9 protein was performed in cultured rat H9c2 cardiomyocytes. B: Scanning densitometry was used for semiquantitative analysis of the expression level of Bcl-2/Bax and cleaved caspase 9 protein; compared with the control group, *P<0.01; compared with the H/R group, #P<0.01; compared with the RA group, ΔP<0.01.
). A: Western blot analysis of Bcl-2/Bax and cleaved caspase 9 protein was performed in cultured rat H9c2 cardiomyocytes. B: Scanning densitometry was used for semiquantitative analysis of the expression level of Bcl-2/Bax and cleaved caspase 9 protein; compared with the control group, *P<0.01; compared with the H/R group, #P<0.01; compared with the RA group, ΔP<0.01.
DISCUSSION
RXR plays key roles in systemic lipid and glucose metabolism, energy homeostasis, and inflammatory control. RXR regulates a wide variety of biological functions though its central role in the regulation of intracellular receptor signaling pathways by acting as a ubiquitous heterodimerization partner of many nuclear receptors.[1,2] Although RXR has been reported to be expressed in the heart, little information is available regarding physiological and pathophysiological roles of this nuclear receptor in cardiovascular diseases and cardiac dysfunction.
Specific genetic knockout approach of all three RXRs has shown that the inactivation of the RXRa gene has more severe consequences than the ablation of RXRp and RXRy. The loss of RXRa is lethal during fetal life, because of hypoplasia of the myocardium and cardiac failure at approximately embryonic day 14.5.[6,7] This suggests that RXRa is essential in the transduction of a retinoid signal required for myocardial development. This supported the idea that RXRa is involved in retinoid signaling in vivo. However, the possible biological function of this nuclear receptor following oxidative stress injury has rarely been reported.[8,9] We have recently demonstrated that pharmacological activation of RXR exerts protective effects against H2O2-induced apoptosis in H9c2 rat ventricular cells.[8] However, the underlying mechanism is still unclear. The present study demonstrates a new role for the nuclear receptor RXR in cardiac myoblasts. RXR agonists, the natural 9-cis RA, prevent apoptosis induced by H/R injury-triggered apoptosis, and this anti-apoptotic effect was inhibited by a RXR antagonist HX531. RXR inhibited the H/ R injury-induced mitochondrial-mediated apoptotic pathway, suggesting that RXR may function as an endogenous apoptosis survival factor against H/R injury in rat cardiomyocytes H9c2 through attenuation of the mitochondria apoptosis signaling pathway.
Evidence from in vivo and in vitro studies suggests that apoptosis plays a central role in the developing heart and the progression of the pathogenesis of cardiovascular diseases. Oxidative stress, such as exposure to H/R injury, causes apoptosis in cardiac myocytes. In this study, the natural RXR ligand 9-cis RA could blunt H/R injury-induced apoptosis demonstrated by the decrease in the number of cells with depolarized mitochondria assessed by JC-1 labeling, a marker of apoptosis at a very early stage. Although the natural RXR ligand 9-cis RA has been reported to bind not only to RXR but also to RAR, we found that a RXR antagonist, HX 531, attenuated the protective effect of 9-cis RA. This indicates that the protective effects of 9-cis RA are mediated by a RXR-mediated pathway.
As mitochondria comprise approximately 30% of the total intracellular volume within a mammalian cardiomyocyte, they can have notable influence on cardiomyocyte energy production and, ultimately, the health of an individual cell.[10]
Mitochondrial membrane permeability transition (mMPT) is a potential early marker for the onset of apoptosis, thereby causing a disruption of ΔΨm, free radical production, declines in respiratory activity, and release of apoptogenic factors such as cytochrome c and apoptosis protease activating factor-1, ultimately, the induction of cell apoptosis.[11] In our studies, the cardiac cells were stained with JC-1 to determine the ΔΨm. JC-1 is a novel membrane-permeable cationic carbocyanine dye that exists as J-aggregates in the mitochondrial matrix and yields red fluorescence with an emission maximum at about 585 nm, while as monomers in the cytoplasm and yields green fluorescence with an emission maximum about 530 nm. Thus, ΔΨm can be measured from the JC-1 red/green fluorescence intensity ratio.[12] In our study, ΔΨm was reduced during H/R injury, and was partly prevented by activating RXR. This finding suggestes that stabilization of ΔΨm may be involved in the protection of activating RXR against H/R injury in H9c2.
To further understand the signaling events involved in the anti-apoptotic effects of activating RXR against H/R injury in rat cardiomyocytes, we examined the effects of RXR agonists on expression of Bcl-2 family proteins, which have been implicated as major regulators of mitochondrial homeostasis in many cellular events.[13] Bcl-2 has been reported to play an antiapoptotic role by preventing cytochrome c release from mitochondria through the stabilization of membrane integrity and the inhibition of the opening of the mMPT pore, whereas proapoptotic member Bax promotes cytochrome c release from mitochondria through the mMPT pore opening. Thus an increased ratio of Bax/Bcl-2 leads to the formation of pores in the mitochondria, and results in an efflux of small molecules inside the mitochondria such as cytochrome c and other proapoptotic factors. We found that activating RXR increased the expression of Bcl-2 and prevented H/R injury-induced increase in the expression of Bax. This indicated that the ratio of Bax/Bcl-2 was involved in the anti-apoptotic effects of activating RXR against H/R injury in rat cardiomyocytes.
The involvement of RXR in the regulation of apoptotic processes has been found in several cell types. Interestingly, RXR was shown to be proapoptotic in several cell types and antiapoptotic in others. For example, RXR agonists inhibited apoptosis in human mesangial cells exposed to H2O2 and naive tlymphocytes.[14,15] However, they facilitated apoptosis in tumor cells, such as lung and breast carcinomas, lymphomas, and leukemias.[16–18] Thus, there is controversy over pro- or anti-apoptotic effect, which may depend on the type of cells and the nature of the apoptotic stimulus.
In summary, RXR ligands may exert their cardioprotective effect by inhibiting oxidative stress-induced mitochondria-mediated apoptosis in cardiac myoblasts, representing a class of novel agents for preventing cardiovascular diseases.
ACKNOWLEDGMENTS
We thank Dr. H. Kagechika from Graduate School of Pharmaceutical Sciences, University of Tokyo, Japan, for synthetic support of the antagonists HX531.
Footnotes
Funding: This study was supported by grants from the National Natural Science Foundation of China (81270282, 81070176, 30600242, 81170192, 81200163), and Wenzhou Science Technology Bureau Foundation (Y20100010) and Education Foundation of Zhejiang Province (Y200906376).
Ethical approval: Not needed.
Conflicts of interest: The authors state that there is no conflict of interests involving the study.
Contributors: Shan PR and Xu WW contributed equally to this study. All authors read and approved the final version of the manuscript.
REFERENCES
- 1.Lefebvre P, Benomar Y, Staels B. Retinoid X receptors: common heterodimerization partners with distinct functions. Trends Endocrinol Metab. 2010;21:676–683. doi: 10.1016/j.tem.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Vaz B, R de Lera A. Advances in drug design with RXR modulators. Expert Opin Drug Discov. 2012;7:1003–1016. doi: 10.1517/17460441.2012.722992. [DOI] [PubMed] [Google Scholar]
- 3.Shan PR, Yuan AC, Wang LM, Shen LH, Chai DJ, Zhou L, et al. The effect of activating retinoid X receptor inhibiting hydrogen peroxide-induced apoptosis in cultured rat neonatal cardiomyocytes. Chin J Emerg Med. 2008;17:464–468. [Google Scholar]
- 4.Fu J, Lin G, Wu Z, Ceng B, Wu Y, Liang G, et al. Anti-apoptotic role for C1 inhibitor in ischemia/reperfusion-induced myocardial cell injury. Biochem Biophys Res Commun. 2006;349:504–512. doi: 10.1016/j.bbrc.2006.08.065. [DOI] [PubMed] [Google Scholar]
- 5.Choudhary R, Baker KM, Pan J. All-trans retinoic acid prevents angiotensin II- and mechanical stretch-induced reactive oxygen species generation and cardiomyocyte apoptosis. J Cell Physiol. 2008;215:172–181. doi: 10.1002/jcp.21297. [DOI] [PubMed] [Google Scholar]
- 6.Kang JO, Sucov HM. Convergent proliferative response and divergent morphogenic pathways induced by epicardial and endocardial signaling in fetal heart development. Mech Dev. 2005;122:57–65. doi: 10.1016/j.mod.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Dyson E, Sucov HM, Kubalak SW, Schmid-Schönbein GW, DeLano FA, Evans RM, et al. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha -/- mice. Proc Natl Acad Sci U S A. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shan P, Pu J, Yuan A, Shen L, Shen L, Chai D, et al. RXR agonists inhibit oxidative stress-induced apoptosis in H9c2 rat ventricular cells. Biochem Biophys Res Commun. 2008;375:628–633. doi: 10.1016/j.bbrc.2008.08.074. [DOI] [PubMed] [Google Scholar]
- 9.Guleria RS, Choudhary R, Tanaka T, Baker KM, Pan J. Retinoic acid receptor-mediated signaling protects cardiomyocytes from hyperglycemia induced apoptosis: role of the renin-angiotensin system. J Cell Physiol. 2011;226:1292–1307. doi: 10.1002/jcp.22457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He A, Wang JA, Gui C, Jiang Y, Sun Y, Chen T. Changes of mitochondrial pathway in hypoxia/reoxygenation induced cardiomyocytes apoptosis. Folia Histochem Cytobiol. 2007;45:397–400. [PubMed] [Google Scholar]
- 11.Zhou SH, Sun YF, Wang G. Effects of hyperbaric oxygen on intestinal mucosa apoptosis caused by ischemia-reperfusion injury in rats. World J Emerg Med. 2012;3:135–140. doi: 10.5847/wjem.j.issn.1920-8642.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brooks MM, Neelam S, Fudala R, Gryczynski I, Cammarata PR. Lenticular mitoprotection. Part A: Monitoring mitochondrial depolarization with JC-1 and artifactual fluorescence by the glycogen synthase kinase-3β inhibitor, SB216763. Mol Vis. 2013;19:1406–1412. [PMC free article] [PubMed] [Google Scholar]
- 13.Orrenius S. Mitochondrial regulation of apoptotic cell death. Toxicol Lett. 2004;149:19–23. doi: 10.1016/j.toxlet.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Manzano VM, Muñoz JC, Jiménez JR, Puyol MR, Puyol DR, Kitamura M, et al. Human renal mesangial cells are a target for the anti-inflammatory action of 9-cis retinoic acid. Br J Pharmacol. 2000;131:1673–1683. doi: 10.1038/sj.bjp.0703728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rasooly R, Schuster GU, Gregg JP, Xiao JH, Chandraratna RA, Stephensen CB. Retinoid x receptor agonists increase bcl2a1 expression and decrease apoptosis of naive T lymphocytes. J Immunol. 2005;175:7916–7929. doi: 10.4049/jimmunol.175.12.7916. [DOI] [PubMed] [Google Scholar]
- 16.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 17.Sun SY, Lotan R. Retinoids and their receptors in cancer development and chemoprevention. Crit Rev Oncol Hematol. 2002;41:41–55. doi: 10.1016/s1040-8428(01)00144-5. [DOI] [PubMed] [Google Scholar]
- 18.Gianni M, Ponzanelli I, Mologni L, Reichert U, Rambaldi A, Terao M, et al. Retinoid-dependent growth inhibition, differentiation and apoptosis in acute promyelocytic leukemia cells. Expression and activation of caspases. Cell Death Differ. 2000;7:447–460. doi: 10.1038/sj.cdd.4400673. [DOI] [PubMed] [Google Scholar]