

Abstract
Background:
Sickle cell disease (SCD) is characterized by chronic hemolytic anemia and vascular occlusion, causing recurrent painful episodes, neuro-cognitive deficits, organ failures and death in early adulthood. Besides the medical consequences, most of the families with a child of SCD have to cope with financial and social crisis. Quality of life (QOL) is a broad multidimensional concept that usually includes subjective evaluations of both positive and negative aspects of life. Other than health; emotional well being, social dysfunction, chronic pain and fatigability are also important aspects of overall quality of life that add to the complexity of its measurement.
Aim:
The present case control study was designed to determine the health related quality of life (HRQoL) in patients of sickle cell disease and to compare it with patients of other chronic non-communicable diseases.
Setting and Design:
Case control study conducted at tertiary health care facility of Central India.
Material and Methods:
The present study conducted to measure HRQoL among patients of SCD and patients of other chronic non-communicable diseases. A translated and pretested version of WHO SF-36 questionnaire was used to measure HRQoL.
Results:
We observed that there was significantly lower HRQoL among SCD patients.
Conclusion:
Besides merely pharmacotherapy, restoration of overall quality of life should be the mainstay of management of patients with SCD.
Keywords: Quality of life, Sickle cell disease, WHO SF-36
INTRODUCTION
Sickle cell disease (SCD) is characterized by chronic hemolytic anemia and vascular occlusion, causing recurrent painful episodes, neurocognitive deficits, organ failures and death in early adulthood. Treatment advances have now transformed SCD into a chronic disease suffered by children and adults. Frequently patients surviving until adulthood experiences significant organ system damage that may include stroke, multiple organ failures, leg ulcers and other many painful events (McClish et al. 2005).[1] Besides the medical consequences, most of the families having a child with SCD have to cope with financial and social crisis. Quality of life (QOL) is a broad multidimensional concept that usually includes subjective evaluations of both positive and negative aspects of life (Ware et al. 1992).[2] What makes it challenging to measure is that, although the term “quality of life” has meaning for nearly everyone and every academic discipline, individuals and groups can define it differently. Although health is one of the important domains of overall quality of life, there are other domains as well, for instance, emotional well being, social dysfunction, chronic pain and fatigability are also key aspects of overall quality of life that add to the complexity of its measurement. WHO SF-36 questionnaire is widely used for assessment of health-related quality of life (HRQoL) in diverse settings (Jenkinson 1994, Thomas 2002, Butterworth 2004, Asnani 2009).[3,4,5,6] The SF-36 has high test-retest reliability, has been shown to predict a number of poor outcomes and has been compared with biological markers for their sensitivity to change in severity of chronic illness (Jenkinson 1994, Butterworth 2004).[3,5] Also this scale is widely used for assessment of HRQoL even in patients of sickle cell disease (Asnani 2009).[6] But the scale is not yet validated in Indian settings. In children, HRQoL is also influenced by factors such as the ability to participate in peer groups and the ability to keep up with developmental activities.
These patients report poor HRQoL in quantitative studies using focus groups and fare worse in their HRQoL compared on health surveys. The factors influencing poor HRQoL will put forth the new dimension in the management of this chronic, painful and debilitating disease.
AIM AND OBJECTIVES
To determine the health-related quality of life in patients of sickle cell disease
To compare HRQoL in SCD and other chronic non-communicable diseases.
MATERIALS AND METHODS
Study design
Case control study.
Settings
The present case control study was conducted in patients of Sickle Cell Disease (SCD) attending pediatric SCD clinic at Acharya Vinoba Bhave Rural Hospital (AVBRH), Sawangi (Meghe), Wardha, Maharashtra.
Ethical approval
The study protocol including the data collection instrument and informed consent form was approved by Institutional Ethical Committee, Jawaharlal Nehru Medical College, Sawangi (Meghe), Wardha, Maharashtra.
Study duration
6 months.
Sample size calculation and assumptions
The present study estimated sample size based on prevalence of poor HRQoL in terms of bodily functions. With 80% power and 5% alfa error the calculated sample size was 100. Therefore 105 patients as cases and similar number of controls were selected for study.
Study participants and data collection procedure
The present case control study was conducted at tertiary health care facility of central India. Cases were selected from Sickle Cell OPD of Dept. of Pediatrics. All the cases had homozygous (HbSS) pattern on hemoglobin electrophoresis. Age and sex matched controls were selected from pediatric outpatient department. Patients of congenital heart disease, nephrotic syndrome, juvenile diabetes mellitus etc., were selected as controls. Both cases and controls were selected such that they had the disease at least since past two years. After selection, cases and controls were invited to participate into the study. The purpose of the study and relevant information were explained to them. Both cases and controls received all routine vaccines. Additional cases received penicillin prophylaxis and pneumococcal vaccine. Standard protocols were followed for penicillin prophylaxis, palliative care and management of acute events for cases as well as for controls (sickle cell crisis, diabetic keto-acidosis, relapse of nephritic syndrome). The treating physician/pediatrician were blinded regarding inclusion of subjects into the study.
A written informed consent of all the study participants were collected before actual data collection.
Selection criteria for cases
Homozygous SCD (HbSS pattern) with confirmed diagnosis on hemoglobin electrophoresis
Should have passed at least two years since diagnosis of the disease.
Selection criteria for controls
Should have inoperable congenital heart disease, nephritic syndrome, juvenile diabetic mellitus diagnosed by standard diagnostic algorithms/modality
Should be matched with cases with respect to age, sex and duration of disease.
Standard management protocols were followed for cases as well as for controls. Patients of SCD received folic acid, hydroxyurea, other vitamins, zinc, pneumococcal vaccination and prophylactic antibiotics (penicillin) at the time of follow up visit and pain crisis wherever appropriate.
The congenital heart disease patients received acute medical care, oxygenation, prophylactic penicillin as per indications. Patients of nephritic syndrome received steroids in appropriate doses. Patients of diabetic mellitus received hydrotherapy and insulin.
The data collection instrument consists of a baseline information proforma including disease history and general physical examination and WHO SF-36 questionnaire.
WHO SF-36 questionnaire (Ware et al. 1992)[2] was translated, pretested and then used for study purpose. The collected information was entered into a spreadsheet program. The data was presented in tables, baseline variables of cases and controls were analyzed with proportions. Chi square test with Yates correction (wherever applicable) were used to compare the findings. Student's t test was applied to compare different domains of HRQoL among cases and controls. Statistical software SYSTAT 12.0 was used for comparison of characteristics of cases and controls.
RESULTS
The present study was conducted to determine the health-related quality of life in patients of Sickle Cell Disease (cases) and age and sex matched patients (controls) of other chronic diseases such as congenital heart disease, nephrotic syndrome, juvenile diabetes mellitus etc.,Table 1 shows the socio-demographic characteristics of study subjects. The mean age of the participants was 11.78 ± 2.67 years of cases and 10.98 ± 4.23 years of controls. The cases and controls were comparable with respect to literacy, socio-economic status and duration of disease. However it was noted that, during previous six months, SCD patients seek greater number of medical consultations, as compared to those by patients suffering from chronic diseases other than SCD.
Table 1.
Socio-demographic characteristics of study subjects
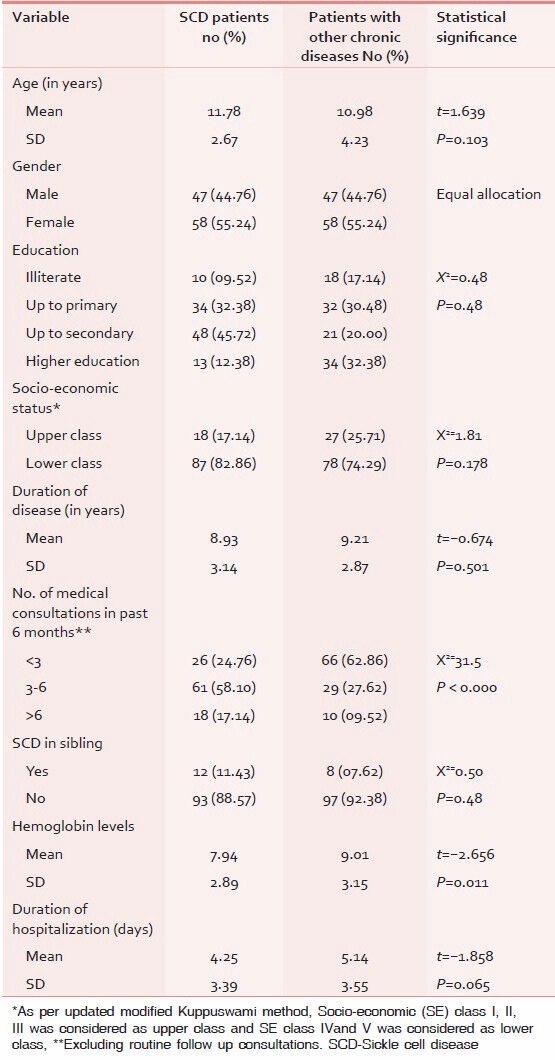
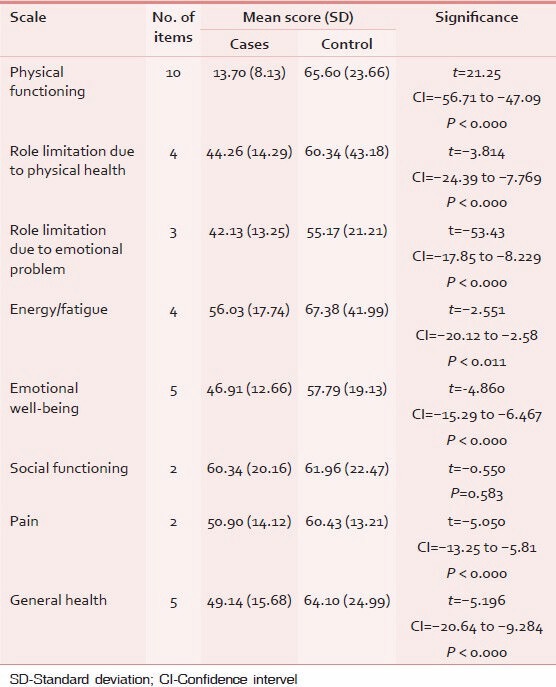
Table 2 shows comparative evaluation of HRQoL among cases and controls. Patients of SCD had significantly lower health-related quality of life as compared with patients of other chronic non-communicable diseases except in the domain of social functions; where HRQoL of SCD patients is lower than controls but statistically non-significant.
Table 2.
Comparison of HRQoL among cases and controls
Besides routine follow-up, all patients consulted the physician/pediatrician for any new or aggravation of existing symptoms e.g. pain crisis in SCD patients, dyspnea and cyanosis in congenital heart disease patients. The emergency consultations were as per requirement of the patients. All the consultations were documented and accessed from patient's records and reconfirmed by hospital information management system.
DISCUSSION
Overall health-related quality of life among SCD patients was observed significantly lower as compared with other chronically ill patients. Panepinto et al. (2005)[7] observed that perceived health-related quality of life is poor in case of children with SCD as well as of their parents. McClish et al. (2005)[1] observed that SCD patients have lower health-related quality of life than the general population. Findings of current study reiterates the poor HRQoL of SCD patients.
With reference to specific domain of quality of life, physical functioning and role limitation due to physical ill health was observed significantly poor in SCD patients. This was also reflected from finding that medical consultations are comparatively more among SCD cases as compared with patients of other chronic diseases [Table 1]. Barakat et al. (2008)[8] also reported significantly diminished physical functioning among patients of SCD as compared with other chronic illness. Tunde-Ayinmode (2011)[9] studied the physical problems in children and psychosocial effects.
According to our study, role limitation due to emotional problems and fatigability was significantly higher in SCD patients. Emotional problems in SCD may be related to chronic fatigue and small physical size, which may divert males with the illness from manifesting difficulties related to aggressive behavior with peers. For females with the illness, the common side effects of the illness may hinder the development of normal social relationships (Noll et al. 1996).[10] Our finding can be justified with above observation.
We observed that pain was reported among greater proportion of individuals as compared with children with other chronic diseases. Fuggle et al. (1996)[11] observed that among children with SCD, the perceived pain is greater than other chronic diseases. This debilitating disease was translated into over seven times increased risk of not attending school and was highly disruptive of social and recreational activities. Social dysfunction among SCD patients was not of higher magnitude in comparison with other non-communicable diseases.
Noll et al. (2010)[12] suggested that social functioning of children with SCD remained stable over time and was not suggestive of emergent social dysfunction. But psychosocial impact of SCD occurs not only on the patient but on entire family itself. Family maladjustment in SCD due to disruptions in the accustomed form and role of social interaction, and accompanying psychological tension in the family and its members, has been well documented (Tunde-Ayinmode 2011).[9] The disruption in family environment may remain concealed within family and may not be revealed to outside world. Therefore psychosocial impact of SCD might not be grossly evident from our study.
But overall general health of SCD patients was poorer than children with other chronic diseases.
These findings implicate the seriousness of poor quality of life among SCD patients, necessitating a national level program and introduce restoration of overall quality of life as mainstay of management of patients with SCD; besides merely symptom based pharmacotherapy. Currently no medical support is provided to SCD patients except free blood transfusion. The current study necessitates the inclusion of palliative care for physical health and functioning. Addressing social domains of QoL (emotional support, social dysfunction); counseling and rehabilitative services are recommended to maintain and improve QoL. Study findings will help stakeholders for making suitable provisions for SCD patients.
CONCLUSIONS
We observed that there was significantly lower health-related quality of life among SCD patients as compared with other chronic non-communicable diseases.
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
REFERENCES
- 1.McClish DK, Penberthy LT, Bovbjerg VE, Roberts JD, Aisiku IP, Levenson JL, et al. Health related quality of life in sickle cell patients: The PiSCES project. Health Qual Life Outcomes. 2005;3:50. doi: 10.1186/1477-7525-3-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ware JE, Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Med Care. 1992;30:473–83. [PubMed] [Google Scholar]
- 3.Jenkinson C, Wright L, Coulter A. Criterion validity and reliability of the SF-36 in a population sample. Qual Life Res. 1994;3:7–12. doi: 10.1007/BF00647843. [DOI] [PubMed] [Google Scholar]
- 4.Thomas VJ, Taylor LM. The psychosocial experience of people with sickle cell disease and its impact on quality of life: Qualitative findings from focus groups. Br J Health Psychol. 2002;7:345–63. doi: 10.1348/135910702760213724. [DOI] [PubMed] [Google Scholar]
- 5.Butterworth P, Crosier T. The validity of the SF-36 in an Australian national household survey: Demonstrating the applicability of the household income and labour dynamics in Australia (HILDA) survey to examination of health inequalities. BMC Public Health. 2004;4:44. doi: 10.1186/1471-2458-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asnani MR, Lipps GE, Reid ME. Utility of WHOQOL-BREF in measuring quality of life in sickle cell disease. Health Qual Life Outcomes. 2009;7:75. doi: 10.1186/1477-7525-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Panepinto JA, O'Mahar KM, DeBaun MR, Loberiza FR, Scott JP. Health-related quality of life in children with sickle cell disease: Child and parent perception. Br J Haematol. 2005;130:437–44. doi: 10.1111/j.1365-2141.2005.05622.x. [DOI] [PubMed] [Google Scholar]
- 8.Barakat LP, Patterson CA, Daniel LC, Dampier C. Quality of life among adolescents with sickle cell disease: Mediation of pain by internalizing symptoms and parenting stress. Health Qual Life Outcomes. 2008;6:60. doi: 10.1186/1477-7525-6-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tunde-Ayinmode MF. Children with sickle cell disease who are experiencing psychosocial problems concurrently with their mothers: A Nigerian study. Afr J Psychiatry (Johannesbg) 2011;14:392–401. doi: 10.4314/ajpsy.v14i5.8. [DOI] [PubMed] [Google Scholar]
- 10.Noll RB, Vannatta K, Koontz K, Kalinyak K, Bukowski WM, Davies WH. Peer relationships and emotional well-being of youngsters with sickle cell disease. Child Dev. 1996;67:423–36. doi: 10.1111/j.1467-8624.1996.tb01743.x. [DOI] [PubMed] [Google Scholar]
- 11.Fuggle P, Shand PA, Gill LJ, Davies SC. Pain, quality of life, and coping in sickle cell disease. Arch Dis Child. 1996;75:199–203. doi: 10.1136/adc.75.3.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noll RB, Kiska R, Reiter-Purtill J, Gerhardt CA, Vannatta K. A controlled, longitudinal study of the social functioning of youth with sickle cell disease. Pediatrics. 2010;125:e1453–9. doi: 10.1542/peds.2009-2996. [DOI] [PubMed] [Google Scholar]