

Abstract
Whole genome sequencing (WGS) studies are uncovering disease-associated variants in both rare and non-rare diseases. Utilizing the next-generation sequencing for WGS requires a series of computational methods for alignment, variant detection, and annotation, and the accuracy and reproducibility of annotation results are essential for clinical implementation. However, annotating WGS with up to date genomic information is still challenging for biomedical researchers. Here we present one of the fastest and highly scalable annotation, filtering, and analysis pipeline –gNOME – to prioritize phenotype-associated variants while minimizing false positive findings. Intuitive graphical user interface of gNOME facilitates the selection of phenotype associated variants, and the result summaries are provided at variant-, gene-, and genome-levels. Moreover, the enrichment results of specific variants, genes, and gene sets between two groups or compared to population scale WGS datasets that is already integrated in the pipeline can help the interpretation. We found a small number of discordant results between annotation software tools in part due to different reporting strategies for the variants with complex impacts. Using two published whole exome datasets of uveal melanoma and bladder cancer, we demonstrated gNOME's accuracy of variant annotation and the enrichment of loss of function variants in known cancer pathways. gNOME web-server and source codes are freely available to the academic community.
Keywords: whole genome sequences, variant annotation, disease gene discovery, analysis pipeline
Introduction
The maturation of ultra-high throughput sequencing technology has opened a new era of personal genome sequencing (Ashley et al., 2010; Chang and Wang, 2012; Cirulli and Goldstein, 2010; Drmanac, 2011; Kidd et al., 2012; Meyerson et al., 2010; Tabor et al., 2011), and shifted the researcher's burden from the identification of genetic variants to the interpretation of large numbers of variants in each individual. Although studies using whole genome sequencing (WGS) in a large disease population might still be a few years away, proof-of-concept studies on WGS and whole exome sequencing (WES) have already proven the technology to be useful in identifying disease-causing mutations in rare Mendelian disorders (Bamshad et al., 2011; Hoischen et al., 2010; Klassen et al., 2011; Lalonde et al., 2010; Lupski et al., 2010; Ng et al., 2010a; Ng et al., 2010b; Roach et al., 2010). Moreover, a few studies using the case-control study design also demonstrated the utility of WGS and WES in identifying disease-associated genomic variants for non-rare diseases (Calvo et al., 2010; Holm et al., 2011; Pelak et al., 2010; Rivas et al., 2011).
An individual human genome has 3-4 million variants, or locations that differ from the human reference genome. Because of the large number of variants, it is essential to filter out those weakly associated with the researchers' target phenotype and to reduce these variants down to a manageable number. This can be done by means of various heuristics such as allele frequencies (AFs) and their impacts on protein functions (Cooper and Shendure, 2011; Goldstein et al., 2013). There is a tremendous need in the biomedical research community for a tool that can filter these millions of variants based on the most up-to-date annotations and utilize the growing arsenal of genome analysis methods.
The number of bioinformatics pipelines for analyzing WGS and WES is rapidly increasing. However, a majority of such tools focus on processing raw sequence data to detect high confidence genomic variants rather than focusing on downstream analyses such as annotation-based variant filtering and statistical analysis (Lam, et al., 2012; McKenna, et al., 2010; Pabinger, et al., 2013). Even available downstream analysis tools are limited by their 1) static filtering methods, 2) insufficient annotation, and 3) absence of multi-genome comparison methods (Cingolani et al., 2012; Ge et al., 2011; MacArthur et al., 2012; San Lucas et al., 2012; Wang et al., 2010; Yandell et al., 2011). Moreover, these tools are difficult to use for most researchers and clinicians due to the lack of an intuitive user interface. To overcome these limitations, we developed gNOME, an interactive downstream analysis pipeline that combines comprehensive genomic annotation sources with statistical analysis in an expandable framework. We demonstrated the accuracy of annotation using the validated genomic variants from published WES datasets. The pipeline is written in C++, Perl, and SQL, and all source codes are freely available to the academic community.
Materials and Methods
Overview of gNOME workflow
The goal of most WGS and WES studies is to find variants that are possibly associated with a phenotype of interest. A common approach toward this goal is to prioritize variants that have deleterious impact on protein function and/or are more frequently observed in cases compared to controls and an ethnicity-matched healthy population (Lim, et al., 2013). Following this strategy, gNOME's streamlined analysis workflow is as follows: 1) creating a project and uploading variant files, 2) annotation, 3) filtering variants using an interactive user interface, 4) statistical analysis, and 5) summarizing the results (Figure 1). We use a double colon (::) to indicate a menu::submenu in the gNOME interface and single quotation marks to denote selected values throughout the description.
Figure 1. A schematic overview of gNOME.

The analysis of whole genome and exome dataset starts with creating a project and uploading it according to project type (Step 1). The uploaded files are annotated with 60 annotation tracks (Step 2), and annotation-based variant filtering can be interactively performed (Step 3). gNOME supports variant-, gene-, and gene set-level association tests between two groups: case vs. ethnicity-matched population data from the 1000 Genomes Project or cases vs. controls (Step 4). Filtering and analysis results are dynamically reported on the web-based interface (Step 5). Steps 3 to 5 can be performed iteratively based on different variant-filtering criteria.
The first step is to define a project with a corresponding experimental design (i.e., ‘Case only’ or ‘Case vs. Control’) and to upload variant files (Step 1). The pipeline supports the variant call format (VCF) (Danecek et al., 2011), genome variation format (GVF) (Reese et al., 2010), and Complete Genomics' VAR file format. Each variant file should be assigned to groups (either ‘case’ or ‘control’) in a project with a specific reference genome build (i.e., ‘hg18’ and ‘hg19’). A set of samples in gNOME is distinguished by the project name and group label. The uploaded variant files are placed in the internal queuing system for annotating with 60 different sources of genomic information collected from 17 publicly available databases (Step 2) (see Materials and Methods and Supp. Table S1 for details). This step takes at most 30 minutes for an individual WGS with 4-5 million variants. For efficient handling of dataset with multiple genomes, it is recommended to upload them as multiple sample VCF files such that gNOME can speed-up the annotation step by processing the entire variants – union of variants found in any of genomes in the file – in a single step. Once the annotation is completed, each genome in the multi-individual VCF file is stored individually. For instance, the merged VCF file for 1,097 samples of the 1000 Genomes Project (1KGP) has only 39,706,715 variants, for which gNOME can complete the annotation in 50 minutes (see Results). The resulting annotated variant files are stored in an internal MySQL database. Once the annotation is completed, users will receive a notification email. The summary statistics for all uploaded variant call files are available on 3.Summary::Genome Level (Supp. Figure S1). This overview is available for a single genome at a time or for multiple genomes. When a group of genomes is selected, gNOME displays the average and range of summary statistics for each variant type.
In Step 3, users can select multiple criteria for annotation-based filtering through an interactive web interface. For instance, one can select rare or novel loss of function (LoF) variants at highly conserved loci that are exclusively found among cases but neither in controls nor in an ethnicity-matched population dataset. Also, during this step, possible false positives are reduced by filtering out low quality variants and variants found in repetitive regions. We grouped filtering options into 4 broad categories in the web interface: 1) Allele Frequency, 2) Functional Impact, 3) Knowledge Enrichment, and 4) Others (Figure 2A). The LoF variants at highly conserved loci that are rare or novel in the European population are selected by setting 1) Allele Frequency::Ancestry ‘European’ (Figure 2A-1), 2) Allele Frequency::Allele Frequency ‘≤ 1% (rare)’ (Figure 2A-2), 3) Functional Impact::Gene impact ‘LoF’ (Figure 2A-3), and 4) Knowledge Enrichment::GERP++ score ≥ ‘2’ (Figure 2A-4). We can exclude the variants with low calling quality scores by setting Other::Variant call score (Figure 2A-5). The selected variants or genes are displayed in a table that can be sorted by column, and are available for download as a tab-delimited text file, which can be used as an input for the other protein-protein interaction network based analysis tools such as DAPPLE (Rossin et al., 2011). 3.Summary::Variant Level lists the detailed annotations for all variants that passed the filtering criteria whereas 3.Summary::Gene Level shows the number of variants that met the criteria for each gene.
Figure 2. Discovering somatic mutations in tumor-blood paired whole exome sequences.

(A) A screenshot for comparing variants from tumor tissue (as ‘case’) and blood sample (as ‘control’), both of which come from a single patient (‘MM56’) (see Finding somatic mutations in uveal melanoma and Materials and Methods for detail). From both tumor tissue and blood sample, allele frequencies were estimated with (1) European ancestry, and (2) rare or novel (3) loss of function variants at (4) highly conserved loci were selected. Low-quality variants were excluded by setting (5) ‘Variant call score ≥ 20’. The potential somatic mutations were selected by choosing variants that were present in tumor sample but not in blood sample (6). (B) The result from the comparison shown in (A). The table can be searched for gene symbol or sorted by the columns. A total of 11 genes including BAP1 (displayed) met the criteria. gNOME performs a gene set enrichment analysis for 5 gene set categories with the genes that passed filtering criteria.
Group comparison, Step 4, is one of the unique features that distinguish gNOME from other WGS and WES annotation tools (Cingolani et al., 2012; Ge et al., 2011;Wang et al., 2010). Group comparison helps to identify a set of variants, genes, and gene sets that are significantly enriched in cases as described in Materials and Methods, and is also useful to identify possible false positive incidental findings such as platform-specific sequencing errors and hypervariable genes and gene sets (Kohane, et al., 2012). These genes can be easily identified in gNOME and filtered out for further analysis if desired (Figure 2A-6). 4.Analyze::Variants and 4.Analyze::Genes (Figure 2) identify interesting variants enriched in case genomes and genes with such variants. Additionally, in 4.Analyze::Genes, gNOME can test whether a set of genes with interesting variants are enriched in precompiled gene sets from the Gene Ontology terms, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and the other disease-gene association databases (Figure 2B). We demonstrated the performance, accuracy, and group comparison features using 3 publicly available WGS and WES datasets.
Ethnicity specific allele frequencies of known variants
To calculate ethnicity specific AFs, we used The NCBI Short Genetic Variations database (dbSNP, http://www.ncbi.nlm.nih.gov/SNP, version 137), the 1KGP (Genomes Project et al., 2012) for European, Asian, and African populations and the Exome Sequencing Project (ESP, http://esp.gs.washington.edu) (Fu, et al., 2013) for European and African populations. The datasets with less than 15 samples in dbSNP were not used due to the inaccuracy in estimating AFs. We categorized AFs into four groups: common (AF ≥ 5%), less common (1% ≤ AF < 5%), rare (AF < 1%), and novel. The numerical codes -1 and -10 are used to represent a reported variant without a known AF and a novel variant, respectively. If AFs from different data sources were inconsistent, the highest value was used to represent the ethnicity specific AF. The same rule was applied for mixed ancestries.
Possible impact on protein function
Predicting the functional impact of amino acid changes resulting from nucleotide changes is an important step for prioritizing disease-associated genes since most known disease-causing variants are in protein-coding regions (Choi, et al., 2009). To provide possible consequences of genomic variants in genic regions, we integrated multiple gene models and prediction algorithms as part of gNOME. The Reference Sequence database (RefSeq, http://www.ncbi.nlm.nih.gov/refseq) (Pruitt et al., 2005), Consensus Coding Sequence (CDS) project (CCDS, http://www.ncbi.nlm.nih.gov/CCDS) (Pruitt, et al., 2009), Ensembl (Hubbard, et al., 2002) (http://www.ensembl.org), and University of California Santa Cruz (UCSC) Known Genes (Hsu et al., 2006) were all implemented in our gene annotation database. The use of multiple transcript models to estimate the functional impact of a variant is essential since possible consequences of a variant can be different across transcript models and an intronic variant in one transcript model can be in the coding region of the other transcript model (see Supp. Figure S2 for an example). Possible impacts of a variant on each transcript are categorized into synonymous, missense, in-frame insertion, in-frame deletion, splice site disruption, nonstop, misstart, frameshift, and nonsense. LoF variants were defined to include splice site disruption, frameshift, and nonsense. A broader category of LoF variants (adding nonstop and misstart) is also provided. We also annotate predicted impacts on protein function using the database for nonsynonymous SNPs' functional predictions (dbNSFP, https://sites.google.com/site/jpopgen/dbNSFP) (Liu et al., 2011) that comprises the predicted impacts estimated using the Sorting Intolerant From Tolerant (SIFT) (Kumar et al., 2009), PolyPhen2 (Adzhubei et al., 2010), MutationTaster (Schwarz et al., 2010), and a likelihood ratio test (LRT) (Chun and Fay, 2009).
Conservation scores, non-coding elements and biomedical knowledge enrichment
Conservation scores according to the Genomic Evolutionary Rate Profiling (GERP++, http://mendel.stanford.org/Sidowlab/downloads/gerp) (Davydov et al., 2010) are used to filter variants per locus, and we used an average score of GERP++ for insertions and deletions (indels). Genotyping errors are more frequently observed in repetitive regions, thus excluding the variants in these regions can reduce false positive findings. We used the RepeatMasker database (http://www.repeatmasker.org) to find any variants in these regions (Smit et al., 1996-2010). The default value for percent overlap with RepeatMasker regions is set to 0 %.
A variant on an important functioning protein domain could have a significant impact on protein function. Known protein domains were collected from the InterPro database (Hunter, et al., 2012) and mapped to the reference genome coordinates in order to facilitate variant annotations. The regulatory regions from the Encyclopedia of DNA Elements (ENCODE) project (Consortium, 2011) (http://encodeproject.org/ENCODE), the conserved transcription factor binding sites from UCSC Table Browser (http://genome.ucsc.edu), and microRNA host genes (Kozomara and Griffiths-Jones, 2011) were included in the annotation database to provide further information for non-coding functioning elements.
The pipeline includes gene sets for diseases, biological processes and canonical pathways. A total of 1,253 disease-associated gene sets were compiled through the gene-to-disease mapping using the literature abstracts annotated with Medical Subject Heading (MeSH) terms and NCBI Genes (Mitchell et al., 2003). The known disease-associated genes and variants from the catalog of genome-wide association studies by National Human Genome Research Institute (Hindorff et al., 2012) (http://www.genome.gov/gwastudies/), Online Mendelian Inheritance in Man (OMIM, http://www.omim.org) (McKusick, 2007), ClinVar (Riggs et al., 2013) (http://www.ncbi.nlm.nih.gov/clinvar), and DisGeNet (http://ibi.imim.es/DisGeNet/web/v02/home) (Bauer-Mehren et al., 2010) were integrated to the annotation database; the Human Gene Mutation Database (HGMD, http://www.hgmd.org) can be used if a user has the license. In addition, we collected 828 biological process gene sets based on the GO terms and 186 KEGG pathways from the Molecular Signatures Database (MSigDB, http://www.broadinstitute.org/gsea/msigdb, c5.bp.v3.0 and c2.cp.v3.0 respectively) (Subramanian, et al., 2005). The original data sources and processing scripts are available on the gNOME website, and the annotation database will be updated with the latest annotation information every 6 months.
Integration of population-scale individual whole genome sequencing data
The purpose of using ethnicity-matched population datasets as a comparison group is twofold. Firstly, the false positive incidental findings can be identified and reduced as previously described (Kohane, et al., 2012). Secondly, the genetic burden due to a set of interesting variants in the ethnicity-matched general population can be estimated and compared with study individuals. The comparison of uploaded data with population-scale data from the 1KGP is one of the unique features of gNOME. Following the categorization of the 1KGP, we included 18 different population categories: 4 by ancestry – European, Asian, African, and admixed American – and 14 by geographical regions.
Statistical comparison at variant-, gene-, and gene set-levels
A set of variants that met a user's annotation filtering criteria can be tested for enrichment in a group. Given two groups of samples (i.e., cases and controls), the interesting variants that are over-represented in cases can be identified as follows. Supposing the existence of N individuals and a total of M LoF variants, we defined M as the number of unique LoF variants across N individuals and set two groups as G0 (for instance, case group) and G1 (for instance, non-case group or ethnicity-matched population from the 1KGP). Then, an M-by-N matrix can be expressed as V = {νi,j}1≤i≤M,1≤j≤N where . The matrix V is illustrated as the colored grid box with 5 cases and 5 non-cases in Figure 3A. Row-wise hypergeometric tests find the variants that are more frequently found in G0. Alternatively, the number of individuals with interesting variants can be set for each group as shown in Figure 2A-6. For instance, by setting Ngroup A genomes ≥ 1 and Ngroup B genomes ≤ 0, a set of interesting variants will be further filtered to a smaller set of variants that are exclusively present in Group A.
Figure 3. Association tests for variants, genes, and gene sets between two groups.

The small number of variants that remain after the annotation-based filtering can be associated with a phenotype in three ways. First, we can test whether a specific variant presents more frequently in cases compared to controls or an ethnicity-matched population (A). Second, an association test can be performed at the gene-level when each case individual may have different variants on the same gene (B). Third, we can expand a gene-level aggregation to gene set-level to find the gene set over-represented with interesting variants among cases (C). The rows marked by x (red) denote “hypervariable” variants, genes, or gene sets that frequently have variants in both cases and controls (see Materials and Methods for details).
With the exception of a few Mendelian disorders, the likelihood of finding the same disease-linked variants across the patients is low (McClellan and King, 2010). Instead, multiple rare LoF and missense variants in the same gene or the same pathway could alter disease risks. Burden tests and kernel-based tests compare the cumulative effects of such variants. In burden tests, each variant is weighted differently according to AF, the impact on protein function, and conservation scores (Han and Pan, 2010; Madsen and Browning, 2009; Morris and Zeggini, 2010; Price et al., 2010; Zawistowski et al., 2010). The most burden tests assume that all variants of interest contribute to the phenotype in the same direction while kernel-based tests such as Sequence Kernel Association Test (SKAT) (Wu et al., 2011) and C-alpha (Neale et al., 2011) combine both protective and deleterious effects as well as variant-variant interactions. In our proposed pipeline, we implemented a burden test with the equal weights for all variants selected for specific criteria. The genes with compound heterozygous variants where each variant met user-defined filtering criteria can be prioritized using 4.Analyze::Genes (Figure 2). gNOME aggregates interesting variants by counting the number of variants in each gene, and perform a gene-level association test. Without loss of generality, we can assume that the M variants are linked to the total of P genes and the membership of each variant to genes can be represented as P-by-M matrix G = {gk,i}1≤k≤P,1≤i≤M, where . The matrix multiplication, B = G · V = {bk,j}1≤k≤P,1≤j≤N, gives us the number of variants in a gene for each individual (Figure 3B). The genes with a significantly different number of variants between two groups are ranked by a row-wise two group comparison test of B. If a gene is hypervariable (Kohane, et al., 2012), it would consistently have non-zero value (bk,j > 0 for all j) and be less significant in group comparison test (marked with a red x in Figure 3B).
A gene-level association test can be further expanded to a set of genes that are functionally related or physically interacting with each other. Even if an individual gene showed weak association in gene-wise test, those genes can collectively contribute to a specific phenotype. Our analysis pipeline provides several options for gene set-level association tests. Firstly, one can perform a gene set enrichment test for the genes with interesting variants in each case (‘Enriched Gene Sets in group A’ function in Figure 2B). Secondly, for each individual in G0 and G1, we can prioritize the gene sets that are more frequently observed as significantly enriched among the individuals in G0. We construct a contingency table T= (t11, t12, t21, t22) for each gene set per individual with the numbers of member genes with the selected variants (t11), member genes without the selected variants (t12), non-member genes with the selected variants (t21), and non-member genes without the selected variants (t22), where the genes in a given gene set are defined as member genes. The relationship between genes and gene sets is defined as Q-by-P matrix S = {Sl,k}1≤l≤Q,1≤k≤P where . Then we can collapse the values in matrix B into 0 or 1 to indicate whether an individual has interesting variants in the gene: Ḃ = {ḃk,j}1≤k≤P,1≤j≤N where ḃk,j = 1 if and only if bk,j > 0. The four values in T are defined as:
with the total number of genes (
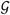 ). The enrichment scores from the contingency tables constitute the Q-by-N matrix C = {cl,j}1≤l≤Q,1≤j≤N (cl,j = the enrichment score of l-th gene sets in j-th individual) (Figure 4C). The row-wise two group comparison tests on C are performed either by using a non-parametric test with odds ratios or hypergeometric p-values or by comparing the proportion of individuals that passed a user-defined statistical threshold. Using the latter option, hypervariable gene sets with interesting variants can be identified (marked with a red x in Figure 3C).
). The enrichment scores from the contingency tables constitute the Q-by-N matrix C = {cl,j}1≤l≤Q,1≤j≤N (cl,j = the enrichment score of l-th gene sets in j-th individual) (Figure 4C). The row-wise two group comparison tests on C are performed either by using a non-parametric test with odds ratios or hypergeometric p-values or by comparing the proportion of individuals that passed a user-defined statistical threshold. Using the latter option, hypervariable gene sets with interesting variants can be identified (marked with a red x in Figure 3C).
Figure 4. Comparison of annotation results from 4 software tools.

For splice site disruption (A), nonsense (B), frameshift insertion and deletion (C), and nonstop (D) variants, we compared the annotation results from 4 different software tools by comparing genomic coordinate, alternative allele, and reported functional impact for each variant. The numbers next to tool names represent the total number of annotated variants in that category, and 4-way Venn diagrams show the concordant and discordant annotation results. Overall, the annotation results are comparable to each other between tools; however, splice site disruption has the most discordant results (A). ANNOVAR reports as frameshift even if such variants are found in canonical splice sites; however, gNOME and SeattleSeq report them as splice site disrupting variants. Supp. Table S3 lists the details on the discordant results.
Evaluation datasets
We used 3 datasets to evaluate gNOME's performance and to compare with other programs. The first dataset consists of 97 patients with transitional cell carcinoma (TCC) (Gui, et al., 2011). For each patient, paired tumor-blood samples were sequenced using WES. We downloaded the raw sequence files from the Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) (Leinonen et al., 2011) (accession number SRA038181), and aligned them to the reference human genome (hg19) using Burrows-Wheeler Aligner (BWA) 0.6.2 (Li and Durbin, 2009). The potential variants were called by the Genome Analysis Toolkit (GATK) 2.3-4 following the guidelines in Best Practice v4 from GATK's website (http://www.broadinstitute.org/gatk). The second WES dataset consisted of tumor-blood pairs from two patients with uveal melanoma (Harbour, et al., 2010). The raw sequences (accession number SRA062369) were processed with the same alignment and variant calling procedure as described in the original paper. This dataset was used to compare the accuracy and performance of gNOME's annotation procedure with the other software tools. Finally, we used all variants from a single individual (NA12889) in 1KGP as well as the entire Phase I integrated call set from the 1KGP to evaluate the scalability with data size.
Comparison with the existing software tools
We compared the performance and annotation results of gNOME with published software tools. The list of software tools included ANNOVAR (http://www.openbioinformatics.org/annovar, latest downloaded on 05/27/2013) (Wang et al., 2010), SnpEff (http://snpeff.sourceforge.net, version 3.2) (Cingolani et al., 2012), and SeattleSeq annotation server (http://snp.gs.washington.edu/SeattleSeqAnnotation137, version 137). We downloaded the latest version and installed on the same workstation if source codes were available, or uploaded the same VCF file to the web server. We used the same RefSeq gene model (downloaded on 05/20/2013 from UCSC Table Browser) for ANNOVAR, SnpEff and gNOME. However, SeattleSeq annotation server was running with a slightly different version of RefSeq model at the time of experiment. We measured the wall clock time to complete annotation for functional consequences of variants using a WGS variant file and 39.7 million variants from 1KGP.
Results
Comparison of performance and annotation results from the other software tools
The annotation speed of gNOME was compared with those of ANNOVAR (Wang et al., 2010) and SnpEff (Cingolani et al., 2012) using variants from a single genome (NA12889) and the integrated variants from 1,092 individuals of the 1000 Genomes Project (1KGP) (Genomes Project et al., 2012). The annotation for functional consequences of variants based on the RefSeq gene model (Pruitt et al., 2005) was performed 10 times for each tool. All 3 tools finished the annotation procedure in a reasonably short amount of time. For a single genome, SnpEff showed the best performance (192.2 ± 4.47 seconds; mean ± standard deviation); however, with 39.7 million variants of 1KGP, gNOME completed the annotation in 484.2 ± 0.92 seconds compared to 3,263.0 ± 65.94 and 1,606.8 ± 43.28 seconds for ANNOVAR and SnpEff (Table 1). The annotation engine of gNOME – gSearch (Song et al., 2012) – was optimized to handle a larger dataset whereas the processing time for ANNOVAR and SnpEff increased linearly with the number of variants. It should also be noted that genomes uploaded to gNOME's web interface will be annotated using 4 popular gene models, taking 4 to 5 times longer than reported in Table 1; at most 30 minutes for a single genome and 50 minutes for 1,092 genomes from 1KGP.
Table 1. Comparison of annotation performance.
We used the variant files from a whole genome sequence and all concatenated variants of 1,092 individuals from the 1000 Genomes Project. For each tool, we repeated the annotation procedure 10 times to calculate the average time required to complete the annotation of variant consequences using RefSeq gene model (the standard deviations are shown in parentheses). All three tools perform reasonably quickly. The annotation time of ANNOVAR and SnpEff linearly increases with the number of variants; however, only 37.8% more processing time is required to complete the annotation of x8.7 larger variant file using gNOME.
| Variant file | ANNOVAR | gNOME | SnpEff |
|---|---|---|---|
| A single genomea | 395.2 (4.78) | 351.4 (3.53) | 192.2 (4.47) |
| 1,092 genomesb | 3,263.0 (65.94) | 484.2 (0.92) | 1,606.8 (43.28) |
All variants found in one individual (NA12889) from CEPH/Utah family (4,581,619 variants).
All variants in the Phase 1 integrated call set from the 1000 Genomes Project (39,706,715 variants).
For the comparison of annotated functional impacts of variants, we included SeattleSeq, a web-based annotation server, as well as ANNOVAR and SnpEff. For this comparison, we concatenated 4 variant files from 2 tumor-blood pairs in the uveal melanoma dataset using the same gene model –RefSeq gene model (downloaded on 05/20/2013 from UCSC Table Browser) – for most tools to preclude discordant annotations due to different gene models (see Materials and Methods). Since each tool uses a different set of terminology to describe functional impacts, we mapped the various description terminologies used by separate tools, as listed in Supp. Table S2. Overall, the annotation results from different tools were similar. However, there were categories of functional impacts that were not reported by ANNOVAR and SeattleSeq (Table 2). For instance, a loss of start codon –categorized as misstart in gNOME – was not reported in ANNOVAR and SeattleSeq, and in-frame insertion and deletions were not listed in SeattleSeq.
Table 2. Comparison of annotation results from various programs.
The functional consequences for all variants found from 2 tumor-blood pairs of whole exome sequencing of uveal melanoma samples are compared (see Materials and Methods). The functional impacts are based on RefSeq gene definitions, and description terms are compared according to the Supp. Table S2. ‘NA’ implies that the corresponding term is not available for the program.
| Category | Variant consequences | ANNOVAR | gNOME | SnpEff | SeattleSeq |
|---|---|---|---|---|---|
| Single nucleotide changes in coding sequence | Disrupt | 125 | 137 | 119 | 92 |
| Missense | 11,687 | 11,729 | 11,825 | 12,042 | |
| Misstart | NA | 22 | 22 | NA | |
| Nonsense | 120 | 118 | 119 | 121 | |
| Nonstop | 14 | 15 | 28 | 22 | |
| Synonymous | 12,970 | 13,051 | 13,105 | 13,206 | |
|
| |||||
| Short insertions and deletions in coding sequence | Frameshift | 113 | 126 | 131 | 148 |
| In-frame insertion | 78 | 78 | 79 | NA | |
| In-frame deletion | 127 | 127 | 126 | NA | |
|
| |||||
| Variants outside of coding sequence | 5′-UTR | 3,677 | 3,941 | 3,941 | 3,879 |
| 3′-UTR | 8,659 | 9,008 | 9,010 | 8,931 | |
| Intron | 262,678 | 267,803 | 267,867 | 253,821 | |
| Intergenic | 255,085 | 265,926 | 265,941 | 264,470 | |
We found differences in the sets of variants for each functional impact category across 4 tools (Figure 4 shows Venn diagrams for splice site disrupting, frameshift, nonsense, and nonstop variants). The differences can be explained partly by the differences in RefSeq versions between SeattleSeq and the other 3 tools (SeattleSeq used genes in Sept. 2012 version from National Center for Biotechnology Information (NCBI), while we used genes in May 2013 version from UCSC Table Browser), by annotation errors in all programs, and by a discrepancy between tools in describing the same variants. Of 92 - 137 splice site disrupting (SSD) variants that were found by 4 tools, 69 were discovered in common. SeattleSeq missed 37 SSD variants that were found by 3 other tools, most of which were suspected from differences in gene models. ANNOVAR and gNOME took different approaches in describing the SSD variants due to insertions and deletions (indels). In gNOME, SSD has priority over other functional consequences if indels were found in canonical splice sites, and vice versa in ANNOVAR. In 21 cases of ambiguous SSD variants, e.g., insertions at exon-intron junctions, gNOME classified all ambiguous cases as SSD, while other programs report only part of them. The proportion of discordant annotations among programs was the smallest for nonsense variants. Among the nonsense variants that were not reported by gNOME, we found 4 erroneous annotations (2 from SnpEff, 1 from ANNOVAR and SeattleSeq each). There were 3 frameshift indels annotated as nonsense by ANNOVAR but as frameshift by the others. The 4 nonsense variants found only by SeattleSeq were due to an outdated gene model. For nonstop variants, 11 out of 15 discordant annotations between gNOME and other tools resulted from the annotation for possible selenocysteine, which was recognized only in gNOME. The other 4 were annotation errors from SnpEff and ANNOVAR. Discordant annotations for frameshift variants were more complex (see Supp. Table S3 for details); however, a majority of discordant annotations was due to the different approaches in classifying functional impacts. The details on discordant annotations between programs are summarized in Supp. Table S3. The functional impact of each variant must be evaluated by experts; however, there is a tremendous need for a standard method to describe variants with complex consequences.
Finding somatic mutations in uveal melanoma
To ensure the annotation accuracy of gNOME, we analyzed a published WES dataset from patients with uveal melanoma (MIM 155720). Harbour and colleagues sequenced two cases of matched tumor and peripheral blood samples using WES to find tumor specific somatic mutations on chromosome 3 (Harbour, et al., 2010). They found an inactivating mutation in each tumor sample on BAP1. One patient (MM56) had a nonsense mutation (p.W196X) whereas the other (MM70) had an 11-bp deletion (p.Q322fsX100) on the same gene. We processed the downloaded data as described in Materials and Methods, and uploaded the variant files from tumor tissues as cases and those from blood as controls to our pipeline and analyzed as depicted in Figure 2. The variant level analysis of gNOME for MM56 revealed 670 possible somatic mutation candidates in protein coding regions including p.W196X in BAP1. After filtering out non-rare (AF >1% in European population, the same ethnic group as the patients) or synonymous variants, 171 candidate variants were found. Of these, four nonsynonymous variants – 3 missense and 1 nonsense (p.W196X in BAP1) – were found on chromosome 3. Similarly, in MM70, the 11bp frameshift deletion in BAP1 was the only high impact tumor-specific nonsynonymous variant on chromosome 3. Interestingly, gene level analysis of gNOME found 11 genes – CEP89, FAM135A, GNAQ, HECTD4, HEXA, KCTD20, RAD17, TAS2R31, THBS3, TTLL1, and WSCD1 – that contain possible somatic mutations in both MM56 and MM70. Of these genes, frequent somatic mutations in GNAQ from patients with uveal melanoma were previously reported (Van Raamsdonk, et al., 2009), and the increased protein expression of HEXA was found in metastatic uveal melanoma (Linge, et al., 2012).
Discovering somatic mutations and enriched pathways in transitional cell carcinoma
Gui and colleagues sequenced 9 tumor-blood pairs from the patients with transitional cell carcinoma (TCC, MIM 109800), and found 465 predicted somatic mutations. Several genes such as ARID1A and CREBBP had different somatic mutations in tumor samples of different patients. Additionally, tumor-blood sample pairs from 88 patients with TCC - 37 non-muscle-invasive (NMI) TCC and 51 muscle-invasive (MI) TCC) were sequenced to find frequently mutated genes in MI-TCC and NMI-TCC. The downloaded data were processed using hg19 (see Materials and Methods). First, all 9 tumors samples were uploaded onto gNOME as cases, and 9 blood samples as controls. We compared the somatic mutation candidates that were identified in gNOME using 4.Analyze::Variants (Ngroup A genomes ≥ 1 and Ngroup B genomes ≤ 0) with the list from the original paper (Supplementary Table 3 in the original paper). Of the 208 somatic substitutions that were validated by genotyping or Sanger sequencing, 195 variants were accurately called and annotated using gNOME, except for 13 variants that were not called by our variant calling approach with hg19. One variant (g.chr1:22186113T>G on hg19) was found in the blood sample of B17 individual, but not in B17's tumor sample. Next, we tested whether the somatic mutation candidates were enriched for any gene sets using 97 tumor-blood pairs. For this analysis, we selected the LoF variants with AF ≤ 1% in Asian population, and p-value threshold ≤ 0.05, Ngroup A genomes ≥ 1, and Ngroup B genomes ≤ 97 in Statistical test parameters in 4.Analyze::Genes. Ninety six genes with the variants that met the criteria were enriched for the cancer related KEGG pathways such as cell cycle (adjusted p-value 0.0014), prostate cancer (adjusted p-value 0.0017), pathways in cancer (adjusted p-value 0.010), arrythmogenic right ventricular cardiomyopathy (adjusted p-value 0.008), and bladder cancer (adjusted p-value 0.013) (Supp. Table S4).
Discussion
gNOME enables users to interactively filter a large number of variants down to a small number of disease/phenotype linked variants dynamically reported at three different levels – variants, genes and gene sets – simultaneously. Additionally, gNOME applies non-parametric statistical tests to variant- and gene-level counts of filtered variants between two groups, as well as to gene set enrichment analysis for biological pathways and known disease-linked genes. With the web interface for interactive annotation-based filtering and statistical tests, we demonstrated our streamlined analysis procedure using two tumor-blood paired datasets – uveal melanoma and TCC. All validated genomic variants in the uveal melanoma dataset and 93% of 208 validated variants in the TCC dataset were accurately identified with gNOME, and new candidate variants and genes were found. Additionally, the cancer related pathways were found to be enriched with LoF variants that were exclusively found in tumor samples.
Precise identification of genomic variants with high accuracy, and the transparent annotation and filtering procedure are essential for clinical sequencing (Gargis, et al., 2012). A combination of the version control system for annotation database and graphical user interface allow gNOME to successfully reproduce results. Furthermore, gNOME can be installed locally as a stand-alone analysis pipeline with a secure storage device behind a firewall since the genome sequence information is confidential. Communications with other servers are not necessary once variant files are transferred. Moreover, enabling the other encryption and security features on MySQL database will make gNOME compatible with the Clinical Laboratory Improvement Amendments.
To further evaluate the annotation accuracy of our proposed pipeline compared to the other genome annotation software tools, we analyzed the same VCF file from the uveal melanoma dataset using ANNOVAR, SnpEff, and SeattleSeq. The results were similar in general, but the number of discordant results varied across functional categories. Aside from cases due to differences in gene models or programming errors, a majority of discordant cases came from variants whose functional consequences can be classified into multiple categories. For instance, the frameshift variants overlapping with canonical splice sites were reported either as frameshift in ANNOVAR or as splice site disrupting in gNOME and SeattleSeq. The ontology for describing the functional consequences of sequence variants and a consensus approach to describe the variants with multiple functional consequences should reduce the number of discordant annotations between tools (Eilbeck et al., 2005).
There are a few limitations of gNOME. First, due to the licensing issue, known disease associated variants in the HGMD were not integrated into gNOME although the integration itself was straightforward. Second, only a limited set of statistical tests were implemented. Adding diverse genetic burden tests such as the methods implemented in PLINK-SEQ (Neale et al., 2011), Efficient and Parallelizable Association Container Toolbox (EPACTS) (Kang et al., 2010), and SKAT (Wu et al., 2011) will improve the flexibility of gNOME. Finally, the scalability of gNOME would be much improved if run on a computing cloud. The current web-based version can process medium-sized datasets of up to 1000 individuals, and a local stand-alone version can be set up to accommodate with a larger dataset or to preserve the confidentiality. However, to analyze a larger dataset (tens of thousands individuals), the gNOME pipeline must be run on a computing cloud. A few WES/WGS tools do support cloud environment as backend: VAT (Habegger et al., 2012) for variant annotation and visualization, Crossbow (Langmead et al., 2009) for variant calling, SIMPLEX (Fischer et al., 2012) for WES analysis, and Galaxy (Goecks et al., 2010) and Taverna (Hull et al., 2006) as general workflow framework. However, these are either difficult to use or do not cover all streamlined process provided by gNOME.
To summarize, we have developed a downstream analysis pipeline for WGS/WES datasets that can perform accurate and reproducible annotation with a graphical user interface for annotation-based filtering. A group comparison is one of unique features that help to reduce possible false positive findings. We have provided a population scale WGS dataset as a part of the pipeline, which enables users to identify variants specific to cases compared to ethnicity-matched generally healthy population. With these strengths, gNOME will be of use to the biomedical research community.
Supplementary Material
Acknowledgments
Grant sponsors: NHGRI U01HG006500, NIMH P50MH094267 and R01MH085143, NRF of Korea 2012R1A1A2039822
Footnotes
Supplemental Data: Supplemental Data include 2 figures and 4 tables.
Conflicts of Interest: Dr. Kohane is a member of the scientific advisory board of the SynapDx (Lexington, MA). All the other authors declare no conflict of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley EA, Butte AJ, Wheeler MT, Chen R, Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Morgan AA, Pushkarev D, Neff NF, et al. Clinical assessment incorporating a personal genome. Lancet. 2010;375(9725):1525–35. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- Bauer-Mehren A, Rautschka M, Sanz F, Furlong LI. DisGeNET: a Cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics. 2010;26(22):2924–6. doi: 10.1093/bioinformatics/btq538. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Tucker EJ, Compton AG, Kirby DM, Crawford G, Burtt NP, Rivas M, Guiducci C, Bruno DL, Goldberger OA, Redman MC, Wiltshire E, et al. High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat Genet. 2010;42(10):851–8. doi: 10.1038/ng.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. Journal of medical genetics. 2012;49(7):433–6. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, Nelson-Williams C, Farhi A, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A. 2009;106(45):19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19(9):1553–61. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat Rev Genet. 2010;11(6):415–25. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- Consortium EP. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS Biol. 2011;9(4):e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12(9):628–40. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6(12):e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R. The advent of personal genome sequencing. Genet Med. 2011;13(3):188–90. doi: 10.1097/GIM.0b013e31820f16e6. [DOI] [PubMed] [Google Scholar]
- Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6(5):R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Snajder R, Pabinger S, Dander A, Schossig A, Zschocke J, Trajanoski Z, Stocker G. SIMPLEX: cloud-enabled pipeline for the comprehensive analysis of exome sequencing data. PLoS ONE. 2012;7(8):e41948. doi: 10.1371/journal.pone.0041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J, Nickerson DA, Bamshad MJ, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493(7431):216–20. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, Lu F, Lyon E, Voelkerding KV, Zehnbauer BA, Agarwala R, Bennett SF, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012;30(11):1033–6. doi: 10.1038/nbt.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge D, Ruzzo EK, Shianna KV, He M, Pelak K, Heinzen EL, Need AC, Cirulli ET, Maia JM, Dickson SP, Zhu M, Singh A, et al. SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27(14):1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11(8):R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, Sunyaev S. Sequencing studies in human genetics: design and interpretation. Nat Rev Genet. 2013 doi: 10.1038/nrg3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habegger L, Balasubramanian S, Chen DZ, Khurana E, Sboner A, Harmanci A, Rozowsky J, Clarke D, Snyder M, Gerstein M. VAT: a computational framework to functionally annotate variants in personal genomes within a cloud-computing environment. Bioinformatics. 2012;28(17):2267–9. doi: 10.1093/bioinformatics/bts368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum Hered. 2010;70(1):42–54. doi: 10.1159/000288704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–3. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff L, MacArthur J, Morales J, Junkins H, Hall P, Klemm A, Manolio T. A Catalog of Published Genome-Wide Association Studies. 2012 Available at: www.genome.gov/gwastudies.
- Hoischen A, van Bon BW, Gilissen C, Arts P, van Lier B, Steehouwer M, de Vries P, de Reuver R, Wieskamp N, Mortier G, Devriendt K, Amorim MZ, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42(6):483–5. doi: 10.1038/ng.581. [DOI] [PubMed] [Google Scholar]
- Holm H, Gudbjartsson DF, Sulem P, Masson G, Helgadottir HT, Zanon C, Magnusson OT, Helgason A, Saemundsdottir J, Gylfason A, Stefansdottir H, Gretarsdottir S, et al. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat Genet. 2011;43(4):316–20. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22(9):1036–46. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, Durbin R, Eyras E, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30(1):38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull D, Wolstencroft K, Stevens R, Goble C, Pocock MR, Li P, Oinn T. Taverna: a tool for building and running workflows of services. Nucleic Acids Res. 2006;34(Web Server issue):W729–32. doi: 10.1093/nar/gkl320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40(Database issue):D306–12. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, Sul JH, Service SK, Zaitlen NA, Kong SY, Freimer NB, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat Genet. 2010;42(4):348–54. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd JM, Gravel S, Byrnes J, Moreno-Estrada A, Musharoff S, Bryc K, Degenhardt JD, Brisbin A, Sheth V, Chen R, McLaughlin SF, Peckham HE, et al. Population genetic inference from personal genome data: impact of ancestry and admixture on human genomic variation. Am J Hum Genet. 2012;91(4):660–71. doi: 10.1016/j.ajhg.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourquin T, Lewis L, Villasana D, Morgan M, Muzny D, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145(7):1036–48. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohane IS, Hsing M, Kong SW. Taxonomizing, sizing, and overcoming the incidentalome. Genet Med. 2012;14(4):399–404. doi: 10.1038/gim.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue):D152–7. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Lalonde E, Albrecht S, Ha KC, Jacob K, Bolduc N, Polychronakos C, Dechelotte P, Majewski J, Jabado N. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum Mutat. 2010;31(8):918–23. doi: 10.1002/humu.21293. [DOI] [PubMed] [Google Scholar]
- Lam HY, Pan C, Clark MJ, Lacroute P, Chen R, Haraksingh R, O'Huallachain M, Gerstein MB, Kidd JM, Bustamante CD, Snyder M. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30(3):226–9. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Schatz MC, Lin J, Pop M, Salzberg SL. Searching for SNPs with cloud computing. Genome Biol. 2009;10(11):R134. doi: 10.1186/gb-2009-10-11-r134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M International Nucleotide Sequence Database C. The sequence read archive. Nucleic Acids Res. 2011;39(Database issue):D19–21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Raychaudhuri S, Sanders SJ, Stevens C, Sabo A, MacArthur DG, Neale BM, Kirby A, Ruderfer DM, Fromer M, Lek M, Liu L, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77(2):235–42. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linge A, Kennedy S, O'Flynn D, Beatty S, Moriarty P, Henry M, Clynes M, Larkin A, Meleady P. Differential expression of fourteen proteins between uveal melanoma from patients who subsequently developed distant metastases versus those who did Not. Invest Ophthalmol Vis Sci. 2012;53(8):4634–43. doi: 10.1167/iovs.11-9019. [DOI] [PubMed] [Google Scholar]
- Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32(8):894–9. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181–91. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335(6070):823–8. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5(2):e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141(2):210–7. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80(4):588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11(10):685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- Mitchell JA, Aronson AR, Mork JG, Folk LC, Humphrey SM, Ward JM. Gene indexing: characterization and analysis of NLM's GeneRIFs. AMIA Annu Symp Proc. 2003:460–4. [PMC free article] [PubMed] [Google Scholar]
- Morris AP, Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 2010;34(2):188–93. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, Kathiresan S, Purcell SM, Roeder K, Daly MJ. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7(3):e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010a;42(9):790–3. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010b;42(1):30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2013 doi: 10.1093/bib/bbs086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelak K, Shianna KV, Ge D, Maia JM, Zhu M, Smith JP, Cirulli ET, Fellay J, Dickson SP, Gumbs CE, Heinzen EL, Need AC, et al. The characterization of twenty sequenced human genomes. PLoS Genet. 2010;6(9) doi: 10.1371/journal.pgen.1001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, Sunyaev SR. Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010;86(6):832–8. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, Hart E, Suner MM, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19(7):1316–23. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33(Database issue):D501–4. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Moore B, Batchelor C, Salas F, Cunningham F, Marth GT, Stein L, Flicek P, Yandell M, Eilbeck K. A standard variation file format for human genome sequences. Genome Biol. 2010;11(8):R88. doi: 10.1186/gb-2010-11-8-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs ER, Wain KE, Riethmaier D, Savage M, Smith-Packard B, Kaminsky EB, Rehm HL, Martin CL, Ledbetter DH, Faucett WA. Towards a Universal Clinical Genomics Database: The 2012 International Standards for Cytogenomic Arrays Consortium Meeting. Hum Mutat. 2013 doi: 10.1002/humu.22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK, Boucher G, Ripke S, Ellinghaus D, Burtt N, Fennell T, Kirby A, et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet. 2011;43(11):1066–73. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AF, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, et al. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328(5978):636–9. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, Daly MJ. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7(1):e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Lucas FA, Wang G, Scheet P, Peng B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics. 2012;28(3):421–2. doi: 10.1093/bioinformatics/btr667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. 1996-2010 http://www.repeatmasker.org.
- Song T, Hwang KB, Hsing M, Lee K, Bohn J, Kong SW. gSearch: a fast and flexible general search tool for whole-genome sequencing. Bioinformatics. 2012;28(16):2176–7. doi: 10.1093/bioinformatics/bts358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabor HK, Berkman BE, Hull SC, Bamshad MJ. Genomics really gets personal: how exome and whole genome sequencing challenge the ethical framework of human genetics research. Am J Med Genet A. 2011;155A(12):2916–24. doi: 10.1002/ajmg.a.34357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21(9):1529–42. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski M, Gopalakrishnan S, Ding J, Li Y, Grimm S, Zollner S. Extending rare-variant testing strategies: analysis of noncoding sequence and imputed genotypes. Am J Hum Genet. 2010;87(5):604–17. doi: 10.1016/j.ajhg.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.