

Abstract
We reported previously a significant linkage signal between psychotic bipolar disorder (BP) and microsatellite markers on chromosome 5q31–34 in the National Institute of Mental Health Bipolar Genetics Initiative (NIMH-BPGI) data set, Wave 1. In an attempt to fine-map this linkage signal we genotyped 1,134 single nucleotide polymorphisms (SNPs) under the linkage peak in 23 informative families (131 individuals) with evidence of linkage. We tested family based association in the presence of linkage with the computer software package FBAT. The most significant association in these families was with a SNP in the second intron of GRIA1 (α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid (AMPA) subunit 1 receptor gene) (rs490922, Z-score = 3.3, P= 0.001). The analysis of 37 additional families with psychotic BP from NIMH-BPGI data sets, Waves 2, 3, and 4 revealed a signal at a SNP in intron 5 of the GRIA1 gene (rs4385264, Z-score = 3.2, P-value = 0.002). A combined analysis of all 60 families continued to support evidence for association of GRIA1 with psychotic BP; however, individual SNPs could not be replicated across datasets. The AMPA1 receptor has been shown to influence cognitive function, such as working memory and reward learning. Our findings suggest that variations in this receptor may contribute to the pathophysiology of BP with psychotic features in some families.
Keywords: genetic, linkage, association, mood disorder, glutamate receptor
Introduction
Bipolar disorder (BP) is characterized by episodes of mania and depression [American Psychiatric Association, 2000]. It is a common disorder with lifetime prevalence between 1% and 2% [Kessler et al, 2005]. Numerous studies suggesting a high heritability for BP have stimulated a wide range of genetic mapping investigations, which so far have failed to identify any genetic variants unequivocally associated with this disorder. The disappointing results of such studies have been attributed in large part to imprecision in the definition of the BP phenotype and have led to the proposal of alternative phenotyping strategies.
About 70–85% of BP patients experience psychotic symptoms, here defined as hallucinations and/or delusions [Morgan et al., 2005]. The susceptibility to psychotic symptoms appears to be heritable and this fact has suggested the possibility that the presence of psychotic symptoms may characterize a specific subtype of BP associated with distinct genomic variations [Potash et al., 2003]. In particular, several lines of evidence suggest a locus for psychosis on distal chromosome 5q. Linkage was observed in this region for psychotic symptoms in families from the Portuguese Islands (non-parametric linkage (NPL) score = 3.28, P = 0.00066) [Sklar et al., 2004] and for families with BP from Costa Rica and Colombia, in which most affected individuals had psychotic symptoms (NPL score = 4.39, P < 0.00004) [Herzberg et al., 2006]. In a previous study, we reported linkage to psychotic symptoms over a 10 centi Morgan (cM) region on chromosome 5q31–34 between markers D5S410 and D5S422 (NPL score = 5.0, P < 0.00001) in 41 families (381 individuals) from the National Institute of Mental Health Bipolar Genetics Initiative (NIMH-BPGI) data set Wave 1 [Kerner et al., 2007]. Based on these findings we custom designed a single nucleotide polymorphism (SNP) panel to carry out a fine-mapping study of this linkage region.
For this study, we selected 131 individuals in 23 informative pedigrees from Waves 1 of the NIMH-BPGI data sets with parental DNA available and added additional families from Wave 2, 3, and 4 of the NIMH-BPGI data sets in order to increase our sample size. Families were chosen based on evidence of linkage to chromosome 5q31–34, psychotic BP phenotype and parental DNA available for genotyping. Here we present the results of association analyses of psychosis in these families using a 5qSNP panel. While we could not definitely map a psychosis locus to a narrow segment of this chromosomal region, the preponderance of evidence was obtained in the vicinity of GRIA1 (α-amino-3-hydroxy-5-methyl-4-isoxa-zole proprionic acid (AMPA) subunit 1 receptor gene).
Materials and Methods
Sample
Our sample consisted of 60 families (330 individuals) from the NIMH-BPGI Waves 1–4. The NIMH-BPGI is a collection of small nuclear families ascertained through a proband with BP type 1 (BP1) by a multi-site collaborative group in two phases with common ascertainment and diagnostic procedures [Nurnberger et al., 1997]. During the first phase (Wave 1 and 2) families were collected at Johns Hopkins University, Indiana University, Washington University in St. Louis, and the NIMH Intramural Program. During the second phase (Wave 3 and 4), the group of collaborating sites was expanded to further include University of California at San Diego, University of Iowa, University of Pennsylvania, University of Chicago, Rush-Presbyterian Medical Center, and University of California at Irvine.
All individuals who consented to be included in the study had been evaluated with the Diagnostic Interview for Genetic Studies (DIGS), Version 2.0 for Wave 1 and 2 and Version 3.0 for Wave 3 and 4 [Nurnberger et al., 1994]. The Family Interview for Genetic Studies (FIGS) and medical records were used for additional information. Psychiatrists reached final diagnoses according to the Diagnostic and Statistical Manual of Mental Disorders (DSM), Version III-Revised (DSM-III-R) (Wave 1 and 2) or DSM-IV diagnostic criteria (Wave 3 and 4) using the Best Estimate Procedure. We selected nuclear families with at least two members affected with psychotic BP or schizoaffective disorder, manic type (SZA) and evidence of linkage to chromosome 5q31–34. We first selected informative nuclear families from Wave 1 of the NIMH-BPGI data sets that had contributed to the original linkage finding, if parental DNA was available for genotyping. Individuals were coded as affected if they had experienced psychotic symptoms (hallucinations and/or delusions) persistently throughout the day for at least one day or intermittently for a period of at least three days independent of underlying diagnoses. All other individuals were coded as unknown. Unaffected siblings were also included when available. This data set consisted of 23 families (131 individuals, 52 males, 79 females). Fifty-five individuals had experienced halluci-nations or delusions, 51 of those individuals were also diagnosed with either BP1 or SZA. Four individuals had received the diagnoses of BP, type 2 (BP2) or major depressive disorder (MDD), 76 individuals had not experienced psychotic symptoms; of those individuals, 10 carried the diagnosis of BP1. In this data set, 22 families were of European descent and one family was African-American.
In our previous analysis of the NIMH-BPGI data sets, we had analyzed families separately by wave, because the families in different waves had been genotyped with different sets of markers. In the entire sample of Waves 2,3, and 4, we did not observe strong linkage to chromosome 5q. From these waves, we selected a subset of families with psychotic BP (n = 37, including 46 nuclear families with 199 individuals (84 males, and 115 females), evidence of linkage to 5q31—34 and parental DNA available for genotyping in the hope of increasing our sample size. In these families, 89 individuals were affected with psychotic symptoms (defined as hallucinations or delusions); 84 of whom were also diagnosed with BP1 or SZA, and five carried either the diagnosis of BP2 or MDD. Hundred and ten individuals had not experienced psychotic symptoms, 10 of whom had been diagnosed with BP1. Thirty-four of these families were of European descent, two were African-American, and one was Asian-American. Overall, the evidence of linkage in those families was weaker than in data set 1 and wider marker spacing (22 Mb) made it difficult to select families with linkage to exactly the same chromosomal region. Both data sets were first analyzed separately and then combined to account for the possibility that unobserved differences existed between the data sets that would have explained the discrepancy in linkage signals. The study was approved by the Institutional Review Board (IRB) at UCLA.
SNP Selection for Association Analysis
A set of 1,134 SNPs across the ∼9.3 mega base (Mb) region between markers D5S410 and D5S422 on chromosome 5q31–34 were selected using publicly available databases (Fig. 1). With the intention of covering the entire region fairly evenly, we selected SNPs based on a minimum linkage disequilibrium (LD) threshold (r2 = 0.7) between the tagging SNP (tSNP) and the captured allele, as well as a minor allele frequency greater than 5%, based on allele frequencies in HapMap individuals of Northern and Western European ancestry using the computer program Haploview Version 3 [Barrett et al., 2005]. Selected SNPs were pre-screened by Illumina using a proprietary algorithm that predicted performance on the Illumina platform. The average spacing between markers was 8.6 kilo base (kb); the maximum spacing was 91.2 kb. Since markers in regions of very low linkage disequilibrium (LD) (here defined as those containing ‘holes’ or gaps of ≥2.5 LD units on linkage disequilibrium unit (LDU) maps may have a limited power to detect association [Service et al., 2006], tSNPs were selected with increased density in such regions (about twofold), which were identified on the LD map constructed from the Phase I release #16 of the HapMap data. The same HapMap data set was used to choose tSNPs for the entire region of interest, except for ∼1.3 Mb comprising three low LD regions, for which tSNPs were selected from a denser map (Phase II release #20). The fraction of SNPs covering GRIA1 was 8.5% of the total number of SNPs genotyped. GRIA1 itself covers 3.4% of base pairs in the region. Fifteen common SNPs were added within or in proximity of candidate genes in this region because they had been included in previous analysis and publications: rs707176 (in GRIA1), rs254664, rs10046055 (in CLINT1), rs187269, rs252944, rs194072, rs1816071 (in GABRB2), rs3219151 (in GABRA6) and rs10056305, rs6862670, rs4478357, rs4260711, rs10051659, rs2279020, rs13171845 (in GABRA1) [Yamada et al., 2003; Horiuchi et al., 2004; Lo et al., 2004; Petryshen et al., 2005; Pimm et al., 2005; Magri et al., 2006]. Figure 1 shows the molecular structure of the region under the linkage peak on chromosome 5q.
Fig. 1.
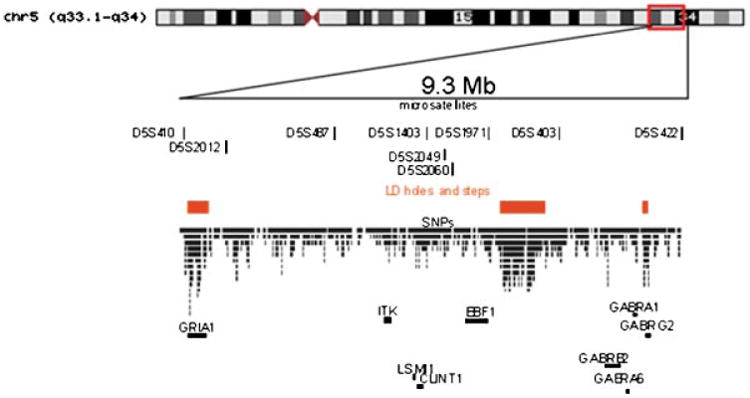
Molecular structure of the region under the linkage peak on chromosome 5q31–34. We show here microsatellite markers, SNPs and localization of linkage disequilibrium holes, and candidate genes in the chromosomal region (GRIA1: ionotropic glutamate receptor 1, AMPA1, #138248; ITK: IL2-inducible T-cell kinase, MIM#186973; LSM11: U7 small nuclear RNA-associated Sm-like protein, gene ID 134353; EBF1: early B-cell factor 1, MIM#164343; GABRB2, gamma-aminobutyric acid (GABA) A receptor, beta 2, MIM#600232; GABRA6, gamma-aminobutyric acid (GABA) A receptor, alpha 6, MIM#137143; GABRA1, gamma-aminobutyric acid (GABA) A receptor, alpha 1, MIM#137160; GABRG2, gamma-aminobutyric acid (GABA) A receptor, gamma 2, MIM#137164). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
SNP Genotyping
Genomic DNA was obtained from the NIMH Cell Repository at Rutgers University. DNA was extracted following standard protocols. All DNA samples were quantified by a Quant-iT PicoGreen double strand DNA (dsDNA) assay (Invitrogen, Carlsbad, CA). Samples were then normalized to a concentrationofabout 50 ng/μl.
One thousand one hundred and thirty-four SNPs were genotyped with the Illumina Goldengate genotyping assay on the Illumina BeadLab 1000 platform following a standard protocol [Fan et al., 2003]. In summary, 250ng of normalized genomic DNA from each sample were biotinylated and captured by avidin-coated paramagnetic particles. Pooled oligonucleotides were then hybridized to the bound genomic DNA and extended by incubation with polymerase. In a ligation step these allele-specific oligonucleotides (ASOs) were joined to locus-specific oligonucleotides (LSOs) and these single-stranded templates were then used in a multiplex polymerase chain reaction (PCR) reaction. Simultaneous PCR amplification of 1,134 loci per sample per oligonucleotide pool was performed using Titanium Taq (BD Biosciences, Franklin Lakes, NJ) and 0.5 units UDG (Invitrogen, Carlsbad, CA) per 60 μl reaction.
We collected and analyzed the fluorescence intensities on a BeadArray Reader (Illumina, San Diego, CA) confocal fluorescence scanner and the BeadScan software package (Illumina, San Diego, CA). The GenCall software package version 6.0.7 (Illumina, San Diego, CA) was used to cluster and call fluorescence intensities. As a quality control, we reviewed the automated clustering by hand. Failed markers, failed samples and individual genotypes with quality scores below the 0.25 threshold were removed from the final data set with the software GTS Reports (Illumina, San Diego, CA). In summary, 31 markers failed to be genotyped; four of these were located adjacent to each other in two chromosomal regions spanning 17,964 base pairs (bp) (at 153,887,439 bp) and 133 bp (at 159,282,900 bp). It is possible that these failures indicate genomic deletions. Two additional markers were located in regions of known copy number variations. These markers showed excessive Mendelian error rates (rs1870738 in four families and rs7353331 in eight families). One additional marker had Mendelian inconsistencies in 11 families indicating possible structural variations around this marker as well. These markers were not included in the analysis. In total, 34 markers were removed. Additional 8 markers had 14 Mendelian errors in 13 families. Genotypes for these markers in these families were set to zero. The average GeneCall scores for markers with Mendelian errors were between 0.992 and 1 and test for Hardy–Weinberg disequilibrium was non-significant. SNPs with Mendelian errors were not located in proximity to each other.
Statistical Analysis in FBAT
We used the Family Based Association Test (FBAT) software package [Horvath et al., 2001] to test each SNP separately for association with BP. This computer program provides a general framework for association testing in small nuclear families. P-Values using the empirical variance were calculated to provide a valid test of association in the presence of linkage. Under these conditions, sibling marker genotypes and multiple nuclear families in a pedigree cannot be considered independent. We therefore corrected the mean of the test statistic under the null hypothesis of “no association and no linkage” using an empirical variance–covariance estimator. This option is implemented in the software package FBAT [Lake et al., 2000].
The Haplotype Based Association Test (HBAT) option of the FBAT software package was used to test three-SNP-haplotypes for association with BP in a sliding window approach [Horvath et al., 2004].
Results
In families from Wave 1 (data set 1), the SNP most significantly associated with the phenotype of psychotic BP was located in the second intron of GRIA1 (rs490922, Z-score = 3.3, P = 0.001, OR = 3). Five additional SNPs were associated with this phenotype at P-values <0.01. Those SNPs were located in the first and second intron of the GRIA1 gene and in two gene poor regions, one close to the GRIA1 gene (rs17553409) and one in the vicinity of the GABA receptor gene cluster (rs885466) (Table I). An additional 29 SNPs out of the 1,134 SNPs throughout the entire chromosomal region under the linkage peak were significant at a P-value<0.05, however, none of those SNPs were within or close to known genes.
Table I. FBAT Results on Chromosome 5q31-33 in Wave 1 of the NIMH-BPGI.
| Wave 1 | Waves 2–4 | Waves 1–4 | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||
| Genes | Location | Marker | Location (bp) | Z | P | Z | P | Z | P |
| Summary | |||||||||
| GRIA1 | Intron 1 | rs472792 | 152,851,377 | 2.6 | 0.009 | 1.1 | 0.3 | 2.8 | 0.006 |
| GRIA1 | Intron 1 | rs524610 | 152,853,173 | 3.3 | 0.001 | ***** | 2.8 | 0.005 | |
| GRIA1 | Intron 2 | rs524905 | 152,862,559 | 2.7 | 0.008 | 0.9 | 0.4 | 2.6 | 0.01 |
| GRIA1 | Intron 2 | rs490922 | 152,864,016 | 3.3 | 0.001 | 0 | 1 | 27 | 0.008 |
| GRIA1 | Intron 2 | rsl864205 | 152,865,023 | 3.2 | 0.002 | 0.4 | 0.7 | 2.3 | 0.02 |
| GRIA1 | Intron 5 | rs4385264 | 153,033,928 | 0.1 | 0.9 | 3.2 | 0.002 | 2.2 | 0.03 |
| n | rs4958751 | 153,975,507 | ***** | ***** | 2.9 | 0.004 | |||
| n | rsl295213 | 154,968,586 | ***** | 2.1 | 0.04 | 2.9 | 0.004 | ||
| ITK | Intron 1 | rs452223 | 156,544,071 | 2.4 | 0.01 | ***** | 3.1 | 0.002 | |
| LSM11 | 3″ UTR | rs6882516 | 157,115,475 | 2.1 | 0.04 | ***** | 2.6 | 0.009 | |
| n | rs4704916 | 157,464,547 | 2.3 | 0.02 | ***** | 2.9 | 0.003 | ||
| EBF1 | Intron 9 | rs6874808 | 158,148,198 | ***** | ***** | 27 | 0.008 | ||
| n | rs7734683 | 159,529,087 | ***** | ***** | 2.6 | 0.01 | |||
| n | rs885466 | 160,263,579 | 2.9 | 0.004 | ***** | 2.9 | 0.004 | ||
| n | rsl914212 | 160,313,280 | 1.7 | 0.09 | 2 | 0.05 | 2.6 | 0.01 | |
| n | rs9284996 | 160,326,927 | ***** | ***** | 2.8 | 0.006 | |||
| Genes | Location | Marker | Allele | afreq | fam# | S | E(S) | Var(S) | Z | P |
|---|---|---|---|---|---|---|---|---|---|---|
| Results Wave 1 | ||||||||||
| GRIA1 | Intron 1 | rs472792 | G(T) | 0.29 | 10 | 26 | 17.33 | 10.94 | 2.6 | 0.009 |
| GRIA1 | Intron 1 | rs524610 | A(G) | 0.4 | 13 | 36 | 26.03 | 9.834 | 3.3 | 0.001 |
| GRIA1 | Intron 2 | rs524905 | C(T) | 0.35 | 10 | 28 | 20.83 | 7.194 | 27 | 0.008 |
| GRIA1 | Intron 2 | rs490922 | C(T) | 0.61 | 14 | 53 | 41.13 | 12.88 | 3.3 | 0.001 |
| GRIA1 | Intron 2 | rs1864205 | A(G) | 0.61 | 13 | 47 | 36.33 | 11.44 | 3.2 | 0.002 |
| GRIA1 | Intron 5 | rs4385264 | A(G) | 0.38 | 11 | 19 | 187 | 8.39 | 0.1 | 0.9 |
| n | rs4958751 | T(C) | 0.21 | 9 | ***** | |||||
| n | rs1295213 | G(T) | 0.64 | 8 | ***** | |||||
| ITK | Intron 1 | rs452223 | G(A) | 0.64 | 10 | 35 | 26.83 | 11.19 | 2.4 | 0.01 |
| LSM11 | 3′ UTR | rsG882516 | A(C) | 0.16 | 10 | 21 | 14.67 | 9.444 | 2.1 | 0.04 |
| n | rs4704916 | A(G) | 0.24 | 10 | 23 | 16 | 9 | 2.3 | 0.02 | |
| EBF1 | Intron 9 | rs6874808 | T(C) | 0.93 | 3 | ***** | ||||
| n | rs7734683 | C(T) | 071 | 8 | ***** | |||||
| n | rs885466 | A(G) | 0.83 | 11 | 47 | 36.5 | 13.25 | 2.9 | 0.004 | |
| n | rs1914212 | A(C) | 0.34 | 10 | 25 | 20.57 | 6.854 | 1.7 | 0.09 | |
| n | rs928499G | C(T) | 0.82 | 7 | ***** | |||||
| Results Waves 2-4 | ||||||||||
| GRIA1 | Intron 1 | rs472792 | G(T) | 0.23 | 12 | 19 | 16.17 | 6.361 | 1.1 | 0.3 |
| GRIA1 | Intron 1 | rs524610 | A(G) | 0.37 | 9 | ***** | ||||
| GRIA1 | Intron 2 | rs524905 | C(T) | 0.23 | 10 | 17 | 14.83 | 5.861 | 0.9 | 0.4 |
| GRIA1 | Intron 2 | rs490922 | T(C) | 0.41 | 11 | 22 | 22 | 7 | 0 | 1 |
| GRIA1 | Intron 2 | rs1864205 | G(A) | 0.41 | 10 | 20 | 19 | 6 | 0.4 | 0.7 |
| GRIA1 | Intron 5 | rs4385264 | G(A) | 0.64 | 14 | 46 | 36.43 | 9.25 | 3.2 | 0.002 |
| n | rs4958751 | T(C) | 0.12 | 9 | ***** | |||||
| n | rs1295213 | T(G) | 0.39 | 13 | 41 | 33.03 | 14.4 | 2.1 | 0.04 | |
| ITK | Intron 1 | rs452223 | A(G) | 0.22 | 9 | ***** | ||||
| LSM11 | 3′ UTR | rsG882516 | C(A) | 0.83 | 5 | ***** | ||||
| n | rs4704916 | G(A) | 0.86 | 7 | ***** | |||||
| EBF1 | Intron 9 | rs6874808 | T(C) | 0.88 | 8 | ***** | ||||
| n | rs7734683 | T(C) | 0.33 | 9 | ***** | |||||
| n | rs885466 | A(G) | 0.93 | 3 | ***** | |||||
| n | rs1914212 | A(C) | 0.36 | 10 | 21 | 16.27 | 5.604 | 2 | 0.05 | |
| n | rs9284996 | T(C) | 0.19 | 6 | ***** | |||||
| Results Wave and Waves 2–4 combined | ||||||||||
| GRIA1 | Intron 1 | rs472792 | G(T) | 0.26 | 22 | 45 | 33.5 | 17.31 | 2.8 | 0.006 |
| GRIA1 | Intron 1 | rs524610 | A(G) | 0.38 | 22 | 55 | 43.03 | 17.83 | 2.8 | 0.005 |
| GRIA1 | Intron 2 | rs524905 | C(T) | 0.28 | 20 | 45 | 35.67 | 13.06 | 2.6 | 0.01 |
| GRIA1 | Intron 2 | rs490922 | C(T) | 0.6 | 25 | 79 | 67.13 | 19.88 | 2.7 | 0.008 |
| GRIA1 | Intron 2 | rs1864205 | A(G) | 0.6 | 23 | 71 | 61.33 | 17.44 | 2.3 | 0.02 |
| GRIA1 | Intron 5 | rs4385264 | G(A) | 0.64 | 25 | 81 | 71.73 | 17.64 | 2.2 | 0.03 |
| n | rs4958751 | T(C) | 0.16 | 18 | 35 | 24.97 | 11.83 | 2.9 | 0.004 | |
| n | rs1295213 | T(G) | 0.38 | 21 | 65 | 50.53 | 25.15 | 2.9 | 0.004 | |
| ITK | Intron 1 | rs452223 | G(A) | 072 | 19 | 66 | 53.83 | 15.69 | 3.1 | 0.002 |
| LSM11 | 3′ UTR | rsG882516 | A(C) | 0.17 | 15 | 37 | 26.17 | 17.19 | 2.6 | 0.009 |
| n | rs4704916 | A(G) | 0.18 | 17 | 34 | 23.5 | 12.81 | 2.9 | 0.003 | |
| EBF1 | Intron 9 | rs6874808 | C(A) | 0.1 | 11 | 17 | 10.5 | 5.93 | 2.7 | 0.008 |
| n | rs7734683 | T(C) | 0.31 | 17 | 47 | 38.67 | 10.43 | 2.6 | 0.01 | |
| n | rs885466 | A(G) | 0.88 | 14 | 57 | 46.17 | 13.86 | 2.9 | 0.004 | |
| n | rs1914212 | A(C) | 0.35 | 20 | 46 | 36.83 | 12.46 | 2.6 | 0.01 | |
| n | rs9284996 | T(C) | 0.19 | 13 | 26 | 17.83 | 8.694 | 2.8 | 0.006 | |
The over-transmitted allele is shown with the alternative allele in brackets; afreq, allele frequency; fam#, number of informative families; S, test statistic in FBAT, corresponding to the number of times an allele is transmitted to an affected offspring; E(S),the expected value of the FBAT test statistic under the null hypothesis of no association; P, empirical P-value;
missing data due to small number of informative families; n, intergenic DNA sequence; ITK, IL2-inducible T-cell kinase, MIM#186973; LSM11, U7 small nuclear RNA-associated Sm-like protein, Gene ID 134353; EBF1, Early B-cell factor 1, MIM#164343.
Analysis of the Wave 1 families for three-marker haplotypes including SNPs in the first and second intron of GRIA1, using a sliding window approach, provided evidence of association with psychotic BP for a haplotype including the markers rs3828595, rs490922, and rs1864205 (nominal P-values = 0.001) (Fig. 2). Five additional haplotypes in the GRIA1 gene were associated with a P-value <0.01.
Fig. 2.
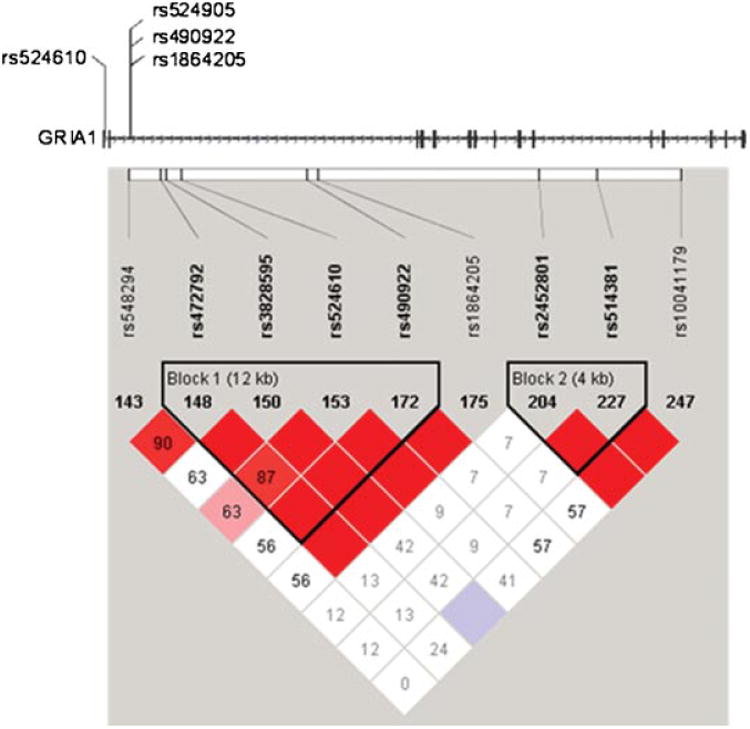
Haplotype structure in the first and second intron of GRIA1. Several SNPs in the first and second intron of the GRIA1 gene are in strong linkage disequilibrium. Localization of the SNPs, which individually or in haplotypes show significant association, is presented on the gene structure (horizontal bars indicate exons, arrows indicated gene orientation). Haploblock structure of the 5′ portion of the GRIA1 gene is shown below. Linkage disequilibriumis expressed in r2 values. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The additional 37 families from Waves 2, 3, and 4 (data set 2) showed association to SNP rs4385264 located in intron 5 of GRIA1 (Z-score = 3.2; empirical P = 0.002, OR = 2.9) (Table I).
A combined analysis of both data sets maintained the evidence for association to the SNPs in intron 1 and 2 of GRIA1 and in addition, revealed signals in the genes ITK (IL2-inducible T-cell kinase), LSM11 (U7 small nuclear RNA associated), EBF1 (early B-cell factor 1) and several SNPs in gene poor regions. Only a small number of families had been informative for those SNPs in the individual data sets, therefore the analysis of all families combined increased the evidence for those loci.
Since our data sets included families of diverse ethnic back-ground, we also performed the analysis without the three African-American families and the Asian family. The results were slightly less significant in data set 1 and did not change significantly in data set 2.
In order to explore the importance of the phenotype for the association signal, we recoded the affected status of our data so that unaffected individuals were coded as affected and vice versa. When the data were reanalyzed in this fashion, no association was found in this region.
Discussion
The original linkage finding to psychosis in BP on chromosome 5q in the NIMH dataset is supported by the results of association analyses in the same pedigrees using densely spaced SNPs. However, while association signals were observed for several SNPs in families from Wave 1 and additional families from Waves 2, 3, and 4, as well as in analyses combining both data sets, these results do not permit localization to a single polymorphism or even to a single narrow segment within this region. Despite a strong linkage signal (NPL-score = 5.0), the evidence for association with individual SNPs was weak and could not be replicated across data sets. There are several alternative explanations for this result: (a) more than one susceptibility variant within the linkage region; (b) locus heterogeneity with some of the signals being false positive; (c) no locus in the region with all association results being false positive; (d) lack of power in the sample to identify variants of low frequency. This study was intended to explore the genomic region under a significant linkage-peak of a genome-wide linkage study in a small sample of families and to create hypotheses for future studies. Our study was under-powered for association testing and therefore, while we corrected for association in the presence of linkage, we did not correct for multiple testing. While we could not definitely map a psychosis locus to a narrow segment of this chromosomal region, the strongest evidence was obtained in or around GRIA1. These results need to be followed up in a well-designed population based case–control association study with sufficient power to detect significant association between the phenotype and genomic variants in GRIA1.
GRIA1 encodes a subunit of the AMPA receptor, a tetrameric ligand-gated ion channel that transmits glutamatergic signals in the brain. The AMPA receptor subunits are expressed in a tissue specific manner with GRIA1 primarily found in the forebrain and hippo-campus, areas that are particularly involved in memory formation and retention of spatial memory tasks. GRIA1 itself has been shown to influence cognitive functions, such as working memory and reward learning [Schmitt et al., 2005]. In addition, several lines of evidence point to the involvement of this receptor in psychiatric disorders, and suggest that GRIA1 could be an attractive candidate gene for psychotic BP. First, polymorphisms in this gene have been associated with schizophrenia (SZ) [Magri et al., 2006]. Second, regional specific abnormalities in gene expression have been re-ported in postmortem brains of individuals with SZ [Beneyto et al., 2007]. Third, it has been reported that ligands to the ionotropic glutamate receptors demonstrate therapeutic effects in animal models of SZ and depression [Black, 2005].
All the associations observed in GRIA1 were to variants in intronic sequences. In this respect it is interesting that mutations in intronic sequences of the GluR2 receptor in mice have been shown to influence the posttranscriptional editing of the protein and change the desensitization rate of the receptor [Higushi et al., 1993; Egebjerg et al., 1994]. Those intronic sequences can pair with exonic sequences and form loop structures that allow for the substitution of one amino acid with another. The resulting amino acid substitutions, for example, altered the Ca2+ permeability of the GluR2 subunit. Future studies would need to explore whether such a mechanism is relevant for the functional significance of the variants that we observed in this study.
Besides signals in the GRIA1 gene, we detected additional polymorphisms that might have potentially been involved in the BP phenotype. None of these genes has been suggested as a candidate for a psychiatric phenotype to date. LSM11 is expressed early in embryonic development and this gene is involved in histone RNA processing [Pillai et al., 2003]. EBF1 [Nutt and Kee, 2007] and ITK [Burbach et al., 2007] are transcription factors involved in early B cell development and T-cell receptor signaling respectively. The clarification of the contribution of variations in these genes to the BP phenotype would need further evaluation.
Our findings suggest that psychotic symptoms in BP can be associated with sequence variations in the GRIA1 gene and possibly other genes in this region in some families. We consider this study exploratory due to our small sample size. Future studies need to evaluate a possible association between genomic variants in GRIA1 and BP or a sub-phenotype of it in a well-designed, population based case–control association study with sufficient power in order to claim significant association between the phenotype and certain genomic variants.
The SNPs that we used in our study were selected to be common. However, because we identified this genomic region through linkage analysis, we cannot exclude that a rare genomic variant in our families in linkage disequilibrium with the common variants accounts for the linkage signal. Family based association studies appear to give an advantage over population-based studies under this scenario, because heterogeneity may be reduced.
We included families of different ethnicities in our sample. Future studies in larger and ethnically homogeneous samples should address the question whether different genomic variants are important in different ethnicities.
The identification of nucleotide polymorphisms that are closely linked to the bipolar phenotype or to specific symptoms of it is of utmost importance for the ultimate identification of risk associated DNA sequence variations. Direct sequencing of this ion channel receptor gene will demonstrate whether functional variation can be found in those families that contributed to the linkage and association signal. The identification of risk alleles for BP will ultimately help to better understand the pathophysiology of the disorder and its diagnostic boundaries to other psychiatric disorders. This may facilitate the development of more specific and effective treatment options.
Acknowledgments
This work was supported by the NIMH grant KO8 MH074057 to B. Kerner and NIMH KO2 MH001374, RO1 MH049499 to N.B. Freimer.
Acknowledgement for Bipolar Disorder Biomaterial and Clinical Data: Data and biomaterial were collected as part often projects that participated in the National Institute of Mental Health (NIMH) Bipolar Disorder Genetics Initiative. From 1991 to 1998, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, U01 MH46282, John Nurnberger, M.D., Ph.D., Marvin Miller, M.D., and Elizabeth Bowman, M.D.; Washington University, St. Louis, MO, U01 MH46280, Theodore Reich, M.D., Allison Goate, Ph.D., and John Rice, Ph.D.; John Hopkins University, Baltimore, MD U01MH46274, J. Raymond DePaulo, Jr., M.D., Sylvia Simpson, M.D., MPH, and Colin Stine, Ph.D.; NIMH Intramural Research Program, Clinical Neurogenetics Branch, Bethesda, M.D., Elliot Gershon, M.D., Diane Kazuba, B.A., and Elizabeth Maxwell, M.S.W. From 1999 to 2003, data and biomaterials were collected as part of ten projects that participated in the National Institute of Mental Health (NIMH) Bipolar Disorder Genetics Initiative. From 1999 to 2003, the Principal Investigators and Co-Investigators were: Indiana University, Indianapolis, IN, R01 MH59545, John Nurnberger, M.D., Ph.D., Marvin J. Miller, M.D., Elizabeth S. Bowman, M.D., N. Leela Rau, M.D., P. Ryan Moe, M.D., Nalini Samavedy, M.D., Rif El-Mallakh, M.D. (at University of Louisville), Husseini Manji, M.D. (at Wayne State University), Debra A.Glitz, M.D.(at Wayne State University), Eric T. Meyer, M.S., Carrie Smiley, R.N., Tatiana Foroud, Ph.D., Leah Flury, M.S., Danielle M. Dick, Ph.D., Howard Edenberg, Ph.D.; Washington University, St. Louis, MO, R01 MH059534, John Rice, Ph.D., Theodore Reich, M.D., Allison Goate, Ph.D., Laura Bierut, M.D.;Johns Hopkins University, Baltimore, MD, R01 MH59533, Melvin McInnis M.D., J. Raymond DePaulo, Jr., M.D., Dean F. MacKinnon, M.D., Francis M. Mondimore, M.D.,James B. Potash, M.D., Peter P. Zandi, Ph.D., Dimitrios Avramopoulos, and Jennifer Payne; University of Pennsylvania, PA, R01 MH59553, Wade Berrettini M.D., Ph.D.; University of California at Irvine, CA R01 MH60068, William Byerley M.D., and Mark Vawter M.D.; University of Iowa, IA, R01 MH059548, William Coryell M.D., and Raymond Crowe M.D.; University of Chicago, IL, R01 MH59535, Elliot Gershon, M.D., Judith Badner Ph.D., Francis McMahon M.D., Chunyu Liu Ph.D.,Alan Sanders M.D.,Maria Caserta, Steven Dinwiddle M.D., Tu Nguyen, Donna Harakal; University of California at San Diego, CA, R01 MH59567, John Kelsoe, M.D., Rebecca McKinney, B.A.; Rush University, IL, R01 MH059556, William Scheftner M.D., Howard M. Kravitz, D.O., M.P.H., Diana Marta, B.S., Annette Vaughn-Brown, MSN, RN, and Laurie Bederow, MA; NIMH Intramural Research Program, Bethesda, MD, 1Z01MH002810-01, Francis J. McMahon, M.D., Layla Kassem, Ph.D, Sevilla Detera-Wadleigh, Ph.D., Lisa Austin, Ph.D., Dennis l. Murphy, M.D. Data and biomaterials for Wave4 were collected and supported by NIMH grant R01 MH59602 (to Miron Baron, M.D.) and by funds from the Columbia Genome Center and the New York State Office of Mental Health. The main contributors to this work were Miron Baron, M.D. (Principal Investigator), Jean Endicott, Ph.D. (Co-Principal Investigator),Jo Ellen Loth, M.S.W., John Nee, Ph.D., Richard Blumenthal, Ph.D. Lawrence Sharpe, M.D., Barbara Lilliston, M.S.W., Melissa Smith, M.A., and Kristine Trautman, M.S.W., all from Columbia University Department of Psychiatry, New York, NY, USA. A small subset of the sample was collected in Israel in collaboration with Bernard Lerer, M.D. and Kyra Kanyas, M.S. from the Hadassah – Hebrew University Medical Center, Jerusalem, Israel. We are grateful to the patients and their family members for their cooperation and support, and to the treatment facilities and other organizations that collaborated with us in identifying families. Most importantly, we thank the families who have participated in and contributed to these studies.
Grant sponsor: National Institute of Mental Health; Grant numbers: KO8 MH074057, KO2 MH001374, RO1 MH049499.
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Black MD. Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology. 2005;179:154–163. doi: 10.1007/s00213-004-2065-6. [DOI] [PubMed] [Google Scholar]
- Burbach BJ, Medeiros RB, Mueller KL, Shimizu Y. T-cell receptor signaling to integrins. Immunol Rev. 2007;218:65–68. doi: 10.1111/j.1600-065X.2007.00527.x. [DOI] [PubMed] [Google Scholar]
- Egebjerg J, Kukekov V, Heinemann SF. Intron sequence directs RNA editing of the glutamate receptor subunit GluR2 coding sequence. PNAS. 1994;91:10270–10274. doi: 10.1073/pnas.91.22.10270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS. Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol. 2003;68:69–78. doi: 10.1101/sqb.2003.68.69. [DOI] [PubMed] [Google Scholar]
- Herzberg I, Jasinska A, Garcia J, Jawaheer D, Service S, Kremeyer B, Duque C, Parra MV, Vega J, Ortiz D, Carvajal L, Polanco G, Restrepo GJ, Lopez C, Palacio C, Levinson M, Aldana I, Mathews C, Davanzo P, Molina J, Fournier E, Bejarano J, Ramirez M, Ortiz CA, Araya X, Sabatti C, Reus V, Macaya G, Bedoya G, Ospina J, Freimer N, Ruiz-Linares A. Convergent linkage evidence from two Latin-American population isolates supports the presence of a susceptibility locus for bipolar disorder in 5q31–34. Hum Mol Genet. 2006;15:3146–3153. doi: 10.1093/hmg/ddl254. [DOI] [PubMed] [Google Scholar]
- Higushi M, Single FN, Köhler M, Summer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: A base-paired intron– exon structure determines position and efficiency. Cell. 1993;75:1361–1370. doi: 10.1016/0092-8674(93)90622-w. [DOI] [PubMed] [Google Scholar]
- Horiuchi Y, Nakayama J, Ishiguro H, Ohtsuki T, Detera-Wadleigh SD, Toyota T, Yamada K, Nankai M, Shibuya H, Yoshikawa T, Arinami T. Possible association between a haplotype of the GABA-A receptor alpha 1 subunit gene (GABRA1) and mood disorders. Biol Psychiatry. 2004;55:40–45. doi: 10.1016/s0006-3223(03)00689-9. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Laird NM. The family based association test method: Strategies for studying general genotype–phenotype associations. Eur J Hum Genet. 2001;9:301–306. doi: 10.1038/sj.ejhg.5200625. [DOI] [PubMed] [Google Scholar]
- Horvath S, Xu X, Lake SL, Silverman EK, Weiss ST, Laird NM. Family-based tests for associating haplotypes with general phenotype data: Application to asthma genetics. Genet Epidemiol. 2004;26:61–69. doi: 10.1002/gepi.10295. [DOI] [PubMed] [Google Scholar]
- Kerner B, Brugman DL, Freimer NB. Evidence of linkage to psychosis on chromosome 5q 33–34 in pedigrees ascertained for bipolar disorder. Am J Med Genet (Neuropsychiatr Genet) 2007;144:74–78. doi: 10.1002/ajmg.b.30402. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- Lake SL, Blacker D, Laird NM. Family-based tests of association in the presence of linkage. Am J Hum Genet. 2000;67:1515–1525. doi: 10.1086/316895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo WS, Lau CF, Xuan Z, Chan CF, Feng GY, He L, Cao ZC, Liu H, Luan QM, Xue H. Association of SNPs and haplotypes in GABAA receptor beta2 gene with schizophrenia. Mol Psychiatry. 2004;9:603–608. doi: 10.1038/sj.mp.4001461. [DOI] [PubMed] [Google Scholar]
- Magri C, Gardella R, Barlati SD, Podavini D, Iatropoulos P, Bonomi S, Valsecchi P, Sacchetti E, Barlati S. Glutamate AMPA receptor subunit 1 gene (GRIA1) and DSM-IV-TR schizophrenia: A pilot case– control association study in an Italian sample. Am J Med Genet (Neuropsychiatr Genet) 2006;141:287–293. doi: 10.1002/ajmg.b.30294. [DOI] [PubMed] [Google Scholar]
- Morgan VA, Mitchell PB, Jablensky AV. The epidemiology of bipolar disorder: Sociodemographic, disability and service utilization data from the Australian National Study of Low Prevalence (Psychotic) Disorders. Bipolar Disord. 2005;7:326–337. doi: 10.1111/j.1399-5618.2005.00229.x. [DOI] [PubMed] [Google Scholar]
- Nurnberger JI, Jr, Blehar MC, Kaufmann CA, York-Cooler C, Simpson SG, Harkavy-Friedman J, Severe JB, Malaspina D, Reich T. Diagnostic interview for genetic studies Rationale, unique features, and training NIMH Genetics Initiative. Arch Gen Psychiatry. 1994;51:849–859. doi: 10.1001/archpsyc.1994.03950110009002. [DOI] [PubMed] [Google Scholar]
- Nurnberger JI, Jr, Depaulo JR, Gershon ES. Genomic survey of bipolar illness in the NIMH genetics initiative pedigrees: A preliminary report. Am J Med Genet. 1997;74:227–237. doi: 10.1002/(sici)1096-8628(19970531)74:3<227::aid-ajmg1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26:715–725. doi: 10.1016/j.immuni.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Petryshen TL, Middleton FA, Tahl AR, Rockwell GN, Purcell S, Aldinger KA, Kirby A, Morley CP, McGann L, Gentile KL, Waggoner SG, Medeiros HM, Carvalho C, Macedo A, Albus M, Maier W, Trixler M, Eichhammer P, Schwab SG, Wildenauer DB, Azevedo MH, Pato MT, Pato CN, Daly MJ, Sklar P. Genetic investigation of chromosome 5q GABAA receptor subunit genes in schizophrenia. Mol Psychiatry. 2005;10:1074–1088. doi: 10.1038/sj.mp.4001739. [DOI] [PubMed] [Google Scholar]
- Pillai RS, Grimmler M, Meister G, Will CL, Luhrmann R, Fischer U, Schumperli D. Unique Sm core structure of U7 snRNPs: Assembly by a specialized SMN complex and the role of a new component, LSM11, in histone RNA processing. Genes Dev. 2003;17:2321–2333. doi: 10.1101/gad.274403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimm J, McQuillin A, Thirumalai S, Lawrence J, Quested D, Bass N, Lamb G, Moorey H, Datta SR, Kalsi G, Badacsonyi A, Kelly K, Morgan J, Punukollu B, Curtis D, Gurling H. The Epsin 4 gene on chromosome 5q, which encodes the clathrin-associated protein enthoprotin, is involved in the genetic susceptibility to schizophrenia. Am J Hum Genet. 2005;76:902–907. doi: 10.1086/430095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potash JB, Chiu YF, MacKinnon DF, Miller EB, Simpson SG, McMahon FJ, McInnis MG, DePaulo JR., Jr Familial aggregation of psychotic symptoms in a replication set of 69 bipolar disorder pedigrees. Am J Med Genet (Neuropsychiatr Genet) 2003;116:90–97. doi: 10.1002/ajmg.b.10761. [DOI] [PubMed] [Google Scholar]
- Schmitt WB, Sprengel R, Mack V, Draft RW, Seeburg PH, Deacon RM, Rawlins JN, Bannerman DM. Restoration of spatial working memory by genetic rescue of Glu-R-A-deficient mice. Nat Neurosci. 2005;8:270–272. doi: 10.1038/nn1412. [DOI] [PubMed] [Google Scholar]
- Service S, DeYoung J, Karayiorgou M, Roos JL, Pretorious H, Bedoya G, Ospina J, Ruiz-Linares A, Macedo A, Palha JA, Heutink P, Aulchenko Y, Oostra B, van Duijn C, Jarvelin MR, Varilo T, Peddle L, Rahman P, Piras G, Monne M, Murray S, Galver L, Peltonen L, Sabatti C, Collins A, Freimer N. Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet. 2006;38:556–560. doi: 10.1038/ng1770. [DOI] [PubMed] [Google Scholar]
- Sklar P, Pato MT, Kirby A, Petryshen TL, Medeiros H, Carvalho C, Macedo A, Dourado A, Coelho I, Valente J, Soares MJ, Ferreira CP, Lei M, Verner A, Hudson TJ, Morley CP, Kennedy JL, Azevedo MH, Lander E, Daly MJ, Pato CN. Genome-wide scan in Portuguese Island families identifies 5q31–5q35asasusceptibility locus for schizophrenia and psychosis. Mol Psychiatry. 2004;9:213–218. doi: 10.1038/sj.mp.4001418. [DOI] [PubMed] [Google Scholar]
- Yamada K, Watanabe A, Iwayama-Shigeno Y, Yoshikawa T. Evidence of association between gamma-aminobutyric acid type A receptor genes located on 5q34 and female patients with mood disorders. Neurosci Lett. 2003;349:9–12. doi: 10.1016/s0304-3940(03)00611-6. [DOI] [PubMed] [Google Scholar]