

Abstract
The adsorption process of the recombinant human growth hormone on organic films, created by self-assembly of octadecyltrichlorosilane, arachidic acid, and dipalmitoylphosphatidylcholine, is investigated and compared to adsorption on silica and methylated silica substrates. Information on the adsorption process of human growth hormone (hGH) is obtained by using total internal reflection fluorescence (TIRF). The intensity, spectra, and quenching of the intrinsic fluorescence emitted by the growth hormone’s single tryptophan are monitored and related to adsorption kinetics and protein conformation. For the various alkylated hydrophobic surfaces with differences in surface density and conformational freedom it is observed that the adsorbed amount of growth hormone is relatively large if the alkyl chains are in an ordered structure while the amounts adsorbed are considerably lower for adsorption onto less ordered alkyl chains of fatty acid and phospholipid layers. Adsorption on methylated surfaces results in a relatively large conformational change in the growth hormone’s structure, as displayed by a 7 nm blue shift in emission wavelength and a large increase in the effectiveness of fluorescence quenching. Conformational changes are less evident for hGH adsorption onto the fatty acid and phospholipid alkyl chains. Adsorption kinetics on the hydrophilic head groups of the self-assembled monolayers are similar to those on solid hydrophilic surfaces. The relatively small conformational changes in the hGH structure observed for adsorption on silica are even further reduced for adsorption on fatty acid head groups.
Introduction
The growing need to control the adsorbed state of proteins in a broad range of biomedical applications requires a fundamental and detailed knowledge of protein/surface interactions. For this purpose, the physical and chemical properties of the system under study have to be well controlled and potentially usable in the biomedical field. Smooth, ordered surfaces with controlled functionality, prepared by self-assembly of organic layers, are useful as models for complex biological systems and have a high potential for use in bioseparation, biosensor, and biocompatibility studies.2–6 Such surfaces are commonly produced on solid supports by self-assembly of alkylsilanes from solution or by transferring Langmuir films of amphiphiles such as fatty acids and phospholipids. Lipid monolayers provide ordered structures with a biomimetic and biocompatible nature. Protein adsorption on the hydrophilic head groups of these amphiphilic molecules can be used as a model to study protein membrane interactions. Surfaces with hydrophobic alkyl chains have a high significance in, for example, bioseparations and intravenous drug delivery systems in which proteins can be incorporated in micelles and liposomes. The use of surfaces constructed of chemically identical alkyl chains but assembled into monolayers with different degrees of intermolecular ordering can also provide more fundamental insight into the substrate’s entropic contribution to protein adsorption.
The present study is focused on the adsorption process of the recombinant human growth hormone (hGH). The growth hormone is a protein with a high relevance in the medical field for its regulatory function in cell growth7 and the metabolism of lipoproteins.8 The pharmaceutical applicability of hGH increased considerably once it became available in the recombinant form.9,10 The human growth hormone is a relatively stable single-chain protein containing 191 residues of which about half are in an α-helical conformation.11,12 The growth hormone activates receptors (hGHbp) present in cell membranes only after two receptors are bound by one hGH molecule. Both the hydrophobic interior and a hydrophobic patch on the exterior of hGH are believed to be responsible for binding the first receptor.13
In this study, self-assembled monolayers (SAMs) are prepared from octadecyltrichlorosilane (OTS), arachidic acid (ArAc), and dipalmitoylphosphatidylcholine (DPPC). Adsorption of hGH is studied on both the head and tail groups of ArAc and DPPC. Results are compared to the adsorption behavior of hGH on silica and methylated silica surfaces. The SAMs are characterized on their hydro-phobicity using the sessile drop and captive bubble techniques. In addition, fluorescent probes are incorporated in the Langmuir-Blodgett layers to monitor the presence and stability of the transferred films during the hGH adsorption experiment.
Total internal reflection fluorescence (TIRF) spectroscopy is used to study adsorption kinetics and conformational changes of hGH adsorbed onto the different substrates. In this technique the single tryptophan of adsorbed hGH molecules is selectively excited by an ultraviolet evanescent wave, created at the solid/liquid interface by a totally internally reflected light beam. By following the fluorescence intensity in time, adsorption and desorption kinetics of hGH are monitored. The adsorbed amount is quantified by calibrating the fluorescence signals using appropriate standards. The use of the intrinsic fluorophore avoids the necessity of labeling, which may exert a distinct effect on the adsorption behavior. Furthermore, the use of tryptophan fluorescence allows one to obtain information on conformational changes in the hGH structure by interpreting the observed fluorescence spectra. In addition, the accessibility of the tryptophan to solutes is investigated by fluorescence quenching using trichloroethanol. Fluorescence spectroscopy is very suitable to study the conformation of the hGH molecule, as its single tryptophan residue (Trp 86) is buried in the hydrophobic interior and is involved in a hydrogen bond with an aspartic acid residue (Asp 169), located in another α-helix strand.12 A high sensitivity for conformational changes in both the secondary and tertiary structure is thus provided. The ability to monitor changes in the tertiary structure of hGH is especially interesting, as it has been reported that a molten globule intermediate exists in the denaturation pathway.11
Experimental Section
Proteins and Chemicals
The recombinant hGH was donated by Pharmacia & Upjohn. Each hGH sample contained 13.8 mg of recombinant hGH, 2.3 mg of glycine, and 14.0 mg of mannitol. hGH has an isoelectric point of 5.1,14 a molecular weight of 22.0 kDa, and crystallographic dimensions of 5.5 × 3.5 × 3.5 nm3.12 Water was purified by double distillation. All experiments were performed at pH 7.0 by using a 0.15 M phosphate-buffered saline (PBS) solution, containing 10 mM phosphate (prepared from sodium phosphate and adjusted to pH 7.0 by means of NaOH) and 0.14 M NaCl. hGH was dissolved in PBS, filtered through a prewashed 0.2 μm pore filter (Acrodisc, Gelman Sci.), and stored at −20 °C in amounts needed for a single experiment. The hGH concentrations were determined using the extinction coefficient: E277 = 0.82 cm2 mg−1.11 The hGH solutions contained the excipients glycine and mannitol with final concentrations of 0.3 and 0.6 mM, respectively. The purity of hGH was investigated using SDS page (Gradient 8-25 Phastgel, Pharmacia) and staining with Coomassie-blue. In this study only one single band with a molecular weight of 22 kDa was observed. Calibration of the TIRF instrumental sensitivity and final alignment were performed using L-5-hydroxy-tryptophan · HCl (TRP) (Calbiochem) in PBS. The fluorescence of the proteins was quenched using trichloroethanol (Aldrich) in PBS. Sorbent surfaces were prepared on fused silica slides (ESCO Products Inc.) and cut into dimensions of 1 × 10 mm2 and lengths varying between 25 and 40 mm. Methylated silica slides were prepared using dichlorodimethylsilane (Hüls Inc.). Self-assembled monolayers were prepared from octadecyltrichlorosilane (United Chemical), arachidic acid (Fluka), and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (Avanti). As fluorescent probes in the transferred Langmuir layers 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramerhylin-docarbocyanine perchlorate (DiI) (Molecular Probes Inc.) were used.
Surface Preparation and Characterization
Prior to surface modification, the silica slides were cleaned in a radio frequency discharge chamber (Plasmod, Tegal Corp.) for 15 min while oxygen (27 Pa) was flowing through. Then they were immersed in a freshly prepared hot acid “piranha” mixture (3:1 v/v of H2SO4 and 30% H2O2) and consecutively rinsed with ethanol and water and dried in a stream of dry nitrogen gas. Silica slides were methylated by immersion for 30 min in 0.3% (v/v) dichlorodimethylsilane (DDS) in trichloroethylene (Aldrich). After silanization the slides were immersed for 5 min in trichloroethylene, rinsed consecutively with ethanol and water, and dried in a jet of nitrogen gas. Alkylsilane (OTS) surfaces were made by immersing the silica slides in a 0.02% (v/v) solution of octadecyltrichlorosilane in dicyclohexyl (Aldrich) for 18 h. After silanization the surfaces were immersed for 30 min in chloroform and rinsed with ethanol and water and dried in a jet of nitrogen gas. The preparation and characterization of the self-assembly process of OTS surfaces is described in more detail by Britt et al.15
Monolayers of ArAc and DPPC were transferred onto OTS-covered silica slides using the Langmuir–Blodgett (LB) technique. In some of the experiments the dyes DiO and DiI were incorporated in the Langmuir layers to monitor the stability of the transferred films during the experiment. The molar fraction of DiO or DiI was 1% for ArAc and 2% for DPPC monolayers, respectively. The Langmuir–Blodgett trough (KSV 5000, KSV Instruments Ltd., Finland) consisted of two baths with dimensions of 12 × 68 cm2. To avoid mechanical distortion and contamination of the films, the trough was placed on an antivibration table and in a dust-free cabinet. Prior to use the trough was rinsed with ethanol and water and filled with 5 × 10−5 M CaCl2 and 3 × 10−4 M PBS. The subphase was aged overnight and kept at a temperature of 19 °C. Before the Langmuir layer was spread, the surface of the subphase was thoroughly cleaned using a suction system. On this subphase, ArAc (1.5 mg mL−1, 50 μL) or DPPC (1.0 mg mL−1, 50 μL) solutions in chloroform were spread and the chloroform was allowed to evaporate for 15 min. The monolayers were symmetrically compressed with two moving barriers at a rate of 6 or 1.2 cm2 min−1 until surface pressures of 35 or 30 mN m−1 were reached for ArAc or DPPC, respectively. The characteristic pressure–area (π–A) isotherms, measured by a platinum Wilhelmy plate attached to a balance, are shown in Figure 1. From the π–A isotherms the cross-sectional areas of the ArAc and DPPC molecules in closely-packed Langmuir layers were obtained from the slope of the condensed phase extrapolated to zero pressure. The molecular areas of 0.21 and 0.54 nm2, determined for ArAc and DPPC, respectively, correspond well with reported literature values.16 The π–Aisotherm of ArAc was not significantly affected by the presence of DiO, which indicates that DiO is miscible in ArAc layers under the present conditions. The π–A behavior for DPPC was somewhat altered by DiO in the two-phase coexistence region wherein domains of a liquid-condensed phase are dispersed in a liquid-expanded phase. An increase in transition pressure to a full condensed phase is generally observed if impurities, less soluble in the condensed phase, are present.17,18
Figure 1.
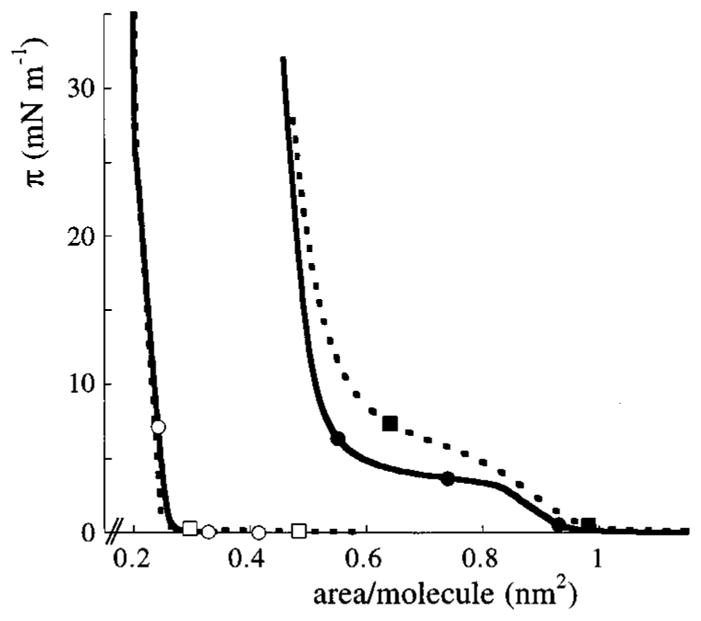
Pressure–area isotherms of ArAc (solid line, ○) and DPPC (solid line, ●). Dotted lines show the isotherms obtained in the presence of a 1% molar fraction of DiO in ArAc (□) and 2% DiO in the DPPC layer (■). All layers were spread on an aqueous subphase containing CaCl2 (5 × 10−5 M) and PBS (3 × 10−4 M), pH 7, T = 19 °C. Compression rates were 6.0 and 1.2 cm2 min−1 for ArAc and DPPC, respectively.
Deposition of the films onto the OTS-covered silica slides was performed by dipping the slides through the air/water interface at a rate of 1 mm min−1 while the film pressures were held at 35 or 30 mN m−1 for ArAc or DPPC, respectively. Surfaces consisting of exposed hydrophilic head groups were obtained after a single dip into the aqueous subphase. To avoid disrupting the LB film, which might occur upon passing through the water/air interface, the slide was transported and mounted to the TIRF cell under water. LB films of exposed hydrophobic alkyl chains were produced by a second dip through the monolayer, returning to air from the aqueous subphase. All film transfer ratios were near unity.
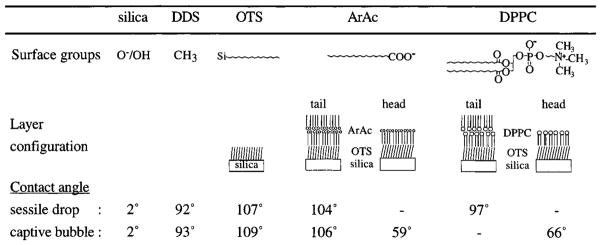
Two important aspects of LB films are their stability and characterization. The surfaces were characterized on their hydrophobicity using the sessile drop and captive bubble methods. Results are given in Table 1 and are presented as the angle between the water phase and the solid substrate. Both sessile drop and captive bubble measurements yielded reproducible results for the silica, DDS, and OTS surfaces with a standard deviation below 1°. The sessile drop method, as applied to the tail groups of transferred ArAc and DPPC layers, also gave reproducible results although some spreading of the water drop was observed at longer contact times. The results of the captive bubble method for LB layers demonstrated a larger variation. Unexpected spreading of the water drop on hydrophobic films or the air bubble on hydrophilic films might be explained by a mechanical distortion of the LB film in the intersection area of the three phases. An upward curvature of the LB layer in the intersection area, favored by a lower interfacial energy, results in a lower apparent contact angle between the drop/bubble and the solid substrate. Other explanations for unexpected spreading of the captive air bubble on hydrophilic films are the adsorption of solutes to the air/water interface thereby, altering the surface tension, or a flipping over of the amphiphilic molecules in the transferred layer, thus exposing their hydrophobic tails to the air phase. Although the captive bubble contact angles were used only after careful interpretation, the given values might be somewhat higher than the actual values. The substrates characterized by both the sessile drop and captive bubble methods show that similar values were obtained although the captive bubble method seems to result in slightly higher contact angles. While it is difficult to make absolute predictions about the monolayer packing densities based only on contact angle data, this technique does provide a reliable means of ensuring consistency among monolayer preparations.
Table 1.
Chemical Surface Groups, Layer Configuration, and Contact Angles between the Water Phase and the Solid Substrate As Determined Using Both Sessile Water Drops for Substrates in Air and Captive Air Bubbles for Substrates Immersed in Water
|
An indication of the high amphiphile packing density in LB films is given by their respective transfer ratios of near unity. However, there is no guarantee that the films will remain on the substrate after transfer. Therefore, experiments were performed in which the first LB film was doped with the dye DiO, and for experiments having a second LB layer, DiI was incorporated. TIRF spectroscopy was used to excite DiO and monitor its fluorescence in the same layer configuration as in the hGH adsorption experiments. If two layers are present, the energy of the excited state of DiO is partially transferred to DiI, depending on their separation. Using this energy transfer system, the amount of dye and the distance between the dyes can be monitored, as described in the Experimental Procedure. The DiO and DiI concentrations were chosen in such a way that the distance between dyes, if uniformly spread in their respective LB films, is less than the distance for which half of the energy is transferred. This half energy transfer distance for the DiO/DiI energy transfer system in the present configuration was estimated to be 6 nm. This value was experimentally inferred from the relative fluorescence intensities of the dyes in samples in which the DiO–DiI separation distance was varied by depositing “spacer” monolayers of ArAc between the respective dye-containing monolayers (not shown here). Fluorescence spectra of the single LB layer with DiO or the double LB layer with DiO/DiI are shown in Figure 2. The single LB layers containing DiO showed similar calibrated fluorescence spectra for each different sample. The double layers containing both DiO and DiI displayed a larger variation in their fluorescence spectra. This variation is partly caused by the overall lower fluorescence intensity for the DiO/DiI energy transfer system and partly because LB double layers increase the scattering of the excitation light. A single LB layer of DiI in arachidic acid did not show significant fluorescent emission in the wavelength region between 490 and 600 nm upon excitation at 460 nm. Furthermore, no significant light absorption or emission by adsorbed hGH was observed in the wavelength region between 450 and 600 nm. No significant changes in the DiO or DiO/DiI fluorescence spectra were observed during hGH adsorption experiments, serving as an indication of the stability of the LB films.
Figure 2.
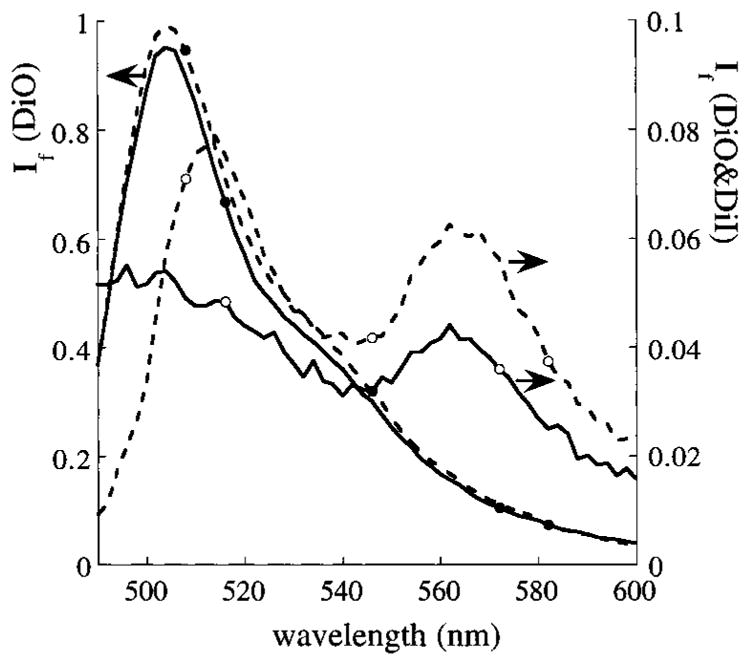
Relative fluorescence intensity, If, of DiO in a single layer (●) or of DiO and DiI in a double layer (○) of ArAc (solid lines) and DPPC (dotted lines). DiO in a single layer has one resolved fluorescence peak with a maximum wavelength of 506 nm. The intensity for these single layers containing DiO is plotted on the left y-axis. For the two-layer system, DiI absorbs part of the energy emitted by the excited DiO molecules and displays fluorescence (intensity on righty-axis) with a maximum wavelength of 564 nm.
Experimental Procedure
The TIRF apparatus and the experimental procedure of alignment and calibration of the instrument and the fluorescence signal were used as previously described.19 All sorbent surfaces were prepared/cleaned within a few hours before use and mounted on top of a silicon rubber gasket which created a 0.5 mm space between the slide and an anodized aluminum support which could be filled with solutions. For slides covered with a surface layer of ArAc or DPPC head groups, this mounting was performed under water. The face of the LB film-covered silica slides which was not used as a sorbent surface was wiped clean using a tissue soaked in ethanol. This procedure effectively removed the LB layers from this face and resulted in a reduction of the intensity of scattered light. Then a dovetail prism was optically coupled to the silica slide using a drop of glycerol. The excitation beam, internally reflected on the solid/liquid interface, was polarized perpendicular to the plane of incidence. After the calibration procedure, which determines the sensitivity of the instrumental setup and the exact wavelength of both the excitation and emission monochromator, the calibration solution in the cell was displaced with the PBS buffer solution which was used throughout the experiment. Next, the background signals for the different types of measurements were recorded. In the experiments for which the fluorescent probes DiO or DiO/DiI were incorporated in the monolayers, the fluorescence of the dyes was monitored in the wavelength region from 490 until 600 nm at 2 nm intervals and 2 s measuring times per point while exciting the dyes at 460 nm. The adsorption experiment started by flowing hGH solutions (0.1 mg mL−1) through the cell at a flow rate of 0.2 mL min−1. The adsorbed hGH molecules were excited using a wavelength of 295 nm, and the adsorption kinetics were followed by monitoring the fluorescence signal at an emission wavelength of 340 nm for 20 min (10 s per point). Then, a fluorescence emission spectrum was recorded at 0.5 nm wavelength intervals and 5 s measuring times per point. Again, if dyes were present in the interface, an additional spectrum as described above was recorded. All the dye spectra showed no significant changes after hGH was adsorbed, indicating that the monolayers were intact. The hGH fluorescence signal during desorption was followed for 8 min while a PBS solution was flowing through the cell (1.0 mL min−1). Fluorescence quenching was measured after gently injecting increasing concentrations of TCE. Between each fluorescence measurement in the presence of a TCE solution, the cell was gently flushed with a pure buffer solution to monitor additional desorption. The additional desorption never exceeded 4% of the adsorbed amount, and it was accounted for by using the average of the nonquenched fluorescence signals as measured before and after monitoring the quenched signal. For some of the experiments an additional check of the background signal was performed by injecting 10 mL of ethanol in the cell, which removed the LB layers and hGH molecules adsorbed onto these layers. Thus recorded background spectra were used for correction of the DiO or DiO/DiI fluorescence spectra.
Results and Discussion
Adsorption and Desorption Kinetics
The adsorption and desorption kinetics of hGH on the different substrates are shown in Figures 3 and 4, respectively. All experiments were performed three times, and the presented results are average values. The standard deviation of the maximum adsorbed amount was less than 0.1 mg m−2 for hGH adsorption on DDS, OTS, and the DPPC heads and less than 0.03 mg m−2 for adsorption onto the other substrates. The initial adsorption rate, limited by the protein flux toward the sorbent surface, is affected by the interactions between hGH and the sorbent. After only a few minutes the adsorption rates level off to near equilibrium adsorbed amounts, depending not only on hGH/sorbent interaction but also on packing geometry, conformation, and lateral interactions. The near steady-state adsorbed amounts, however, are well below a complete monolayer coverage, which is at least 1 mg m−2 for hGH, randomly packed and having the crystallographic dimensions. Furthermore, all curves with a low initial adsorption rate are also followed by a low steady-state adsorption level, indicating that the hGH/sorbent interaction might be the main parameter determining not only the initial adsorption rates but also the equilibrium amounts adsorbed.
Figure 3.
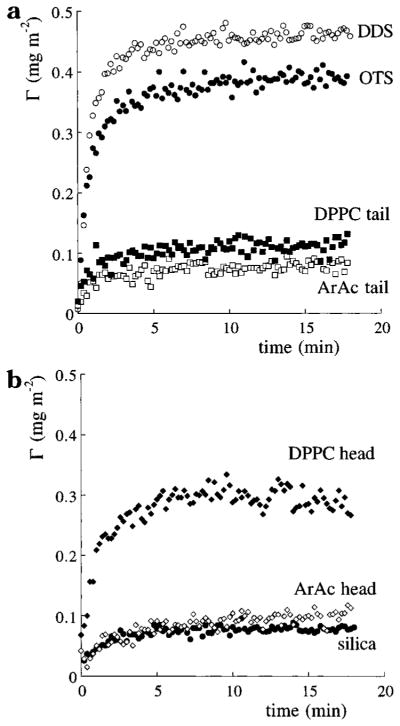
Adsorption kinetics of hGH on substrates of (a) alkyl groups and (b) the more hydrophilic substrates and head groups. The sorbent surfaces are depicted in Table 1.
Figure 4.
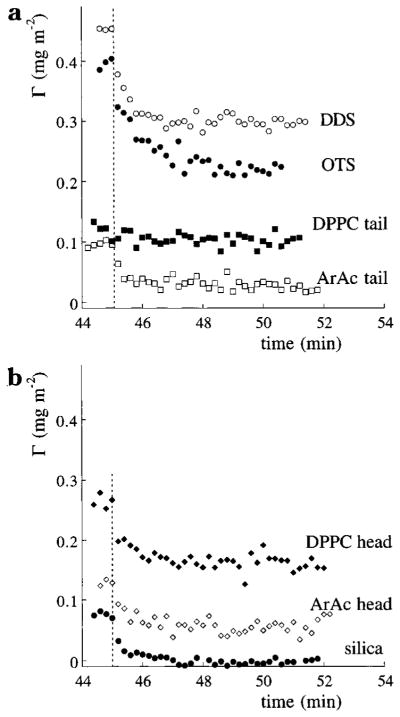
Desorption kinetics of hGH on substrates of (a) alkyl groups and (b) the more hydrophilic substrates and head groups. The sorbent surfaces are depicted in Table 1. Buffer flow started at t = 45 min, as indicated by the vertical dotted lines.
In a previous publication19 we reported that adsorption of hGH is dominated by hydrophobic interactions, as, for example, can be concluded when the hGH adsorption on the hydrophobic DDS is compared to that on hydrophilic silica. However, as shown in Figure 3a, both adsorption rates and amounts adsorbed are strongly reduced going from surfaces on which the alkyl chains are covalently attached (DDS and OTS) to surfaces on which the alkyl chains are physisorbed (ArAc and DPPC). As these alkylated surfaces have similar chemical compositions (terminal methyl groups) and hydrophobicities, the question arises as to why there are such large differences in hGH adsorption. The main difference between these surfaces is the alkyl chain packing density. A variation in packing density might cause differences in hGH adsorption by, for example, changing the number of contacts point between hGH and the sorbent surface. This theory, however, is contradicted by the relatively high adsorbed amounts of hGH on DDS; DDS is expected to have the least densely packed methyl groups according to its water contact angle.20 A more likely explanation is that long chain alkylated layers, although more hydrophobic than DDS, have more conformational freedom, which is reduced upon hGH adsorption, thereby creating a positive contribution to the overall adsorption free energy. A similar deduction has been made to explain the relatively low affinity observed for protein adsorption onto a film of polymerized 10,12-pentacosadiyonic acid.21
The strength and rigidity of the packing in self-assembled monolayers depend mainly on van der Waals attraction between the alkyl chains together with some geometric considerations, such as the size of the polar head groups. It should also be noted that the chlorosilanes of OTS are cross-linked on the surface (polysiloxane), which restricts their lateral and rotational movement, while ArAc and DPPC are only physically adsorbed. Self-assembled monolayers form closely packed parallel arrangements of the tails in which van der Waals attractions can be increased by tilting their angles away from the normal to the surface. OTS and to a lesser extent ArAc have small head groups which allow the alkyl chains to assemble closely together, and in general, relatively stable and dense monolayers are obtained.3 The larger head group of ArAc resulted in a tilt angle of 26°, as found for a condensed film in air transferred from a subphase containing CaCl2,6 while for OTS, self-assembled on silica, a tilt angle of 10° has been observed.22 Even in air, crystalline structures have been observed for ArAc tails6 and OTS on silica.23 The larger size of the DPPC head group will geometrically obstruct a close packing, and to our knowledge no crystalline structure has been reported for DPPC tails. Although crystalline structures of OTS and ArAc tails are observed, all alkyl chains in the monolayers have cross-sectional areas which allow them some rotational freedom in the transferred films24 as well as considerable chain flexibility.2,25 Experimentally, the differences in ordering between the different alkyl chains can, for example, be accessed by the infrared absorption peak position of the CH2 symmetric υs(CH2) and antisymmetric υa(CH2) stretching modes, polarized perpendicular to the alkyl chains.22,26 Disordered chains have peak positions of 2856 and 2928 cm−1 for υs(CH2) and υa-(CH2), respectively.22 Reported literature values on the υs(CH2) and υa(CH2) modes for self-assembled monolayers similar or identical to those used here yield 2852 and 2921 cm−1 for condensed DPPC,26 2853 and 2920 cm−1 for docosanoic acid,27 and 2850 and 2917 cm−1 for OTS on silica.22 These values correlate well with the adsorption behavior of hGH on the different alkyl chains if it is assumed that adsorption is reduced as the less ordered alkyl chains lose entropy upon hGH adsorption. It is important to realize these results indicate that the entropy of alkyl chains, which are considered to be closely packed in self-assembled monolayers, can counteract the effect of hydrophobic attraction of the hGH molecule.
Adsorption of hGH onto the hydrophilic silica and ArAc head groups (Figure 3b) results in small amounts adsorbed. Apparently, hGH has a low affinity for substrates with a relatively high negative charge density. For adsorption onto the phosphatidylcholine head group, however, a higher affinity is observed. One obvious reason is that the negatively charged hGH is not electrostatically repelled by the overall neutral DPPC layer, as is the case for silica and ArAc. The hGH can even be electrostatically attracted, as the location of the charged groups in the DPPC layer produces an electric dipole moment. Another possibility is that hGH displays a higher affinity for the exposed methyl groups of DPPC than for the exposed carboxyl groups of ArAc or the silanol groups of silica. Although a previous study on hGH adsorption showed that adsorption kinetics of hGH are only weakly affected by long range electrostatic interactions,19 it might still be possible that the affinity of hGH for sorbent surfaces is reduced by sorbent surfaces bearing a high negative charge density.
One may expect that hGH adsorption on ArAc and DPPC head groups is reduced due to surface entropy reasons similar to those for adsorption on the tails. However, it is expected that the charged head groups of ArAc have less conformational freedom, as they are closely associated in a nearly crystalline array with their counterions. Similarly, the zwitterionic DDPC head groups are expected to pack tightly, forming a charged lattice network in which the head groups are conformationally constrained due to ionic interactions with nearest neighbors. Indeed, atomic force microscope experiments reveal a crystalline structure for DPPC head groups on OTS surfaces (D. W. Britt et al., submitted).
The desorption patterns of hGH (Figure 4) all show a fast initial decrease, the extent of which varies strongly for the different types of surfaces, followed by a more or less stable amount adsorbed. Apparently, there are two fractions of hGH molecules, of which one is weakly bound and readily desorbs while the other fraction is irreversibly adsorbed in the time scale investigated. It is likely that hGH molecules can adsorb in two different states, where one state is, for example, adsorbed in aggregates or in a different conformation, resulting in a higher adsorption energy. This type of desorption behavior was also observed in a previous study of hGH at solid surfaces.19 Also, other studies have demonstrated that proteins can adsorb in two states with substantial differences in mobility28 and biological activity.5 The only exception to this two-state desorption process is seen for hGH on the DPPC tail surface, for which no fast initial desorption is observed. Apparently, all hGH molecules are strongly adsorbed. A possible explanation is that the less dense DPPC tail packing allows the hGH molecules to squeeze partly into the layer, thereby increasing the surface area for interactions with the sorbent surface.
Conformational Changes
Information about the conformation of hGH in the adsorbed state is obtained by monitoring the emission maximum, λm, and by performing fluorescence quenching experiments. The maxima in fluorescence intensity from hGH molecules in solution and in the adsorbed state are given in Table 2. As quenching occurs upon molecular contact, it reveals the accessibility of the single tryptophan residue for TCE, which is a polar uncharged quencher. It should be noted, however, that quenching is enhanced if the lifetime of the excited state increases. Collisional or dynamic quenching of fluorescence is described by the Stern–Volmer equation
Table 2.
Peak Fluorescence Wavelength (λm) and Stern–Volmer Quenching Constants (K) for hGH in Solution and Adsorbed to the Different Sorbent Surfaces
| λm (nm) | K (M−1) | |
|---|---|---|
| solution | 338.1 | 7.8 |
| OTS | 330.4 | 62.9 |
| DDS | 330.8 | 46.9 |
| ArAc tail | 332.6 | 24.6 |
| DPPC tail | 336.7 | 24.2 |
| silica | 334.0 | |
| ArAc head | 336.1 | |
| DPPC head | 333.4 |
| (1) |
where F0 and F are the fluorescence intensities in the absence and presence of quencher, respectively, Q is the concentration of the quencher, and K is the Stern–Volmer quenching constant. Quenching of adsorbed proteins was measured after the desorption step, which means that only the more tightly bound proteins are still adsorbed. Quenching constants are determined for hGH adsorbed onto the hydrophobic alkylated substrates only. The results of the quenching measurements are shown as Stern–Volmer plots in Figure 5, and the calculated quenching constants are given in Table 2. It should be taken into account that the surface can influence fluorescence quenching by blocking access of TCE to the tryptophan.
Figure 5.
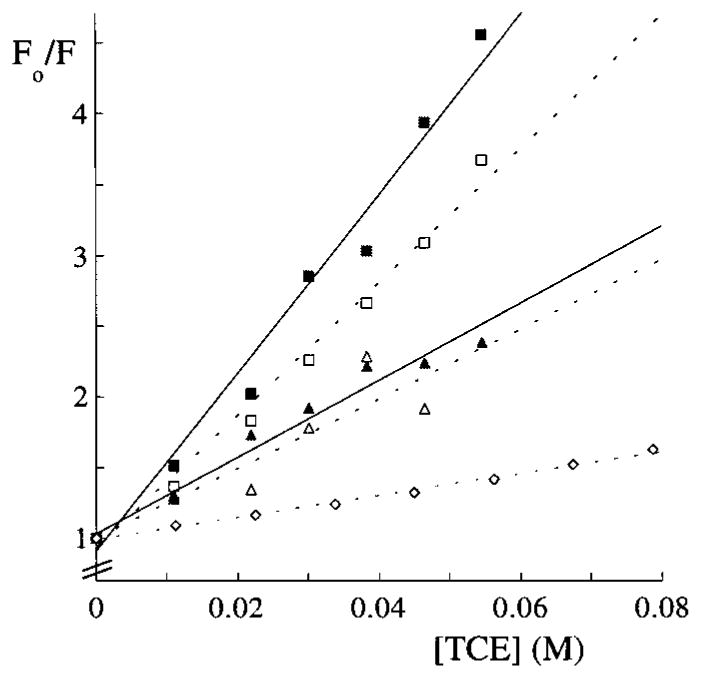
Stern–Volmer plots of fluorescence quenching of hGH by trichloroethanol (TCE). Quenching results for proteins in solution or adsorbed on the different substrates are as indicated: solution (◇); ArAc tail (△); DPPC tail (▲); DDS (□); OTS (■).
Tryptophan fluorescence of proteins generally shows a red-shift if the tryptophan becomes exposed to polar water molecules. However, as seen in a previous study,19 the fluorescence wavelength decreases upon adsorption although a generally more expanded structure is expected. Evidence that the hGH structure is expanded upon adsorption is clearly supported by the results of the quenching experiments. There are two effects which will cause a reduction in λm: a change to a less polarizable environment and/or a decrease of the lifetime of the excited state, giving less time for the relaxation process which takes the energy from the excited state. Although quenching shows an increase in accessibility of the tryptophans for molecules in solution and thus for water molecules, the first effect is still very reasonable. The tryptophan is buried in the hydrophobic interior of hGH, and an emission wavelength peak around 330 nm can be expected. The observed maximum at 338 nm for hGH in solution might be the result of the polarity which arises from an H-bond of the tryptophan with an aspartic acid residue.29
As can be seen from both the λm shifts and the quenching constants, relatively large changes are observed for adsorption onto the OTS and DDS surfaces. These were also the surfaces for which hGH showed the highest affinity. Conversely, hGH adsorption onto the low-affinity substrates results in relatively small shifts in fluorescence emission maxima. These results indicate a correlation between hGH adsorption affinity and conformational changes. Note that the more mobile surfaces lead to smaller conformational changes in the adsorbed hGH structure. For example, the effect of the substrate mobility can be seen when hGH adsorption on ArAc tails is compared with adsorption on DPPC tails, or when hGH adsorption on ArAc heads is compared to that on silica. These systems show similar adsorption kinetics, but smaller conformational changes are noted for the surfaces having a higher degree of possible configurations. The relatively low extent of conformational changes in hGH upon adsorption onto more mobile surfaces might be useful for bioseparation purposes as adsorption-induced conformational changes limit the applicability of the substrates. Preference of surfaces with a lower, more liquid like, alkyl chain density for chromatography purposes has been demonstrated.4
Conclusions
The hGH is strongly adsorbed onto the methyl- and alkyl-silanized hydrophobic surfaces, and this process is accompanied by a relatively large alteration of the hGH structure. A much lower affinity is observed for hGH adsorption onto sorbent surfaces with a similar chemical composition but a lower packing density of the alkyl chains and presumably a higher degree of conformational entropy. It is inferred that the entropy of a sorbent surface is an important parameter and should be considered in describing the protein adsorption process. Adsorption of hGH on the more hydrophilic surfaces results in low amounts adsorbed for surfaces having a relatively large negative charge density. In general, it was found that adsorption of hGH onto low-affinity surfaces results in a relatively small extent of conformational changes. This leads to the conclusion that the affinity for adsorption is an important parameter determining the hGH conformation in the adsorbed state. The comparison of fluorescence emission spectra and quenching results among surfaces for which hGH displays a similar adsorption pattern indicates that the extent of conformational changes is less if hGH is adsorbed onto more mobile surfaces. This suggests that hGH adsorption onto surfaces with a higher entropy but similar total adsorption energy renders hGH in a less altered conformation.
Acknowledgments
J.B. gratefully acknowledges the fellowship from the Netherlands Organization for Scientific Research (NWO). This work was supported by NIH Grant RO1 HL 44538.
References
- 2.Swalen JD, Allara DL, Andrade JD, Chandross EA, Garoff S, Israelachvili J, McCarthy TJ, Murray R, Pease RF, Rabolt JF, Wynne KJ, Yu H. Langmuir. 1987;3:932. [Google Scholar]
- 3.Ulman A. Chem Rev. 1996;96:1533. doi: 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 4.Wirth MJ, Fairbank RWP, Fatunmbi HO. Science. 1997;275:44. doi: 10.1126/science.275.5296.44. [DOI] [PubMed] [Google Scholar]
- 5.Marron-Brignone L, Morélis RM, Coulet PR. Langmuir. 1996;12:5674. doi: 10.1006/jcis.1997.4958. [DOI] [PubMed] [Google Scholar]
- 6.Zasadzinski JA, Viswanathan R, Madsen L, Garnaes J, Schwartz DK. Science. 1994;263:1726. doi: 10.1126/science.8134836. [DOI] [PubMed] [Google Scholar]
- 7.Wells JA, De Vos AM. Annu Rev Biophys Biomol Struct. 1993;22:329. doi: 10.1146/annurev.bb.22.060193.001553. [DOI] [PubMed] [Google Scholar]
- 8.Russell-Jones DL, Watts GF, Weissberger A, Naoumova R, Myers J, Thompson GR, Sönksen PH. Clin Endocrinol. 1994;41:345. doi: 10.1111/j.1365-2265.1994.tb02555.x. [DOI] [PubMed] [Google Scholar]
- 9.Chang CN, Rey M, Bochner B, Heyneker H, Gray G. Gene. 1987;55:189. doi: 10.1016/0378-1119(87)90279-4. [DOI] [PubMed] [Google Scholar]
- 10.Bell R. Science. 1996;380:661. doi: 10.1038/380661a0. [DOI] [PubMed] [Google Scholar]
- 11.Wicar S, Mulkerrin MG, Bathory G, Khundkar LH, Karger BL. Anal Chem. 1994;66:3908. doi: 10.1021/ac00094a011. [DOI] [PubMed] [Google Scholar]
- 12.De Vos AM, Ultsh M, Kosiakoff AA. Science. 1992;255:306. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 13.Clackson T, Wells JA. Science. 1995;267:383. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 14.Ettori C, Righetti PG, Chiesa C, Frigerio F, Galli G, Grandi G. J Biotechnol. 1992;25:307. doi: 10.1016/0168-1656(92)90163-4. [DOI] [PubMed] [Google Scholar]
- 15.Britt DW, Hlady V. J Colloid Interface Sci. 1996;178:775. doi: 10.1006/jcis.1996.0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mingotaud A-F, Mingotaud C, Patterson LK. Handbook of Monolayers. Vol. 1. Academic Press; San Diego, CA: 1993. [Google Scholar]
- 17.Klopfer KJ, Vanderlick TK. J Colloid Interface Sci. 1996;182:220. [Google Scholar]
- 18.This immiscible behavior of DiO (and DiI) in DPPC affected the experiments as a reduction in the dyes’ fluorescence intensity, as was observed for films transferred after the Langmuir layer was held compressed for longer times. DiO is miscible in the liquid-expanded phases, as observed by fluorescence microscopy. However, upon increasing the pressure above the transition to the liquid-condensed phase, it is likely that the DiO and DiI were slowly squeezed out of the monolayer. Consequently, to use the dye system as a control for the stability of transferred films, identical experimental conditions including the duration of each step are required.
- 19.Buijs J, Hlady V. J Colloid Interface Sci. 1997;190:171. doi: 10.1006/jcis.1997.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cassie ABD. Discuss Faraday Soc. 1952;75:5041. [Google Scholar]
- 21.Norde W, Giesbers M, Pingsheng H. Colloids Surf, B. 1995;5:255. [Google Scholar]
- 22.Allara DL, Parikh AN, Rondelez F. Langmuir. 1995;11:2357. [Google Scholar]
- 23.Fujii M, Sugisawa S, Fukada K, Kato T, Shirakawa T, Seimiya T. Langmuir. 1994;10:984. [Google Scholar]
- 24.Jyoti A, Prokop RM, Li J, Vollhardt D, Kwok DY, Miller R, Möhwald H, Neumann AW. Colloids Surf, A. 1996;116:173. [Google Scholar]
- 25.Taut C, Pertsin AJ, Grunze M. Langmuir. 1996;12:3481. [Google Scholar]
- 26.Okamura E, Umemura J, Takenaka T. Biochim Biophys Acta. 1985;812:139. [Google Scholar]
- 27.Bonnerot A, Chollet PA, Frisby H, Hoclet M. Chem Phys. 1985;97:365. [Google Scholar]
- 28.Tilton RD, Gast AP, Robertson CP. Biophys J. 1990;58:1321. doi: 10.1016/S0006-3495(90)82473-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oroszlan P, Wicar S, Teshima G, Wu SL, Hancock WS, Karger B. Anal Chem. 1992;64:1623. doi: 10.1021/ac00038a021. [DOI] [PubMed] [Google Scholar]