

Abstract
Oral delivery remains a challenge for poorly permeable hydrophilic macromolecules. Poly(amido amine) (PAMAM) dendrimers have shown potential for their possible oral delivery. Transepithelial transport of carboxyl-terminated G3.5 and amine-terminated G4 PAMAM dendrimers was assessed using isolated rat jejunal mucosae mounted in Ussing chambers. The 1 mM FITC-labeled dendrimers were added to the apical side of mucosae. Apparent permeability coefficients (Papp) from the apical to the basolateral side were significantly increased for FITC when conjugated to G3.5 PAMAM dendrimer compared to FITC alone. Minimal signs of toxicity were observed when mucosae were exposed to both dendrimers with respect to transepithelial electrical resistance changes, carbachol-induced short circuit current stimulation, and histological changes. [14C]-mannitol fluxes were not altered in the presence of 1 mM dendrimers, suggesting that the paracellular pathway was not affected at this concentration in this model. These results give insight into the mechanism of PAMAM dendrimer transepithelial rat jejunal transport, as well as toxicological considerations important for oral drug delivery.
Introduction
Polymeric drug delivery can improve solubility, biodistribution, and bioavailability of insoluble and highly toxic drugs.1 Poly(amido amine) (PAMAM) dendrimers are a highly branched class of polymers that can increase solubility and intestinal permeability of drugs.2−4 These versatile carriers have multiple surface groups that can be functionalized with imaging agents, drugs, labels, and targeting ligands.5−8 PAMAM dendrimers can also be surface-engineered to tune their toxicity and pharmacokinetic profiles, allowing them to be tailored for specific biomedical applications.9,10
PAMAM dendrimers penetrate the intestinal barrier in vitro and in vivo, suggesting a rationale for use in oral drug delivery.11−19 The oral route of drug delivery has the distinct advantage of increased patient compliance, reduced risk of needle-borne infections and improved pharmacokinetic profiles compared to parenteral dosing.20 Many chronic diseases including cancer treatments require years of regular injections, which could be circumvented if the drug was absorbed orally. Many other small molecules require injection due to their poor intestinal solubility and/or low or variable intestinal absorption, especially when the drug has a narrow therapeutic index. PAMAM dendrimers have shown to increase the intestinal permeability of camptothecin,21 propanolol,22 naproxen,23 SN38,24 and silybin.25 This provides rationale for the development of PAMAM dendrimers as an oral drug delivery system for poorly absorbed drugs. In this study we evaluated the ability of PAMAM dendrimers to be absorbed through the isolated rat jejunal epithelium.
Caco-2 cell cultures have been used for the majority of evaluations of PAMAM dendrimer transport through the intestinal epithelium.10,12−15,17,18,26,27 Such studies provided analysis of dendrimer intestinal penetration and toxicities. They revealed that dendrimer permeation is a function of the dendrimer generation, concentration, and incubation time.18 These studies have also provided mechanistic insights into the routes via which PAMAM dendrimers can penetrate the intestinal epithelium. Previous work demonstrated that specific pharmacologic endocytosis inhibitors reduced the flux of G4 PAMAM dendrimers across Caco-2 monolayers.17 Further work showed that PAMAM dendrimer transport was clathrin, dynamin and energy-dependent in Caco-2 cells. While this indicates that endocytic mechanisms are involved in dendrimer transport, these studies also showed that dendrimers facilitated tight junction opening, as evidenced by occludin staining and increased mannitol transport.10,13,14,28,29 Thus, the route of dendrimer penetration across Caco-2 monolayers appears to be via a combination of the transcellular and paracellular route. Alternative data achieved in CD-1 mice indicated no increase in mannitol permeability or tight junction opening when PAMAM dendrimers were administered orally at concentrations greater than those used in Caco-2 cell cultures (e.g., G4-NH2 (2.1 mM), G3.5-COOH (7.7 mM)).21 This apparent discrepancy may be due to the differences between the models used. Cell culture models lack some of properties that native tissues contain. In comparison to in vivo models, Caco-2 cell monolayers lack mucus layers, extracellular matrix proteins, supportive mixed cell populations, basement membranes, and metabolic protein expression. Indeed, Caco-2 cell cultures can give a wide range of transport data based on differences between source,30,31 selection pressure,30 passage number,32 and tissue culture conditions.33−35 Caco-2 cells have been noted for their increased sensitivity to penetration enhancers, excessively high resistant tight junctions and increased indications of cytotoxicity upon exposure to enhancers compared to native isolated rat and pig intestinal tissue.36,37 Thus, to account for such deficiencies in Caco-2 cells, we evaluated the mechanism of dendrimer permeability and toxicity in an isolated rat jejunal model in Ussing chambers.
The Ussing chamber model has been used to study the mechanisms of transport and toxicity of drugs across isolated intestinal tissue.38 In this study we utilized isolated rat intestinal epithelium to test PAMAM dendrimer transport due to its higher correlation to human jejunum effective permeability (Peff) than Caco-2 cell cultures (R2 = 0.95 vs 0.79, respectively).39 The concentration, incubation time, and molecular weight of dendrimers used were chosen to exceed typical limits of cytotoxicity in Caco-2 cultures previously observed in our lab, in order to observe if isolated tissue histology would similarly be affected. Due to the hypersensitivity of Caco-2 cell cultures to penetration enhancers, we hypothesized that a supra-toxic concentration of PAMAM dendrimers would yield reduced evidence of toxicity in an isolated tissue model.36,37,40 Thus, concentrations of 1.0 mM with an incubation time of 120 min with G4 dendrimers were used for this study as these have previously exhibited cytotoxicity to Caco-2 cells (Table 1).10,14,15,18
Table 1. Intestinal Toxicity in the Presence of Dendrimersa.
| G3.5 PAMAM Dendrimers | ||||
|---|---|---|---|---|
| 0.01 mM | 0.1 mM | 1 mM | 10 mM | |
| 90 min | –b | –b | +b | |
| 120 min | –c | –c | –d; –f | –e |
| 150 min | +b | +b | +b | |
| 180 min | –l | –l | ||
| 210 min | +b | +b | +b | |
| G4 PAMAM Dendrimers | ||||
|---|---|---|---|---|
| 0.01 mM | 0.1 mM | 1 mM | 10 mM | |
| 90 min | +g | +g | ||
| 120 min | –f; –i; +h | +f; +h | –d; –k; +f; +h | |
| 150 min | +g | +g | ||
| 180 min | –j | +j; +l | +j | |
| 210 min | +g | +g | +g | |
(+) = Indication of toxicity; (−) = No indication of toxicity.
Caco-2, LDH release.14
Caco-2, WST-1 assay.27
This study: rat jejunum; histology; carbachol response.
Mice, 7.7 mM dose; histology, TEM.21
Caco-2, TEM.15
Caco-2, LDH release.13
Caco-2, WST-1 assay.18
Caco-2, WST-1 assay.17
Caco-2, WST-1 assay.10
Mice, 0.9 mM dose; animal wt, blood chemistry.9
Caco-2, MTT assay.29
In addition the hypothesis that PAMAM dendrimers induce tight junction opening was explored in this study in order to reconcile results of previous mice- and in vitro Caco-2 (Table 2).14,21 Concentrations, incubation times, and generations of PAMAM dendrimers were selected that have previously been reported to increase mannitol permeability in Caco-2 monolayers.14 The 1.0 mM G3.5 and G4 PAMAM dendrimers, incubated with tissue for 90 min, increased mannitol permeability in Caco-2.14,18 With this concentration, we planned to probe the differences between isolated tissue and Caco-2 models as influenced by PAMAM dendrimers. This study provides evidence for the role of isolated tissue studies in oral drug discovery and the limitations of Caco-2 cells for toxicity screening.
Table 2. Mannitol Permeability in the Presence of Dendrimers in Different Bioassays.
Thus, the aim of this study was to evaluate the intestinal permeability of PAMAM dendrimers G3.5 and G4 in isolated rat jejunum, using concentrations that probe the limits of toxicity in isolated tissue versus Caco-2 cell culture. Additionally, transepithelial transport of PAMAM dendrimers was monitored to explore the feasibility of dendrimer oral drug delivery for future biomedical use.
Experimental Section
Materials
PAMAM dendrimers (G4.0 and G3.5) were purchased from Dendritech, Inc. (Michigan, U.S.A.). FlTC and FITC-dextran were obtained from Sigma-Aldrich (Dorset, U.K.). Acetone was obtained from VWR (Ireland). Carbachol was obtained from Calbiochem, Inc. (Massachusetts, U.S.A.). Disposable size exclusion PD-10 columns were obtained from GE Lifesciences (Buckinghamshire, U.K.). 14C Mannitol (56.5 mCi/mmol) was obtained from Perkin Elmer (U.S.A.). All other reagents were obtained from Sigma-Aldrich (Ireland).
Methods
Synthesis of FITC-Labeled PAMAM Dendrimers
FITC-dendrimer conjugates were synthesized using previous methods with some modifications.15 Briefly, FITC was dissolved in acetone (<5 mg/mL) and added to amine-terminated (G4.0) dendrimers at a ratio of 1:1.2, at pH 7.4 in PBS. The reaction proceeded overnight with stirring at room temperature and the product was then dialyzed for 24 h.
The carboxylic groups of G3.5 were activated with N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide (EDC), and then tert-butyl N-(2-aminoethyl) carbamate (molar ratio 1:12:4) was added in PBS at a pH of 7.4. The reaction was then dialyzed for 24 h. The tert-butyl (Boc) protecting group was removed by adding 1 mL of trifluoroacetic acid to the dialysate and stirring for 4 h, followed by further dialysis (24 h, four water changes). These slightly amine-modified dendrimers were then reacted with FITC similar to G4.0 dendrimers above.
FITC conjugated dendrimers were fractionated by size exclusion chromatography using a Fast Protein Liquid Chromatography (FPLC) system to remove small molecular weight impurities. Fractions were taken from 161 to 232 mL elution volume. FITC conjugated dendrimers were fractionated using a XK 26/70 column packed with Superdex 200 prep grade media (GE Lifesciences, Buckinghamshire, U.K.) at a flow rate of 2.5 mL/min of PBS (pH 7.4 PBS). They were then further dialyzed and lyophilized. The FITC-dendrimer conjugates were analyzed by FPLC to assess for small molecular weight impurities. FITC loading was quantified spectrophotometrically (Figure S1).
Ussing Chamber Experiments
Isolated jejunal tissue was obtained from male Wistar rats (Charles River, U.K.) of weight 250–500g in accordance with the UCD Animal Research Ethics Committee policy on use of tissue post-mortem. Rats were sacrificed by cervical dislocation, followed by immediate removal of the jejunum (up to 20 cm proximal from cecum). Tissue was immediately immersed in fresh Krebs-Henseleit (KH) buffer maintained at 37 °C, pH 7.4, and oxygenated with carbogen gas. Tissue was then opened along the mesenteric border and was pinned mucosal-side down on a corkboard. The external muscularis layer was then gently stripped away from the submucosa using a watchmaker’s size 5 fine forceps leaving an intact epithelium with lamina propria. Tissue was mounted between the two halves of an Ussing chamber (World Precision Instruments, U.K.) with a 5 mL bath volume each side, a gas air-lift system and an 0.63 cm2 exposed tissue area.41 Chambers were bilaterally filled with fresh oxygenated KH buffer. Following mounting, mucosae were equilibrated in oxygenated buffer for 15 min followed by 30 min of voltage clamping in order to calculate transepithelial electrical resistance (TEER) values and to ensure that levels were above minimum acceptable values (30 Ω·cm2).42 Test probes were added to apical side of tissue and sampling occurred (200 μL) every 20 min for 120 min from the basolateral side, and at 0 and 120 min from the apical side. The chamber volume was maintained on the basolateral side by replacing sample volume with fresh oxygenated KH buffer after each sampling point.
Apparent Permeability (Papp) Measurement
Permeability of FITC labeled-PAMAM Dendrimers (G3.5, G4.0), FITC, and FITC-dextran (4 kDa;10 kDa) were tested across mucosae. FITC-dextrans were used as macromolecular control markers for paracellular flux. 0.5 μCi of 14C-Mannitol was also added to the apical side of all experiments to serve as an additional paracellular marker of low molecular weight hydrophilic molecule permeation. Fluorescence was detected in samples using a spectrophotometer (λex/λem of 495/525 nm, MD Spectramax Gemini). Samples were then transferred to vials and mixed with 3 mL of scintillation cocktail (Ecoscint, National Diagnostics). Scintillation counting was performed on a Packard Tricarb 2900 TR (PerkinElmer, Ireland).
TEER Measurement
Following permeability experiments, the electrogenic chloride secretory responses of mucosae were tested to ascertain retention of intestinal function. A cholinomimetic, carbachol, was added to the basolateral side of the chamber at concentrations from 0.1 to 10 μM. The change in short circuit current (ΔISC) was measured relative to the baseline current.43,44 Tissue was then gently removed and fixed in 10% buffered formaldehyde for 24 h in preparation for histological staining with hematoxylin and eosin (H&E) or alcian blue and neutral red (AB/NR).45
The potential difference (PD) and ΔISC across the epithelial layer was monitored by Ag/AgCl electrodes using an EVC-4000 amplifier (WPI, U.K.) and Pro-4 timer (WPI, UK). A 3 M KCl solution in 3% agar (w/v) was used as an electrode bathing solution. Electrical signals were converted from analogue to digital using Powerlab data acquisition unit. Data was recorded with Chart software (AD instruments, U.K.) and TEER was calculated indirectly using Ohm’s Law from the ISC and PD values. Voltage clamping to zero was performed using a cyclical 30 s voltage clamp to 0 mV followed by three seconds open circuit period using the Pro-4 timer.
Apparent permeability coefficients (Papp) of fluorescent and radioactive compounds was calculated using the equation:
where dQ/dt is the rate of appearance of sample on the basolateral side, C0 is the apical concentration, and A is the exposed surface area of the tissue.
Statistical Analysis
Permeability data was analyzed using GraphPad Prism (version 6.0c). Analysis was performed using one-way analysis of variance (ANOVA) with Tukey’s post-test (pooled variance). Statistics on TEER values was performed using a two-tailed Student’s t test comparing TEER at t = 0 to the later time points. Values with P < 0.05 were considered significant.
Results
FITC-labeled dendrimers were synthesized and fractionated to remove free FITC prior to testing in Ussing chambers. Purification by FPLC resulted in removal of small molecular weight peaks occurring after 232 mL elution volume on the XK 26/70 column (Figure 1).
Figure 1.

Size exclusion chromatograms of G3.5-FITC and G4-FITC conjugates before and after fractionation. Gray line = before fractionation, black line = after fractionation.
Papp values for FITC-labeled PAMAM dendrimers through isolated rat jejunum was obtained in Ussing chambers. The Papp of FITC-G3.5 PAMAM dendrimers was significantly increased over that of free FITC. The Papp of FITC-G4.0 dendrimers was not statistically increased compared to free FITC, although there was a trend. Importantly, the Papp of FITC-dextran (both 4 kDa and 10 kDa) was not different than that of free FITC (Figure 2), so it is not the case that conjugation to any molecule increases the FITC Papp per se. The [14C]-mannitol Papp was not significantly increased in the presence of either of the two dendrimer conjugates compared to untreated (Figure 3).
Figure 2.

Papp of FITC-PAMAM (1.0 mM), FITC-dextran 4 kDa (0.625 mM), FITC-dextran 10 kDa (0.25 mM), and free FITC (0.02 mM) across isolated rat jejunum. FITC-G3.5 dendrimers had significantly increased Papp compared to free FITC (*p < 0.05).
Figure 3.

Papp of [14C]-mannitol through isolated rat jejunum. No significant difference was observed for G4.0 and G3.5 dendrimer (1.0 mM) treatments vs free FITC, indicating no enhanced paracellular transport in the presence of either dendrimer.
The average basal TEER for jejunal segments was 57 ± 20 Ω·cm2 (n = 45). This is consistent with previously reported rat jejunal TEER values.40,44,46 TEER values of the control, G4.0 dendrimer, and FITC dextran treatments were significantly reduced after 40–60 min in Ussing chambers compared to baseline TEER at t = 0. The TEER of the G3.5 treatment was observed to drop much more rapidly, reaching significantly reduced levels at t = 5 min and onward (P < 0.05). Decreases in TEER were not reflected in increased Papp of mannitol (Figure 3), so the relevance of transient TEER decreases to overall paracellular permeability is questionable.
Figure 4.

Percent TEER changes of isolated jejunal tissue: control (●), G4 dendrimers 1.0 mM (■), G3.5 dendrimers 1.0 mM (◆), FITC-dextran 4 kDa (Δ), FITC-dextran 10 kDa (∇). Percent TEER values were calculated as a percentage of the initial TEER at t = 0 in each group. Significant differences from TEER of each individual group at t = 0 are marked with open circles (G3.5 dendrimers), star (control, G3.5 dendrimers, FITC-dextran 4 kDa), and asterisk (all groups).
The electrogenic chloride transport secretory response to carbachol showed similar concentration-dependent, large ISC increases in jejunal mucosae exposed to apical additions of both unconjugated dendrimers and FITC dextrans for 120 min (Figure 5). Carbachol-stimulated ISC increases in the presence of dendrimers were similar to untreated controls and demonstrated that tissue secretory function was retained.
Figure 5.

ΔISC response to basolateral additions of carbachol to jejunal mucosae. No significant difference in response was observed for test groups: control (●), G4 dendrimers 1.0 mM (■), G3.5 dendrimers 1.0 mM (◆), FITC-dextran 4 kDa (Δ), FITC-dextran 10 kDa (∇).
Histological evaluation at 120 min postmounting in chambers showed mild edema in all samples including untreated controls, likely due to the severance of lymphatic drainage, but there was no significant membrane disruption due to dendrimer treatments (Figure 6). All tissues therefore showed an intact barrier, consistent with the retention of secretory ion transport capacity.
Figure 6.
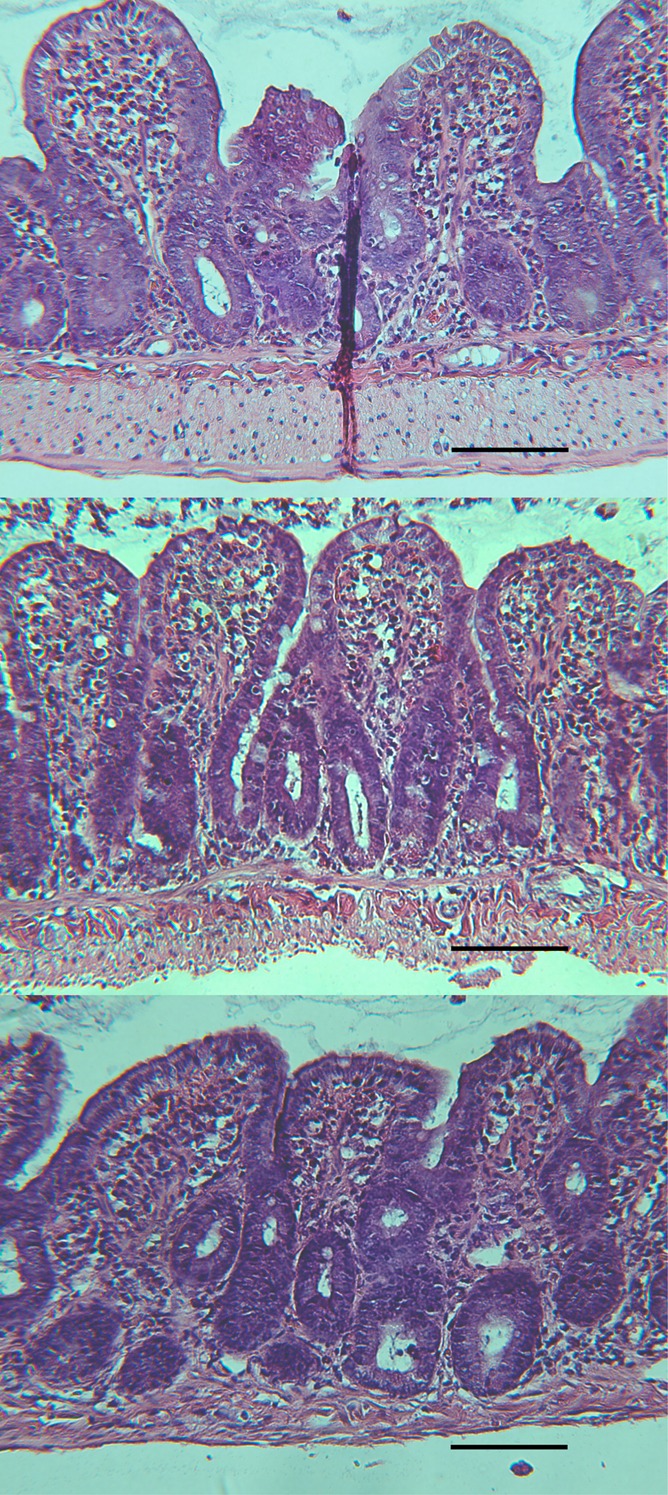
H&E staining of isolated jejunal tissue after 120 min incubation in Ussing chambers. No difference in histology was observed between controls and dendrimer-treated tissue: 1 mM G3.5 dendrimer (upper), 1 mM G4 dendrimer (middle), control (lower). Scale bar = 100 μm for all figures.
The absence of small molecular weight FITC of the FITC–dendrimer conjugate was monitored through size exclusion chromatography on PD-10 columns after each experiment. FITC-dendrimers collected from the basolateral chamber remained stable, with no appearance of the free label peak (30 mL elution volume) compared to the dendrimer peak (6 mL elution volume). This signifies that detected fluorescence on the basolateral side of the chamber was not due to free FITC cleaved from the dendrimer, but rather due to the FITC-labeled dendrimer. This result is critical to validating the stability of the conjugate during the 120 min flux period (Figure S2).
Discussion
The primary goal of this study was to observe the suitability of PAMAM dendrimers for oral drug delivery, and therapeutic application. PAMAM dendrimers have shown the capacity to permeate the small intestinal epithelium and to increase the solubility of copresented drugs in vitro and in vivo.2 Their size and surface functionality makes them capable of a variety of biomedical functions.3 The potential for PAMAM dendrimers in oral drug delivery has been validated in cell culture models and in animals.9,11−14,21 It is remarkable that selected PAMAM dendrimers penetrate the rat small intestinal epithelium in spite of their large molecular weight, complex macromolecular structure and hydrophilic nature as shown here. In particular, G3.5 dendrimers permeated the rat jejunum very well compared to free FITC, as indicated by a Papp in excess of 7 × 10–6 cm/s. Although the probes would still be classified into BCS class III for low permeability/high solubility molecules based on the results of this study, such a Papp value is associated with an in vivo human fad of 40–60%.47 Indeed, anionic G6.5 PAMAM dendrimers, that have a molecular weight 8-fold larger than the probes in this study have been observed to have a fa of 9.4% in mice.11 Since larger molecules are generally more slowly absorbed than smaller ones, it is reasonable to expect a fa of 40–60% for the lower generation dendrimers studied here. Future clinical applications of PAMAM dendrimers rests upon careful evaluation of these parameters.
The permeability of G3.5 and G4 dendrimers in Caco-2 cells has been previously studied in Caco-2 cell cultures and isolated tissue, facilitating comparison of results obtained from isolated tissue and cell culture models.10,16,18 Kolhatkar et al. observed G4 dendrimers with a Papp 1.5 × 10–6 cm/s in Caco-2 cultures at 120 min incubation time.10 This value of Papp was much less than the value of Papp (4.47 × 10–6 cm/s) we observed in this study, but the apical concentration was also 100-fold less. Kitchens et al. observed a Papp value for G4 dendrimers ranging from 20 to 35 × 10–6 cm/s at 1.0 mM for 60–120 min incubation times.18 These results far exceed the values obtained in our study, but the dendrimers used in their study had a ratio of dendrimer to FITC of 1:8, potentially altering the physiochemical properties (the dendrimers in this study had a dendrimer FITC ratio of 1:1.2).18
G3.5 PAMAM dendrimer Papp values in previous studies were less variable in comparison (Table 1). Results from multiple Caco-2 studies observed Papp values ranging from 2.5 × 10–6 cm/s at 0.1 mM to 6 × 10–6 cm/s at 1 mM and 120 min time points.16,18 The values of Papp for G3.5 dendrimers obtained in this study were in the same range as previous results from Caco-2. These results add to the debate on whether hydrophilic macromolecules and nanoparticles can penetrate the intestinal epithelium.48,49 Similar molecular weight dextran molecules (4 and 10 kDa) did not cross the intestinal barrier to the same extent as the G3.5 (12.9 kDa) and G4 (14 kDa) dendrimers, indicating the unique physiochemical characteristics and structure of dendrimers facilitate their higher than expected transepithelial transport.12
In an initial study, Wiwattanapatapee et al. evaluated the transepithelial transport of G4 and G3.5 PAMAM dendrimers in everted rat intestinal sacs.19 This study compared the endocytic index (EI) of the dendrimers (ng dendrimer transferred/mg intestinal tissue protein), bovine serum albumin (BSA), and other polymers. G3.5 and G4 dendrimers were noted for their higher EI than BSA and poly(vinylpyrrolidone) polymers. Comparison to this study is not facilitated by the nature of the permeability data obtained (EI vs Papp). Although the permeability data in that study was not entirely linear, this study indicated that PAMAM dendrimers penetrate the intestinal epithelium.19
The secondary aim of this study was to compare mechanistic information obtained from mannitol permeability in this study to previous results obtained in Caco-2 models. Interestingly, mannitol transport did not increase when tissues were exposed to 1 mM G3.5 and G4 PAMAM dendrimers. These results are in contrast to previous studies in Caco-2 where increased transport of mannitol in the presence of 1.0 mM G3.5 and G4 dendrimers was observed. G3.5 dendrimers at 120 min were observed to increase mannitol Papp 6-fold, while G4 dendrimers at 120 min incubation time showed a 12-fold increase (Table 2).14 This discrepancy may be due to the differences between isolated tissue models and Caco-2 cell culture. Isolated rat jejunal mucosae contain properties that map to the human jejunum.39 These include mucus layers, extracellular matrix proteins, host enzyme levels,47 supportive cells, and basement muscle layers.50−52 Lacking mucus and supportive cells underlying the epithelial barrier, Caco-2 cell cultures may be sensitive to PAMAM dendrimer induced mannitol permeability enhancement compared to rat jejunal mucosae. Other studies have noted the increased sensitivity of Caco-2 cells to penetration enhancers, compared to isolated tissue.36,37 A recent study in CD-1 mice orally gavaged with G3.5 or G4 dendrimers formulated with [14C]-mannitol also showed no induction of paracellular transport of mannitol, confirming the results of this study.21 Since mannitol is an indicator of tight junction opening and enhanced paracellular transport, this suggests that PAMAM dendrimers do not increase paracellular transport in isolated tissue and oral gavage in vivo, at the concentrations, generations, surface modifications, and incubation times studied.
The alternative possibility would be a transcellular route via an endocytic pathway. In parallel with the tight junction route, it has been confirmed in Caco-2 cells where Papp of radiolabeled G4 was observed to decrease in the presence of endocytic inhibitors.17 Further work showed a decrease in G3.5 PAMAM Papp during incubation with clathrin inhibitor (mondansyl cadaverine) or dynamin inhibitor (dynasore). The caveolin inhibitor, genistein, did not have a significant affect, indicating that transport across the membrane may depend primarily on clathrin-mediated transcytosis.28
The effect of PAMAM dendrimers on the viability of epithelial layers was compared to controls. 1.0 mM G3.5 and G4 PAMAM dendrimers retained functional electrogenic chloride secretory pathways after 120 min incubation, indicating a lack of apical membrane disruption. Also, histological evaluation of the tissue showed intact villous structure. The signs of toxicity differ significantly in comparison to Caco-2 cell culture studies. Caco-2 studies have observed significant toxicity at concentrations >0.1 mM and >90 min incubation times for G4 PAMAM dendrimers13,15,18 and >1 mM and >150 min incubation times for G3.5 dendrimers, respectively.14 Indications of reduced proliferation,10,17,18,28 mitochondrial damage,29 and cellular membrane damage13,14 have been apparent. These effects were dependent on concentration, incubation time, and assay, as reviewed in Table 1. Suffice to say that the results of this study concord with in vivo studies performed in CD-1 mice that showed no toxicity for both PAMAM generations by TEM and morphological evaluation of microvilli, as well as histological evaluation of intestinal epithelium.21 The concentrations used in the in vivo study were actually higher than those in the present one. Similar oral toxicity studies have shown no signs of toxicity in CD-1 mice when dosed at 300 mg/kg for G4 and G3.5 dendrimers.53 More studies in the area of toxicity are needed to understand the upper limit of safe oral PAMAM dendrimer administration. These studies give rationale for the potential use of PAMAM dendrimers to carry poorly bioavailable drugs across the intestine to their site of action. Future clinical application of PAMAM dendrimers rests upon careful evaluation of their permeability and toxicity profile.
Conclusion
The elevated transport of the G3.5 PAMAM dendrimers through isolated tissue gives rationale for dendrimer usage in oral drug delivery. The high permeability may indicate that PAMAM dendrimers can carry payloads into systemic circulation (such as poorly permeable and poorly soluble drugs (BCS Class IV). PAMAM dendrimers’ transport through isolated intestinal epithelia was greater than controls and greater than similar size FITC-labeled dextrans. Our results show minimal indication of toxicity to the epithelial barrier when treated with PAMAM dendrimers at concentrations of 1.0 mM. This provides evidence that PAMAM dendrimers may be able to be dosed at nontoxic concentrations yet still penetrate to a sufficient extent for increasing drug absorption. Payloads could potentially be delivered as a mixture or as a conjugated moiety. Future work should be aimed at further understanding of the physiochemical characteristics that promote increased transepithelial transport and specific therapies for oral drug delivery. Results of this study indicate the potential of PAMAM dendrimers as an oral drug delivery system.
Acknowledgments
University College Dublin Seed Funding Grant for visiting scientists. Science Foundation Ireland Cluster Grant 07 SRC B1154 and NIH R01 EB007470. D.H. would like to acknowledge the assistance from staff of the School of Veterinary Medicine and the Conway Institute, University College Dublin, especially from Fiona McCartney. D.H. also acknowledges assistance of Jay Olsen of the University of Utah NMR core facility, Krishna Parsawar of the mass spectrometry core facility, and Chris Rodesch of the confocal microscopy core facility for their assistance.
Supporting Information Available
FITC loading of PAMAM dendrimers, stability of FITC-labeled PAMAM dendrimers, MALDI-TOF and NMR characterization of dendrimer materials, and fluorescent images of FITC and FITC-dendrimer treated tissue. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Duncan R.; Vicent M. J. Adv. Drug Delivery Rev. 2013, 65, 60–70. [DOI] [PubMed] [Google Scholar]
- Sadekar S.; Ghandehari H. Adv. Drug Delivery Rev. 2012, 64, 571–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchens K. M.; El-Sayed M. E. H.; Ghandehari H. Adv. Drug Delivery Rev. 2005, 57, 2163–2176. [DOI] [PubMed] [Google Scholar]
- D’Emanuele A.; Attwood D. Adv. Drug Delivery Rev. 2005, 57, 2147–2162. [DOI] [PubMed] [Google Scholar]
- Yang W.; Cheng Y.; Xu T.; Wang X.; Wen L. Eur. J. Med. Chem. 2009, 44, 862–868. [DOI] [PubMed] [Google Scholar]
- Majoros I. J.; Myc A.; Thomas T.; Mehta C. B.; Baker J. R. Biomacromolecules 2006, 7, 572–579. [DOI] [PubMed] [Google Scholar]
- Mignani S.; El Kazzouli S.; Bousmina M.; Majoral J. P. Adv. Drug Delivery Rev. 2013, 10, 1316–1330. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; MacKay J. A.; Fréchet J. M. J.; Szoka F. C. Nat. Biotechnol. 2005, 23, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Greish K.; Thiagarajan G.; Herd H.; Price R.; Bauer H.; Hubbard D.; Burckle A.; Sadekar S.; Yu T.; Anwar A.; Ray A.; Ghandehari H. Nanotoxicology 2012, 6, 713–723. [DOI] [PubMed] [Google Scholar]
- Kolhatkar R. B.; Kitchens K. M.; Swaan P. W.; Ghandehari H. Bioconjugate Chem. 2007, 18, 2054–2060. [DOI] [PubMed] [Google Scholar]
- Thiagarajan G.; Sadekar S.; Greish K.; Ray A.; Ghandehari H. Mol. Pharmacol. 2013, 10, 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed M.; Rhodes C. A.; Ginski M.; Ghandehari H. Int. J. Pharm. 2003, 265, 151–157. [DOI] [PubMed] [Google Scholar]
- El-Sayed M.; Ginski M.; Rhodes C.; Ghandehari H. J. Controlled Release 2002, 81, 355–365. [DOI] [PubMed] [Google Scholar]
- El-Sayed M.; Ginski M.; Rhodes C. A.; Ghandehari H. J. Bioact. Compat. Polym. 2003, 18, 7–22. [Google Scholar]
- Kitchens K. M.; Foraker A. B.; Kolhatkar R. B.; Swaan P. W.; Ghandehari H. Pharm. Res. 2007, 24, 2138–2145. [DOI] [PubMed] [Google Scholar]
- Sweet D. M.; Kolhatkar R. B.; Ray A.; Swaan P.; Ghandehari H. J. Controlled Release 2009, 138, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchens K. M.; Kolhatkar R. B.; Swaan P. W.; Ghandehari H. Mol. Pharmaceutics 2008, 5, 364–369. [DOI] [PubMed] [Google Scholar]
- Kitchens K. M.; Kolhatkar R. B.; Swaan P. W.; Eddington N. D.; Ghandehari H. Pharm. Res. 2006, 23, 2818–2826. [DOI] [PubMed] [Google Scholar]
- Wiwattanapatapee R.; Carreño-Gómez B.; Malik N.; Duncan R. Pharm. Res. 2000, 17, 991–998. [DOI] [PubMed] [Google Scholar]
- Kermode M. Health Promot. Int. 2004, 19, 95–103. [DOI] [PubMed] [Google Scholar]
- Sadekar S.; Thiagarajan G.; Bartlett K.; Hubbard D.; Ray A.; McGill L. D.; Ghandehari H. Int. J. Pharm. 2013, 456, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Emanuele A.; Jevprasesphant R.; Penny J.; Attwood D. J. Controlled Release 2004, 95, 447–453. [DOI] [PubMed] [Google Scholar]
- Najlah M.; Freeman S.; Attwood D.; D’Emanuele A. Int. J. Pharm. 2006, 308, 175–182. [DOI] [PubMed] [Google Scholar]
- Kolhatkar R. B.; Swaan P.; Ghandehari H. Pharm. Res. 2008, 25, 1723–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Wu Z.; Gao W.; Chen Q.; Yu B. Drug Dev. Ind. Pharm. 2011, 37, 419–427. [DOI] [PubMed] [Google Scholar]
- Grass G. M.; Sweetana S. A. Pharm. Res. 1988, 5, 372–376. [DOI] [PubMed] [Google Scholar]
- Goldberg D. S.; Vijayalakshmi N.; Swaan P. W.; Ghandehari H. J. Controlled Release 2011, 150, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg D. S.; Ghandehari H.; Swaan P. W. Pharm. Res. 2010, 27, 1547–1557. [DOI] [PubMed] [Google Scholar]
- Jevprasesphant R.; Penny J.; Attwood D.; McKeown N. B.; D’Emanuele A. Pharm. Res. 2003, 20, 1543–1550. [DOI] [PubMed] [Google Scholar]
- Vachon P. H.; Beaulieu J. F. Gastroenterology 1992, 103, 414–423. [DOI] [PubMed] [Google Scholar]
- Walter E.; Kissel T. Pharm. Res. 1994, 11, 1575–1580. [DOI] [PubMed] [Google Scholar]
- Walter E.; Kissel T. Eur. J. Pharm. Sci. 1995, 3, 215–230. [Google Scholar]
- Nicklin P.; Irwin B.; Hassan I.; Williamson I.; Mackay M. Int. J. Pharm. 1982, 83, 197–209. [Google Scholar]
- Wilson G.; Hassan I. F.; Dix C. J.; Williamson I.; Shah R.; Mackay M.; Artursson P. J. Controlled Release 1990, 11, 25–40. [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Adv. Drug Delivery Rev. 2001, 46, 27–43. [DOI] [PubMed] [Google Scholar]
- Legen I.; Salobir M.; Kerč J. Int. J. Pharm. 2005, 291, 183–188. [DOI] [PubMed] [Google Scholar]
- Tarirai C.; Viljoen A. M.; Hamman J. H. Pharm. Biol. 2012, 50, 254–263. [DOI] [PubMed] [Google Scholar]
- Clarke L. L. Am. J. Physiol. 2009, 296, G1151–G1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennernäs H.; Nylander S.; Ungell A.-L. Pharm. Res. 1997, 14, 667–671. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Taki Y.; Sakane T.; Nadai T.; Sezaki H.; Yamashita S. Pharm. Res. 1995, 12, 523–528. [DOI] [PubMed] [Google Scholar]
- Brayden D. J.; Bzik V. A.; Lewis A. L.; Illum L. Pharm. Res. 2012, 29, 2543–2554. [DOI] [PubMed] [Google Scholar]
- Ungell A. -L; Nylander S.; Bergstrand S.; Sjöberg Å.; Lennernäs H. J. Pharm. Sci. 1998, 87, 360–366. [DOI] [PubMed] [Google Scholar]
- Maher S.; Feighery L.; Brayden D. J.; McClean S. Pharm. Res. 2007, 24, 1346–1356. [DOI] [PubMed] [Google Scholar]
- Petersen S. B.; Nolan G.; Maher S.; Rahbek U. L.; Guldbrandt M.; Brayden D. J. Eur. J. Pharm. Sci. 2012, 47, 701–712. [DOI] [PubMed] [Google Scholar]
- Carleton H. M.; Drury R. A. B.; Wallington E. A.. Carleton’s Histological Technique; Oxford University Press: New York, 1980. [Google Scholar]
- Tsutsumi K.; Li S. K.; Ghanem A.-H.; Ho N. F. H.; Higuchi W. I. J. Pharm. Sci. 2003, 92, 344–359. [DOI] [PubMed] [Google Scholar]
- Peternel L.; Kristan K.; Petruševska M.; Rižner T. L.; Legen I. J. Pharm. Sci. 2012, 101, 1436–1449. [DOI] [PubMed] [Google Scholar]
- Morishita M.; Peppas N. A. Drug Discovery Today 2006, 11, 905–910. [DOI] [PubMed] [Google Scholar]
- Florence A. T. Pharm. Res. 1997, 14, 259–266. [DOI] [PubMed] [Google Scholar]
- Hilgendorf C.; Ahlin G.; Seithel A.; Artursson P.; Ungell A.-L.; Karlsson J. Drug Metab. Dispos. 2007, 35, 1333–1340. [DOI] [PubMed] [Google Scholar]
- Sambuy Y.; De Angelis I.; Ranaldi G.; Scarino M. L.; Stammati A.; Zucco F. Cell Biol. Toxicol. 2005, 21, 1–26. [DOI] [PubMed] [Google Scholar]
- Van Breemen R. B.; Li Y. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [DOI] [PubMed] [Google Scholar]
- Thiagarajan G.; Greish K.; Ghandehari H. Eur. J. Pharm. Biopharm. 2013, 84, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.