

Abstract
On the basis of our recently resolved first cocrystal structure of Mdm4 in complex with a small molecule inhibitor (PDB ID 3LBJ), we devised an approach for the generation of potential Mdm4 selective ligands. We performed the Ugi four-component reaction (Ugi-4CR) in 96-well plates with an indole fragment, which is specially designed to mimic Trp23, a key amino acid for the interaction between p53 and Mdm4. Generally the reaction yielded mostly precipitates collected by 96-well filter plates. The best hit compound was found to be active and selective for Mdm4 (Ki = 5 μM, 10-fold stronger than Mdm2). This initial hit may serve as the starting point for designing selective p53-Mdm4 inhibitor with higher affinity.
Keywords: peptidomimetic p53-Mdm4 inhibitor, Ugi four-component reaction, library generation
The protein–protein interaction between the transcription factor p53 and the negative regulator Mdm2 is an important recent oncology target.1 The interaction is crystallographically characterized and druggable and several compounds are in late preclinical and early clinical evaluation.2 The Mdm4 protein is a closely related protein to Mdm2 and it also binds to the same epitope of p53. The sequence homology, the shape, dimension and size is similar. Nevertheless all current compound scaffolds characterized by cocrystal structure analysis are highly specific for Mdm2 and show no or very little Mdm4 binding which is surprising and not well understood regarding the great similarity between the two proteins (Figure 1).3,4 Nutlin-3a 1, for example, binds to Mdm4 too but with a roughly 1000-fold lower affinity of about 25 μM.5 Several Novartis compounds 2-4 show weak low μM Mdm4 affinity while being very potent binders to Mdm2 (again ∼1000-fold difference, Figure 2).6,7 Other described Mdm4 selective compounds are either covalent binders or not validated (5, 6).8,9 Surprisingly, pyrazolone compound65 loses activity to Mdm4 in the presence of a reducing reagent, dithiothreitol (DTT). Incubation of these compounds with Mdm4 under nonreducing conditions lead to a time dependent change of Mdm4 structure determined by NMR; concomitantly, the MS analysis showed the presence of covalent adducts of the compound with Mdm4. Additionally, we have found out, by 1H NMR, that the pyrazolone 5 reacts with β-mercaptoethanol in chloroform.
Figure 1.
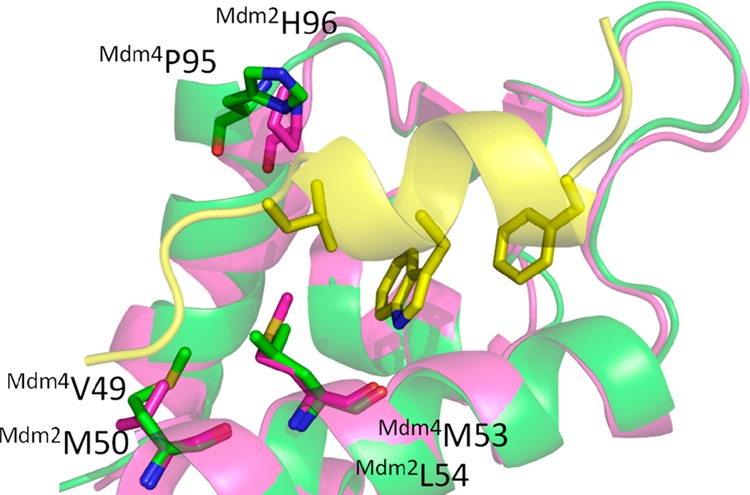
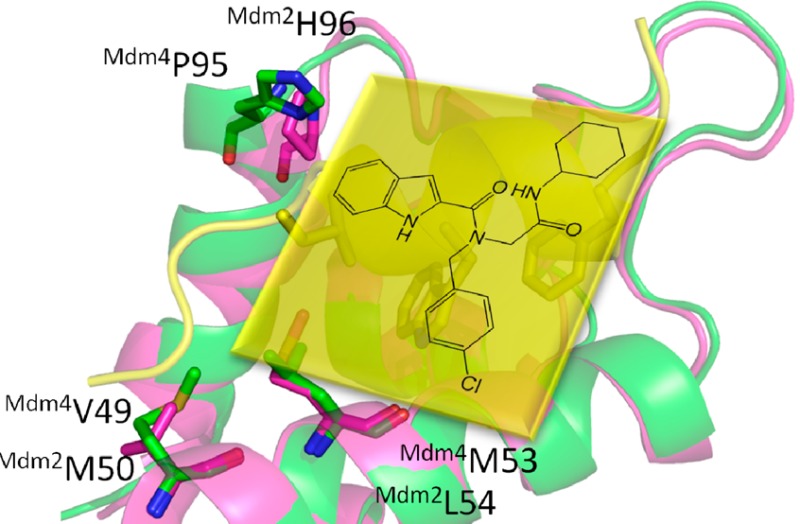
Alignment of Mdm4 (green cartoon, PDB 3DAB) and Mdm2 (magenta cartoon PDB 1YCR) with the Mdm2 peptide (yellow cartoon and sticks) highlighting the similarities and differences of the two binding sites (the Mdm4 binding p53 peptide is omitted for clarity). The main differences between the two receptor p53 binding sites are in the p53L26 pocket. Because of the L > M and the H > P exchanges the Mdm4 pocket is more narrow and less deep. An additional difference is at the outer rim of the p53L26 pocket the M > V exchange. The key amino acids of Mdm2, Mdm4, and p53 are shown as sticks.
Figure 2.

Some known Mdm2 antagonists together with their Mdm4 activity.
Selective Mdm4 antagonists are highly sought after since Mdm4 and Mdm2 proteins are differentially overexpressed in several cancers and both play a prominent but presumably different role in apoptosis induction.10 Herein, we describe the discovery of B1, a selective p53-Mdm4 inhibitor (with ∼5 μM affinity to Mdm4 but 54 μM affinity to Mdm2) with reversed selectivity compared with most p53-Mdm2 inhibitors, which we believe is a good starting point to elaborate Mdm4 selective compounds.
Based on our previously generated insight into the binding of small molecules into Mdm2 and Mdm4 and our recently developed Mdm2 and Mdm4 fluorescence polarization assay, we planned to synthesized libraries of potential Mdm2/4 binding compounds.5,11−23 Thus, we generated a 96-member library of peptidomimetic small molecules via Ugi four-component reaction (Ugi-4CR) (Scheme 1). Each compound contains the indole or p-halobenzyl fragment to mimic the Trp23 “anchor”, which is the key anchor residue in the p53 Mdm2 and Mdm4 protein–protein interaction interface, respectively. Figure 3 illustrates the structure of amine and isocyanide inputs, as well as the experimental setting in a 96-well microliter plate. Since the reaction products regularly precipitated, the compounds were collected by a 96-well filter plate, and washed with ether to remove unreacted starting materials. The yields of the isolated products were between low (6%) and good (56%) with an average of 28% over all 96 wells (Supporting Information). In addition, the purities of the isolated materials were considered sufficient for an initial screening. The collected samples were dissolved as a 10 mM stock solution in DMSO for the screening.
Scheme 1. Ugi-4CR for High-Throughput Synthesis.
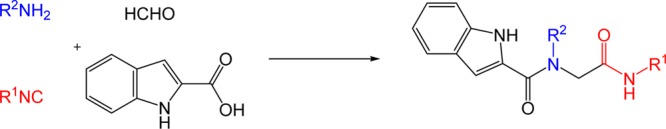
Figure 3.

Parallel synthesis of “anchor” biased compound library via Ugi-4CR. Above: Structures of the amine A and isocyanide B starting materials used.
Compound B1 was identified as a p53-Mdm4 inhibitor (Ki = 19 μM) via our recently reported fluorescence polarization assay. The hit compound was resynthesized and purified by flash chromatography, which was further confirmed by the binding with Mdm4 (Ki = 5.5 μM), as shown in Figure 4.5 Although the p53-binding sites within the Mdm4 and Mdm2 proteins are closely related, known Mdm2 small-molecule inhibitors have been shown experimentally not or very poorly to bind to its homologue Mdm4. This hit compound may provide a new avenue for the design of potential selective inhibitors of the p53-Mdm4 interaction. Further studies are ongoing in our lab to uncover the puzzle of the Mdm2 and Mdm4 selectivity.
Figure 4.

Hit compound as p53-Mdm4 inhibitor. A: Structure of B1. B: Ki = 54 μM (Mdm2). C: Ki = 5.5 μM (Mdm4) as determined by FP.
For further optimizing purposes a second library was synthesized, that follows the structure of hit compound B1, yielding a total of 38 new compounds. Minor changes were made in the indole moiety (from the carboxylic acid component) and different halogen substituted benzylamines were employed, keeping the cyclohexyl fragment intact, as shown in Figure 5. This time a sequential approach was used, which made it possible to run 1 mmol scale reactions as opposed to 0.2 mmol scale in the 96-well plate. Increased yields up to 79% were observed, in average 46%, which confirms that larger scale Ugi reactions in general give better yields.24 Unfortunately, all the other compounds synthesized in Figure 5 showed worse (>50 μM) or no activity in the FP assay.
Figure 5.

Starting materials for the second library of compounds with the structures of A the carboxylic acids and B the amines.
In summary, this work demonstrates that the Ugi four-component reaction (Ugi-4CR) can be used to address the requirements for efficient high-throughput synthesis of diverse compounds in a cost- and time-effective manner. Integrated with biochemical screening assays, a peptidomimetic p53-Mdm4 inhibitor B1 was identified from a 96-membered library generated via Ugi-4CR of an indole fragment. This approach provides an efficient strategy for the discovery of small molecule probes selectively targeting protein–protein interactions. Further optimization studies on B1 are ongoing and will be reported in due course.
Materials and Methods
High-Throughput Synthesis of a 96-Member Library
Add indole-2-carboxylic acid (200 μL, 1 M methanol stock solution; 0.2 mmol), amine (200 μL, 1 M methanol stock solution; 0.2 mmol), isocyanide (200 μL, 1 M methanol stock solution; 0.2 mmol), formaldehyde (120 μL, 2 M methanol stock solution; 0.24 mmol) into each well of 96 deep well plate with printed labeling (VWR D108839, 1.2 mL). The plated was sealed by aluminum foil sealing film and was ultrasounded for 1h, then stand overnight under RT. After the partial evaporation of the solvent, the product was collected by filtration and washed by ether (AcroPrep 96 filter plate from PALL (0.2 um GHP, 1 mL well) was used, 96 deep well plate was used as the receiver). For the second library equal conditions were used, but performed in 4 mL vials.
Characterization of N-(4-Chlorobenzyl)-N-(2-(cyclohexylamino)-2-oxoethyl)-1H-indole-2-carboxamide (B1)
White solid, 20.6 mg. Yield: 24%. HPLC/MS: tR = 11.61 min; m/z = 424.2 [M + H]+. HRMS: C24H26N3O2ClNa, 446.1611 (calcd.), 446.1634 (found). 1H NMR (600 MHz, d6-DMSO): 1.18–1.27 (m, 5H), 1.53–1.74 (m, 4H), 3.58 (m, 1H), 3.94 (m, 1H), 4.18 (m, 1H), 4.66 (m, 1H), 5.00 (m, 1H), 6.53 (m, 1H), 6.75 (m, 1H), 7.03 (m, 1H), 7.18 (m, 1H), 7.37–7.56 (m, 4H), 7.84 (m, 1H), 8.05 (m, 1H), 11.70 (m, 1H). 13C NMR (150 MHz, d6-DMSO): 24.4, 25.1, 32.2, 47.5, 49.7, 51.2, 54.9, 103.9, 111.99, 112.04, 119.8, 121.4, 123.5, 126.8, 128.3, 129.8, 131.8, 135.9, 136.3, 163.8, 166.9.
Acknowledgments
This work was supported by the NIH grant P41 GM094055-02 and 1R01GM097082-01 (to AD), European Lead Factory (IMI under grant agreement no. 115489, to AD) and by a Marie Curie FP7-Reintegration-Grants within the seventh European Community Framework Programme and by a Project operated within the Foundation for Polish Science TEAM Programme, cofinanced by the EU European Regional Development Fund (to T.A.H.).
Supporting Information Available
Methods, summary of compounds, and characterization of selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): A.D. is shareholder in the biotech company Carmolex, Inc.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cheok C. F.; Verma C. S.; Baselga J.; Lane D. P. Translating p53 into the clinic. Nat. Rev. Clin Oncol 2011, 8, 568–568. [DOI] [PubMed] [Google Scholar]
- Khoury K.; Popowicz G. M.; Holak T. A.; Dömling A. The p53-MDM2/MDMX axis —A chemotype perspective. MedChemComm 2011, 2, 246–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popowicz G. M.; Dömling A.; Holak T. A. The structure-based design of Mdm2/Mdmx–p53 inhibitors gets serious. Angew. Chem., Int. Ed. 2011, 50, 2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carry J. C.; Garcia-Echeverria C. Inhibitors of the p53/hdm2 protein–protein interaction-path to the clinic. Bioorg. Med. Chem. Lett. 2013, 23, 2480–5. [DOI] [PubMed] [Google Scholar]
- Popowicz G. M.; Czarna A.; Rothweiler U.; Szwagierczak A.; Krajewski M.; Weber L.; Holak T. A. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle 2007, 6, 2386–2392. [DOI] [PubMed] [Google Scholar]
- Berghausen J.; Buschmann N.; Furet P.; Gessier F.; Hergovich L. J.; Holzer P.; Jacoby E.; Kallen J.; Masuya K.; Pissot S. C.; Ren H.; Stutz S.. Isoquinolinone and quinazolinone derivatives as MDM2 and MDM4 inhibitors and their preparation and use for the treatment of diseases. Int. Patent WO2011076786A1.
- Bold G.; Furet P.; Gessier F.; Kallen J.; Hergovich L. J.; Masuya K.; Vaupel A.. Preparation of tetrasubstituted heteroaryl compounds and their use as MDM2 and/or MDM4 modulators. Int. Patent WO2011023677A1.
- Reed D.; Shen Y.; Shelat A. A.; Arnold L. A.; Ferreira A. M.; Zhu F.; Mills N.; Smithson D. C.; Regni C. A.; Bashford D.; Cicero S. A.; Schulman B. A.; Jochemsen A. G.; Guy R. K.; Dyer M. A. Identification and characterization of the first small molecule inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. H.; Zhang Q.; Jo S.; Chai S. C.; Oh M.; Im W.; Lu H.; Lim H.-S. Novel pyrrolopyrimidine-based α-helix mimetics: Cell-permeable inhibitors of protein–protein interactions. J. Am. Chem. Soc. 2010, 133, 676–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine J.-C. W.; Dyer M. A.; Jochemsen A. G. MDMX: From bench to bedside. J. Cell Sci. 2007, 120, 371–378. [DOI] [PubMed] [Google Scholar]
- Srivastava S.; Beck B.; Wang W.; Czarna A.; Holak T. A.; Dömling A. Rapid and efficient hydrophilicity tuning of p53/mdm2 antagonists. J. Comb. Chem. 2009, 11, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popowicz G. M.; Czarna A.; Wolf S.; Wang K.; Wang W.; Dömling A.; Holak T. A. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wolf S.; Bista M.; Meireles L.; Camacho C.; Holak T. A.; Dömling A. 1,4-Thienodiazepine-2,5-diones via MCR (I): Synthesis, virtual space and p53-Mdm2 activity. Chem. Biol. Drug Des. 2010, 76, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarna A.; Beck B.; Srivastava S.; Popowicz G. M.; Wolf S.; Huang Y.; Bista M.; Holak T. A.; Dömling A. Robust generation of lead compounds for protein–protein interactions by computational and MCR chemistry: p53/Hdm2 antagonists. Angew. Chem., Int. Ed. 2010, 49, 5352–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista M.; Kowalska K.; Janczyk W.; Dömling A.; Holak T. A. Robust NMR screening for lead compounds using tryptophan-containing proteins. J. Am. Chem. Soc. 2009, 131, 7500–7501. [DOI] [PubMed] [Google Scholar]
- Rothweiler U.; Czarna A.; Krajewski M.; Ciombor J.; Kalinski C.; Khazak V.; Ross G.; Skobeleva N.; Weber L.; Holak T. A. Isoquinolin-1-one inhibitors of the MDM2–p53 interaction. ChemMedChem. 2008, 3, 1118–1128. [DOI] [PubMed] [Google Scholar]
- Popowicz G.; Czarna A.; Holak T. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle 2008, 7, 2441–2443. [DOI] [PubMed] [Google Scholar]
- Rothweiler U.; Czarna A.; Weber L.; Popowicz G. M.; Brongel K.; Kowalska K.; Orth M.; Stemmann O.; Holak T. A. NMR screening for lead compounds using tryptophan-mutated proteins. J. Med. Chem. 2008, 51, 5035–5042. [DOI] [PubMed] [Google Scholar]
- Czarna A.; Popowicz G. M.; Pecak A.; Wolf S.; Dubin G.; Holak T. A. High affinity interaction of the p53 peptide-analogue with human Mdm2 and Mdmx. Cell Cycle 2009, 8, 1176–1184. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Wolf S.; Koes D.; Popowicz G. M.; Camacho C. J.; Holak T. A.; Dömling A. Exhaustive fluorine scanning toward potent p53–Mdm2 antagonists. ChemMedChem. 2012, 7, 49–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koes D.; Khoury K.; Huang Y.; Wang W.; Bista M.; Popowicz G. M.; Wolf S.; Holak T. A.; Dömling A.; Camacho C. J. Enabling large-scale design, synthesis and validation of small molecule protein–protein antagonists. PLoS One 2012, 73e32839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Wolf S.; Beck B.; Köhler L. M.; Khoury K.; Popowicz G. M.; Goda S. K.; Subklewe M.; Twarda A.; Holak T. A.; Dömling A. Discovery of highly potent p53-MDM2 antagonists and structural basis for anti-acute myeloid leukemia activities. Chem. Biol. 2014, 9, 802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista M.; Wolf S.; Khoury K.; Kowalska K.; Huang Y.; Wrona E.; Arciniega M.; Popowicz G. M.; Holak T. A.; Dömling A. Transient protein states in designing inhibitors of the MDM2–p53 interaction. Structure 2014, 21, 2143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H.; Liu H.; Dömling A. Efficient multicomponent reaction synthesis of the schistosomiasis drug praziquantel. Chem.—Eur. J. 2010, 16, 12296–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.