

Abstract
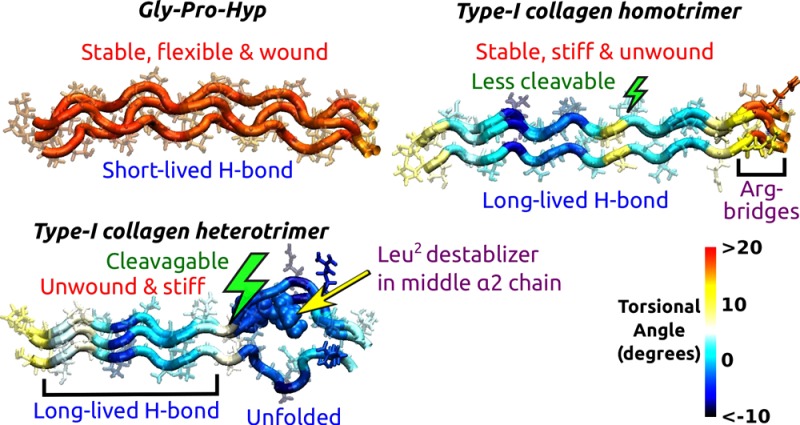
Degradation of fibrillar collagen is critical for tissue maintenance. Yet, understanding collagen catabolism has been challenging partly due to a lack of atomistic picture for its load-dependent conformational dynamics, as both mechanical load and local unfolding of collagen affect its cleavage by matrix metalloproteinase (MMP). We use molecular dynamics simulation to find the most cleavage-prone arrangement of α chains in a collagen triple helix and find amino acids that modulate stability of the MMP cleavage domain depending on the chain registry within the molecule. The native-like state is mechanically inhomogeneous, where the cleavage site interfaces a stiff region and a locally unfolded and flexible region along the molecule. In contrast, a triple helix made of the stable glycine-proline-hydroxyproline motif is uniformly flexible and is dynamically stabilized by short-lived, low-occupancy hydrogen bonds. These results provide an atomistic basis for the mechanics, conformation, and stability of collagen that affect catabolism.
Introduction
Collagens possess distinct properties as the main building blocks of the extracellular matrix. They assemble hierarchically into near-macroscopic order, making up both soft and hard tissues.1,2 Of 28 known types, fibrillar collagens, including types I, II, and III, are dominant.3 To achieve structural diversity and larger-scale compliance while maintaining precise local order within the extracellular matrix, fibrillar collagens adopt residue-specific interactions1 as well as other less specific interactions such as water-mediated force that also exists in other biopolymers.4−6 Such balance between crystallinity and disorder2 likely applies to other types of collagens as well. The “order-and-disorder” features are based on the domain organization within a single collagen molecule, which consists of the stable imino-rich (Pro or Hyp) domains for which the representative structural motif is GPO (Gly-Pro-Hyp; O is the single-letter code for hydroxyproline), and “labile” domains where X and Y in the GXY triplet are not imino acids.7 The imino-rich domain is thermally stable due to the constraint on the backbone dihedral angle imposed by the imino rings, which prefers the polyproline type II conformation of the α chain in a collagen triple helix.8−10 Hydroxyproline in the Y position of the GXY motif provides further stabilization due to a stereoelectronic effect that favors the α chain backbone dihedral angles in the triple helical conformation11,12 and also via possible water-mediated hydrogen bond (H-bond) formation.8,10,13,14 Labile domains are thought to be more loosely wound and flexible compared to the imino-rich domain.7,15,16 The collagen cleavage site hydrolyzed by MMP is located in a labile domain, about 3/4 along the length of the molecule.17,18 Since a well-folded collagen triple helix is highly resistant to protease cleavage,17,19 local unfolding of the labile domain is critical for cleavage by MMP.17,20,21 Our earlier study using molecular dynamics (MD) simulation showed that, in the case of an imino-poor domain of type III collagen, unwinding initiates at a typical cleavage bond (Gly-Ile) at temperatures as low as 300 K.16 Spontaneous unwinding of the labile domain is likely implicated in the instability of isolated type I collagen molecules at body temperature,22 and it may also contribute to recognition and additional disruption of the triple helix by MMPs.20,21
Apart from the general picture for the collagen molecule as a whole, far less is known regarding the mechanism by which the individual α chains forming a collagen triple helix affect the conformational behavior. This is especially important for type I collagen, a heterotrimer comprised of two α1 chains and one α2 chain. Herein, we call the α1 and α2 chains simply as α1 and α2, respectively. Compared to the native heterotrimer, a homotrimer comprised of three α1 is more stable,23 assembles less efficiently,24,25 and is more resistant to MMP cleavage.23 The α1 homotrimer is found in fetal tissues, fibrotic tissues, and carcinomas23,26 and is implicated in osteogenesis imperfecta.27 Comparing the primary structure, α1 has a net charge of +11e (e = 1.6 × 10–19 C) and 64 large nonpolar residues (Ile, Leu, Met, Phe, Tyr, and Val). α2 has +31e and 106 large nonpolar residues (for comparison, sequences for the triple helix part in the Uniprot P02452 for α1 and P08123 for α2 were used). More nonpolar residues in α2 would mean greater hydrophobic attraction, promoting assembly, whereas a higher net charge may keep the molecule hydrated, thus, it may allow axial sliding of collagen molecules in a bundle that is crucial for proper ordering.5,6 On the other hand, α2 has a smaller number of imino acids, which supports its destabilizing role. However, beyond the sequence-level information, structural mechanisms for different α chains in modulating the stability and conformation of a collagen triple helix are unclear. Since the three α chains in a collagen triple helix are staggered by one residue,8 three isomers of type I collagen are possible depending on whether α2 is in the leading (the most N-terminal side), middle, or trailing position. While a modest resolution (5.16 Å) X-ray fiber diffraction structure of rat tail tendon suggests that α2 is in the middle,28 a systematic study of the dependence of the conformational properties on chain registry is lacking. A related issue is the load-dependent cleavage of collagen by MMP. It is generally accepted that collagen fibrils under tensile load are more resistant to cleavage.29−36 However, single-molecule experiments yielded conflicting results, with cleavage rate either decreased37 or increased by as much as 100-fold.38 While one suggestion was the difference in the behavior of hetero- versus homotrimers used in the two experiments,39 it has subsequently been shown that the cleavage rate increases in both cases.40 One of the difficulties in studying collagen is its long length (∼300 nm) that is organized into different domains for numerous ligand bindings and signalings.18 Model collagen mimetic peptides (also called triple helical peptides) have thus been instrumental for analyzing behaviors of specific subdomains or chain registry.41−46 They also have potential for biomedical applications.47,48
Here we use MD simulations of various collagen mimetic peptides containing the MMP cleavage domain of type I collagen to systematically analyze its properties. We find that chain registry plays a critical role for the stability and flexibility of the triple helix. A heterotrimer with α2 in the leading position behaves similar to the stable α1 homotrimer, despite the general destabilizing role of α2. The interchain H-bond formed by the arginine side chain, together with clustering of nonpolar residues, is a major determinant for the registry dependence, in agreement with experiment.43 The heterotrimer with α2 in the middle is mechanically the most labile at and downstream to the MMP cleavage site, suggesting that this isomer may be the most prone to cleavage. The imino-rich domain upstream to the MMP cleavage site is unwound but is stiff, supported by long-lived H-bonds. The MMP cleavage domain is thus characterized by a rapid transition in stiffness and stability. In contrast, the backbone H-bond occupancy and lifetime for the stable GPO peptide is much smaller. The rapidly forming H-bonds allow the GPO peptide to remain flexible while maintaining the triple helical structure. We also find that the conformational behavior and mechanical response of the triple helix depend sensitively on how loads are applied to the ends of the molecule. The loading-condition dependence addresses recent debates about whether mechanical load increases38,40 or decreases37 the MMP cleavage rate of a monomer. Present results elucidate dynamic versus static mechanisms for stabilizing the collagen triple helix and their relation to mechanics. Furthermore, simple “rules of thumb” such as regarding α2 as generally destabilizing, or the stabilizing role of arginine, should be exercised with caution.
Computational Methods
Peptide Generation
We used 30-residue long α chains to build collagen-like peptides. Residues 7–24 have the corresponding sequence from the MMP cleavage domain of human type-I collagen (residues 766–783,49 with the cleavage site between 775 and 776; Table 1). Residues 1–6 and 25–30 are GPO triplets that stabilize the ends.15 For comparison, we also considered α chains made only of the GPO triplet. The mutant chains α1(R21O) and α2(O21R) had Arg21 of α1 and Hyp21 of α2 switched, to investigate the role of arginine for the triple helix stability. The five α chains in Table 1 were used to build the triple helices in Table 2. Backbones of the triple helical structures were built using the THeBuScr program50 and side chains were added by using CHARMM.51
Table 1. Sequences of α Chains Used49a.
| triad | 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| residue | 1 | 3 | 5 | 7 | 9 | 11 | 13 | 15 | 17 | 19 | 21 | 23 | 25 | 27 | 29 | |||||||||||||||
| α1 | G | P | O | G | P | O | G | A | O | G | T | P | G | P | Q | G | I | A | G | Q | R | G | V | V | G | P | O | G | P | O |
| α2 | G | P | O | G | P | O | G | P | O | G | T | O | G | P | Q | G | L | L | G | A | O | G | I | L | G | P | O | G | P | O |
| gpo | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O | G | P | O |
| α1(R21O) | G | P | O | G | P | O | G | A | O | G | T | P | G | P | Q | G | I | A | G | Q | O | G | V | V | G | P | O | G | P | O |
| α2(O21R) | G | P | O | G | P | O | G | P | O | G | T | O | G | P | Q | G | L | L | G | A | R | G | I | L | G | P | O | G | P | O |
Table 2. Composition and Chain Registry of Triple Helices Used in This Study.
| name | leading | middle | trailing |
|---|---|---|---|
| huco1 | α2 | α1 | α1 |
| huco2 | α1 | α2 | α1 |
| huco3 | α1 | α1 | α2 |
| homo | α1 | α1 | α1 |
| homo2 | α2 | α2 | α2 |
| gpo10 | gpo | gpo | gpo |
| homom | α1(R21O) | α1(R21O) | α1(R21O) |
| homo2m | α2(O21R) | α2(O21R) | α2(O21R) |
| huco1m | α2(O21R) | α1(R21O) | α1(R21O) |
Basic Simulation Procedure
For simulation, we used the CHARMM program51 with the param27 all-atom force field53 and additional parameters for Hyp.54 Before solvation, a brief energy minimization (2000 steps) was carried out in the generalized Born with a simple switching (GBSW) implicit solvent model of CHARMM.55 The peptide was solvated in an orthorhombic box of about 135 × 55 × 55 Å3. The box size was chosen so that there is at least 20-Å gap between the molecule and the boundary of the box, which is larger than the 12-Å nonbond interaction cutoff. Ions were added to neutralize the system, at approximately 150 mM NaCl and 10 mM MgCl2.26 The solvated system was energy minimized again by 1600 steps. Simulation proceeded by heating from 0 to 300 K for 30 ps followed by equilibration at 300 K for 170 ps. Production runs were either 8 or 24 ns, with most measurements done during the last 12 ns of the 24 ns runs. Coordinates were saved every 5 ps. The total simulation time was over 2.5 μs.
Triad-Based Description of the Triple Helix Conformation
We used local coordinate bases {e1, e2, e3} (triads) to describe torsional and bending motion of the molecule along its length.16 Triads were assigned based on adjacent backbone carbonyl C atoms from each α chain. We chose C since its radial position from the axis of the initial straight triple helix varies less compared to Cα or N atoms, so that the resulting triads are more uniformly aligned. To eliminate end effects, we only considered the region spanning residue 6 to 25 on the leading chain. Due to chain staggering, C atoms of residue 6, 5, and 4 from the leading, middle, and trailing chains, respectively, constitute triad 1 and so on. The three C atoms for each triad make a triangle, whose centroid is the origin of the triad and the unit vector normal to the triangle and pointing to the C-terminus is set as e3. The unit vector pointing from the centroid to the midpoint of the C atoms of the leading and middle chains is e1, which fixes e2 = e3 × e1. Local torsional angle was measured as the Euler angle between two successive triads relative to e3.16
Calculation of Mechanical Properties
Force–Extension Relationship
To control extension of the molecule, harmonic potentials were applied to the Cα atoms of G4 and G28 in the leading chain at a given distance (Supporting Information, Figure S1). By restraining only one α chain, rotation or unwinding of the triple helix is allowed. In some cases, we restrained ends of all three α chains to study the effect of the loading condition on the conformational behavior of the triple helix. To avoid large abrupt changes in extension, we gradually changed it. Starting with 72 Å (the distance between the restrained atoms in the initially built triple helix), the extension was either increased or decreased in 4 Å intervals with 100 ps equilibration for each, covering 60–84 Å. At each extension, the production run was 8 ns (Figure 1a, open symbols). In the physiologically more relevant region (see Results), we carried out another set of 24 ns simulations in 0.8 Å intervals (Figure 1a,b, solid symbols).
Figure 1.
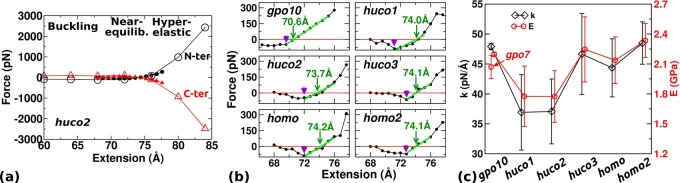
Extensional behavior. (a) Overview of the force–extension relation with huco2 as an example (see Table 2 for peptide names). Open/solid symbol: 8 ns/24 ns simulation in 4 Å/0.8 Å steps. The last half of each simulation period was used to calculate force. N-ter/C-ter: Axial forces exerted by the restrained Cα atoms at 4th/28th residues of the leading chain. Transverse components of the force are very small (less than 10 pN), isotropic, and are independent of extension. (b) Force–extension relations from 24 ns simulations as solid symbols in (a). Sign of the C-terminal force is reversed and averaged with the N-terminal force. Arrowhead: extension below which buckling occurs. Arrow: Leq where local linear fit to the near-equilibrium regime (thick green line) crosses the zero-force point. (c) Extensional stiffness k (diamond) and Young’s modulus E (circle). Young’s modulus of gpo7 is shown in the gpo10 column.
The force exerted by the molecule at a given extension was calculated by using the tug-of-war sampling method.56 Briefly, if we denote the i-th Cartesian component of the deviation of the restrained atom from the center of the potential during the simulation by δri (i = 1, 2, 3; Supporting Information, Figure S1), the i-th component of the force Fi exerted by the restrained atom at the center of the potential is given by
| 1 |
where ⟨·⟩ denotes an average over coordinate frames, kB is the Boltzmann constant, and T (=300 K) is temperature. The harmonic potential had a spring constant 10 kcal/(mol·Å2). This choice does not affect the measured force since eq 1 is independent of the spring constant,56 which we confirmed by performing simulations using 5 kcal/(mol·Å2) (Supporting Information, Figure S2). For simulations with all three chains restrained, the spring constant was reduced by 1/3.
Bending Stiffness
Local bending stiffness was measured by analyzing the fluctuation in the polar angle of e3 between two triads. Let the deviation of this angle from its average during the simulation by δθ, and the distance between two triads by s. The bending stiffness κf between these triads is given by57
| 2 |
For eq 2, we carried out simulations without any positional restraints. To prevent self-interaction through the periodic boundary by the freely diffusing peptide, we used a larger cubic box of side length 125 Å. For each peptide, we carried out a 24 ns simulation and used the last 12 ns for calculation.
Results
Peptide Design
Among the peptides tested (Table 2), gpo10 serves as a stable control. huco1, huco2, and huco3 are the three isomers of the human type I collagen cleavage domain. The homo is an α1 homotrimer where the corresponding full-length molecule is known to be stable and resists cleavage, homo2 is an α2 homotrimer that is expected to be less stable, and homom, homo2m, and huco1m are designed to test the role of Arg21 on α1 (Table 1).
Extensional Behavior
Our initial force–extension curve based on 8 ns simulation in 4 Å steps possesses approximately three regimes of behavior: buckling, near-equilibrium, and hyper-elastic (open symbols in Figure 1a). In the buckling regime, the molecule takes a bent conformation. The near-equilibrium regime is around the region where the force is close to zero. In the hyper-elastic regime, force increases sharply, beyond physiologically relevant levels (see Discussion). Based on the initial characterization, we carried out 24 ns simulations to obtain refined force–extension curves surrounding the near-equilibrium regime in 0.8-Å steps. They cover 66.4–76.0 Å for gpo10 and 68.0–77.6 Å for other peptides. We used the last 12 ns of these simulations for calculating forces (Figure 1b). Compared with 8 ns simulations, forces decrease in magnitude in small and large extensions due to conformational relaxation. Taking huco2 as an example, in 8 ns simulation the force in the buckling regime is nonzero (open symbols in Figure 1a), while it decreases to zero in 24 ns simulation (Figure 1b). In the compressed state, there is an extension below which the force does not increase in magnitude (arrowheads in Figure 1b). Below this extension, conformational change occurs, such as breakage of existing hydrogen bonds and/or formation of new contacts. Only gpo10 maintains a nonzero force (Figure 1b) as it remains stably wound even in the buckling regime (explained below).
In the near-equilibrium regime, the point where the force–extension curve crosses the zero-force point defines the equilibrium length Leq (Figure 1b, arrows). Except for gpo10 (Leq = 70.6 Å), it is similar among other peptides (∼74 Å) with huco2 being the shortest (73.7 Å). Compared to the initial canonical triple helix (72 Å), gpo10 wound more tightly thus became shorter, whereas others containing the labile domain became longer due to unwinding. In the hyperelastic regime, in addition to conformational relaxation, more extensive unfolding can occur. For example, the reduced force of huco1 at the largest extension (Figure 1b, 77.6 Å) is due to splaying of one of α chains on its C-terminal end at 16.1 ns.
For gpo10, the force–extension curve is fairly linear in the near-equilibrium regime. Other peptides show less linear behavior (Figure 1b). Nevertheless, it is informative to measure the extensional stiffness k and Young’s modulus E to compare with previous estimates. Linear fit to the force–extension curve around Leq gives k (Figure 1c, diamond). Using r = 7.0 Å as the radius of a hydrated collagen molecule,16 Young’s modulus is given by58E = k(Leq/πr2) (Figure 1c, circle). While k depends on the system size, E is a material property. The calculated E (1.77–2.34 GPa) lies on the lower end of previous experiments59,60 and simulations,61,62 2.4–9 GPa. The large variation in previous works is due to different experimental methods used and choices for the radius r. The largest value, 9.0 GPa was obtained using inelastic light scattering, which the authors suggested to be an overestimate.59 Ref (60) used X-ray diffraction and obtained E = 2.9 GPa. They used r = 6.15 Å. If we use this radius, our estimate becomes 2.3–3.0 GPa, which agrees well with their result. In simulations, steered molecular dynamics (pulling the molecule with a constant speed) is frequently used.62,63 In this case, E tends to be overestimated due to the lack of conformational relaxation and E increases with the pulling speed.63 Relaxation can be seen in our simulation by measuring k in 4 ns intervals, which generally decreases with time before 8 ns (Supporting Information, Figure S3). If we use the 4–8 ns period, the calculated E indeed increases to 2.2–2.5 GPa. To test independence of E on the length of the peptide, we carried out another set of 24 ns simulations using 7 GPO repeats, gpo7, whose E is comparable to that for gpo10 (Figure 1c).
Among the peptides tested, huco1 and huco2 possess the smallest k, which is also seen in calculations over 4 ns intervals (Supporting Information, Figure S3). Since MMP locally unwinds or deforms collagen for cleavage,20,64huco1 or huco2 may possess the native registry of α chains among the three type I collagen isomers, which we examine further below.
RMSD
At each extension, we calculated the root-mean-square deviation (RMSD) of the positions of backbone heavy atoms in the triad region from those at the beginning of simulation (Supporting Information, Figure S4). In most cases, RMSD increases during the first few nanoseconds and stays fluctuating after about 6 ns, which partly supports making measurements during 12–24 ns (Supporting Information, Figure S4a). Additional analysis of time scale is in Discussion. As expected, RMSD generally decreases with extension (Supporting Information, Figure S4b), although the trend is not strictly monotonic. Comparing different peptides, RMSD of gpo10 is the smallest, reflecting its stability.
Local Bending Stiffness
In simulations for measuring local bending stiffness, no restraint was applied. During 12–24 ns, distances between the 4th and 28th Cα atoms of the leading chains were 70.9 ± 0.9 Å (gpo10; average ± standard deviation), 74.4 ± 0.6 (huco1), 74.4 ± 2.3 (huco2), 72.6 ± 1.0 (huco3), 74.0 ± 0.8 (homo), and 72.7 ± 1.0 (homo2), which are comparable to Leq in Figure 1b. To use eq 2, the interval s between two triads needs to be chosen. If it is too short (e.g., between two immediately neighboring triads), κf may reflect properties of atomic-level covalent bonds rather than representing a local average for the peptide as a filament. On the other hand, if s is too long, fluctuations of all atoms within this interval will contribute to the measurement, so that the meaning of κf as a local property will be unclear. Due to the staggering of chains, MMP cleavage bonds (boldface in Table 1) occur over three triads. We thus used triad i and i + 3 (i = 1–17) for calculating s. For each pair of triads, we took s as an average over 12–24 ns and used it for calculating κf. Averaged over all triads in each peptide, s follows the same trend as the average end-to-end distance, which is the shortest for gpo10 (8.94 ± 0.02 Å) and the longest for huco1 (9.49 ± 0.16 Å).
For gpo10, κf is nearly constant (34900 ± 2600 pN·Å2; Figure 2a). In other peptides, κf in the imino-rich domain (triad 1–7) is overall higher. This is because this region unwinds to make the three α chains rather parallel and suppresses bending motion (cf, Figure 3). Among peptides other than gpo10, huco2, and huco3 have the two lowest κf values in the cleavage domain (Figure 2c,d). As discussed above, taking compliance of the cleavage domain as an attribute utilized by MMP, huco2, and huco3 may be better choices than huco1 with regard to bending. Combined with the results for the extensional stiffness, huco2 is mechanically the most compliant in both extension and bending, thus, it may be the best candidate for MMP binding and cleavage. As explained below, this is due to the arrangement of residues in huco2 that destabilizes the labile domain and leads to unfolding (row Free in Figure 3c).
Figure 2.
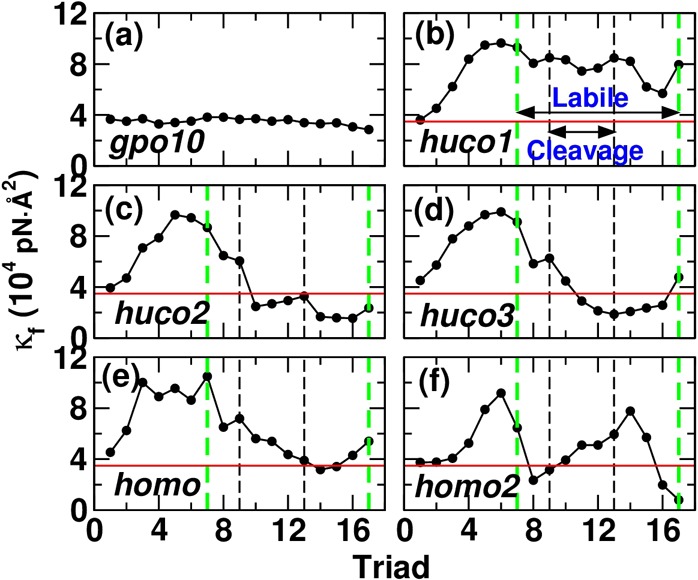
Local bending stiffness κf: (a) gpo10, (b) huco1, (c) huco2, (d) huco3, (e) homo, and (f) homo2 (see Table 2 for peptide names). Horizontal line (red) is the average κf for gpo10 (3.49 × 104 pN·Å2), as a guide. While there are 20 triads (Table 1), since triads i and i + 3 are used to calculate κf, the last data point ends at triad 17. Likewise, the MMP cleavage bond appears across triads 9–13. In triad 11, all three α chains contain the cleavage bond. The cleavage and the imino-poor labile (triads 7–17) domains are marked by vertical lines (noted in b).
Figure 3.

Torsional angles between successive triads averaged over 12–24 ns, displayed on conformations at the end of each simulation. Two GPO triplets at each end of the peptide are not shown. (a) gpo10, (b) huco1, (c) huco2, (d) huco3, (e) homo, (f) homo2, and (g) huco2 with ends of all three α chains restrained (see Table 2 for peptide names). Buckling, Near-eq , and Hyper-ela are representative structures from respective regimes, where the extensions are 66.4, 70.4, and 76.0 Å for gpo10, 68.0, 73.6, and 77.6 Å for huco2, and 68.0, 74.4, and 77.6 Å for all other peptides. These are based on differences in Leq (Figure 1b). Free is for simulation without any restraint. The same extensional regimes are used in Figures 4 and 5. For the torsional angle measured between triad i and i + 1, residues of triad i are colored (marked triad 1–19 in (a)). Free: simulation without any restraint. Ile17 in α1 and Leu17 in α2 at the cleavage site (Table 1) are shown in van der Waals representation to show their location (marked in (b)). Molecular structures are rendered using VMD.67
When κf is calculated in 4 ns intervals, gpo10 shows no time dependence (Supporting Information, Figure S5). In other peptides, κf varies over time to different degrees, reflecting their conformational motion. Yet, huco2 and huco3 are still more flexible in the cleavage domain than huco1 and homo. To compare our measurement with experiment, we calculated the persistence length lp = (κf/kBT). It ranges between 84.2 (gpo10) to 181 nm (huco1), which lie well within the experimental estimates, 14.5–180 nm.65 For our estimation, κf in each peptide was averaged over triads. For a full-length collagen, the apparent lp may be dictated by highly flexible, locally unfolded regions such as the cleavage domain of huco2, which may be smaller. A recent study using atomic force microscopy reports 12–40 nm.66
Torsional Behavior
Twist of a triple helix is an important descriptor of collagen conformation,15,16,68 which may also be functionally important as it affects binding of MMP and cleavage of collagen.16,17,20,21 In simulations where the ends of only the leading chain are restrained, torsional angle decreased with extension, indicative of unwinding (Figure 3a–f). Consistent with its stability, gpo10 unwound the least (Figure 3a). In other peptides, the region around the MMP cleavage site underwent the greatest unwinding (darker color in Figure 3b–f). In the buckling regime, kinking of huco1, huco2, and huco3 was observed at the cleavage site. These results further corroborate its labile nature. In simulations without any restraint, cleavage domains of huco2 and huco3 disrupt compared to that of huco1 (row Free in Figure 3b–d). The extent of disruption is the greatest in huco2, which supports it as the most cleavable isomer. The imino-rich domain upstream to the cleavage site unwinds, likely due to Ala8 in α1 and Thr11 in both α1 and α2 (Table 1). However, further unfolding of this region does not occur and the three α chains stay aligned, resulting in elevated bending stiffness (left of the cleavage site in Figure 3b–f).
Dependence on Loading Condition
We restrained the ends of only one α chain when studying the extensional behavior, which allowed conformational (especially torsional) motion under load. To test the effect of disallowing torsional motion of the end, for huco2, we applied restraints to three Cα atoms of residues 4, 3, and 2, respectively, from the leading, middle, and trailing chain, and likewise restrained residues 28, 27, and 26. In this case, the extensional stiffness was k = 186.4 ± 6.6 pN/Å (Supporting Information, Figure S6), which is about 5× greater than the case with only one α chain restrained. The corresponding Young’s modulus, 8.75 ± 0.31 GPa, is comparable to the maximum among previous estimates.59 Furthermore, the triple helix unwinds far less, with much reduced dependence on extension (Figure 3g). These results highlight the sensitivity of the conformational behavior on the loading condition. Its implication for MMP cleavage is considered in Discussion.
Hydrogen Bonding Events
H-bonds are critical for the stability of the collagen triple helix.1,2,69 We classified them into “native” and “non-native”. Native H-bonds are formed between backbone amide hydrogen of glycine in a GXY triplet to the backbone carbonyl oxygen of the residue at the X position of a neighboring α chain.1 They are thus formed in a helical manner, between leading-middle, middle-trailing, and trailing-leading chains. Since atoms forming native H-bonds are present in any GXY sequence, native H-bonds can form in both imino-rich and imino-poor domains.15 Non-native H-bonds refer to all others, including those between backbone to backbone, backbone to side-chain, and side-chain to side-chain. For identification of a H-bond, a cutoff distance of 2.4 Å between hydrogen and oxygen atoms was used.70 H-bonding events were quantified by occupancy (number of coordinate frames where a H-bond is formed, divided by the total number of frames), average lifetime (average duration of consecutive frames where a H-bond is formed), and standard deviation of the lifetime. The H-bond occupancy and lifetime can together provide a dynamic picture of the H-bonding events. For example, two bonds may have the same occupancy but differ in lifetimes, as one bond may rapidly form and break, while the other may be longer-lived but forms more sparsely. The converse may also hold, with similar lifetimes but different occupancies depending on the frequency of H-bond formation.
Strikingly, the stable gpo10 has the lowest native H-bond occupancy compared to those of other peptides (Figure 4a). Its average native H-bond lifetime and fluctuation (standard deviation) are also the shortest (Figure 4b and Supporting Information, Figure S7a). This suggests that the native H-bonds of gpo10 stabilize the structure dynamically, by rapid formation and breakage in a uniform manner. The native H-bond occupancy of gpo10 becomes uneven along its length in the buckling and hyper-elastic regimes as strain builds up in the structure. In the hyper-elastic regime, the native H-bond occupancy overall increases, which is also observed in other peptides (Figure 4a). An exception is huco2 with all three α chains restrained (“huco2 (three)” in Figure 4a), where the native H-bond occupancy is lower in the hyper-elastic compared to the near-equilibrium regime. In this case, the peptide becomes more wound with extension (Figure 3g), becoming conformationally closer to gpo10 whose native H-bond occupancy is low. These results indicate that unwinding of the triple helix actually promotes native H-bond formation. Consistent with this, triads 5–10 that are upstream to the MMP cleavage site, have elevated occupancy and longer lifetime (Figure 4). As explained earlier, this region unwinds without α chains falling apart (Figure 3). The higher occupancy of native H-bonds in this region likely contributes to its larger bending stiffness (Figure 2).
Figure 4.

Dynamics of native backbone H-bonds (see Table 2 for peptide names, and Figure 3 for Buckling, Near-eq , Hyper-ela, and Free). (a) Occupancy and (b) average lifetime. Standard deviations of lifetimes are in Supporting Information, Figure S7a. Measured values with glycines as H-bond donors are averaged for each triad and represented in color. Triads 11–14 contain cleavage sites (solid box). The imino-poor domain spans triad 10–20. Compared to other peptides, H-bonds of gpo10 have notably smaller occupancy, lifetime, and standard deviation (Supporting Information, Figure S7a), suggesting a dynamic stabilization mechanism.
Non-native H-bonds show more punctate behavior (Figure 5). The well-folded gpo10 has very few non-native H-bonds. This is also the case in other peptides upstream to the MMP cleavage domain (triads 5–10) that are unwound without falling apart. Non-native H-bonds occur downstream to the cleavage site (triads 15–20), as this region is more disrupted (Figure 3). In particular, triads 17–18 of homo have high-occupancy non-native H-bonds in all extensional regimes and also in the restraint-free case (Figure 5). They mainly involve a H-bond between Arg21 of α1 (Table 1) and the backbone oxygen atom in a neighboring chain (Figure 6a). We call it the Arg-bridge. Although several other very short-lived non-native H-bonds in these triads caused the average lifetime below 50 ps, the Arg-bridge can persist beyond 100 ps, so it can play a substantial role in local stabilization.
Figure 5.

Dynamics of non-native H-bonds (see Table 2 for peptide names, and Figure 3 for Buckling, Near-eq, Hyper-ela, and Free). (a) Occupancy and (b) average lifetime. Standard deviations of lifetimes are in Supporting Information, Figure S7b. For each triad, H-bonds are counted only when residues in the triad serve as H-bond donor, to avoid double counting across different triads. Triads 11–14 contain cleavage sites (solid box). High-occupancy bonds in triads 17–18 in homo (also in huco1) are due to Arg-bridges (Figure 6).
Figure 6.

Role of the Arg-bridge and chain registry on the conformation of the imino-poor domain (see Table 2 for peptide names). Structures are taken after 24 ns MD without any restraint. (a) homo: Arg-bridges are marked by arrows. Arg21 in the leading chain does not form a bridge. (b) huco1: Leu17 and Leu18 of the leading α2 are held by Ile17 in middle and Gln15 in trailing chains. (c) huco2: Leu18 inserts between α chains and the trailing chain separates. Arg-bridges are absent. (d) huco3: Leu17 and Leu18 of the trailing α2 are held by residues in the leading chain and by Gln15 of α2. The longitudinal compaction causes the middle chain to bend severely. (a) is rendered larger than (b–d).
Molecular Origin of the Dependence on Chain Registry
In addition to the Arg-bridge, we found that two bulky nonpolar residues Leu17 and Leu18 in α2, located right next to the cleavage bond (Table 1), play a critical role in determining registry-dependent conformational behavior. In huco1, since α2 is in the leading position, Arg21 of α1, being farther downstream, can form a bridge, while Leu17 and Leu18 interact with surrounding residues (Figure 6b). In huco2, placement of α2 in the middle separates Arg21 in two α1, resulting in the greatest destabilization (Figure 6c). In huco3, since the two leucines of α2 are close to Arg21, their hydrophobic stabilization requires local deformation of the molecule and interferes with Arg-bridge formation, which again have a destabilizing effect, but to a less extent compared to huco2 (Figure 6d).
To test the stabilizing role of the Arg-bridge, we constructed models of three mutant peptides, huco1m, homom, and homo2m, where Hyp21 and Arg21 in respective chains are switched (Table 2). For each peptide, we carried out 24 ns MD simulation without any restraint applied, whereas triads 16–19 in huco1 and homo remained wound (Figure 3b,e; “Free”), this region in huco1m and homom unwound, with very low occupancy of non-native H-bond (Figure 7a,b). The homo2m behaves oppositely, where triads 16–19 are wound more compared to homo2 and have high-occupancy non-native H-bonds (Figure 7c), mainly Arg-bridges. These results corroborate the importance of the Arg-bridge, which may contribute to the stability and cleavage resistance of the type-I collagen homotrimer.23,26
Figure 7.
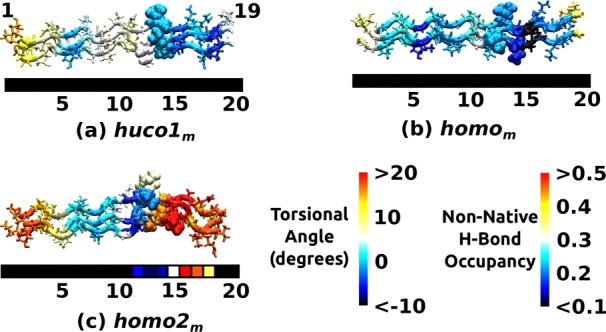
Conformational behavior of mutant peptides in Table 2. (a) huco1m, (b) homom, (c) homo2m. Structures are taken after 24 ns MD without any restraint. Coloring schemes are the same as in Figure 3b,e,f (torsional map) and Figure 5a (non-native H-bond occupancy). Since the molecular structure is 3-dimensional, its triads do not align exactly with the triad numbers of the color strip below. Without the Arg-bridge, triads 16–19 of huco1m and homom undergo unwinding. Conversely, homo2m stays wound due to the presence of the Arg-bridge that manifests as a high-occupancy non-native H-bond.
Discussion
Present results elucidate mechanical and conformational differences between homo- vs heterotrimers of collagen, or between isomers with different registry of α chains. Although our calculation shows that huco1 and huco2 (see Table 2 for peptide names) have the two lowest extensional stiffness, variation in extensional stiffness among peptides tested (37–49 pN/Å) is well within 2-fold (Figure 1c). By comparison, the local bending stiffness κf varies by as much as 5-fold (Figure 2). It is thus a more sensitive measure of local conformational properties. For gpo10 that is uniform in flexibility, we can calculate Young’s modulus using bending stiffness, E = κf/I, where I = (π/4)r4 (r = 7 Å) is the second moment of inertia of cross section for a circular cylinder of radius r.58 Using the average κf for gpo10, 3.49 × 104 pN·Å2, we get E = 1.85 GPa, which is comparable to the value based on extensional stiffness (Figure 1c). This reflects consistency of our measurements in simulations with and without restraints.
Among the three extensional regimes, the near-equilibrium regime is likely the most physiological. In tissues, collagen bundles form macroscopic crimps71 so that molecular-level buckling is unlikely to happen under compression. On the extensional side, we can estimate a typical tensile load by considering tendon. The cross sectional area of a human tendon is on the order of cm2, and it bears ∼kN forces. Assuming that the entire cross section of a tendon consists of collagen molecules 7 Å in radius, there are about 6.5 × 1013 collagen molecules, so that each molecule will bear about 15 pN. Thus, up to 100 pN in Figure 1b will be physiological, which lies within the near-equilibrium regime.
In addition to force, we examine the relevant time scale. Lifetimes of contacts are at most a few hundred picoseconds, majority of which being less than 100 ps (Figures 4 and 5). Thus, the 12 ns measurement time during the later half of 24 ns simulation was sufficient for monitoring the dynamics of contacts, We also observed transient formation of β-sheets, consistent with experiment.72 They are mostly short, formed by two backbone H-bonds between two α chains in parallel and are rarer than individual contacts. Additional analysis would be necessary to elucidate the role of transient β-sheets in conformational dynamics of the molecule.
Even though individual bonds form and break rapidly, the overall conformational fluctuation of the peptide may be slower. The RMSD undulates on the order of a few nanoseconds (Supporting Information, Figure S4). We estimate the slowest relaxation time of the peptide as an elastic rod suspended in a viscous medium.58 For a rod of length L, diameter d, and bending stiffness κf, its slowest relaxation time τ in a solution of viscosity η is given by τ = (c⊥/κf)(L/ω1)4, where c⊥ = 4πη/[ln(L/d) + 0.84] is the transverse drag coefficient per unit length of the rod, and ω1 is a constant of order 1 for the slowest vibrational mode, which depends on the boundary condition of motion. For gpo10, we have L = 87.5 Å, d = 14 Å, and κf = 3.49 × 104 pN·Å2 (Figure 2). This gives τ ≃ 140–330 ps, which is comparable to the longest H-bond lifetimes. This shows that the 24 ns simulation time was much longer than the equilibrium fluctuation time of the peptide. However, large deviations from the triple helical conformation can occur over a longer time scale, such as formation and breakage of β-sheets mentioned above, and water molecules can break in and out between α chains in the locally unfolded labile domain. A more detailed analysis of such events would require at least an order of magnitude longer simulation, which would be impractical. Nevertheless, the 24 ns simulation time employed in the present study was sufficient for distinguishing between the relative stability and region-specific conformational behavior of the triple helical peptides, which is further supported by the agreement of our calculations with available experimental data. On the other hand, although biasing potentials, for example, in umbrella sampling, may further drive conformational changes,73 unless reaction coordinates are properly chosen, it is difficult to interpret the observed changes.74
A fundamental aspect that we revealed is the dynamic stabilization mechanism for the GPO repeat: gpo10 has low-occupancy, short-lived native backbone H-bonds (Figure 4), which is an effective strategy to maintain the triple helical structure while remaining flexible (Figure 2a). This is reminiscent of the stabilization of single α-helical domains by dynamic and “malleable” contacts between appropriately located charged side chains that hold the “brittle” α-helix backbone.75−77 Analogously, in the case of the collagen GPO domain, dynamic backbone H-bonds hold the triple helix tertiary structure.
For the MMP cleavage domain, there are stronger regional variations in flexibility and stability. We found that huco2 is the isomer that is likely the most cleavage-prone, as it has the highest mechanical compliance and the greatest unfolding of the region at and downstream the cleavage site. Instability of huco2 in the labile domain is due to the axial separation of Arg21 in the leading and trailing α1, together with the two tandem leucines of α2 that locally destabilize the region and hamper the Arg-bridge formation. Although configurations in Figure 6 may not be the only way how these residues organize locally, they illustrate the unfavorable arrangement of arginines and leucines in huco2 compared to other isomers. The stabilizing role of the Arg-bridge has been shown experimentally in a model heterotrimer,43 and similar roles for lysine and glutamic acid were also suggested for type IV collagen.42 However, as Figure 6 shows, placement of these residues within the molecule affects the extent of stabilization.
The high stiffness of the imino-rich domain N-terminal to the cleavage site (Figure 2) is a result of unwinding without separation of α chains (Figure 3) that appears to promote native backbone H-bond formation (Figure 4). While there are many sites along the collagen molecule whose amino acid sequences are partially similar to the actual MMP cleavage site, the latter is distinguished by local imino-rich GXY repeats followed by an imino-poor domain.17,49 This suggests that the abrupt transition in local bending stiffness may be unique to the MMP cleavage site, thus it may provide a mechanical recognition signal as MMP diffuses over collagen and searches for the cleavage site.78
The present analysis also makes a testable prediction: huco1, possessing the Arg-bridge (Figures 5 and 6b) behaved similarly to homo. While the presence of α2 may make huco1 not as cleavage-resistant as homo, compared to huco2 or huco3, it is likely to be so and also be more stable, which will be an interesting subject for experiments. The possible stabilizing role of arginine in type-III collagen has been previously proposed,79 although the atomistic basis was unclear. Our analysis shows that Arg-bridges are dynamic, whose strength depends on their location relative to other residues (Figure 6).
Last, we discuss the conflicting reports of the cleavage rate of single collagen molecules, that either increased38,40 or decreased37 with load in similar magnetic tweezer experiments. In the former case, a homotrimeric peptide containing the MMP cleavage domain38 (similar to homo) or a full-length type-I collagen heterotrimer40 (corresponding to huco2) was linked between a glass coverslip and a magnetic bead. Since the bead can rotate, unwinding of the molecule is possible regardless of the number of α chains in a molecule linked to the bead or to the coverslip. As in our simulation (Figure 3), stretching will result in more unwinding, which may assist with cleavage by MMP. In comparing between homotrimer and heterotrimer, cleavage of the former was more sensitive to load, which was interpreted to be due to its higher propensity to unwind under load while the heterotrimer is unwound even without load.40 Our simulation supports this, as homo unwound substantially in the hyper-elastic regime compared to near-equilibrium or load-free case (Figure 3e), whereas huco2 is already unwound (near-equilibrium) or unfolded (load-free; Figure 3c). It should be noted that unwinding can either stabilize or destabilize the triple helix, depending on whether the domain is imino-rich or imino-poor, as seen in our analysis of bending stiffness and H-bonds.
In another study, cleavage rate of collagen decreased by nearly 10-fold with load.37 In this case, antibody-functionalized beads were exposed to a large volume of type-I collagens to achieve conjugation. This may result in multiple collagen molecules attaching to a single bead. Even though a single collagen tether may be formed between the bead and the surface, neighboring collagen molecules can bind to the tethered collagen, affecting its conformational motion. Likewise, in tissues, other neighboring molecules in a bundle may limit conformational motion of the cleavage domain under load, thereby protect it from cleavage. While presence of many other factors makes it difficult to directly apply analysis of a single triple helix to a tissue, our study demonstrates that susceptibility to MMP cleavage depends sensitively on the loading condition and the local arrangement of molecules, and not simply on the magnitude of load. Experimentally, when studying load-dependent cleavage of collagen by MMP, it would thus be necessary to probe or control the mechanical environment around collagen molecules in limiting or promoting local unfolding.
Conclusions
Fibrillar collagen is the major load-bearing component of the tissue, so that continuum mechanical description of a collagen molecule as a biopolymer is needed. On the other hand, its biological function involves residue-specific behaviors, as in any globular proteins. Our study elucidates the atomistic origin for the mechanical and conformational properties of the MMP cleavage domain of type I collagen. Fundamental aspects that we found, such as the local conformational behavior of the triple helix under load, static versus dynamic origin for the flexibility and stability, and the effect of chain registry, will also be useful for understanding the behaviors of other domains or other types of fibrillar collagens.
Acknowledgments
We acknowledge the Texas A&M Supercomputing Facility and the Texas Advanced Computing Center (TACC) at University of Texas at Austin for providing computing resources. This work was supported in part by the U.S. NIH Grant R01GM087677.
Supporting Information Available
Figure S1, Illustration of tug-of-war sampling for force–extension relation; Figure S2, Testing force–extension relation with two different spring constants of the sampling potential; Figure S3, Extensional stiffness versus time; Figure S4, Analysis of RMSD; Figure S5, Bending stiffness vs time; Figure S6, Force–extension relation for the case when all three chains are restrained; Figure S7, Standard deviation of H-bond lifetimes. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Brodsky B.; Persikov A. V. Adv. Protein Chem. 2005, 70, 301–339. [DOI] [PubMed] [Google Scholar]
- Wess T. J. Adv. Protein Chem. 2005, 70, 341–374. [DOI] [PubMed] [Google Scholar]
- Ricard-Blum S. Cold Spring Harbor Perspect. Biol. 2011, 3, a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikin S.; Parsegian V. A.; Rau D. C.; Rand R. P. Annu. Rev. Phys. Chem. 1993, 44, 369–395. [DOI] [PubMed] [Google Scholar]
- Leikin S.; Rau D. C.; Parsegian V. A. Nat. Struct. Biol. 1995, 2, 205–210. [DOI] [PubMed] [Google Scholar]
- Ravikumar K. M.; Hwang W. J. Am. Chem. Soc. 2011, 133, 11766–11773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles C. A.; Bailey A. J. Micron 2001, 32, 325–332. [DOI] [PubMed] [Google Scholar]
- Bella J.; Eaton M.; Brodsky B.; Berman H. M. Science 1994, 266, 75–81. [DOI] [PubMed] [Google Scholar]
- Kramer R. Z.; Vitagliano L.; Bella J.; Berisio R.; Mazzarella L.; Brodsky B.; Zagari A.; Berman H. M. J. Mol. Biol. 1998, 280, 623–638. [DOI] [PubMed] [Google Scholar]
- Sundar R. S.; Gopalakrishnan R.; Wade R. C.; Subramanian V. J. Phys. Chem. B 2011, 115, 2593–2607. [DOI] [PubMed] [Google Scholar]
- Shoulders M. D.; Raines R. T. Annu. Rev. Biochem. 2009, 78, 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Satyshur K. A.; Forest K. T.; Raines R. T. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran G. N.; Bansal M.; Bhatnagar R. S. Biochim. Biophys. Acta 1973, 322, 166–171. [DOI] [PubMed] [Google Scholar]
- Nishi Y.; Uchiyama S.; Doi M.; Nishiuchi Y.; Nakazawa T.; Ohkubo T.; Kobayashi Y. Biochemistry 2005, 44, 6034–6042. [DOI] [PubMed] [Google Scholar]
- Kramer R. Z.; Bella J.; Mayville P.; Brodsky B.; Berman H. M. Nat. Struct. Biol. 1999, 6, 454–457. [DOI] [PubMed] [Google Scholar]
- Ravikumar K. M.; Hwang W. Proteins: Struct., Funct., Bioinf. 2008, 72, 1320–1332. [DOI] [PubMed] [Google Scholar]
- Fields G. B. J. Theor. Biol. 1991, 153, 585–602. [DOI] [PubMed] [Google Scholar]
- Sweeney S. M.; Orgel J. P. R. O.; Fertala A.; McAuliffe J. D.; Turner K. R.; Di Lullo G. A.; Chen S.; Antipova O.; Perumal S.; Ala-Kokko L.; Forlino A.; Cabral W. A.; Barnes A. M.; Marini J. C.; Antonio J. D. S. J. Biol. Chem. 2008, 283, 21187–21197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; McBride D. J.; Losert W.; Leikin S. J. Mol. Biol. 2008, 383, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L.; Dinakarpandian D.; Yoshida N.; Lauer-Fields J. L.; Fields G. B.; Visse R.; Nagase H. EMBO J. 2004, 23, 3020–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manka S. W.; Carafoli F.; Visse R.; Bihan D.; Raynal N.; Farndale R. W.; Murphy G.; Enghild J. J.; Hohenester E.; Nagase H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leikina E.; Mertts M. V.; Kuznetsova N.; Leikin S. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 1314–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makareeva E.; Han S.; Vera J. C.; Sackett D. L.; Holmbeck K.; Phillips C. L.; Visse R.; Nagase H.; Leikin S. Cancer Res. 2010, 70, 4366–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride D. J. Jr.; Kadler K. E.; Hojima Y.; Prockop D. J. Matrix 1992, 12, 256–263. [DOI] [PubMed] [Google Scholar]
- McBride D. J. Jr.; Choe V.; Shapiro J. R.; Brodsky B. J. Mol. Biol. 1997, 170, 275–284. [DOI] [PubMed] [Google Scholar]
- Han S.; Makareeva E.; Kuznetsova N. V.; DeRidder A. M.; Sutter M. B.; Losert W.; Phillips C. L.; Visse R.; Nagase H.; Leikin S. J. Cell Biol. 2010, 285, 22276–22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipman S. D.; Sweet H. O.; McBride D. J. Jr.; Davisson M. T.; Marks S. C. Jr.; Shuldiner A. R.; Wenstrup R. J.; Rowe D. W.; Shapiro J. R. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 1701–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel J. P. R. O.; Irving T. C.; Miller A.; Wess T. J. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 9001–9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.; Yannas I. V. J. Biomed. Mater. Res. 1977, 11, 137–154. [DOI] [PubMed] [Google Scholar]
- Nabeshima Y.; Grood E. S.; Sakurai A.; Herman J. H. J. Orthop. Res. 1996, 14, 123–130. [DOI] [PubMed] [Google Scholar]
- Ruberti J. W.; Hallab N. J. Biochem. Biophys. Res. Commun. 2005, 336, 483–489. [DOI] [PubMed] [Google Scholar]
- Lotz J. C.; Hadi T.; Bratton C.; Reiser K. M.; Hsieh A. H. Eur. Spine J. 2008, 17, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karla E. K. W.; Bourne J. W.; Torzilli P. A. J. Biomech. Eng. 2009, 131, 051004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhole A. P.; Flynn B. P.; Liles M.; Saeidi N.; Dimarzio C. A.; Ruberti J. W. Philos. Trans. R. Soc., A 2009, 367, 3339–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn B. P.; Bhole A. P.; Saeidi N.; Liles M.; DiMarzio C. A.; Ruberti J. W. PLoS One 2010, 5, e12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadi M. F.; Sander E. A.; Ruberti J. W.; Barocas V. H. Mech. Mater. 2012, 44, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camp R. J.; Liles M.; Beale J.; Saeidi N.; Flynn B.; Moore E.; Murthy S. K.; Ruberti J. W. J. Am. Chem. Soc. 2011, 133, 4073–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari A. S.; Chai J.; Dunn A. R. J. Am. Chem. Soc. 2011, 133, 1686–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S.-W.; Flynn B. P.; Ruberti J. W.; Buehler M. J. Biomaterials 2012, 33, 3852–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari A. S.; Glassey E.; Dunn A. R. J. Am. Chem. Soc. 2012, 134, 13259–13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer R. Z.; Venugopal M. G.; Bella J.; Mayville P.; Brodsky B.; Berman H. M. J. Mol. Biol. 2000, 301, 1191–1205. [DOI] [PubMed] [Google Scholar]
- Saccà B.; Renner C.; Moroder L. J. Mol. Biol. 2002, 324, 309–318. [DOI] [PubMed] [Google Scholar]
- Russell L. E.; Fallas J. A.; Hartgerink J. D. J. Am. Chem. Soc. 2010, 132, 3242–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Mo X.; Kim D.; Yu S. M. Biopolymers 2011, 95, 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan A. A.; Hartgerink J. D. Curr. Opin. Chem. Biol. 2013, 17, 960–967. [DOI] [PubMed] [Google Scholar]
- Fields G. B. J. Biol. Chem. 2013, 288, 8785–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S. M.; Li Y.; Kim D. Soft Matter 2011, 7, 7927–7938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary L. E. R.; Fallas J. A.; Bakota E. L.; Kang M. K.; Hartgerink J. D. Nat. Chem. 2011, 3, 821–828. [DOI] [PubMed] [Google Scholar]
- Xiao J.; Addabbo R. M.; Lauer J. L.; Fields G. B.; Baum J. J. Biol. Chem. 2010, 285, 34181–34190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey J. K.; Goh M. C. Bioinformatics 2004, 20, 2458–2459. [DOI] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr.; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S.; Caisch A.; Caves L.; Cui Q.; Dinner A. R.; Feig M.; Fischer S.; Gao J.; Hodoscek M.; Im W.; Kuczera K.; Lazaridis T.; Ma J.; Ovchinnikov V.; Paci E.; Pastor R. W.; Post C. B.; Pu J. Z.; Schaefer M.; Tidor B.; Venable R. M.; Woodcock H. L.; Wu X.; Yang W.; York D. M.; Karplus M. J. Comput. Chem. 2009, 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese S.; Jung M. C.; Ihling C.; Heinz A.; Neubert R. H. H.; Schmelzer C. E. H. Biochim. Biophys. Acta 2010, 1804, 731–739. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D. Jr.; Bashford D.; Bellott M.; Dunbrack R. L. Jr.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E. III; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Anderson D.Ph.D. Thesis, University of Toronto, Toronto, Canada, 2005. [Google Scholar]
- Im W.; Lee M. S.; Brooks C. L. J. Comput. Chem. 2003, 24, 1691–1702. [DOI] [PubMed] [Google Scholar]
- Hwang W. J. Chem. Phys. 2007, 127, 175104. [DOI] [PubMed] [Google Scholar]
- Lakkaraju S. K.; Hwang W. Cell. Mol. Bioeng. 2009, 2, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J.Mechanics of Motor Proteins and the Cytoskeleton; Sinauer Associates: Sunderland, MA, 2001. [Google Scholar]
- Harley R.; James D.; Miller A. Nature 1977, 267, 285–287. [DOI] [PubMed] [Google Scholar]
- Sasaki N.; Odajima S. J. Biomech. 1996, 29, 655–658. [DOI] [PubMed] [Google Scholar]
- Vesentini S.; Fitié C. F.; Montevecchi F. M.; Redaelli A. Biomech. Model. Mechanobiol. 2005, 3, 224–234. [DOI] [PubMed] [Google Scholar]
- Lorenzo A. C.; Caffarena E. R. J. Biomech. 2005, 38, 1527–1533. [DOI] [PubMed] [Google Scholar]
- Gautieri A.; Buehler M. J.; Redaelli A. J. Mech. Behav. Biomed. Mater. 2009, 2, 130–137. [DOI] [PubMed] [Google Scholar]
- Bertini I.; Fragai M.; Luchinat C.; Melikian M.; Toccafondi M.; Lauer J. L.; Fields G. B. J. Am. Chem. Soc. 2011, 134, 2100–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumara L.; Agarwal G. Biomaterials 2010, 31, 4802–4808. [DOI] [PubMed] [Google Scholar]
- Lovelady H. H.; Shashidhara S.; Matthews W. G. Biopolymers 2014, 101, 329–335. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Okuyama K.; Miyama K.; Mizuno K.; Bächinger H. P. Biopolymers 2012, 97, 607–616. [DOI] [PubMed] [Google Scholar]
- Rich A.; Crick F. H. C. J. Mol. Biol. 1961, 3, 483–506. [DOI] [PubMed] [Google Scholar]
- Loof H. D.; Nilsson L.; Rigler R. J. Am. Chem. Soc. 1992, 114, 4028–4035. [Google Scholar]
- Fratzl P.; Misof K.; Zizak I.; Rapp G.; Amenitsch H.; Bernstorff S. J. Struct. Biol. 1997, 122, 119–122. [DOI] [PubMed] [Google Scholar]
- Gullekson C.; Lucas L.; Hewitt K.; Kreplak L. Biophys. J. 2011, 100, 1837–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerenberg P. S.; Stultz C. M. J. Mol. Biol. 2008, 382, 246–256. [DOI] [PubMed] [Google Scholar]
- Bartels C.; Karplus M. J. Comput. Chem. 1997, 18, 1450–1462. [Google Scholar]
- Sivaramakrishnan S.; Spink B. J.; Sim A. Y.; Doniach S.; Spudich J. A. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13356–13361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommese R. F.; Sivaramakrishnan S.; Baldwin R. L.; Spudich J. A. Protein Sci. 2010, 19, 2001–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich J. A.; Sivaramakrishnan S. Nat. Rev. Mol. Cell Biol. 2010, 11, 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar S. K.; Marmer B.; Goldberg G.; Neuman K. C. Curr. Biol. 2012, 22, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salsas-Escat R.; Stultz C. M. Proteins: Struct., Funct., Bioinf. 2010, 78, 325–335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.