

Abstract
Silkworm silk has been widely used as a textile fiber, as biomaterials and in optically functional materials due to its extraordinary properties. The β-sheet-rich natural nanofiber units of about 10–50 nm in diameter are often considered the origin of these properties, yet it remains unclear how silk self-assembles into these hierarchical structures. A new system composed of β-sheet-rich silk nanofibers about 10–20 nm in diameter is reported here, where these nanofibers formed into “flowing hydrogels” at 0.5–2% solutions and could be transformed back into the solution state at lower concentrations, even with a high β-sheet content. This is in contrast with other silk processed materials, where significant β-sheet content negates reversibility between solution and solid states. These fibers are formed by regulating the self-assembly process of silk in aqueous solution, which changes the distribution of negative charges while still supporting β-sheet formation in the structures. Mechanistically, there appears to be a shift toward negative charges along the outside of the silk nanofibers in our present study, resulting in a higher zeta potential (above −50 mV) than previous silk materials which tend to be below −30 mV. The higher negative charge on silk nanofibers resulted in electrostatic repulsion strong enough to negate further assembly of the nanofibers. Changing silk concentration changed the balance between hydrophobic interactions and electrostatic repulsion of β-sheet-rich silk nanofibers, resulting in reversible hydrogel–solution transitions. Furthermore, the silk nanofibers could be disassembled into shorter fibers and even nanoparticles upon ultrasonic treatment following the transition from hydrogel to solution due to the increased dispersion of hydrophobic smaller particles, without the loss of β-sheet content, and with retention of the ability to transition between hydrogel and solution states through reversion to longer nanofibers during self-assembly. These reversible solution-hydrogel transitions were tunable with ultrasonic intensity, time, or temperature.
1. Introduction
Silks spun by silkworms and spiders represent some of the strongest and toughest biological materials.1−3 It is generally believed that the outstanding mechanical properties of silks are due to its sophisticated hierarchical structures involving amorphous and crystalline protein phases and their interplay, which are controlled by a combination of the chemistry and the spinning process.4−6 Computational experiments have shown that the geometric confinement of silk fibrils to diameters of 50 ± 30 nm is critical to enable the utilization of weak hydrogen bonds and simple material building blocks to achieve outstanding mechanical properties.7,8 Yet the recapitulation of native silk fibroin spinning solution and formation of fiber properties in vitro from reconstituted silk solutions remains a challenge,9,10 in part because the self-assembly process at the nanoscale remains unclear.
From an engineering perspective, silk has been utilized in various technological fields including tissue regeneration, drug release, optical components, and electronic applications.11−14 Beyond the recapitulation of native fiber properties in vitro, the effective control of silk nanostructures, mechanical properties, degradation behavior and surface properties offers further tuning and applications. It is evident that understanding and controlling the self-assembly process of silk is pivotal for the design and preparation of functional silk-based materials.15,16 Several mechanisms for silk processing and self-assembly have been proposed in order to gain insight into the formation of the structural hierarchy of silk.5,17,18 Recently, an improved mechanism to control the formation of silk nanofibrous structures implied that the self-assembly of silk in aqueous solution was a thermodynamically driven process where kinetics also play a key role.19 Four factors including molecular mobility, charge, hydrophilic interactions, and concentration underlie the process, suggesting the possibility to control nanostructure formation in solution. Although several critical developments in self-assembly mechanisms have been achieved in recent years,20−23 there remain few studies of regulating the structural hierarchy of silk toward the design of novel functional materials using such a bottom-up approach.
In our recent study, silk nanofibers with different sizes and secondary conformations have been prepared in aqueous solution.24 However, most of the nanofibers were metastable and easily changed their nanostructures and secondary conformations spontaneously, making it difficult to study their properties and possible applications. Therefore, in the present study, stable silk nanofibers in aqueous solution were prepared through further controlling the self-assembly of silk fibroin. Unlike prior approaches, the silk nanofibers with high β-sheet content in the present study could still be dispersed in aqueous solution at low concentrations (below 0.5 wt %) and transformed into hydrogels with increasing concentration. The transition between solution and hydrogel was reversible, and could be further regulated through changed the nanostructures of silk fibroin under controllable process, which is first reported to the best of our knowledge. Films and porous scaffolds derived from the nanofibers showed distinct properties associated with charge distribution, implying the feasibility to design functional silk-based materials with this bottom-up approach.
2. Experimental Section
2.1. Preparation of Aqueous Silk Fibroin Solutions
Production of silk fibroin solution was previously described.25Bombyx mori cocoons were boiled for 20 min in an aqueous solution of 0.02 M Na2CO3, and then rinsed thoroughly with distilled water to extract the sericin proteins. The extracted silk was dissolved in 9.3 M LiBr solution (Sigma-Aldrich, St. Louis, MO) at 60 °C, yielding a 20% (w/v) solution. This solution was dialyzed against distilled water, using Slide-a-Lyzer dialysis cassettes (Pierce, MWCO 3500) for 72 h to remove the salt. The solution was optically clear after dialysis and was centrifuged at 9000 rpm for 20 min at 4 °C to remove silk aggregates formed during the process. The final concentration of aqueous silk solution was about 6 wt %, determined by weighing the remaining solid after drying.
2.2. Silk Nanofiber Formation
To prepare silk nanofibers, fresh silk fibroin solution was treated by a concentration-dilution process.26 The solution (6 wt %) was slowly concentrated to about 20 wt % over 24 h at 60 °C to form metastable nanoparticles, and then diluted to below 2 wt % with distilled water. The diluted silk solution was incubated for about 24 h at 60 °C to induce the nanofiber formation.
2.3. Ultrasonic Treatment
A SL-650D Sonifier (Shunliu Instrument Co., Nanjing, China), which consisted of the (Model 650D) with output power supply 650 W, was used to sonicate the silk solutions.27 The silk concentration was varied from 0.5 wt % to 2 wt %, and ultrasonic intensity was varied from 100 to 500 W for 10 min to generate silk nanofibers with different lengths. Then the sonicated solutions were incubated at different temperatures to monitor the hydrogel–solution transition process.
2.4. Electrogelation of Silk Fibroin Solutions
Electrodes were immersed in an aqueous solution of silk fibroin, and 25 VDC was applied over a 3 min period to a pair of conductive electrodes.28 Within seconds of the application of the voltage, a visible gel formed at the positive electrode and is termed e-gel.
2.5. Ions-Induced Hydrogel–Solution Transition
To determine the effect of ions on the hydrogel–solution transition process, an equal weight of a 50 mM CaCl2 aqueous solution was added dropwise into the silk nanofiber solution without stirring. Gelation occurred rapidly upon the addition of the metal salt solution.
2.6. Preparation of Solid State Materials from Silk Nanofibers
To prepare silk nanofiber films, 1.5 mL of silk nanofiber hydrogel (2 wt %) was cast on a polystyrene Petri dish (diameter 30 mm) and then dried to form film at room temperature. Silk nanofiber scaffolds were prepared through a lyophilization process.26 Three milliliters of silk nanofiber hydrogel (2 wt %) was poured into a cylindrically shaped container (diameter 15 mm), frozen at −20 °C for 12 h, and then lyophilized for 48 h.
2.7. Zeta Potential
Surface charges of silk solutions were determined via zeta potential measurement.24 One milliliter of the solution was loaded to a Zetasizer (Nano ZS, Malvern, Worcesteshire, UK) for the zeta potential measurement at 25 °C.
2.8. Circular Dichroism (CD)
The secondary structures of the silk hydrogels were measured with a Jasco-815 CD spectrophotometer (Jasco Co., Japan).24 CD spectra were recorded from 250 to 190 nm wavelengths with an accumulation of five scans at a scanning rate of 100 nm min–1 at 25 °C. The results were averaged from three repeated experiments.
2.9. Dynamic Oscillatory Rheology
Rheological studies were run on a Rheometer (AR2000, TA Instruments, New Castle, USA) fitted with a 20 mm cone plate (Ti, 20/1°).28 Prior to each experimental day, the rheometer underwent a torque map with a 10 Pa s calibration oil. Frequency sweeps were collected continuously over a wide frequency range from 100 to 1 rad s–1 at 25 °C. All samples were stabilized for 20 min before the measurement.
2.10. Fourier Transform Infrared (FTIR) Spectroscopy
The secondary structures of the various samples in solid state were analyzed by FTIR on a Nicolet FTIR 5700 spectrometer (Thermo Scientific, FL, USA).29 For each measurement, 64 scans were coded with a resolution of 4 cm–1, with the wavenumber ranging from 400 to 4000 cm–1.
2.11. X-ray Diffraction (XRD)
Crystal structure of samples was determined by XRD.29 The experiments were conducted with an X-ray diffractometer (X’Pert-Pro MPD, PANalytical, Almelo, Holland) with Cu Kα radiation at 40 kV and 30 mA and scanning rate of 0.6° min–1. Before examination, the dried samples were pressed into sheets with a hydraulic compressor.
2.12. Scanning Electron Microscopy (SEM)
The morphology of samples was observed using an SEM (S-4800, Hitachi, Tokyo, Japan) at 3 kV to avoid the destruction of silk structure.29 Before SEM examination, the dried samples were coated with platinum.
2.13. Atomic Force Microscopy (AFM)
For AFM experiments, silk solutions were diluted to below 0.1 wt % to avoid masking the original morphology by multilayers of silk.29 A total of 2 μL of the diluted silk solution was dropped onto freshly cleaved 4 × 4 mm2 mica surfaces. The morphology of silk fibroin was observed by AFM (Nanoscope V, Veeco, NY, USA) in air. A 225 μm long silicon cantilever with a spring constant of 3 N m–1 was used in tapping mode at 0.5–1 Hz scan rate.
3. Results and Discussion
Previous studies implied that nanofiber formation and nanoscale confinement of β-sheet nanocrystals of silk had fundamental roles in achieving stiffness, resilience, and fracture toughness at the microscale.4,19,30−33 Silk nanofibers with different sizes and secondary conformations have been prepared in aqueous solution by regulating the self-assembly process of silk fibroin in order to provide structural units for further assembly.24,34 However, unlike silk in vivo that can maintain a metastable state in the spinning dope,1,35,36 the nanostructures and secondary conformations prepared in vitro undergo changes easily in aqueous solution, making it difficult to design functional materials from nanofibril building blocks.
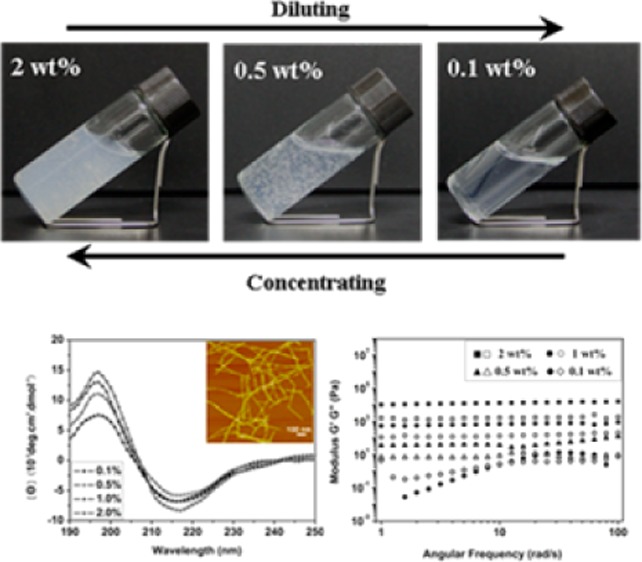
The basis for the current studies comes from recent observations where silk nanofibers could be formed from metastable silk nanoparticles by regulating concentration, incubation temperature and time. To prepare these materials, the key was to use freshly prepared silk solution (6 wt %) and utilize slow concentration processes to generate about 20% over 24 h at 60 °C to form the metastable nanoparticles (Figure S1, Supporting Information). These particles were then diluted to below 2 wt % with distilled water to induce disassembly. After further incubation for about 24 h at 60 °C, the diluted silk solution (0.5–2 wt %) transformed into hydrogel because of nanofiber formation with lengths of about 1 μm (Figure 1). Although silk fibroin in the system showed typical hydrogel viscoelastic behaviors (Figure 2), it still maintained flowing, making it difficult to define the state. Therefore, this feature is termed a “flowing hydrogel” in the present our study to distinguish with typical solution and hydrogel states. This observation prompted further investigation into the conditions and responses of the solution under different concentrations. Diluted fresh silk solution (0.5 wt %) without metastable nanoparticles maintained the solution state after incubation for more than 7 days at 60 °C, implying a critical effect of the preformation of the nanoparticles for the transformation to the hydrogel state. When the metastable nanoparticles were present in the silk solution, hydrogel formation depended on silk concentration, temperature, and time. Although silk solutions with different concentrations could transform into hydrogels at different temperatures if enough time was provided for the self-assembly process from random coil to silk II (β sheet) structure,37,38 the hydrogels composed of the aggregated silk nanoparticles did not exhibit flow (Figure S2). Concentrations from 0.5 wt % to 2 wt % and temperatures above 60 °C were conditions for the transformation of the solution to “flowing hydrogel”. Based on our previous study,19 silk II formation is a spontaneous process whose rate can be influenced by temperature, concentration, or original silk conformation. In the present conditions, disassembly from nanoparticle to nanofiber and conformational transitions from random coil to silk II have suitable rates since nanofibers formed when silk fibroin transformed into silk II structure.
Figure 1.

Hydrogel–solution reversible transformation of silk nanofibers in concentrating-diluting process (A), SEM images of silk in the reversible process (B), and CD spectra of silk fibroin in the reversible process (C). Silk keeps nanofiber structure and stable beta-sheet secondary conformation in the concentrating-diluting process.
Figure 2.

Storage modulus (G′, filled symbols) and loss modulus (G″, open symbols) versus frequency of different concentration of silk in the solution-hydrogel transition process (A); and storage modulus (G′, filled symbols) and loss modulus (G″, open symbols) versus frequency of silk gels with concentration of 2 wt % prepared through ultrasonication treatment (ultragel, hydrogels prepared by ultrasonicating fresh silk solution at the 20% amplitude setting for 30 s), electric field treatment (e-gel), and self-assembled nanofiber transition in our present study (nanofiber-gel) (B). The secondary structures of silk in the different gels were investigated with FTIR spectra (C) and XRD peak (D).
Remarkably, once the “flowing hydrogel” formed, this state was retained at different temperatures (from 4 to 90 °C) without a transition to solid hydrogel (Figure S3). Importantly, the “flowing hydrogel” formed returned to the solution state by dilution of the hydrogel to below 0.5%. The diluted solution was also stable without a transition to a hydrogel even after incubation at 90 °C for more than 1 month, very different behavior than fresh silk solution at the same concentration (Figure S3). The “flowing hydrogel” could be formed again if the diluted solution was concentrated to above 0.5%. The reversible system could be cycled many times. The solution and hydrogel states were also confirmed by dynamic oscillatory shear rheology (Figure 2).
CD curves of silk fibroin in the hydrogel and solution states indicated that β-sheet was the dominant secondary structure in both states (Figure 1). Lyophilization and air-drying were subsequently used to prepare porous matrices and films from the hydrogels and dilute solutions, respectively. The nanofiber structures in the hydrogel and solution states remained unchanged after the two processes and FTIR and XRD data indicated that β-sheet structure was maintained in these solid state samples (Figure S4).
Based on previous studies, β-sheet and nanofiber structures are critical to achieve water-insolubility and improve mechanical properties of silk materials.39−42 However, in the present study, silk fibroin nanofibers composed of β-sheet structures were maintained in solution at low concentrations (below 0.5%) and changed to “flowing hydrogels” at higher concentrations (0.5–2%). These hydrogels could be used to generate materials albeit with fragile mechanical properties in film and porous matrix states. These films and porous matrices redissolved in water, a very different behavior than previous studies (Figure S4).
The solution-hydrogel transitions following changes in concentration were confirmed by dynamic oscillatory shear rheology (Figure 2). The silk nanofiber solution at low concentration (0.1%) behaved as a viscoelastic fluid (sol), with loss modulus (G″) higher than storage modulus (G′). Both G″ and G′ increased with increased concentration. When the concentration was above 0.5%, G′ became larger than G″ and presented nearly frequency-independent behavior in the entire frequency range tested, which was a characteristic of solid (gel)-like behavior. Solution-hydrogel transitions could be maintained upon repeated dilution-concentration processes.
In order to clarify structure–mechanical property relationships, silk gels prepared by other methods were also studied. Dynamic shear strain sweeps of G′ collected from e-gels (hydrogel gels prepared under electric fields),28 ultrasonication-induced gels (hydrogels prepared by ultrasonicating fresh silk solution at the 20% amplitude setting for 30 s),27 and the new nanofiber-gels were all prepared at the same concentration (2 wt %). Previous studies indicated that the formation of β-sheet and nanofiber structures were responsible for enhanced mechanical properties of silk hydrogels, mainly because these features improved physical cross-links.43−46 Although the nanofiber gels and the ultrasonication-induced gels had similar β-sheet contents, significantly higher than that of e-gels (Figure 2), the storage moduli of the nanofiber-gels were similar to that of e-gels at high frequencies, but significantly lower than the ultrasonication-induced gels. Since the storage moduli of silk hydrogel depended on the physical cross-links of hydrophobic regions of silk, the results implied fewer physical cross-links between nanofibers in the new hydrogels. The hydrogel–solution transition following dilution also confirmed the formation of weaker physical cross-links between nanofibers, since these interactions were easily disrupted during the dilution process.
The goal was to understand why the nanofiber hydrogel system showed such unusual properties. In recent studies, critical repulsive effects of the negative charges on silk nanofibers was described.19,24 The possible repulsive action of these charges on physical cross-links of nanofibers was therefore considered important in terms the mechanisms related to nanofiber-hydrogel transitions. Silk fibroin molecules act as hydrophilic–hydrophobic–hydrophilic polymers, with the formation of micelles or aggregates in water in which hydrophilic domains may still be present inside the assemblies.5,19 The hydrophilic regions inside the assemblies could migrate to the surface of the assemblies to reduce the free energy of the silk–water system,19 resulting in the increase in negative charge on the surface of the assemblies. The process was confirmed by the increased zeta potential of the silk fibroin solution and hydrogel at different conditions.24 Therefore, the groups with negative charges distribute both inside and on the surface of the assemblies and can change their distribution following self-assembly.
The high zeta potential (−50 mV) with silk nanofiber hydrogels (0.5%) was maintained in diluted nanofiber solutions, a different outcome than observed with previous silk solutions and hydrogels (−10 to −25 mV).24,47,48 The formation of nanofibers about 10–20 nm in diameter and β-sheet structure both facilitated the distribution of negative charges outside the surface. This difference, which implied that more negative charges were distributed on the surface of nanofibers, resulted in higher repulsive forces between the nanofibers. Therefore, it is reasonable to hypothesize that the higher electrostatic repulsion due to the negative charges on the surface of the nanofibers, with the hydrophobic properties of the β-sheet-rich nanofibers, reached a balance to control the assembly behavior of silk fibroin. Since β-sheet-rich nanofibers provided enough hydrophobicity for hydrogel formation, while high electrostatic repulsion restrained physical cross-links, silk nanofiber solutions at higher concentrations (0.5–2%) represented typical viscoelasticity of hydrogels (Figure 2) but could still flow, unlike typical silk usual hydrogels. Also, the result was the formation of silk assemblies with high β-sheet content and negative charges, allowing the nanofibers to achieve viscoelastic properties and reversable hydrogel–solution features.
Reduced silk nanofiber aggregation was evident in AFM images where spaces were observed between nanofibers, failing to form compact layers in dried samples (Figure S5). Calcium chloride was added to the “flowing hydrogel” to neutralize the negative charges on the nanofibers, and, as a result, the hydrogel–solution transition following dilution disappeared (Figure S5). Therefore, because of the only limited aggregation of nanofibers, solid state materials (silk films and porous matrices) derived from the silk nanofibers were so fragile that they became powers under tiny pressure, making it impossible to prepare samples for mechanical test. This was in contrast to computational experiments in which silk nanofiber formation was predicted to result in improved mechanical properties.30−32,42 This difference implied the pivotal need to control assembly of the nanofibers to regulate the properties of silk-based materials. More importantly, since a significant increase of negative charges in silk hydrogels or solutions depended on β-sheet formation,23,24,49 it was concluded that maintaining amorphous states before spinning is a critical step in controlling assembly of silk to achieve outstanding mechanical properties.
Since nanostructures from silk fibroin are important in regulating hydrogel formation,31,38,48,50 it is possible to design other hydrogel–solution reversible systems. Ultrasonic treatment previously used to prepare silk hydrogels by inducing β-sheet formation was applied to change the nanostructures of silk fibroin,27,44 achieving a transition from hydrogel to solution. The hydrogel–solution transition rate was affected by ultrasonic intensity and silk concentration, where faster transitions occurred under higher ultrasonic intensity and lower silk concentrations. AFM and SEM indicated that the silk nanofibers ruptured under ultrasonic treatment, gradually shortening following increased ultrasonic intensity and became nanoparticles of several tens of nanometers in diameter when ultrasonic intensity was above 450 W (Figure 3). Since β-sheet structure and zeta potential of silk fibroin remained stable under ultrasonic treatment (Figure 3B and Table 1), the change in nanostructures was a primary reason for the hydrogel–solution transition. The repulsive force of negative charge restrained aggregation of silk fibroin with high β-sheet content, allowing the silk nanofibers flow in aqueous environments. The disassembly of silk nanofibers after ultrasonic treatment improved dispersion, resulting in the transition from hydrogel to solution state. By controlling the nanostructures with ultrasonic treatment silk fibroin with high beta-sheet content was maintained in the solution state until the concentration was above 2%, unlike previous studies.27,38 The concentration for the hydrogel–solution transition negatively correlated with the length of the silk nanofibers. For example, the solution-hydrogel transition point at 0.5% silk nanofiber with length of about 1 μm increased to 2% when the nanofibers were disassembled into nanoparticles tens of nanometers in size. The short nanofibers or particles formed after ultrasonic treatment reassembled to form longer nanofibers when incubated at different temperatures, accompanied by an inverse transition from solution to hydrogel (Figure S6). Different factors such as the length and concentration of silk nanofibers/particles in the solution state, as well as the incubation temperature, influenced the transition rate from solution to hydrogel, implying that the hydrogel formation time was controllable (Figure S6).
Figure 3.

(A) Reversible hydrogel–solution transition of silk nanofiber-based system and their nanostructure changes through ultrasonic-culturing treatment cycle. The samples were as follows: (a) untreated 2 wt % hydrogels or the recycled hydrogels after ultrasonic-culturing process after 7 days (similar nanofiber structures were achieved again after the cycle, so the typical image is shown in a); (b) hydrogels after ultrasonic treament for 10 min with the intensity of 100 W; (c) transformed solutions after ultrasonic treatment for 10 min with the intensity of 200 W; (d) transformed solutions after ultrasonic treatment for 10 min with the intensity of 500 W; (e) solutions after the sample in panel d was cultured at 60 °C for 1 day; (f) reversed hydrogels after the sample in panel d was cultured at 60 °C for 5 days. The cycle process could be repeated many times. (B) The secondary structures of silk in the reversible hydrogel–solution transition process (CD spectra, FTIR spectra, and XRD patterns). Both CD spectra derived from silk gel or solution, and FTIR and XRD spectra derived from freeze-dried samples showed that silk kept stable β-sheet structures in the reversible process. (C) Schematic models for the reversible hydrogel–solution transition of silk with high β-sheet content. The repulsive force derived from negative charge of silk could restrain further assembly of silk nanofibers with high β-sheet content, resulting in the flowing behaviors of hydrogels in higher concentrations and further the transformation from hydrogel to solution states when the solutions were diluted or the nanofibers were broken into shorter fiber/particles.
Table 1. Zeta Potential of Silk Hydrogels after Ultrasonic Treatment at Different Ultrasonic Intensities.
| hydrogels (no treatment) | 100 W | 200 W | 500 W | |
|---|---|---|---|---|
| zeta potential (mV) | –52.7 ± 1.76 | –52.2 ± 2.86 | –53.1 ± 1.76 | –53.8 ± 2.05 |
Many potential applications for reversible systems such as those reported here can be envisioned. For example, an injectable system for filling tissue voids may be useful, when considering the biocompatibility of silk-based biomaterials. Cells or drugs can be included in the ultrasonic-treated silk solutions and then transformed into nanofiber hydrogels for delivery. The reversing transition rate between hydrogel and solution states could be regulated by ultrasonic intensity, treatment time and silk concentration at specific temperature such as 37 °C, which would improve feasibility in biomedical applications. The dispersion in water would allow the use of silk fibroin nanofibers with high beta-sheet content to be useful as templates or nucleation sites for morphology and growth control of different nanomaterials. For example, silk nanofibers could be added into fresh silk solution to induce nanofiber growth and transition to stable silk II structures (Figure 4). The mechanical properties of the nanofibrous scaffolds could further be regulated with different annealing processes. Functional nanomaterials such as gold nanoparticles could be grown along the silk nanofibers to achieve control of particle size. As a template, silk could be used to coat different nanomaterials to form core–shell structures (Figure 4).
Figure 4.

Possible applications of the reversible hydrogel–solution system: (a) Injectable property of silk hydrogel composed of nanofibers. (b,c) Water-insoluble scaffold composed of silk nanofibers through directly freeze-drying process. The silk nanofibers in our present system could be blended with fresh silk solution, and induce more nanofiber growth and stable conformation formation in fresh solution, resulting in water-insoluble nanofibrous scaffold formation. (d,e) Silk-modulated morphology control of CaCO3 using silk with various nanostructures as templates. Sample d was regulated with ultrasonic-treated short nanofibers with length of about 200 nm, while sample e was controlled with ultrasonic-treated nanoparticles with length of about 50 nm. (f) The oriented growth of gold nanoparticles following silk nanofibers. The size of gold nanoparticles was easily controlled through adjusting gold forming conditions such as pH. (g) One-step synthesis of core–shell structure of Fe3O4–silk composite nanoparticles. The negative charge of silk shell provided enough repulsive force to restrain the aggregation of nanoparticles, endowing the nanoparticles excellent stability in aqueous solutions.
4. Conclusions
In this study, a new reversible hydrogel–solution system composed of β-sheet-rich silk nanofibers is reported, depending on silk concentration and the length of the nanofibers. The reversible solution-hydrogel transitions are regulated by changing the balance between hydrophobic interactions and electrostatic repulsion of the silk nanofibers. These nanofiber-based hydrogel–solution systems represent a new view of silk proteins as tunable reversible material systems.
Acknowledgments
We thank the National Basic Research Program of China (973 Program 2013CB934400, 2012CB22302), and NSFC (21174097, 81272106) for support of this work. We also thank the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), the Excellent Youth Foundation of Jiangsu Province (BK2012009), the NIH (EB002520), and the Key Natural Science Foundation of the Jiangsu Higher Education Institutions of China (11KGA430002) for support of this work.
Supporting Information Available
Additional details of “flowing hydrogels” synthesis and characterization are introduced. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Omenetto F. G.; Kaplan D. L. Science 2010, 329, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z. Z.; Vollrath F. Nature 2002, 418, 741–741. [DOI] [PubMed] [Google Scholar]
- Vollrath F.; Knight D. P. Nature 2001, 410, 541–548. [DOI] [PubMed] [Google Scholar]
- Keten S.; Xu Z. P.; Ihle B.; Buehler M. J. Nat. Mater. 2010, 9, 359–367. [DOI] [PubMed] [Google Scholar]
- Jin H. J.; Kaplan D. L. Nature 2003, 424, 1057–1061. [DOI] [PubMed] [Google Scholar]
- Chae S. K.; Kang E.; Khademhosseini A.; Lee S. H. Adv. Mater. 2013, 25, 3071–3078. [DOI] [PubMed] [Google Scholar]
- Giesa T.; Arslan M.; Pugno N. M.; Buehler M. J. Nano Lett. 2011, 11, 5038–5046. [DOI] [PubMed] [Google Scholar]
- Du N.; Liu X. Y.; Narayanan J.; Li L.; Lim M. L. M.; Li D. Biophys. J. 2006, 91, 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland C.; Terry A. E.; Porter D.; Vollrath F. Polymer 2007, 48, 3388–3392. [Google Scholar]
- Vollrath F.; Porter D.; Holland C. Soft Matter. 2011, 7, 9595–9600. [Google Scholar]
- Kasoju N.; Bora U. Adv. Healthcare Mater. 2012, 1, 393–412. [DOI] [PubMed] [Google Scholar]
- Lammel A. S.; Hu X.; Park S. H.; Kaplan D. L.; Scheibel T. R. Biomaterials 2010, 31, 4583–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansil N. C.; Koh L. D.; Han M. Y. Adv. Mater. 2012, 24, 1388–1397. [DOI] [PubMed] [Google Scholar]
- Kim D. H.; Viventi J.; Amsden J. J.; Xiao J. L.; Vigeland L.; Kim Y. S.; Blanco J. A.; Panilaitis B.; Frechette E. S.; Contreras D.; Kaplan D. L.; Omenetto F. G.; Huang Y. G.; Hwang K. C.; Zakin M. R.; Litt B.; Rogers J. A. Nat. Mater. 2010, 9, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q.; Wang X. L.; Lu S. Z.; Li M. Z.; Kaplan D. L.; Zhu H. S. Biomaterials 2011, 32, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seib F. P.; Pritchard E. M.; Kaplan D. L. Adv. Funct. Mater. 2013, 23, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee S.; Slotta U.; Scheibel T.; Bausch A. R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 6590–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martel A.; Burghammer M.; Davies R. J.; Cola E. D.; Vendrely C.; Riekel C. J. Am. Chem. Soc. 2008, 130, 17070–17074. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Zhu H. S.; Zhang C. C.; Zhang F.; Zhang B.; Kaplan D. L. Biomacromolecules 2012, 13, 826–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagn F.; Eisoldt L.; Hardy J. G.; Vendrely C.; Coles M.; Scheibel T.; Kessler H. Nature 2010, 465, 239–242. [DOI] [PubMed] [Google Scholar]
- Askarieh G.; Hedhammar M.; Nordling K.; Saenz A.; Casals C.; Rising A.; Johansson J.; Knight S. D. Nature 2010, 465, 236–238. [DOI] [PubMed] [Google Scholar]
- Greving I.; Cai M. Z.; Vollrath F.; Schniepp H. C. Biomacromolecules 2012, 13, 676–682. [DOI] [PubMed] [Google Scholar]
- Wong Po Foo C.; Bini E.; Hensman J.; Knight D. P.; Lewis R. V.; Kaplan D. L. Appl. Phys. A: Mater. Sci. Process. 2006, 82, 223–233. [Google Scholar]
- Bai S. M.; Liu S. S.; Zhang C. C.; Xu W. A.; Lu Q.; Han H. Y.; Kaplan D. L.; Zhu H. S. Acta Biomater. 2013, 9, 7806–7813. [DOI] [PubMed] [Google Scholar]
- Rockwood D. N.; Preda R. C.; Yucel T.; Wang X. Q.; Lovett M. L.; Kaplan D. L. Nat. Protoc. 2011, 6, 1612–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S. S.; Dong C. F.; Lu G. Z.; Lu Q.; Li Z. X.; Kaplan D. L.; Zhu H. S. Acta Biomater. 2013, 9, 8991–9003. [DOI] [PubMed] [Google Scholar]
- Wang X. Q.; Kluge J. A.; Leisk G. G.; Kaplan D. L. Biomaterials 2008, 29, 1054–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisk G. G.; Lo T. J.; Yucel T.; Lu Q.; Kaplan D. L. Adv. Mater. 2010, 22, 711–715. [DOI] [PubMed] [Google Scholar]
- Yao D. Y.; Dong S.; Lu Q.; Hu X.; Kaplan D. L.; Zhang B. B.; Zhu H. S. Biomacromolecules 2012, 13, 3723–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nova A.; Keten S.; Pugno N. M.; Redaelli A.; Buehler M. J. Nano Lett. 2010, 10, 2626–2634. [DOI] [PubMed] [Google Scholar]
- Guo H.; Zhang J. M.; Xu T.; Zhang Z. D.; Yao J. R.; Shao Z. Z. Biomacromolecules 2013, 14, 2733–2738. [DOI] [PubMed] [Google Scholar]
- Du N.; Yang Z.; Liu X. Y.; Li Y.; Xu H. Y. Adv. Funct. Mater. 2011, 21, 772–778. [Google Scholar]
- Zhang F.; Zuo B. Q.; Fan Z. H.; Xie Z. G.; Lu Q.; Zhang X. G.; Kaplan D. L. Biomacromolecules 2012, 13, 798–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golinska M. D.; Pham T. T. H.; Werten M. W. T.; Wolf F. A. D.; Stuart M. A. C.; Gucht J. V. D. Biomacromolecules 2013, 14, 48–55. [DOI] [PubMed] [Google Scholar]
- Kratky O.; Schauenstein E.; Sekora A. Nature 1950, 165, 319–320. [DOI] [PubMed] [Google Scholar]
- He S. J.; Valluzzi R.; Gido S. P. Int. J. Biol. Macromol. 1999, 24, 187–195. [DOI] [PubMed] [Google Scholar]
- Kim U. J.; Park J.; Li C. M.; Jin H. J.; Valluzzi R.; Kaplan D. L. Biomacromolecules 2004, 5, 786–792. [DOI] [PubMed] [Google Scholar]
- Matsumoto A.; Chen J. S.; Collette A. L.; Kin U. J.; Altman G. H.; Cebe P.; Kaplan D. L. J. Phys. Chem. B 2006, 110, 21630–21638. [DOI] [PubMed] [Google Scholar]
- Kim U. J.; Park J.; Kim H. J.; Wada M.; Kaplan D. L. Biomaterials 2005, 26, 2775–2785. [DOI] [PubMed] [Google Scholar]
- Nazarov R.; Jin H. J.; Kaplan D. L. Biomacromolecules 2004, 5, 718–726. [DOI] [PubMed] [Google Scholar]
- Zhang X. H.; Baughman C. B.; Kaplan D. L. Biomaterials 2008, 29, 2217–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F. F.; Liu S. S.; Liu X.; Pei Y. Z.; Bai S. M.; Zhao H. J.; Lu Q.; Ma F. G.; Kaplan D. L.; Zhu H. S. Acta Biomater. 2014, 10, 921–930. [DOI] [PubMed] [Google Scholar]
- Numata K.; Yamazaki S.; Naga N. Biomacromolecules 2012, 13, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Hu X.; Lu Q.; Sun L.; Cebe P.; Wang X. Q.; Zhang X. H.; Kaplan D. L. Biomacromolecules 2010, 11, 3178–3188. [DOI] [PubMed] [Google Scholar]
- Guziewicz N.; Best A.; Perez-Ramirez B.; Kaplan D. L. Biomaterials 2011, 32, 2642–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammensee S.; Huemmerich D.; Hermanson K. D.; Scheibel T.; Bausch A. R. Appl. Phys. A: Mater. Sci. Process. 2006, 82, 261–264. [Google Scholar]
- Wu X. L.; Hou J.; Li M. Z.; Wang J. N.; Kaplan D. L.; Lu S. Z. Acta Biomater. 2012, 8, 2185–2192. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Huang Y. L.; Li M. Z.; Zuo B. Q.; Lu S. Z.; Wang J. N.; Zhu H. S.; Kaplan D. L. Acta Biomater. 2011, 7, 2394–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicko C.; Kenney J. M.; Knight D.; Vollrath F. Biochemistry 2004, 43, 14080–14087. [DOI] [PubMed] [Google Scholar]
- Yucel T.; Cebe P.; Kaplan D. L. Biophys. J. 2009, 97, 2044–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.