

Abstract
Detailed investigation of variation in genes involved in pathogen recognition is crucial for understanding co-evolutionary processes between parasites and their hosts. Triggering immediate innate response to invading microbes, Toll-like receptors (TLRs) belong presently among the best-studied receptors of vertebrate immunity. TLRs exhibit remarkable interspecific variation and also intraspecific polymorphism is well documented. In humans and laboratory mice, several studies have recently shown that single amino acid substitution may significantly alter receptor function. Unfortunately, data concerning polymorphism in free-living species are still surprisingly scarce. In this study, we analyzed the polymorphism of Toll-like receptor 4 (Tlr4) over the Palearctic range of house mouse (Mus musculus). Our results reveal contrasting evolutionary patterns between the two recently (0.5 million years ago) diverged house mouse subspecies: M. m. domesticus (Mmd) and M. m. musculus (Mmm). Comparison with cytochrome b indicates strong directional selection in Mmd Tlr4. Throughout the whole Mmd western Palaearctic region, a single variant of the ligand-binding region is spread, encoded mainly by one dominant haplotype (71% of Mmd). In contrast, Tlr4 in Mmm is much more polymorphic with several haplotypes at intermediate frequencies. Moreover, we also found clear signals of recombination between two principal haplogroups in Mmm, and we identified eight sites under positive selection in our dataset. Our results suggest that observed differences in Tlr4 diversity may be attributed to contrasting parasite-mediated selection acting in the two subspecies.
Keywords: Adaptive evolution, arms race, directional selection, host–pathogen interaction, MAMPs, Mus musculus, parasite-mediated selection, pattern-recognition receptors
Introduction
Selective forces imposed by parasites can affect various traits of their hosts, including population dynamics, life histories, mating systems, sexual dimorphism etc. (Schmid-Hempel 2011). The detrimental effects of parasites are countered by function of immune system, which in vertebrates comprises both innate and acquired immunity (Danilova 2006). Study of evolution in immune-related genes is, therefore, of paramount importance for comprehension of dynamics in parasite–host relationships (see e.g., Woolhouse et al. 2002; Carlton 2003). Despite the complexity of the immune system, most studies in free-living vertebrates have focused on genes involved in acquired immunity, namely the major histocompatibility complex (MHC; e.g., Milinski 2006). However, mapping and association studies have revealed that at least half of the genetic variation responsible for resistance to various infections is attributable to non-MHC genes (Acevedo-Whitehouse and Cunningham 2006). Most of these genes seem to be associated with innate immunity and there is an increasing evidence that variation in these genes may have a fundamental effect on the host fitness in free-living populations (e.g., Turner et al. 2011; Tschirren et al. 2013).
Innate immunity receptors that directly detect and bind to parasite structures (microbe-associated molecular patterns, MAMP), the pattern-recognition receptors (PRR), stand in the first line of immune defense (Medzhitov and Janeway 2002; Akira et al. 2006). Their fast and effective functioning is thus crucial for host survival (O'Neill 2004; Akira et al. 2006). Among PRRs, the Toll-like receptors (TLR) have been shown to be particularly important (Akira et al. 2001). These receptors form a group of membrane-bound, noncatalytic proteins present in most immune cells, especially in macrophages. Distinct MAMPs (e.g., lipopolysaccharides [LPS] and lipoproteins in bacterial cell walls, zymosan of yeast, bacterial flagellin or viral nucleic acids) are recognized by distinct TLRs and the set of TLR types varies substantially among vertebrate lineages (Janssens and Beyaert 2003; Akira et al. 2006; Vinkler and Albrecht 2009; Kawai and Akira 2010). The potential action of TLRs in the context of host–parasite interactions in free-living organisms is increasingly drawing attention of evolutionary biologists and immunologists (Medzhitov et al. 1997; Pasare and Medzhitov 2004; Takeda and Akira 2005; Vinkler and Albrecht 2009). Contradicting the previous assumption of evolutionary conservatism of these receptors, evolution-focused immunogenetic investigations yielded a clear evidence that at the interspecific level diversifying selection has significantly increased diversity of orthologous Tlr genes, mainly in the ligand-binding region (LBR, Poltorak et al. 1998; Smirnova et al. 2000; Downing et al. 2010; Park et al. 2010; Wlasiuk and Nachman 2010; Areal et al. 2011; Tschirren et al. 2011; Fornuskova et al. 2013).
Information regarding the structure and variation of TLRs in free-living rodents is still relatively scarce. Interspecific comparisons of European and Asian rodents confirmed purifying selection as a prevalent evolutionary force shaping these genes (namely Tlr2, 4, and 7), probably due to functional constraints posing on the receptor molecules (Tschirren et al. 2011; Fornuskova et al. 2013). However, signatures of positive selection have also been revealed in all studied genes (mainly in the extracellular domain [ECD], containing LBRs responsible for pathogen recognition, see below), with a more intense signal in the bacterial-sensing Tlr2 and Tlr4 than in the viral-sensing Tlr7 gene (Tschirren et al. 2011; Fornuskova et al. 2013). Following study of Tschirren et al. (2012) showed that TLRs are polymorphic even within species and that intraspecific variation may strikingly differ even between two sympatric species of rodents inhabiting the same environment. In one of these species, the bank vole (Myodes glareolus), a particular group of alleles was shown to be significantly associated with resistance to Borrelia infection, suggesting an on-going evolution in the receptor (Tschirren et al. 2013). These results illustrate the urgent need for further research focused on polymorphism in PRRs at the intraspecific level, as the genetic variability in PRRs might represent an important missing element for understanding the effects of a host genotype on individual fitness.
TLR4 is one of the most essential bacterial-sensing PRRs, binding, among others, bacterial endotoxins (i.e., LPS) as ligands (Poltorak et al. 1998). At the interspecific level, this cell-surface receptor has the highest number of positively selected sites among all mammalian TLRs (Areal et al. 2011). Most of these sites are localized in the ECD which is responsible for LPS binding (Poltorak et al. 1998; Kim et al. 2007; Vinkler et al. 2009; Fornuskova et al. 2013). This domain consists of several leucine-rich repeat motifs and includes the LBR, which is in direct physical contact with MAMP structures. The ECD is followed by the transmembrane domain (TMD), anchoring the receptor into the cell membrane, and the intracellular domain (ICD). The ICD comprises the Toll/interleukin-1 (TIR) domain responsible for signal transduction and cell activation triggering the immune responses (Werling et al. 2009; Botos et al. 2011).
Genetic research in laboratory mice enabled identification of the Tlr4 gene function and assessment of the level of its polymorphism among laboratory strains (Poltorak et al. 1998; Smirnova et al. 2000; Stephan et al. 2007). However, artificial genetic variation occurring in “classical” laboratory strains (Yang et al. 2011) hampers understanding variation present in wild mice displaying much wider ranges of immunoresponsivity (Piálek et al. 2008; Abolins et al. 2011; Babayan et al. 2011; Pedersen and Babayan 2011; Riley and Viney 2011). Several house mouse subspecies have been described. Divergence of house mice is usually located to northern India and/or Pakistan and dated to about 0.5 million years ago (Boursot et al. 1993; Suzuki et al. 2004; Geraldes et al. 2008; Macholán et al. 2012). Two subspecies, M. m. musculus (Mmm) and M. m. domesticus (Mmd), have colonized Europe where they met along a secondary hybrid zone running across the continent (Boursot et al. 1993; Macholán et al. 2007; Bonhomme et al. 2011; Cucchi et al. 2012, 2013). Although the two subspecies might come into contact at least once during the expansions and contractions of their ranges (Duvaux et al. 2011), allowing them to exchange beneficial mutations, they remained for most of the colonization time in allopatry. As their westward expansions followed different routes (Mmm north of the Black Sea, Mmd through the Middle East and Mediterranean region), the two subspecies may have experienced different histories leaving distinct genetic footprints in PRR genes, including Tlr4. A recent study of the gastrointestinal tract microbiota in western European mouse populations showed geography to be the most significant factor explaining the composition of bacterial communities in this species (Linnenbrink et al. 2013). Even though gastrointestinal bacteria may have not necessarily been the pathogenic agents selecting for immunological divergence in the two subspecies, we may expect similar geographic or subspecies-specific variation also among other microbes. Genetic differences between non-bacterial parasites of the two house mouse subspecies and the lack of their significant introgression in the hybrid zone have been described recently (Kváč et al. 2013).
In this study, we have analyzed free-living specimens of the two European Mus musculus subspecies across a wide geographic range to answer the question whether the distinct recent evolutionary histories of the subspecies have left any footprints in Tlr4 variation. Based on preliminary data from classical laboratory strains (CLS) and wild-derived strains (WDS), we expect significant differences between the two house mouse subspecies. These potentially contrasting patterns could be explained either by different selection forces mediated by pathogens or simply by differences in demographic histories of the taxa (e.g., population expansions and/or bottlenecks). Given scarcity of data on pathogen background in the sampled regions, we tested the two plausible explanations by analyzing also the mitochondrial cytochrome b (mt-Cytb) gene widely used as a selectively neutral marker for assessing demographic histories of species and populations. Whereas similar patterns observed in both mt-Cytb and Tlr4 would support the effect of demographic changes, distinct patterns in the two genes would suggest the effect of selection on Tlr4. By genotyping Tlr4 and mt-Cytb in 44 Mmd and 30 Mmm sampled across the Western Palaearctic region, we document (1) a subspecies-specific distribution of genetic variation, (2) different selection patterns operating on Tlr4 gene in the two subspecies, and (3) important role of recombination increasing the polymorphism of the Tlr4 gene.
Materials and Methods
Sampling
We sampled 28 and 42 populations (1–2 individuals per site) of free-living Mmm and Mmd, respectively, scattered across the Western Palaearctic region (with exception of two localities from central Asia; Fig. 1, Table S1). In addition, we included also mice of three CLSs of predominantly Mmd origin (C3Ha, A/J, C57BL/6J; see Yang et al. (2011) for their genomic composition), 15 WDSs of the Mmd origin, and nine WDSs of the Mmm origin (Piálek et al. 2008; Vyskocilová et al. 2009; for the origin of WDSs, see Table S1). In comparison with laboratory strains, WDSs encompass more natural polymorphism and, at the same time, the homozygote variants are useful for distinguishing heterozygote sequences of natural populations (Guénet and Bonhomme 2003; Stephan et al. 2007; Piálek et al. 2008).
Figure 1.
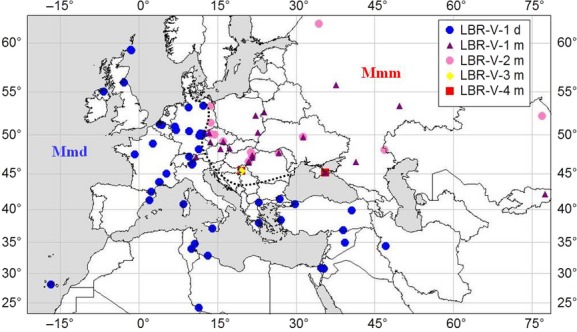
Distribution of samples analyzed in this study. Blue circles: Mus musculus domesticus (Mmd); yellow diamond, red squares, violet triangles and pink circles: M. m. musculus (Mmm). Different symbols represent distinct protein variants of the ligand-binding region (LBR). Individuals were assigned to the subspecies using hybrid index based on five X-linked loci. Dashed line represents the hybrid zone between two subspecies. Besides sampling localities of free-living populations, the localities of wild-derived strains origin are shown.
Assignment of specimens to subspecies
Assigning each analyzed individual to one of the two subspecies was based on the combination of X-linked and mtDNA diagnostic markers proven to display low levels of introgression across the European house mouse hybrid zone (Ďureje et al. 2012). The first set of markers consisted of five X-linked SINE and/or LINE insertions chosen to be distributed along the whole chromosome: X332, X65, X347, Btk, and Syap1 (Macholán et al. 2011). For each individual, a hybrid index (HI) was calculated as the mean frequency of Mmm-specific alleles over all five loci (10 for a female and five for a male). While the majority of mice displayed HI = 1.00 (Mmm) or HI = 0.00 (Mmd), 13 individuals were not fixed for all Mmm or all Mmd alleles (Table S1). This may be due to introgression and/or incomplete lineage sorting of the X-linked markers. Note that also C57BL/6, that is, one of the most “classical” laboratory strains of predominately Mmd origin (Yang et al. 2011), harbors Mmm alleles (Table S1). However, regardless underlying causes, in all these cases, admixture was negligible, allowing reliable subspecific identification.
The mitochondrial marker was a BamHI restriction site in the Nd1 gene shown to discriminate between the subspecies (Božíková et al. 2005). Mice were assigned to Mmm when the site was absent and to Mmd when the site was present. All 62 mice (wild, CLS, and WDS) assigned on the basis of the HI to Mmd also carried the BamHI restriction site. Of the remaining 39 mice assigned to Mmm according to the HI, three individuals (two wild individuals from Lindhorst and Lauchhammer in Germany, and one from a still not fully inbredized WDS established from Lindhorst) carried the Mmd-specific restriction site, suggesting introgression of Mmd mtDNA across the hybrid zone into Mmm range (Table S1). These three specimens (SLINT-WDS, SK843 and SK837) were analyzed as Mmm in the Tlr4 dataset and as Mmd in the mtDNA dataset (see below for details).
Genetic variation within subspecies
In total, 101 specimens (free-living mice together with WDSs and CLSs) were successfully sequenced for both Tlr4 and mt-Cytb genes. We sequenced exon 3 of Tlr4 (2244 bp), encompassing 90% (748 of 835 amino acid residues) of the gene coding sequence, following the protocol described in Fornuskova et al. (2013). Almost complete mt-Cytb (1123 bp) was sequenced after amplification by universal primers L14724 and H15915 (Lecompte et al. 2002). Sequences were manually checked and aligned using seqscape v.2.5 (Applied Biosystems, Forster city, CA) and bioedit v.7.1.3 (Hall 1999).
Individual Tlr4 alleles (thereafter called haplotypes for simplicity of comparison with mt-Cytb) were reconstructed from the complete alignment using the Bayesian PHASE routine implemented in DnaSP v.5.10 (Stephens and Donnelly 2003; Librado and Rozas 2009). This analysis was carried out using 1000 iterations, 10 thinning intervals and 1000 burn-in iterations. Four heterozygous Tlr4 sequences resolved with low support were checked by cloning using the protocol of pGEM®-T Easy Vector System II (Promega Madison, WI). Initially, two clones from each individual were sequenced and this number was later increased until we obtained both sequences of each heterozygote (identification of the four cloned cases can be found in Table S1). Positions of TLR4 domains (ECD, TMD, ICD/TIR) were determined using the on-line program SMART according to Fornuskova et al. (2013). Amino acids were numbered according to a GenBank M. musculus TLR4 protein sequence (GenBank Number: AGA16686.1).
The numbers of nucleotide haplotypes (N) and amino acid variants (A) for both Tlr4 and mt-Cytb genes were estimated using Fabox DNA collapser (Villesen 2007). Nucleotide diversity (π), average number of nucleotide differences (k), number of polymorphic sites (S) and haplotype diversity (Hd) were computed in DnaSP v.5.10. Haplotypes were assigned to haplogroups (HG) based on their phylogenetic interrelationships inferred with MrBayes v. 3.1 (Huelsenbeck and Ronquist 2001) and according to a median joining network constructed with Network v. 4.6.1.1. (Bandelt et al. 1999). The HKY+Γ (Hasegawa et al. 1985) and GTR+Γ (Tavaré 1986) models, determined using jModelTest v. 0.1.1. (Posada 2008), were applied to Tlr4 and mt-Cytb data, respectively. For both genes, we ran 10,000,000 MCMC generations of which 2,500,000 generations were discarded as burn-in. Geographical distribution of the HG was projected onto a map using the PanMap software (http://www.pangaea.de/software/PanMap/). All these computations were based on a subset of wild and WDS mice (i.e., we excluded all sequences from CLSs).
Analysis of molecular evolution of Tlr4
For detection of recombination breakpoints in the Tlr4 gene, we used two algorithms, the single breakpoint recombination (SBP) and genetic algorithm recombination detection (GARD), provided on the DataMonkey web server (Pond and Frost 2005a,b2005b; Pond et al. 2006a,b2006b; Delport et al. 2010). The Tlr4 dataset was partitioned according to the breakpoints detected with the SBP and GARD methods. Because it is now widely recognized that the evolutionary process is not homogeneous across sites, we performed also an analysis partitioned by three codon positions.
Selection on Tlr4 was analyzed at the intersubspecific level. We aimed to identify codons subject to positive or negative selection using test implemented in the DataMonkey program (Pond and Frost 2005a; Pond et al. 2006a): random effects likelihood (REL). The REL test tends to be somewhat susceptible to Type 1 errors, especially for small datasets, where parameter estimates are likely to have large associated errors (Pond and Frost 2005b). The Bayes factor was set up to 50. Finally, we employed the McDonald–Kreitman test (MKT), which compares variation within species to the amount of divergence between species at putatively neutral (synonymous) and nonsynonymous sites (McDonald and Kreitman 1991). Four types of comparisons were used in the MKT: Mmm/Mmd versus rats of the tribe Rattini; Mmm/Mmd versus R. norvegicus; Mmm/Mmd versus southeastern-Asian mouse species M. caroli, M. cooki, M. cervicolor; and Mmm versus Mmd (results available upon request). All selection tests were applied to a set of wild and WDS mice (i.e., excluding CLSs). Sequences of Asiatic species of Mus and Rattini were taken from Fornuskova et al. (2013).
The crystal structure of mouse TLR4 ECD (PDB 2z64) was adopted and modified from the RCSB PDB Protein Data Bank (http://www.rcsb.org/pdb/explore.do?structureId=2z64; Kim et al. 2007). Subsequently, nonsynonymous substitutions, sites under positive and negative selection detected by REL, and previously described binding sites for LPS and MD-2 (lymphocyte antigen 96; Kim et al. 2007; Park et al. 2009; Ohto et al. 2012) were visualized using PyMOL, v. 1.6 (The PyMOL Molecular Graphics System, Schrödinger, LLC; available at http://www.pymol.org/, accessed January 25, 2013).
Results
Genetic diversity of Tlr4
We successfully amplified Tlr4 sequences of 44 wild Mmd (27 homozygotes and 17 heterozygotes) and 30 wild Mmm (17 homozygotes and 13 heterozygotes; see Table S1 for the number of heterozygous sites for each individual). We found neither heterozygotes between Mmd and Mmm subspecific variants nor trans-subspecific polymorphism. Phylogenetic analysis of amplified sequences of both genes (Tlr4 and mt-Cytb) showed divergence of genetic diversity into two clades corresponding to the Mmm and Mmd subspecies (Table S1, Figs. S1, S2). In total, we found 18 and 15 Tlr4 haplotypes for Mmm and Mmd, respectively (including WDSs, CLSs and wild mice). Similarly, we identified 23 and 37 haplotypes of mt-Cytb, for Mmm and Mmd, respectively. All Mmd with the present BamHI restriction site harbored an Mmd-related mt-Cytb haplotype, and the same holds for Mmm mice (Table S1, Figs. S1B, S2B).
Genetic variation in the Tlr4 locus was considerably higher in Mmm (NMmm = 18, AMmm = 15, πMmm = 0.0025 ± 0.00016 SD) than in Mmd (NMmd = 15, AMmd = 7, πMmd = 0. 0009 ± 0.00007 SD). This is even more noticeable for the ECD with fourfold nucleotide diversity and twofold number of segregating sites in Mmm relative to Mmd (Table 1). Contrary to Tlr4, genetic variation in mt-Cytb was comparable for both subspecies (NMmm = 23, NMmd = 37; πMmm = 0.0047 ± 0.00046 SD, πMmd = 0.0046 ± 0.00022 SD; Table 1).
Table 1.
Genetic diversity of Tlr4 and mt-Cytb in two house mouse subspecies
| N/N1 | A/A1 | π ± SD1 | k1 | S1 | Hd ± SD1 | |
|---|---|---|---|---|---|---|
| Tlr4-exon 3 2244 bp | ||||||
| Mmd | 15/14 | 7/6 | 0.0009 ± 0.00007 | 1.929 | 10 | 0.736 ± 0.052 |
| Mmm | 18/16 | 15/13 | 0.0025 ± 0.00016 | 5.595 | 18 | 0.882 ± 0.028 |
| Tlr4-ECD 1644 bp | ||||||
| Mmd | 9/8 | 5/4 | 0.0005 ± 0.00007 | 0.845 | 6 | 0.554 ± 0.066 |
| Mmm | 12 | 7/7 | 0.0020 ± 0.00015 | 3.267 | 12 | 0.800 ± 0.043 |
| Tlr4-LBR 666 bp | ||||||
| Mmd | 2/1 | 2/1 | 0.0000 | 0.000 | 0 | 0.000 |
| Mmm | 7/7 | 4/4 | 0.0022 ± 0.00022 | 1.473 | 6 | 0.627 ± 0.063 |
| Tlr4-ICD 531 bp | ||||||
| Mmd | 5/5 | 2/2 | 0.0020 ± 0.00014 | 1.085 | 4 | 0.568 ± 0.039 |
| Mmm | 8/7 | 8/7 | 0.0026 ± 0.00016 | 1.398 | 4 | 0.784 ± 0.028 |
| Tlr4-TIR 435 bp | ||||||
| Mmd | 4/4 | 2/2 | 0.0024 ± 0.00015 | 1.052 | 3 | 0.551 ± 0.038 |
| Mmm | 3/2 | 3/2 | 0.0001 ± 0.00010 | 0.047 | 1 | 0.047 ± 0.044 |
| Cyt b 1123 bp | ||||||
| Mmd | 37/36 | 15/15 | 0.0046 ± 0.00022 | 5.105 | 49 | 0.983 ± 0.009 |
| Mmm | 23/20 | 9/9 | 0.0047 ± 0.00046 | 5.254 | 36 | 0.974 ± 0.016 |
Mmd, Mus musculus domesticus; Mmm, Mus musculus musculus; 62 and 39 specimens were analyzed for Mmd and Mmm, respectively; N, number of nucleotide haplotypes; A, number of amino acid variants; π, nucleotide diversity; k, average number of nucleotide differences; S, number of polymorphic sites; Hd, haplotype diversity; SD, standard deviation.
Indicate analysis without wild-derived strains and classical laboratory strains.
Moreover, in all but one Mmd samples, we identified a single protein variant of the LBR. The only exception was the A/J laboratory strain which possessed the conservative substitution V254I. This lack of polymorphism is in contrast to variation in Mmm where four different variants of LBR were found, with two of them being equally frequent in the Mmm distribution area (Fig. 1). These variants differed at three codons (F350C, D462N, and I464V; Table 2). Nevertheless, all substitutions in the LBR brought about exchanges between biochemically similar amino acids. An overview of all amino acid substitutions, their physicochemical properties and distribution are presented in Fig. 2 and Table 3.
Table 2.
Description of ligand-binding region (LBR) variants. Colored symbols correspond to Fig. 1
| LBR variants | I254V | F350C | D462N | I464V |
|---|---|---|---|---|
LBR-V-1d 
|
V | F | D | I |
| LBR-V-2d A/J | I | F | D | I |
LBR-V-1 m 
|
V | F | D | I |
LBR-V-2 m 
|
V | F | N | I |
LBR-V-3 m 
|
V | C | D | I |
LBR-V-4 m 
|
V | F | D | V |
The distribution of particular variants among sampled specimens is shown in Table S1.
Figure 2.
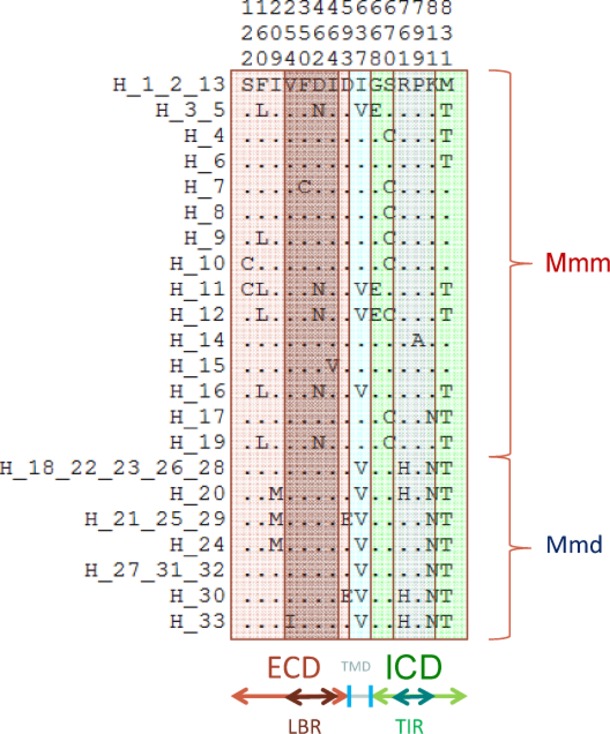
Overview of Tlr4 nonsynonymous substitutions in Mmd and Mmm. Numbers above alignment indicate amino acid position. ECD, extracellular domain; TMD, transmembrane domain; ICD, intracellular domain; LBR, ligand-binding region; TIR, Toll/interleukin-1 domain. Distribution of individual haplotypes (=alleles) among sampled specimens is presented in Table S1.
Table 3.
Physicochemical properties of the amino acids involved in nonsynonymous substitutions of Tlr4
| Position | aa1 | Properties | aa2 | Properties |
|---|---|---|---|---|
| 122 | S | SM, P, NEU | C | SM, NP, NEU |
| 160 | F | NP, NEU | L | NP, NEU |
| 209 | I | NP, NEU | M | NP, NEU |
| 2541 | V | NP, NEU | I | NP, NEU |
| 3501 | F | NP, NEU | C | SM, NP, NEU |
| 4621 | D | SM, P, NEG | N | SM, P, NEU |
| 4641 | I | NP, NEU | V | NP, NEU |
| 593 | D | SM, P, NEG | E | P, NEG |
| 637 | I | NP, NEU | V | NP, NEU |
| 668 | G | SM, NP, NEU | E | P, NEG |
| 670 | S | SM, P, NEU | C | SM, NP, NEU |
| 7612 | R | P, POS | H | P, POS |
| 7992 | P | SM, NP, NEU | A | SM, NP, NEU |
| 8112 | K | P, POS | N | SM, P, NEU |
| 831 | M | NP, NEU | T | SM, P, NEU |
SM, small; NP, nonpolar; P, polar; NEU, neutral; POS, positively charged; NEG, negatively charged.
Sites placed in ligand-binding region.
Sites placed in Toll/interleukin-1 domain.
Haplotype network analysis and distribution of genetic groups
The haplotype networks based on nucleotide sequences of exon 3 of Tlr4 were strikingly different in the two mouse subspecies. In Mmd, there was a single most frequent haplotype (H_18; Fig. 3A). It was present in 71% of all individuals (including CLSs and WDSs) and in 66% of wild mice only (in wild mice it was present in 18 specimens in the homozygote state and in 11 specimens as heterozygotes). Conversely, in Mmm, individual haplotypes were more evenly represented, none of them occurring in more than 39% of all specimens. The most common Mmm haplotype (H_5) was found in 33% of wild mice only. Based on the phylogenetic analysis and topology of the haplotype network (Figs. S1, S2), we defined one and two HG for each subspecies, respectively (HG-Idfor Mmd and HG-Im and HG-IIm for Mmm). Notwithstanding the absence of distinct genetic structuring of HG-Id, a subgroup of three haplotypes (for clarity hereafter denoted as S-I) appears rather basal to other two subgroups (S-II and S-III, respectively; Fig. 3A) and restricted to the eastern Mediterranean region and northern Tunisia, while haplotypes of the latter two subgroups either have a wide distribution (e.g., H_18, H_24) or have arisen in situ after westward spread of ancestral haplotypes (see the inset in Fig. 3A). This geographic distribution suggests a recent expansion accompanied by a loss of variation. This is especially exemplified by the star-like pattern of S-III haplotypes centered on haplotype H_18 (Fig. 3A).
Figure 3.
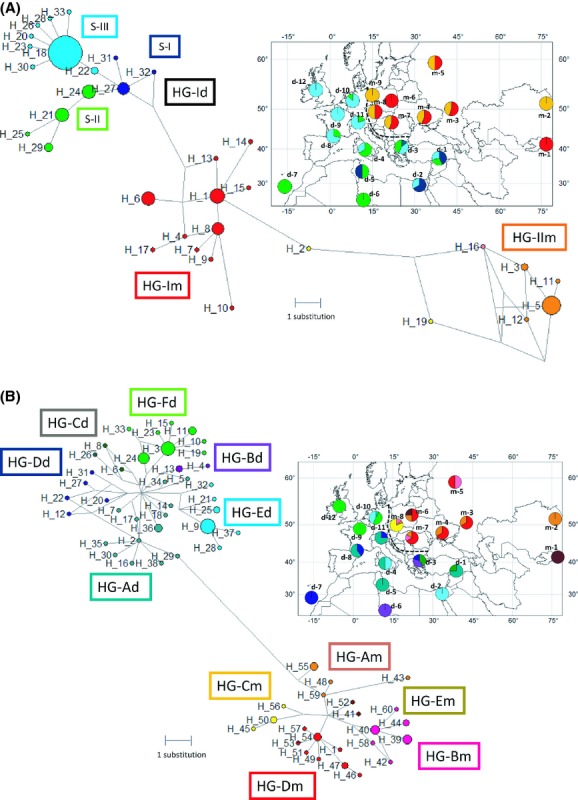
(A) Haplotype network and haplogroup distribution of Tlr4, H_haplotypes, HG-, haplogroups, S- subgroups identified in Figs. S1a and S2a. The size of circles corresponds to the frequency of haplotypes; length of lines is related to the number of substitutions. More detailed information can be found in Table S1. The inset figure represents the geographical distribution of HGs. Color circles on the map represent the proportion of particular HG or S (colors correspond to the haplotype network), labels indicate geographic assignment to population groups detailed in Table S1; dashed line shows the position of the house mouse hybrid zone. H_2 and H_19 were excluded from HG due to recombination (see the text for more details). (B) Haplotype network and haplogroup distribution of mt-Cytb, H_ identified haplotypes, HG-, identified haplogroup. The size of circles corresponds to the frequency of haplotypes; length of lines is related to the number of substitutions. More detailed information can be found in Table S1. The inset figure represents the geographical distribution of HGs. Color circles on the map represent the proportion of particular HG (colors correspond to the haplotype network); labels indicate geographic assignment to population groups detailed in Table S1; dashed line shows the position of the house mouse hybrid zone.
In Mmm, there were two distinct haplotype clouds separated at least by eight substitutions (HG-Im and HG-IIm). Both groups were interconnected by H_2 (CZ, Buškovice) and H_19 (WDS, DE, Lindhorst) which were not included in any HG (see below). The geographical distribution of HG-Im and HG-IIm is very wide, from central Asia to central Europe and they are largely overlapping in most of the Mmm distribution area. Interestingly, the distance between HG-Im and HG-IIm is higher (minimum eight substitutions) than the distance between HG-Im and HG-Id (minimum four substitutions). In contrast to Tlr4, the pattern of the mt-Cytb haplotype network was very similar for both subspecies with several star-like branching patterns suggesting local spatial/demographic expansions (Fig. 3B). The geographic distribution of both Mmm and Mmd HG seems to be more intermingled than that of Tlr4 HGs (see the inset in Fig. 3B). Identification of haplotypes in particular specimens is detailed in Table S1.
Recombination and selection in the Tlr4 gene
A recombination breakpoint between Mmd and Mmm at position 1779 bp was detected by both tests implemented in DataMonkey. This breakpoint was recognized in one Mmm individual (ST8335, H_13) sampled in Poland. However, it is based only on a single synonymous substitution at position 849 and homoplasy seems equally plausible explanation. At the intrasubspecific level, we detected recombination in two individuals of Mmm. This breakpoint was identified in a conserved region between the LBR and ICD (the SBP algorithm located the recombination breakpoint to nucleotide position 1587 = AA 529, while GARD placed it to position 1611 = AA 537). Haplotypes H_2 and H_19 likely represent recombinant haplotypes between two main Mmm HG (Fig. S3).
The REL test detected eight positively and 14 negatively selected sites (Table 4). Four of the positively selected sites were placed in the ECD; however, none of them was in the LBR (Fig. 4, Table 4). Ten of the 14 negatively selected sites were located in the ECD, three of these codons being in LBR (Fig. 4, Table 4). The MK test revealed mostly signs of negative selection (not shown).
Table 4.
Selection tested by random effects likelihood (REL) in both subspecies together, including wild-derived strains (WDSs); classic laboratory strains (CLSs) were excluded for this analysis
| REL (Mmm+Mmd+WDSs) | ECD (88–635) | TMD (636–658) | ICD (659–835) |
|---|---|---|---|
| Positively selected sites | 122, 160, 209, 593 | 637 | 670, 811, 831 |
| Negatively selected sites | 104, 132, 139, 192, 370, 416, 463, 529, 537, 575 | 647 | 690, 719, 833 |
ECD, extracellular domain, TMD, transmembrane domain, ICD, intracellular domain. Underlined sites in ECD are placed in ligand-binding region (248–469). Underlined sites in ICD are placed in Toll/interleukin-1 domain (671–816). Numbers in brackets indicate position of domains in protein (ECD start with codon 88, first 87 codons are in exon 1 and 2). All sites detected by REL had pp = 0.99.
Figure 4.
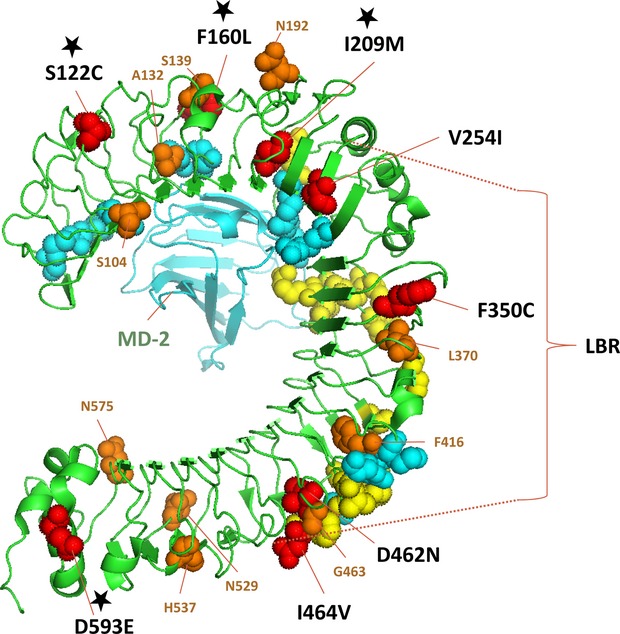
Ribbon diagram of the TLR4 extracellular domain (ECD) 3D structure (PDB 2z64 from RCSB PDB Protein Data Bank, http://www.rcsb.org/pdb/explore.do?structureId=2z64, functional sites were described according to (Kim et al. 2007; Park et al. 2009; Ohto et al. 2012) and important substitutions were visualized as amino acid space-fill models: cyan corresponds to binding positions for MD-2, yellow represents binding sites for lipopolysaccharides (LPS), red represents nonsynonymous amino acid changes, orange represents sites under negative selection detected between subspecies by random effects likelihood (REL); black stars represent detected sites under positive selection differing between the subspecies as revealed by REL; the TLR4 ECD is represented by green color, MD-2 is represented by cyan color, LBR, Ligand-Binding Region is marked by dashed lines. Description of sites responsible for LPS binding and MD-2 binding is in Table S2, modeling of different sites and design correction made in PyMOL Version 1.5.
Discussion
Tlrs are generally believed to evolve mainly under purifying selection and, thus, it has been predicted that these genes are relatively uniform within species (e.g., Mukherjee et al. 2009). Contrary to this expectation, we found a moderate intrasubspecific level of Tlr4 polymorphism. With 15 protein variants in Mmm and 7 protein variants in Mmd, this finding holds true more for Mmm than for Mmd. Indeed, we revealed decreased variation in Mmd Tlr4 both at the nucleotide and amino acid levels (Table 1), especially in the LBR where we found only a single variant in Mmd, whereas higher polymorphism level (four variants of LBR) is maintained in Mmm populations. Given the crucial function of TLR4 in mammalian innate immune defense, we may assume that the single LBR variant of TLR4 was advantageous in the past, before or during expansion of Mmd into Western Mediterranean and western/north Europe. On the other hand, we observed similar levels and geographic patterns of genetic variation of mt-Cytb in both subspecies. This indicates that the observed pattern does not result from a generally decreased level of genetic polymorphism in Mmd. Altogether, our results may imply the action of contrasting types of selection acting specifically on Tlr4 in the two house mouse subspecies. A similar contrast in selection on TLR4 across geographically distinct populations is known also in other species. For instance, in humans, it has been shown that different haplotypes are positively selected in Sub-Saharan Africa and Eurasia (Ferwerda et al. 2007). Identifying selective forces differentiating subspecies and populations thus appears an intriguing question of current evolutionary biology.
The role of TLR4 in LPS signaling is indisputable and molecular mechanisms of LPS binding were very well described in human and/or mouse (Kim et al. 2007; Park et al. 2009; Resman et al. 2009; Ohto et al. 2012). LPSs are present in the outer membrane of Gram-negative bacteria and immunologically act as endotoxins, that is, substances eliciting a strong immune response in animals. Variability of LPS may affect adhesive properties of a microorganism to the cells of its host but also the induced release of inflammatory mediators. Modifications of LPSs (mainly acylation in the lipid A region) play an important role in the infection process, evasion of the host immune response, and serotypification of Gram-negative bacteria (Robinson et al. 2008). Polymorphism of LPSs has been already shown to be associated with differences in virulence of bacterial strains, for example, Francisella tularensis, Pseudomonas aeruginosa or Yersinia pestis (Day and Marrceau-Day 1982; Ray et al. 1991; Hajjar et al. 2006; Knirel et al. 2006; Montminy et al. 2006), and as such may be responsible also for evolution and maintenance of recognition mechanisms. This applies especially to Tlr4 variation. As the genetic variation of human and livestock TLR4 is associated with susceptibility to various infectious and inflammatory diseases (e.g., Leveque et al. 2003; Hawn et al. 2005; Achyut et al. 2007; Sentitula Kumar and Yadav 2012; Zaki et al. 2012) and several nonsynonymous single nucleotide substitutions (nsSNP) has been identified as immunologically relevant (Ferwerda et al. 2007), we focused on physical properties of the nsSNPs we detected in the house mouse Tlr4. In total, we detected 15 nsSNP positions, which were distributed evenly across the whole sequenced region including the ECD, TMD, and ICD. Of these 15 nsSNPs we found four (V254I, F350C, D462N and I464V) that were located in the LBR close to the ligand-binding site of LPSs (Fig. 4). Out of these, the substitution V254I has been identified only in the LBR of the A/J laboratory strain and not in any WDS and/or free-living mice (see also Smirnova et al. 2000). We, therefore, suggest that this substitution does not represent a naturally occurring polymorphism and may have originated in laboratory breeds. On the other hand, particularly functionally important might be the residues 462 and 464 that lie in immediate topological proximity to site F461, which has been previously identified as a residuum essential for LPS binding through hydrophobic interactions in mammals (Park et al. 2009; Resman et al. 2009). We, therefore, hypothesize that these nsSNPs can influence the protein function. Our tests of selection, however, did not support this view as no positively selected sites were identified in the LBR. This suggests that D462N and I464V substitutions either have no functional impact or, at least, that there is no selection differentiating these sites in Mmm and Mmd. Nonetheless, the selection analysis showed that three of eight sites positively selected on the intersubspecific level were present in the MD-2-binding region, indicating selection differentiating Mmm and Mmd in the TLR4-MD-2 co-evolution. Recent data have shown that mouse subspecies harbor genetically different parasites (e.g., Cryptosporidium tyzzeri; Kváč et al. 2013). Both subspecies may therefore differ in immune response to specific genetic lineages of pathogens. Preliminary laboratory experiments have already shown differences in immunological response between two WDSs derived from both subspecies (Mmm BULS and Mmd STRA) by stimulating in vitro by Concanavaline A and a B-cell mitogen bacterial LPS (Piálek et al. 2008).
Although most substitutions identified in the present study involve physically very similar amino acids, it has been shown that even subtle changes in the topological proximity of the binding interface may have substantial impact on the protein function and binding affinity (Zhang et al. 2012). Further studies are, however, needed to test the functional significance of the nsSNPs for recognition of LPS variants.
Previous studies showed that genes encoding TLRs exhibit moderate levels of polymorphism even at intraspecific level (Smirnova et al. 2000; Tschirren et al. 2011; Bergman et al. 2012; Grueber et al. 2012) and that this can have important fitness consequences. In free-living populations, it was documented that selection linked with presence of pathogens can vary across different geographic regions and over time (Tschirren et al. 2012). Polymorphism in immune receptors is thought to be primarily maintained by pathogen-evoked balancing selection. This may be viewed as an evolutionary key-and-lock process described by the Matching alleles model (Frank 1993). Applied to receptor-ligand co-evolution, this model proposes that polymorphism in ligands protecting parasites from recognition is mirrored by adaptive host polymorphism allowing detection of ligand-variants by specifically matching receptor alleles (Agrawal and Lively 2002, 2003).
In addition to nucleotide substitutions, also intragenic recombination can very quickly create new allele variants. In house mouse, the effect of recombination in the evolution of immune genes is well documented, for example, in the MHC genes (Cizkova et al. 2011). However, in most recent studies on intraspecific TLR polymorphism the relevant tests of recombination have not been performed. Using two alternative approaches, our study detected recombination events in the ECD located close to the boundary with the TMD in Mmm. This finding adds another piece of information to the puzzle of PRR polymorphism in free-living rodents showing that recombination might be an important factor increasing TLR variability. Our results are consistent with studies of several other mammals reporting signals of recombination in the ECD in human TLR4 (Zaki et al. 2012) or bovine TLR3, TLR4 and TLR10 (Seabury et al. 2010). Detailed analysis of our sequences suggests that haplotypes H_2 and H_19 are recombinants composed of the ECD from haplogroup HG-IIm and ICD of HG-Im. These two Mmm haplotypes are genetically dissimilar and were found in two specimens separated by 500 km (see Table S1). Assuming that they represent two independent recombination events, we suggest that recombination in this genic region may be relatively frequent in nature. On the other hand, the recombinant haplotypes were found only in two individuals and the estimation of real selective advantage of recombination remains unknown. Because the recombination breakpoints combine different ECDs and ICDs, the WDS SLINT bearing H_19 (in combination with other WDSs from Tlr4 haplogroups HG-Im and HG-IIm) provides a unique opportunity to discriminate the role of LBR-mediated LPS recognition from the transduction of the signal by the TIR domain.
Although pathogens likely play an important role in evolution of Tlr4 variability, it may be admitted that the observed difference between the subspecies in TLR4 polymorphism might have arisen as a result of nonadaptive evolutionary processes during mouse colonization of the Western Palearctic. For example, in some avian populations affected by bottlenecks the dominant force influencing evolution of TLRs seems to be genetic drift, outweighing the effect of selection (Grueber et al. 2012, 2013). Similarly, genetic drift also shaped the genetic history of human TLR4 during population expansion out of Africa (Netea et al. 2012). Thus, the pattern observed in mice might result, for example, from differences between subspecies in historical demographic processes (quick expansion of Mmd and two founder populations or refuges for Mmm). In such a case we would, however, expect similar contrasting patterns in mt-Cytb. As this was not the case, we may consider the explanation of the observed pattern of Tlr4 by genetic drift as unlikely. Finally, we must also bear in mind that Mmd Tlr4 may not be the positively selected gene itself but only a gene involved in gene hitchhiking. Nevertheless, this hypothesis is in contradiction with results of selection analysis, which have detected eight positively selected sites in ECD of free-living Mus musculus.
Acknowledgments
We thank the following colleagues for donating mouse samples: S. J. E. Baird, J. Goüy de Bellocq, N. Bulatova, J. Burgstaller, D. Čížková, B. Dod, Ľ. Ďureje, J. Forejt, S. Gryseels, H. C. Hauffe, S. Ihle, A. Konečný, S. Martínek, N. Martínková, F. Matur, P. Mikulíček, G. Mitsainas, A. Mishta, M. Mrkvicová, P. Munclinger, J. Piálková, A. Ribas, J. Svobodová, V. Volobouev, M. Vujošević, J. M. Wójcik, and J. Zima. L. Vlčková genotyped X-linked loci and the Nd1 gene. This research was supported by the Czech Science Foundation (project 206/08/0640) to JP and MM and by Ministry of Education, Youth and Sports of the Czech Republic, NextGenProject: Next-generation technologies in evolutionary genetics (CZ.1.07/2.3./20.0303). We thank S. J. E. Baird for language corrections and significant improvement of previous versions of the manuscript and J.-F. Cosson for general support and commentaries. We also thank two anonym reviewers for their constructive suggestions. Phylogenetic reconstructions were computed at the CBGP HPC computational platform located at Centre de Biologie et Gestion des Populations, Montferrier-sur-Lez.
Conflict of Interest
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Summary of sampled specimens, identification of haplotypes, and NCBI GenBank accession numbers.
Table S2. Binding sites between TLR4/LPS/MD-2.
Figure S1. (A) Tlr4, Phylogeny based on Bayesian inference. (B) mt-Cytb, Phylogeny based on Bayesian inference.
Figure S2. (A) Tlr4, Haplogroup definition. (B) mt-Cytb, Haplogroup definition.
Figure S3. Evidence of recombination between HG-Im and HG-IIm of Mmm.
References
- Abolins SR, Pocock MJO, Hafalla JCR, Riley EM, Viney ME. Measures of immune function of wild mice, Mus musculus. Mol. Ecol. 2011;20:881–892. doi: 10.1111/j.1365-294X.2010.04910.x. [DOI] [PubMed] [Google Scholar]
- Acevedo-Whitehouse K, Cunningham AA. Is MHC enough for understanding wildlife immunogenetics? Trends Ecol. Evol. 2006;21:433–438. doi: 10.1016/j.tree.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Achyut BR, Ghoshal UC, Moorchung N, Mittal B. Association of Toll-like receptor-4 (Asp299Gly and Thr399Ileu) gene polymorphisms with gastritis and precancerous lesions. Hum. Immunol. 2007;68:901–907. doi: 10.1016/j.humimm.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Agrawal AF, Lively CM. Infection genetics: gene-for-gene versus matching-alleles models and all points in between. Evol. Ecol. Res. 2002;4:79–90. [Google Scholar]
- Agrawal AF, Lively CM. Modelling infection as a two-step process combining gene-for-gene and matching-allele genetics. Proc. Biol. Sci. 2003;270:323–334. doi: 10.1098/rspb.2002.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Areal H, Abrantes J, Esteves PJ. Signatures of positive selection in Toll-like receptor (TLR) genes in mammals. BMC Evol. Biol. 2011;11:368. doi: 10.1186/1471-2148-11-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babayan SA, Allen JE, Bradley JE, Geuking MB, Graham AL, Grencis RK, et al. Wild immunology: converging on the real world. Ann. N. Y. Acad. Sci. 2011;1236:17–29. doi: 10.1111/j.1749-6632.2011.06251.x. [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- Bergman I-M, Edman K, Ekdahl KN, Rosengren KJ, Edfors I. Extensive polymorphism in the porcine Toll-like receptor 10 gene. Int. J. Immunogenet. 2012;39:68–76. doi: 10.1111/j.1744-313X.2011.01057.x. [DOI] [PubMed] [Google Scholar]
- Bonhomme F, Orth A, Cucchi T, Rajabi-Maham H, Catalan J, Boursot P, et al. Genetic differentiation of the house mouse around the Mediterranean basin: matrilineal footprints of early and late colonization. Proc. Biol. Sci. 2011;278:1034–1043. doi: 10.1098/rspb.2010.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Structure. 2011;1993:447–459. doi: 10.1016/j.str.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursot P, Auffray JC, Britton-Davidian J, Bonhomme F. The evolution of house mice. Annu. Rev. Ecol. Syst. 1993;24:119–152. [Google Scholar]
- Božíková E, Munclinger P, Teeter KC, Tucker PK, Macholán M, Piálek J. Mitochondrial DNA in the hybrid zone between Mus musculus musculus and Mus musculus domesticus: a comparison of two transects: mtDNA in the house mouse hybrid zone. Biol. J. Linn. Soc. 2005;84:363–378. [Google Scholar]
- Carlton JM. Genome sequencing and comparative genomics of tropical disease pathogens. Cell. Microbiol. 2003;5:861–873. doi: 10.1046/j.1462-5822.2003.00331.x. [DOI] [PubMed] [Google Scholar]
- Cizkova D, de Bellocq JG, Baird SJE, Pialek J, Bryja J. Genetic structure and contrasting selection pattern at two major histocompatibility complex genes in wild house mouse populations. Heredity. 2011;106:727–740. doi: 10.1038/hdy.2010.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucchi T, Auffray JC, Vigne JD. On the origin of the house mouse synanthropy and dispersal in the Near East and Europe: zooarchaeological review and perspectives. In: Macholán M, Baird SJE, Munclinger P, Piálek J, editors. Evolution of the house mouse. Cambridge: Cambridge Univ. Press; 2012. pp. 65–93. [Google Scholar]
- Cucchi T, Kovács ZE, Berthon R, Orth A, Bonhomme F, Evin A, et al. On the trail of Neolithic mice and men towards Transcaucasia: zooarchaeological clues from Nakhchivan (Azerbaijan) Biol. J. Linn. Soc. 2013;108:917–928. [Google Scholar]
- Danilova N. The evolution of immune mechanisms. J. Exp. Zool. B Mol. Dev. Evol. 2006;306B:496–520. doi: 10.1002/jez.b.21102. [DOI] [PubMed] [Google Scholar]
- Day DF, Marrceau-Day ML. Lipopolysaccharide variability in Pseudomonas aeruginosa. Curr. Microbiol. 1982;7:93–98. [Google Scholar]
- Delport W, Poon AFY, Frost SDW, Pond SLK. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics. 2010;26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing T, Lloyd AT, O'Farrelly C, Bradley DG. The differential evolutionary dynamics of avian cytokine and tlr gene classes. J. Immunol. 2010;184:6993–7000. doi: 10.4049/jimmunol.0903092. [DOI] [PubMed] [Google Scholar]
- Ďureje L, Macholán M, Baird SJE, Piálek J. The mouse hybrid zone in Central Europe: from morphology to molecules. Folia Zool. 2012;61:308–318. [Google Scholar]
- Duvaux L, Belkhir K, Boulesteix M, Boursot P. Isolation and gene flow: inferring the speciation history of European house mice. Mol. Ecol. 2011;20:5248–5264. doi: 10.1111/j.1365-294X.2011.05343.x. [DOI] [PubMed] [Google Scholar]
- Ferwerda B, McCall MBB, Alonso S, Giamarellos-Bourboulis EJ, Mouktaroudi M, Izagirre N, et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc. Natl Acad. Sci. USA. 2007;104:16645–16650. doi: 10.1073/pnas.0704828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornuskova A, Vinkler M, Pagès M, Galan M, Jousselin E, Cerqueira F, et al. Contrasted evolutionary histories of two Toll-like receptors (Tlr4 and Tlr7) in wild rodents (Murinae) BMC Evol. Biol. 2013;13:194. doi: 10.1186/1471-2148-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SA. Specificity versus detectable polymorphism in host-parasite genetics. Proc. Biol. Sci. 1993;254:191–197. doi: 10.1098/rspb.1993.0145. [DOI] [PubMed] [Google Scholar]
- Geraldes A, Basset P, Gibson B, Smith KL, Harr B, Yu H-T, et al. Inferring the history of speciation in house mice from autosomal, X-linked, Y-linked and mitochondrial genes. Mol. Ecol. 2008;17:5349–5363. doi: 10.1111/j.1365-294X.2008.04005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, King TM, Jamieson IG. Variation at innate immunity Toll-like receptor genes in a bottlenecked population of a New Zealand Robin. PLoS One. 2012;7:e45011. doi: 10.1371/journal.pone.0045011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber CE, Wallis GP, Jamieson IG. Genetic drift outweighs natural selection at toll-like receptor (TLR) immunity loci in a re-introduced population of a threatened species. Mol. Ecol. 2013;22:4470–4482. doi: 10.1111/mec.12404. [DOI] [PubMed] [Google Scholar]
- Guénet J-L, Bonhomme F. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet. 2003;19:24–31. doi: 10.1016/s0168-9525(02)00007-0. [DOI] [PubMed] [Google Scholar]
- Hajjar AM, Harvey MD, Shaffer SA, Goodlett DR, Sjostedt A, Edebro H, et al. Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 2006;74:6730–6738. doi: 10.1128/IAI.00934-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- Hasegawa M, Kishino H, Yano T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985;22:160–174. doi: 10.1007/BF02101694. [DOI] [PubMed] [Google Scholar]
- Hawn TR, Verbon A, Janer M, Zhao LP, Beutler B, Aderem A. Toll-like receptor 4 polymorphisms are associated with resistance to Legionnaires' disease. Proc. Natl Acad. Sci. USA. 2005;102:2487–2489. doi: 10.1073/pnas.0409831102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- Janssens S, Beyaert R. Role of Toll-like receptors in pathogen recognition. Clin. Microbiol. Rev. 2003;16:637–646. doi: 10.1128/CMR.16.4.637-646.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kim HM, Park BS, Kim J-I, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130:906–917. doi: 10.1016/j.cell.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Knirel YA, Dentovskaya SV, Senchenkova SN, Shaikhutdinova RZ, Kocharova NA, Anisimov AP. Structural features and structural variability of the lipopolysaccharide of Yersinia pestis, the cause of plague. J. Endotoxin Res. 2006;12:3–9. doi: 10.1179/096805105X67283. [DOI] [PubMed] [Google Scholar]
- Kváč M, McEvoy J, Loudová M, Stenger B, Sak B, Květoňová D, et al. Coevolution of Cryptosporidium tyzzeri and the house mouse (Mus musculus. Int. J. Parasitol. 2013;43:805–817. doi: 10.1016/j.ijpara.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecompte E, Granjon L, Peterhans JK, Denys C. Cytochrome b-based phylogeny of the Praomys group (Rodentia, Murinae): a new African radiation? C. R. Biol. 2002;325:827–840. doi: 10.1016/s1631-0691(02)01488-9. [DOI] [PubMed] [Google Scholar]
- Leveque G, Forgetta V, Morroll S, Smith AL, Bumstead N, Barrow P, et al. Allelic variation in TLR4 is linked to susceptibility to Salmonella enterica serovar Typhimurium infection in chickens. Infect. Immun. 2003;71:1116–1124. doi: 10.1128/IAI.71.3.1116-1124.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Linnenbrink M, Wang J, Hardouin EA, Künzel S, Metzler D, Baines JF. The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Mol. Ecol. 2013;22:1904–1916. doi: 10.1111/mec.12206. [DOI] [PubMed] [Google Scholar]
- Macholán M, Vyskocilová M, Bonhomme F, Krystufek B, Orth A, Vohralík V. Genetic variation and phylogeography of free-living mouse species (genus Mus) in the Balkans and the Middle East. Mol. Ecol. 2007;16:4774–4788. doi: 10.1111/j.1365-294X.2007.03526.x. [DOI] [PubMed] [Google Scholar]
- Macholán M, Baird SJE, Dufková P, Munclinger P, Bímová BV, Piálek J. Assessing multilocus introgression patterns: a case study on the mouse X chromosome in central Europe. Evolution. 2011;65:1428–1446. doi: 10.1111/j.1558-5646.2011.01228.x. [DOI] [PubMed] [Google Scholar]
- Macholán M, Pialek J, Baird SJE, Munclinger P. Evolution of the house mouse. 1st ed. Cambridge, U.K: Cambridge Univ. Press; 2012. [Google Scholar]
- McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- Milinski M. The major histocompatibility complex, sexual selection, and mate choice. Annu. Rev. Ecol. Evol. Syst. 2006;37:159–186. [Google Scholar]
- Montminy SW, Khan N, McGrath S, Walkowicz MJ, Sharp F, Conlon JE, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 2006;7:1066–1073. doi: 10.1038/ni1386. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Sarkar-Roy N, Wagener DK, Majumder PP. Signatures of natural selection are not uniform across genes of innate immune system, but purifying selection is the dominant signature. Proc. Natl Acad. Sci. 2009;106:7073–7078. doi: 10.1073/pnas.0811357106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Wijmenga C, O'Neill LAJ. Genetic variation in Toll-like receptors and disease susceptibility. Nat. Immunol. 2012;13:535–542. doi: 10.1038/ni.2284. [DOI] [PubMed] [Google Scholar]
- Ohto U, Fukase K, Miyake K, Shimizu T. Structural basis of species-specific endotoxin sensing by innate immune receptor TLR4/MD-2. Proc. Natl Acad. Sci. USA. 2012;109:7421–7426. doi: 10.1073/pnas.1201193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LAJ. TLRs: Professor Mechnikov, sit on your hat. Trends Immunol. 2004;25:687–693. doi: 10.1016/j.it.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi B-S, Lee H, Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- Park S, Park D, Jung Y, Chung E, Choi S. Positive selection signatures in the TLR7 family. Genes Genomics. 2010;32:143–150. [Google Scholar]
- Pasare C, Medzhitov R. Toll-like receptors and acquired immunity. Semin. Immunol. 2004;16:23–26. doi: 10.1016/j.smim.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Pedersen AB, Babayan SA. Wild immunology. Mol. Ecol. 2011;20:872–880. doi: 10.1111/j.1365-294X.2010.04938.x. [DOI] [PubMed] [Google Scholar]
- Piálek J, Vyskočilová M, Bímová B, Havelková D, Piálková J, Dufková P, et al. Development of unique house mouse resources suitable for evolutionary studies of speciation. J. Hered. 2008;99:34–44. doi: 10.1093/jhered/esm083. [DOI] [PubMed] [Google Scholar]
- Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics. 2005a;21:2531–2533. doi: 10.1093/bioinformatics/bti320. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005b;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006a;23:1891–1901. doi: 10.1093/molbev/msl051. [DOI] [PubMed] [Google Scholar]
- Pond SLK, Posada D, Gravenor MB, Woelk CH, Frost SDW. GARD: a genetic algorithm for recombination detection. Bioinformatics. 2006b;22:3096–3098. doi: 10.1093/bioinformatics/btl474. [DOI] [PubMed] [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Ray A, Redhead K, Selkirk S, Poole S. Variability in LPS composition, antigenicity and reactogenicity of phase variants of Bordetella pertussis. FEMS Microbiol. Lett. 1991;79:211–218. doi: 10.1016/0378-1097(91)90088-r. [DOI] [PubMed] [Google Scholar]
- Resman N, Vasl J, Oblak A, Pristovsek P, Gioannini TL, Weiss JP, et al. Essential roles of hydrophobic residues in both MD-2 and Toll-like receptor 4 in activation by endotoxin. J. Biol. Chem. 2009;284:15052–15060. doi: 10.1074/jbc.M901429200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EM, Viney ME. Wild mice provide insights into natural killer cell maturation and memory. Mol. Ecol. 2011;20:4827–4829. doi: 10.1111/j.1365-294X.2011.05315.x. [DOI] [PubMed] [Google Scholar]
- Robinson RT, Khader SA, Locksley RM, Lien E, Smiley ST, Cooper AM. Yersinia pestis evades TLR4-dependent induction of IL-12(p40)2 by dendritic cells and subsequent cell migration. J. Immunol. 2008;181:5560–5567. doi: 10.4049/jimmunol.181.8.5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Hempel P. Evolutionary parasitology: the integrated study of infections, immunology, ecology, and genetics. New York: Oxford Univ. Press, Oxford; 2011. [Google Scholar]
- Seabury CM, Seabury PM, Decker JE, Schnabel RD, Taylor JF, Womack JE. Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle. Proc. Natl Acad. Sci. USA. 2010;107:151–156. doi: 10.1073/pnas.0913006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sentitula Kumar R, Yadav BR. Molecular analysis of TLR4 gene and its association with intra-mammary infections in Sahiwal cattle and Murrah buffaloes. Indian J. Biotechnol. 2012;11:267–273. [Google Scholar]
- Smirnova I, Poltorak A, Chan EK, McBride C, Beutler B. Phylogenetic variation and polymorphism at the Toll-like receptor 4 locus (TLR4) Genome Biol. 2000;1:research002.1–research002.10. doi: 10.1186/gb-2000-1-1-research002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan K, Smirnova I, Jacque B, Poltorak A. Genetic analysis of the innate immune responses in wild-derived inbred strains of mice. Eur. J. Immunol. 2007;37:212–223. doi: 10.1002/eji.200636156. [DOI] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Shimada T, Terashima M, Tsuchiya K, Aplin K. Temporal, spatial, and ecological modes of evolution of Eurasian Mus based on mitochondrial and nuclear gene sequences. Mol. Phylogenet. Evol. 2004;33:626–646. doi: 10.1016/j.ympev.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int. Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Tavaré S. Some probabilistic and statisical problems on the analysis of DNA sequences. Lectures Math. Life Sci. 1986;17:57–86. [Google Scholar]
- Tschirren B, Råberg L, Westerdahl H. Signatures of selection acting on the innate immunity gene Toll-like receptor 2 (TLR2) during the evolutionary history of rodents. J. Evol. Biol. 2011;24:1232–1240. doi: 10.1111/j.1420-9101.2011.02254.x. [DOI] [PubMed] [Google Scholar]
- Tschirren B, Andersson M, Scherman K, Westerdahl H, Råberg L. Contrasting patterns of diversity and population differentiation at the innate immunity gene Toll-like receptor 2 (TLR2) in two sympatric rodent species. Evolution. 2012;66:720–731. doi: 10.1111/j.1558-5646.2011.01473.x. [DOI] [PubMed] [Google Scholar]
- Tschirren B, Andersson M, Scherman K, Westerdahl H, Mittl PRE, Råberg L. Polymorphisms at the innate immune receptor TLR2 are associated with Borrelia infection in a wild rodent population. Proc. Biol. Sci. 2013;280:20130364. doi: 10.1098/rspb.2013.0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AK, Begon M, Jackson JA, Bradley JE, Paterson S. Genetic diversity in cytokines associated with immune variation and resistance to multiple pathogens in a natural rodent population. PLoS Genet. 2011;7:e1002343. doi: 10.1371/journal.pgen.1002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villesen P. FaBox: an online toolbox for fasta sequences. Mol. Ecol. Notes. 2007;7:965–968. [Google Scholar]
- Vinkler M, Albrecht T. The question waiting to be asked: innate immunity receptors in the perspective of zoological research. Folia Zool. 2009;58:15–28. [Google Scholar]
- Vinkler M, Bryjová A, Albrecht T, Bryja J. Identification of the first Toll-like receptor gene in passerine birds: TLR4 orthologue in zebra finch (Taeniopygia guttata. Tissue Antigens. 2009;74:32–41. doi: 10.1111/j.1399-0039.2009.01273.x. [DOI] [PubMed] [Google Scholar]
- Vyskocilová M, Prazanová G, Piálek J. Polymorphism in hybrid male sterility in wild-derived Mus musculus musculus strains on proximal chromosome 17. Mamm. Genome. 2009;20:83–91. doi: 10.1007/s00335-008-9164-3. [DOI] [PubMed] [Google Scholar]
- Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ. Variation matters: TLR structure and species-specific pathogen recognition. Trends Immunol. 2009;30:124–130. doi: 10.1016/j.it.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Wlasiuk G, Nachman MW. Adaptation and constraint at Toll-like receptors in primates. Mol. Biol. Evol. 2010;27:2172–2186. doi: 10.1093/molbev/msq104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse MEJ, Webster JP, Domingo E, Charlesworth B, Levin BR. Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet. 2002;32:569–577. doi: 10.1038/ng1202-569. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang JR, Didion JP, Buus RJ, Bell TA, Welsh CE, et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat. Genet. 2011;43:648–655. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki HY, Leung KH, Yiu WC, Gasmelseed N, Elwali NEM, Yip SP. Common polymorphisms in TLR4 gene associated with susceptibility to pulmonary tuberculosis in the Sudanese. Int J Tuberc Lung Dis. 2012;16:934–940. doi: 10.5588/ijtld.11.0517. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Miteva MA, Wang L, Alexov E. Analyzing effects of naturally occurring missense mutations. Comput. Math. Methods Med. 2012;2012:e805827. doi: 10.1155/2012/805827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of sampled specimens, identification of haplotypes, and NCBI GenBank accession numbers.
Table S2. Binding sites between TLR4/LPS/MD-2.
Figure S1. (A) Tlr4, Phylogeny based on Bayesian inference. (B) mt-Cytb, Phylogeny based on Bayesian inference.
Figure S2. (A) Tlr4, Haplogroup definition. (B) mt-Cytb, Haplogroup definition.
Figure S3. Evidence of recombination between HG-Im and HG-IIm of Mmm.