

Abstract
The bacteriophage T4 baseplate is the control center of the virus, where the recognition of an E. coli host by the long tail fibers is translated into a signal to initiate infection. The short tail fibers unfold from the baseplate for firm attachment to the host, followed by shrinkage of the tail sheath that causes the tail tube to enter and cross the periplasmic space ending with injection of the genome into the host. During this process, the 6.5 MDa baseplate changes its structure from a “dome” shape to a “star” shape. An in vitro assembled hubless baseplate has been crystalized. It consists of six copies of the recombinantly expressed trimeric gene product (gp) 10, monomeric gp7, dimeric gp8, dimeric gp6 and monomeric gp53. The diffraction pattern extends, at most, to 4.0 Å resolution. The known partial structures of gp10, gp8, and gp6 and their relative position in the baseplate derived from earlier electron microscopy studies were used for molecular replacement. An electron density map has been calculated based on molecular replacement, single isomorphous replacement with anomalous dispersion data and 2-fold non-crystallographic symmetry averaging between two baseplate wedges in the crystallographic asymmetric unit. The current electron density map indicates that there are structural changes in the gp6, gp8, and gp10 oligomers compared to their structures when separately crystallized. Additional density is also visible corresponding to gp7, gp53 and the unknown parts of gp10 and gp6.
Keywords: Bacteriophage T4, baseplate, X-ray crystallography, low resolution
1. INTRODUCTION
Bacteriophage T4 is one of the most studied bacterial viruses (Karam, 1994). It consists of a prolate head capsid that encapsidates a 172 kbp DNA molecule, representing a 168kbp concatenated genome, a tail tube surrounded by a contractile sheath that terminates with a baseplate and long tail fibers with which the virus recognizes potential host cells. At least 15 structural protein species contribute to a total of 134 protein subunits in the ~6.5 MDa baseplate. The hexagonal baseplate assembles from six wedges surrounding a central hub.
The baseplate is the communication center and receives signals from the long tail fibers on host recognition. The receptor recognition by the long tail fibers triggers the rearrangement of the baseplate proteins, causing the short tail fibers to unfold from the bottom of the baseplate. The irreversible binding of the short tail fibers to the host cell results in a drastic conformational change of the dome-shaped baseplate to a star-shaped structure. Both the dome-shaped and star-shaped conformations of the baseplate have hexagonal symmetry (Crowther et al., 1977). The dome-shaped structure has a maximum diameter of ~520 Å and is ~270 Å high. The star-shaped baseplate is ~610 Å in diameter and ~120 Å in height. The relaxation of the high-energy metastable dome-shaped baseplate to a low-energy star-shaped baseplate induces the contraction of the tail sheath which then drives the tail tube to penetrate the host cell membrane and triggers the ejection of the phage DNA into the host cell.
These conformational changes have been studied using cryo-electron microscopy (cryoEM) that, in turn, was interpreted by fitting the independently determined crystal structures of eight individual proteins (gp5, gp6, gp8, gp9, gp10, gp11, gp12, gp27) into the cryoEM density of both the dome- (Kostyuchenko et al., 2003) and star-shaped (Leiman et al., 2004) cryoEM maps of the baseplate. The currently available resolution of these electron density maps is 12 Å and 16 Å, respectively. The assumption of these studies was that the individual protein structures remain rigid during the conformational transition. Furthermore, these interpretations lack detailed structural knowledge to provide information about the mechanisms that are involved in this process. These studies suggested that the individual protein domains act as rigid bodies that slide over each other during the conformational change of the baseplate.
The assembly of bacteriophage T4 is an ordered sequential event. In particular the assembly pathway of the tail was studied using mutant phages, in vitro complementation tests and electron microscopy (Kikuchi and King, 1975a; Kikuchi and King, 1975b; Kikuchi and King, 1975c). This well-studied sequential assembly pathway of T4 was examined to determine how a baseplate could be assembled with a minimal number of protein species (Yap et al., 2010). Five wedge proteins, not including the central hub proteins and the peripheral short tail fibers, were shown to be required for the assembly of a hexagonal baseplate-like post infection structure. We report here an approximately 5.0 Å resolution X-ray crystallographic electron density map of an in vitro assembled, hubless, star-shaped baseplate of T4 phage. We compare this structure with the earlier cryoEM results and independent structures of some of the component proteins.
2. MATERIALS AND METHODS
2.1. Purification, crystallization and derivative preparation
T4 baseplate-like protein complexes were assembled in vitro using recombinant proteins as previously described in detail (Yap et al., 2010). In brief, seven T4 wedge proteins (gp10, gp7, gp8, gp6, gp53, gp25 and gp11) were expressed independently using an E. coli expression system. Among the seven proteins, gp6 was expressed with a His6 tag at the C-terminus. Harvested cells were resuspended in buffer 1 (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM DTT, 0.5 mM EDTA), mixed and lysed by sonication. The cell lysates were subjected to centrifugation at 30,000g for 20 minutes. The supernatant fraction was loaded onto a HisTrap 5ml column (GE Healthcare). Targeted protein complexes were eluted by a gradient of 20 to 500 mM imidazole in buffer 1. The eluted peak fractions were subjected to a HiTrap Q HP-5ml column (GE Healthcare). Targeted protein complexes were eluted by a gradient of 0 to 1 M NaCl in buffer 1. Protein complexes were further purified using size-exclusion column Sephacryl 16/60 HR S-400 (GE Healthcare) in buffer 1. The seven proteins were then successfully incorporated into the assembly of the baseplate-like structure. Five proteins are essential for the efficient assembly, namely gp10, gp7, gp8, gp6, and gp53. These proteins follow the sequential assembly pathway that had been elucidated previously (Kikuchi and King, 1975a).
The T4 baseplate intermediate complexes (gp10-gp7-gp8-gp6-gp53, gp10-gp7-gp8-gp6-gp53-gp25, and gp10-gp7-gp8-gp6-gp53-gp25-gp11) were assembled in vitro and purified. Purified protein complexes were concentrated to 15 mg/ml for protein crystal screening using the sitting drop vapor-diffusion method. Various Hampton Research screens and Emerald screens were tested to obtain the initial crystallization conditions. The gp10-gp7-gp8-gp6-gp53 complex crystallized as hexagonal plates, whereas the other complexes crystallized as thin needles. Only the hexagonal crystal form gave any X-ray diffraction pattern. After several rounds of optimization using the hanging drop vapor-diffusion method, the best condition for obtaining well-shaped hexagonal crystals was 6.5 % PEG 6000, 2 M NaCl, and 0.1 M Bis-Tris buffer pH5.5 with a protein (12 mg/ml) to reservoir solution ratio of 2:1. The crystals grew to have diameters of between 100 μm to 300 μm and a thickness of about 50 μm after about 10 days at 20 ° C.
2.2 X-Ray data collection
Initially the crystals were examined on a home Rigaku rotating anode X-ray generator and were found to give reasonable X-ray diffraction to about 10 Å resolution. However, simple soaks in cryo-solvent were found to be harmful to the diffraction quality of the crystals. Therefore, prior to data collection the crystals were soaked in stepwise increasing amounts of glycerol (5 % to 25 %) in pre-equilibrated buffer followed by flash-freezing in liquid nitrogen. Crystals treated with this stepwise freezing strategy diffracted consistently beyond 7 Å resolution but never better than 4 Å resolution. Both crystal annealing and dehydration were tested without obtaining any improvement in the diffraction quality of the crystals.
A tantalum bromide cluster Ta6Br122+ derivative was used to determine the phases of the X-ray reflections. The Ta6Br122+ was added as a powder to the solution containing grown crystals. These crystals were soaked for 24 hours before flash freezing. A single-wavelength dataset was collected to 6.8 Å resolution using radiation with a wavelength of 1.2548 Å corresponding to the maximum anomalous signal from the cluster. All native and derivative datasets were collected on a MAR 300 CCD detector at APS Sector 23 GM/CA-CAT at 100 K.
2.3 X-Ray Free-Electron Laser (XFEL) data
An attempt was made to improve the resolution of the diffraction data collection using the XFEL source at Linac Coherent Light Source (LCLS) at Stanford University. Crystals were tested using two sample delivery systems, namely the X-ray Pump-Probe (XPP) and the Coherent X-ray Imaging (CXI) (DePonte et al., 2008) instruments. Cryo-cooled crystals were used at the XPP instrument and warm microcrystals were used at the CXI instrument. The goniometer-mounted cryo-cooled-crystal method on the XPP instrument was found to be more useful because the amount of crystalline material was more limited. The beam size at XPP was varied from 30 to 3 μm. The crystals had diameters varying from about 100 to 200 μm. About five different successful exposures could be made from different volumes of a crystal when using a 30 μm beam diameter, but the crystal was completely annihilated even after only one exposure using a 3 μm beam diameter. The Bragg reflections were better defined using the smallest beam size. The resolution of the data at best was marginally as good as that collected at the APS synchrotron. Data processing was problematic as the procedures for limited data collections using XFEL radiation are still under development (Hattne et al., 2014).
3. RESULTS
3.1 Data collection and processing
The baseplate crystals were extremely radiation sensitive. Only about twenty 0.5° oscillation images could be collected before the diffraction patterns displayed obvious signs of radiation damage. Different spots on the same crystal could sometimes be used with a beam size of 100 × 50 μm for data collection (Table 1). All data were processed, scaled and post-refined with the HKL2000 program (Otwinowski and Minor, 1997). The merged data was scaled and post refined using the program HKL2000. Reflections with structure amplitudes less than 0.8σ were omitted for all phase determinations. The value of Rsym and the resultant scaled data showed that the crystals were hexagonal and belonged to space group P622 with a=b= 469.7 Å, c= 457.1 Å. The final Rmerge for native and the Ta6Br122+ heavy atom derivative were 21.6 % and 12.1 % to 4.0 Å and 6.8 Å resolution, respectively (Table 1). As the crystals used for data collection were randomly orientated, the decline of completeness of the data with increasing orientation was due to the high “temperature” factor of the reflections.
Table 1.
Crystallographic data collection statistics.
| Data set | Native | Ta6Br122+(peak) |
|---|---|---|
| Wavelength (Å) | 1.0332 | 1.2548 |
| Space group | P622 | P622 |
| Cell dimensions | ||
| a, b, c (Å) | 469.7, 469.7, 457.1 | 472.4, 472.4, 463.0 |
| α, β, γ (°) | 90.0, 90.0, 120 | 90.0, 90.0, 120 |
| Resolution (Å)a | 4.00 (4.07-4.00) | 6.80 (6.92-6.80) |
| Rmerge | 0.216 (0.251) | 0.121 (0.507) |
| I/σ (I) | 4.5 (1.1) | 6.2 (1.0) |
| CC½b | (0.443) | (0.103) |
| Completeness (%) | 66.6 (5.3) | 86.3 (21.0) |
| Completeness >90%, to resolution (Å) | 5.40 | 8.00 |
| Redundancy | 9.2 (1.0) | 3.0 (1.3) |
| Number of images | 834 | 204 |
| Number of reflection | 1452299 | 135465 |
| Number of unique reflection | 162723 | 45212 |
Highest resolution shell is shown in parenthesis
Correlation coefficient between half-datasets
3.2 Crystal packing
CryoEM images of the in vitro assembled baseplate shows a star-shaped particle with a diameter of about 480 Å (Fig. 1). The expected diameter of the star-shaped baseplate is 610 Å (Leiman et al., 2004). However, the in vitro assembled baseplates are missing the peripheral gp12 (short tail fibers) and gp11 proteins, suggesting that the in vitro assembled baseplate corresponds to the central portion of the star-shaped structure, which has a diameter of about 480Å. As the a and b dimensions of the crystallographic cell are 469 Å-long and since the crystals have hexagonal symmetry, there is a baseplate sitting on a 6-fold crystallographic axis at the corners of the hexagonal unit cell, resulting in a two-dimensional close packing of the baseplates.
Fig. 1. CryoEM of in vitro assembled baseplates.
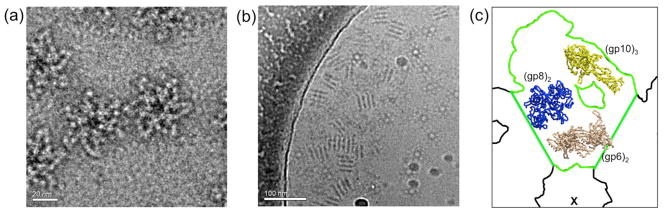
The five recombinant wedge proteins (gp10, 7, 8, 6, and 53) self-assemble into a hubless baseplate-like structure. The diameter of a particle is about 480 Å. (a) Top view and (b) side view. Note that in (b) many of the particles can be seen stacking on top of each other in a manner similar to their packing on the 6-fold crystallographic axis in the hexagonal crystals. (c) The molecular replacement search model constructed from the independently determined partial structures of trimeric gp10 (yellow), dimeric gp6 (beige) and the complete structure of dimeric gp8 (blue), positioned into a wedge (outlined in green) of the star-shaped baseplate (outlined in black) as determined from the cryoEM analysis of a T4 tail. The center of the baseplate is indicated by a cross.
The presence of 2-fold crystallographic axes parallel to a and b intersecting the 6-fold axis will require two baseplates sitting on each 6-fold axis back-to-back. However, the thickness of the star-shaped baseplate was previously reported to be only 120 Å (Leiman et al., 2004). Thus, the two baseplates positioned on the 6-fold axis would have a total thickness of about 240 Å. Yet the unit cell length of the c axis is 457 Å, almost twice the expected length if there were only two baseplates in the unit cell. Therefore, two additional baseplates must be positioned on each 6-fold axis, rotated and perhaps inverted relative to each other giving a total of four baseplates in the unit cell. With two crystallographically independent baseplates in the crystal unit cell there must be the equivalent of two independent wedges per crystallographic asymmetric unit. This was confirmed by calculating a rotation function (Tong and Rossmann, 1990) (Fig. 2) that showed a non-crystallographic symmetry (NCS) 2-fold axes perpendicular to the c axis at an angle of +3.8° and 30.0°+3.8° relative to the b* axis. This also implied that the two independent baseplates were inverted (“back-to-back”) with respect to each other. Hence, the baseplates stacked on each 6-fold axis are facing alternatively up and down.
Fig. 2. The κ=180.0° rotation function for the in vitro assembled baseplate.

The non-crystallographic 2-fold axis (indicated by an arrow) is perpendicular to the c axis. The function was calculated using 6 Å resolution data. The 10% of the reflections with the largest intensities were used for calculating the comparison Patterson. The continuous contours are at intervals of 250 starting at 250 arbitrary units of rotation function heights. These show the position of the crystallographic 2-fold axes. The dashed contours are drawn in intervals of 25 starting at a level of 25 and stop at 250 in units of height. These show the non-crystallographic symmetry axis at 30.0°+3.8°, relative to the b* axis and equivalent symmetry related axes.
The calculated molecular weight of the in vitro assembled baseplate is 3.3 MDa. That gives an unusually large Matthews coefficient of 8.8 Å3/Da corresponding to there being solvent in 81 % of the unit cell volume. However, the molecular weight of an in vivo assembled baseplate would be about 6.5 MDa, which would give a more reasonable Matthews coefficient of 4.4 Å3/Da. Thus, omission of some of the proteins creates holes in the baseplate which is probably the cause of the poor diffraction quality of the crystals.
3.3 Molecular Replacement
The in vitro assembled baseplate was assumed to have a similar structure as the star-shaped conformation. The latter was, therefore, used as a molecular replacement search model. The search model (Fig. 1c) was constructed from the independently determined partial structures of trimeric gp10 ((Leiman et al., 2006), PDB ID code 2FKK), dimeric gp6 ((Aksyuk et al., 2009), PDB ID 3H2T) and the complete structure of dimeric gp8 ((Leiman et al., 2003), PDB ID 1N80), placed into the cryoEM density of the star-shaped baseplate ((Kostyuchenko et al., 2005), EMDB ID EMD-1086). Missing from this model were the structures of gp7, gp53, about two thirds of gp10, and about half of gp6, amounting to about two thirds of the total mass of the in vitro assembled baseplate. Although no structures of gp7 and gp53 are known, gp7 probably connects together some of the other proteins within a wedge and gp53 helps to associate the different wedges within the baseplate (Kikuchi and King, 1975d; Kostyuchenko et al., 2003). An R-factor search was performed over reasonable limits for the position of one of the two baseplates. The refinable search parameters were therefore the model’s rotation about and translation along the c axis as well as the intersection of the NCS 2-fold axis with the crystallographic 6-fold axis. However, no position and orientation of the baseplate with convincingly low R-factor was found.
In view of this failure, an attempt was made to use the program PHASER (McCoy et al., 2007) to solve the structure using not the whole baseplate but only one wedge. The initial cross rotation function at 5.0 Å resolution gave 130 resolved peaks above the mean value. The three top translation functions had Z scores of 9.7, 9.0 and 7.9. The first of these solutions gave a reasonable packing result. The other solutions were discarded. When a second wedge was added to the structure there was only one significant result that had a Z score of 20.1. Operating with the crystallographic 6-fold axis on the position and orientation of these two wedges generated two back-to-back baseplates similar to each other and to the cryoEM derived baseplate structure (Leiman et al., 2004). Furthermore, these two baseplates were related to each other by an approximate 2-fold NCS axis within 0.1° of that observed in the self-rotation function. The R-factor of this solution was 55.9 %. Analysis of the relationship between the two independent wedges showed that they were related by a rotation axis with an orientation defined by the polar angles ψ=4.0°, φ=19.0° (Rossmann and Blow, 1962). As the baseplates had been self-assembled in vitro and were related by a NCS axis shown to be orthogonal to the crystallographic c axis by the rotation function, the Phaser solution was adjusted to be based on a ψ=3.8° and φ=0.0° NCS axis. The distance of the NCS axis from the origin along the c axis had to be at about 0.25c and was refined by the program IMP (Jones, 1992) to minimize the density difference between NCS related positions within the volume defined by a wedge.
The failure to solve the structure using the whole baseplate as a search model requiring only three independent parameters, as opposed to the successful solution when using a wedge as a search model with essentially 12 parameters, was likely due to changes in the structural components that constituted the search model. This was also supported by the larger R-factor (60.0%) obtained by using the model of the complete baseplate, as compared to the 55.9% R-factor using independent wedges as reported above.
Although phases could be calculated once the molecular replacement solution had been determined, these phases would be biased by the assumption implicit in the construction of the atomic model. Furthermore, the fraction of atoms whose position had been used in constructing the phasing model was only about one third of the total number of atoms in the baseplate assembly. Thus, the starting phase solution would be rather poor and strongly biased by the known components of the baseplate. For this reason a Ta6Br122+ cluster compound (Table 1) was used to help improve the phases in conjunction with NCS 2-fold averaging and solvent flattening.
3.4 Phasing by isomorphous replacement, anomalous dispersion and NCS averaging
A difference electron density map between the Ta6Br122+ derivative and native data, using the molecular replacement phases, showed that there were two peaks per crystallographic asymmetric unit related to each other by the observed NCS 2-fold axis. These peaks were about 9σ above the mean density, whereas the next highest peak was only 4σ high. The heavy atom parameters were then refined with the MLPHARE program (Argos and Rossmann, 1974) while combining the molecular replacement phases with the single isomorphous phases (Rossmann and Blow, 1961). Two additional low occupancy Ta6Br122+ sites were found during this process. One of these sites was situated on the 2-fold NCS axis. The other site did not have a related 2-fold NCS site (Table 2). After convergence had been reached, the anomalous dispersion Bijovet differences for the Ta6Br122+ data were included in the refinement. Eventually joint phase probabilities of the molecular replacement phases to 4.0 Å resolution, single isomorphous replacement phases to 6.7 Å resolution and anomalous dispersion phases to 6.7 Å resolution were used to compute an improved electron density map to 4.0 Å resolution. This map was improved using the Uppsala software factory programs (Kleywegt and Jones, 1994; Kleywegt and Jones, 1996) and the CCP4 suite of programs (Collaborative Computational Project, 1994) by iterative cycling while modifying the electron density using 2-fold NCS averaging and solvent flattening within an envelope defined by the cryoEM structure of the star-shaped baseplate for the proteins in the in vitro assembled baseplate. The overall converged correlation coefficient and R-factor between the observed amplitudes and the calculated amplitudes of the Fourier back transformed density modified map were 0.70 and 0.21, respectively (Fig. 3).
Table 2.
Heavy atom refinement analysis.
| Ta6Br122+ cluster sites (fractional coordinates)
|
Occ. | Anom. Occ. | Temperature factor Å2 | |||
|---|---|---|---|---|---|---|
| Atom | x | y | z | |||
| 1 | 0.155 | 0.148 | 0.416 | 0.948 | 1.377 | 169 |
| 2 | 0.280 | 0.320 | 0.362 | 0.582 | 0.145 | 48 |
| 3 | 0.155 | 0.142 | 0.088 | 0.822 | 1.706 | 58 |
| 4 | 0.140 | 0.151 | 0.246 | 0.304 | 0.359 | 60 |
| Resol | Nref_aa | DISOb_aa | LOCc_aa | PhPd_aa | CullRe_aa | Nref_cf | DISOb_cf | LOCc_cf | PhP d_cf | CullRe_cf |
|---|---|---|---|---|---|---|---|---|---|---|
| 36.86 | 136 | 966.4 | 948.8 | 0.22 | 0.98 | 70 | 1432.5 | 1379.0 | 0.18 | 0.96 |
| 22.59 | 847 | 903.1 | 883.2 | 0.27 | 0.98 | 273 | 1326.3 | 1213.6 | 0.24 | 0.92 |
| 16.29 | 1928 | 819.6 | 801.6 | 0.30 | 0.98 | 409 | 1216.0 | 1157.5 | 0.20 | 0.95 |
| 12.73 | 3457 | 792.7 | 777.7 | 0.30 | 0.98 | 513 | 1046.6 | 1010.2 | 0.23 | 0.97 |
| 10.45 | 5329 | 827.5 | 817.5 | 0.26 | 0.99 | 606 | 1035.8 | 1016.5 | 0.19 | 0.98 |
| 8.87 | 7544 | 698.8 | 689.7 | 0.28 | 0.99 | 655 | 926.2 | 898.7 | 0.21 | 0.97 |
| 7.70 | 9344 | 524.1 | 516.6 | 0.33 | 0.99 | 631 | 708.6 | 689.8 | 0.23 | 0.97 |
| 6.80 | 5295 | 401.7 | 396.6 | 0.38 | 0.99 | 229 | 582.1 | 559.8 | 0.28 | 0.96 |
|
| ||||||||||
| total | 33880 | 647.1 | 637.5 | 0.30 | 0.99 | 3386 | 978.0 | 941.4 | 0.22 | 0.96 |
_a: acentric
Isomorphous difference (DISO) = | FPhi − FP|
Lack of closure (LOC) = | FPH| − | FP+FH|
Phasing power (PhP) = <FH/|lack_of_closure|) >
Cullis R factor (CullR) = <lack_of_closure>/<isomorphous difference>
_c: centric
Fig. 3. Comparison between observed and calculated structure factors.

The final converged correlation coefficient (dashed line) and R-factor (solid line) between the observed amplitudes and the calculated amplitudes determined by the Fourier transform of the modified map are shown as a function of reciprocal resolution.
In an attempt to assess the quality of the phasing provided by the “Cullis R factor” we can compare the phasing power of Hg atoms in the determination of the structure of horse oxyhemoglobin, where the Cullis R-factor was originally defined (Cullis et al., 1961), with that of the Ta6Br122+ clusters in the present case. The “Cullis R factor” was about 0.8 for the various heavy atom compounds used to determine the hemoglobin structure at 6.5 Å resolution. In that case the mass of protein per asymmetric unit was about 32 kDa and the heavy atom component was two Hg atoms (160 electrons) per asymmetric unit. Thus, the protein mass per electron was about 200 Da/electron. Making a similar calculation for the Ta6Br122+ clusters in the present case gives about 425 Da/electron. The Cullis R factor was 0.9 in the present case for a phase determination extending to about 4.5 Å resolution. Thus, the slightly poorer Cullis R factor is readily justified by the far greater mass/electron.
3.5 Structure analysis
It was not possible to make a detailed structural interpretation of the resultant electron density map. However, this was not surprising, considering that the data was less than 50% complete beyond 5 Å resolution and the protein structures contained mostly β-strands with only a few short α-helices. Therefore, the program EMfit (Rossmann et al., 2001) was used to verify the orientation and position of the known structures (gp6, gp8, gp10) fitted as rigid units into the electron density map. The best fit was determined for each of the known structures in the baseplate using a complete three-dimensional search. Two good fits, significantly greater than any of the next best fits, were found for the dimeric gp6 and gp8 proteins and three good fits were found for the trimeric gp10 protein (Table 3). Furthermore, the center of mass of each of the oligomers superimposed on itself for the local 2- and 3- fold related positions, demonstrating that the oligomer symmetry axis corresponded to the local symmetry of the electron density map. However, these results might be merely the result of biased phasing from molecular replacement.
Table 3.
Quality of fitting (sumf) for the gp10, gp8, and gp6 molecules into the electron density maps using the EMfit program.
Since gp10 is a trimer there are always three equivalent fitting results. Similarly, since gp8 and gp6 are dimers there are always two equivalent fitting results.
| Molecule | Electron density map
|
|||
|---|---|---|---|---|
| MR | MR + SIR + SAD | MR + SIR + SAD + 2-fold averaging | MR + SIR + SAD + 2-fold averaging + local symmetry averaging | |
| gp10 (trimer) | 23.4, 23.4, 23.1 | 9.4, 9.2, 9.0 | 6.7, 6.5, 6.2 | 9.6, 9.3, 8.4 |
| gp8 (dimer) | 24.9, 21.9 | 9.2, 8.6 | 9.1, 8.6 | 7.5, 7.2 |
| gp6 (dimer) | 21.5, 15.8 | 9.6, 8.2 | 9.1, 8.6 | 6.5, 6.4 |
| Molecule | Electron density map
|
|||
|---|---|---|---|---|
| MR | SIR + SAD | SIR + SAD + 2-fold averaging | SIR + SAD + 2-fold averaging + local symmetry averaging | |
| gp10 (trimer) | 23.4, 23.4, 23.1 | 2.7, 2.3, 2.2 | 4.4, 4.5, 4.2 | 6.7, 6.6, 6.7 |
| gp8 (dimer) | 24.9, 21.9 | 3.2, 2.9 | 5.5, 5.3 | 4.9, 4.3 |
| gp6 (dimer) | 21.5, 15.8 | 3.8, 3.4 | 6.5, 6.1 | 4.5, 4.6 |
In order to avoid model bias, a map was calculated based only on single isomorphous replacement (SIR) and single wavelengths anomalous dispersion (SAD). First, this map was examined for the presence of the 2-fold NCS symmetry relating the two independent baseplates to each other. A correlation map was calculated that determined the correlation of the electron density within a small volume with the 2-fold NCS related density (Kleywegt and Jones, 1999). Thus, the correlation should be higher for positions within the envelope covering the volume occupied by the 2-fold NCS related baseplates. The resultant envelope (Fig. 4a) corresponded roughly to the boundary that had been determined by the earlier cryoEM studies (Fig. 4b).
Fig. 4. Segmentation of the crystallographic electron density map.
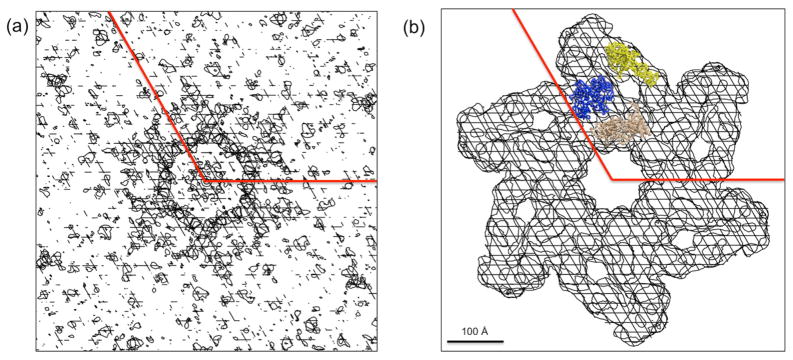
(a) An NCS 2-fold symmetry correlation coefficient map of the Ta6Br122+ phased map. (b) The molecular boundary (white shaded area) of the in vitro assembled hubless baseplate based on the cryoEM model. The position of gp10 (yellow), gp8 (blue) and gp6 (beige) are shown in (b).
The single isomorphous replacement and anomalous dispersion (SIRAS) phased map was further examined by fitting the known proteins, using “sumf”, the average density of the fitted atoms, as a measure of quality. The sumf values were significantly above noise level, but less than when the molecular replacement phases had been included in the phasing (Table 3). This map was then improved by cycles of averaging using the NCS 2-fold axis. Fitting the three known proteins into the resultant map gave good fits, albeit not quite as good as the map that had included the molecular replacement phasing (Table 3). However, gp10 had a slightly worse fit than gp6 or gp8 suggesting that it might have changed its structure slightly in the assembled baseplate relative to its independent crystal structure. Nevertheless, these results verified the general correctness of the assumed structure as the phasing was unbiased by any previous knowledge of the protein structures.
Further improvement could be made to the electron density maps by local 2-fold averaging of gp6 and of gp8 as well as local 3-fold averaging of gp10, followed by iterative 2-fold averaging refinement. This procedure improved the fitting of gp10 but reduced the quality of fit of the gp6 and gp8 molecules. However, as mentioned above, gp10 started off with a worse fit than the other two molecules, presumably because the structure of gp10 in the baseplate is a little different than in the crystal structure. Thus, local 3-fold averaging improved the quality of the map giving an overall better fit of the atomic crystal structure to the map. In contrast gp6 and gp8 have not changed their structures as much. Thus, the original fitting of gp6 and gp8 into the maps is better than gp10. However, when the maps are further improved by local averaging, the difference between the atomic structure and the maps is enhanced resulting in a loss of fitting quality.
In summary, it would appear that gp10 has some significant changes in structure when in the baseplate as compared to its crystal structure, whereas gp6 and gp8 have some changes of structure but these are much smaller. These observations are consistent with the observation that gp10 (in the absences of gp11 and the folded short tail fibers, gp12) forms the periphery of the baseplate and is therefore less restricted in its possible conformational changes. The smaller changes of structure in gp6 are undoubtedly due in part to the clash between the neighboring ends (Fig. 5) of the molecules that form an inner ring within the baseplate.
Fig. 5. Association of the proteins in the in vitro assembled baseplate.

(a) The hexameric ring of the gp10 (yellow), gp8 (blue) and gp6 (beige) proteins, based on the earlier cryoEM reconstruction of the star shape baseplate and the crystal structures of trimeric gp10, dimeric gp8, and dimeric gp6, around the 6-fold crystallographic axis. A wedge corresponds roughly to the green triangle. (b) There is a clash (black rectangle) between neighboring ends (brown and beige) of the 6-fold related gp6 molecules when fitting the independently determined crystal structures into the cryoEM density, indicating that there are at least some small conformational differences in the crystal structures.
4. DISCUSSION
It is clear that at least some of the protein structures in the baseplate undergo small perturbations when assembled into the baseplate. This is hardly surprising in view of the large conformational changes that occur in the baseplate when bacteriophage T4 infects an E. coli cell. However, to characterize these further and hence to be able to follow the pathway of the baseplate conformational changes, it will require a better resolved map. The lack of higher resolution data is probably the result of intrinsic conformational flexibility and hence lack of homogeneity of the baseplate. Crystal size was not a limitation and the use of more intense X-rays provided by the Stanford University Linac Coherent Light Source (LCLS) X-ray free electron laser did not improve the resolution limit of the baseplate crystals. The quality of the map based on the calculated structure factors of the assumed model can be seen in Fig. 6 at 5.0 Å and 4.0 Å resolutions. These “perfect” maps are compared in Fig. 6c with the present, unbiased map based on isomorphous replacement, anomalous dispersion and NCS averaging at 6.8 Å resolution. Although some α-helices can be recognized, the β-sheet regions are still very poor. However, Fig. 6b shows that a “perfect” map at 4.0 Å resolution would be sufficient to resolve the polypeptide chains. Thus, if better phasing and more of the unobserved reflections could be measured, it should be possible to obtain an interpretable map with the available crystals. This might be achievable with one or more additional isomorphous heavy atom cluster derivatives and by using a Pilatus type single photon counting detector for a greater signal-to-noise ratio in measuring the less intense reflections.
Fig. 6. Map quality possibilities.

Maps at different resolution limits were generated using calculated structure factors for all observed and unobserved reflections assuming the EM based model. A temperature factor of 200 Å2 was used for this calculation, corresponding to the effective temperature factor of the observed data (a) at 5.0 Å and (b) at 4.0 Å resolution. In (c) is shown the current, model unbiased map calculated to 6.8 Å resolution based on isomorphous replacement, anomalous dispersion and 2-fold NCS averaging using only the reflections greater than 0.8σ.
Acknowledgments
We thank Sheryl Kelly for help in the preparation of this manuscript. We are very grateful to the staff of APS sector 23 (GM/CA) for their help in setting up the station for diffraction data collection, including attending to an errant robot while we were comfortable in our remote offices. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357. We also thank many individuals at LCLS where we attempted to improve our data. In particular we thank Nicholas Sauter, Michael Soltis, Sébastien Boutet, Axel Brunger, Aina Cohen, Marc Messerschmidt, Robert Shoeman, Sabine Botha, Aaron Brewster, Johan Hattne, Mona Uervirojnangkoorn, and Oliver Zeldin. The work was supported by NIH grant (AI081726) to MGR.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aksyuk AA, Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG. The structure of gene product 6 of bacteriophage T4, the hinge-pin of the baseplate. Structure. 2009;17:800–808. doi: 10.1016/j.str.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Argos P, Rossmann MG. Determining heavy-atom positions using non-crystallographic symmetry. Acta Crystallogr sect A. 1974;30:672–677. [Google Scholar]
- Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Lenk EV, Kikuchi Y, King J. Molecular reorganization in the hexagon to star transition of the baseplate of bacteriophage T4. J Mol Biol. 1977;116:489–523. doi: 10.1016/0022-2836(77)90081-x. [DOI] [PubMed] [Google Scholar]
- Cullis AF, Muirhead H, Perutz MF, Rossmann MG, North ACT. The structure of haemoglobin. VIII. A three-dimensional Fourier synthesis at 5.5 Å resolution: determination of the phase angles. Proc Roy Soc Lond. 1961;A265:15–38. [Google Scholar]
- DePonte DP, Weierstall U, Schmidt K, Warner J, Spence JCH, Doak RB. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J Phys D: Appl Phys. 2008;41:195505. [Google Scholar]
- Hattne J, Echols N, Tran R, Kern J, Gildea RJ, Brewster AS, Alonso-Mori R, Glockner C, Hellmich J, Laksmono H, Sierra RG, Lassalle-Kaiser B, Lampe A, Han G, Gul S, DiFiore D, Milathianaki D, Fry AR, Miahnahri A, White WE, Schafer DW, Seibert MM, Koglin JE, Sokaras D, Weng TC, Sellberg J, Latimer MJ, Glatzel P, Zwart PH, Grosse-Kunstleve RW, Bogan MJ, Messerschmidt M, Williams GJ, Boutet S, Messinger J, Zouni A, Yano J, Bergmann U, Yachandra VK, Adams PD, Sauter NK. Accurate macromolecular structures using minimal measurements from X-ray free-electron lasers. Nat Methods. 2014;11:545–548. doi: 10.1038/nmeth.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA. A set of averaging programs. In: Dodson EJ, Gover S, Wolf W, editors. Molecular Replacement. SERC Daresbury Laboratory; Warrington: 1992. pp. 91–105. [Google Scholar]
- Karam JD. Molecular Biology of Bacteriophage T4. American Society for Microbiology; Washington, D.C: 1994. [Google Scholar]
- Kikuchi Y, King J. Genetic control of bacteriophage T4 baseplate morphogenesis. I Sequential assembly of the major precursor, in vivo and in vitro. J Mol Biol. 1975a;99:645–672. doi: 10.1016/s0022-2836(75)80178-1. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, King J. Genetic control of bacteriophage T4 baseplate morphogenesis. II Mutants unable to form the central part of the baseplate. J Mol Biol. 1975b;99:673–694. doi: 10.1016/s0022-2836(75)80179-3. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, King J. Genetic control of bacteriophage T4 baseplate morphogenesis. III Formation of the central plug and overall assembly pathway. J Mol Biol. 1975c;99:695–716. doi: 10.1016/s0022-2836(75)80180-x. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, King J. Genetic control of bacteriophage T4 baseplate morphogenesis. I Sequential assembly of the major precursor, in vivo and in vitro. J Mol Biol. 1975d;99:645–672. doi: 10.1016/s0022-2836(75)80178-1. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Halloween … masks and bones. In: Bailey S, Hubbard R, Waller D, editors. From First Map to Final Model. SERC Daresbury Laboratory; Daresbury, Warrington, U. K: 1994. pp. 59–66. [Google Scholar]
- Kleywegt GJ, Jones TA. xdlMAPMAN and xdlDATAMAN - programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr D Biol Crystallogr. 1996;52:826–828. doi: 10.1107/S0907444995014983. [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA. Software for handling macromolecular envelopes. Acta Crystallogr sect D. 1999;55:941–944. doi: 10.1107/s0907444999001031. [DOI] [PubMed] [Google Scholar]
- Kostyuchenko VA, Leiman PG, Chipman PR, Kanamaru S, van Raaij MJ, Arisaka F, Mesyanzhinov VV, Rossmann MG. Three-dimensional structure of bacteriophage T4 baseplate. Nat Struct Biol. 2003;10:688–693. doi: 10.1038/nsb970. [DOI] [PubMed] [Google Scholar]
- Kostyuchenko VA, Chipman PR, Leiman PG, Arisaka F, Mesyanzhinov VV, Rossmann MG. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat Struct Mol Biol. 2005;12:810–813. doi: 10.1038/nsmb975. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Shneider MM, Kostyuchenko VA, Chipman PR, Mesyanzhinov VV, Rossmann MG. Structure and location of gene product 8 in the bacteriophage T4 baseplate. J Mol Biol. 2003;328:821–823. doi: 10.1016/s0022-2836(03)00366-8. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Chipman PR, Kostyuchenko VA, Mesyanzhinov VV, Rossmann MG. Three-dimensional rearrangement of proteins in the tail of bacteriophage T4 on infection of its host. Cell. 2004;118:419–429. doi: 10.1016/j.cell.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Leiman PG, Shneider MM, Mesyanzhinov VV, Rossmann MG. Evolution of bacteriophage tails: structure of T4 gene product 10. J Mol Biol. 2006;358:912–921. doi: 10.1016/j.jmb.2006.02.058. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology Macromolecular Crystallography Part A. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Blow DM. The refinement of structures partially determined by the isomorphous replacement method. Acta Crystallogr. 1961;14:641–647. [Google Scholar]
- Rossmann MG, Blow DM. The detection of sub-units within the crystallographic asymmetric unit. Acta Crystallogr. 1962;15:24–31. [Google Scholar]
- Rossmann MG, Bernal R, Pletnev SV. Combining electron microscopic with X-ray crystallographic structures. J Struct Biol. 2001;136:190–200. doi: 10.1006/jsbi.2002.4435. [DOI] [PubMed] [Google Scholar]
- Tong L, Rossmann MG. The locked rotation function. Acta Crystallogr sect A. 1990;46:783–792. doi: 10.1107/s0108767390005530. [DOI] [PubMed] [Google Scholar]
- Yap ML, Mio K, Leiman PG, Kanamaru S, Arisaka F. The baseplate wedges of bacteriophage T4 spontaneously assemble into hubless baseplate-like structure in vitro. J Mol Biol. 2010;395:349–360. doi: 10.1016/j.jmb.2009.10.071. [DOI] [PubMed] [Google Scholar]