

Abstract
Purpose of review
There has been tremendous progress in the approach to childhood interstitial lung diseases (ILD), with particular recognition that ILD in infants is often distinct from forms that occur in older children and adults. Diagnosis is challenging due to the rarity of ILD and the fact that presenting symptoms of ILD often overlap those of common respiratory disorders. This review summarizes newly published recommendations for diagnosis and management and highlights recent scientific advances in several specific forms of childhood ILD.
Recent findings
Clinical practice guidelines emphasize the role for chest CT, genetic testing, and lung biopsy in the diagnostic evaluation of children with suspected ILD. Recent studies have better defined the characteristics and molecular understanding of several different forms of ILD, including Neuroendocrine cell Hyperplasia of Infancy (NEHI) and ILD due to mutations in genes affecting surfactant production and metabolism. Despite significant progress, definitive therapies are often lacking.
Summary
Childhood ILD encompasses a collection of rare, diffuse lung diseases. Timely recognition of children with suspected ILD and initiation of appropriate diagnostic evaluations will facilitate medical management. Systematic approaches to clinical care and further study are needed to improve the outcomes of children with these rare disorders.
Keywords: Interstitial lung disease, ABCA3, NKX2.1, SFTPC, NEHI
Introduction
The term childhood interstitial lung disease (chILD) encompasses a broad group of pulmonary disorders that are associated with significant morbidity and sometimes mortality. Historically, these diseases have been defined based on lung biopsy histopathologic findings. However, recent advances have facilitated increased noninvasive diagnosis through genetic testing and use of chest computed tomography (CT) scans. In this review, we summarize newly published recommendations for diagnosis and management of chILD and highlight recent progress in specific diseases such as NEHI and ILD due to mutations in genes affecting surfactant production and metabolism.
Definitions
Interstitial lung disease (ILD) is a term that refers to a heterogeneous collection of disorders characterized by abnormal gas exchange due to altered structure of the interstitial region of the lung. As many entities also affect the distal bronchioles and alveolar spaces, the term “diffuse lung disease” is probably a more accurate description. ILD occurs in a variety of clinical contexts, including as isolated pulmonary disorders, due to environmental exposures, as a consequence of chemotherapy or radiation therapy, and as part of systemic processes, such as autoimmune diseases. Although some of the conditions that cause ILD in children and adults are similar, there are many important differences, including distinct forms only seen in infants. Therefore, as detailed in Table 1 and summarized in a prior review on this topic[2], a classification system has been developed specifically for childhood ILD (diffuse lung disease)[1], and an expert panel recently developed an official American Thoracic Society clinical guideline on the classification, evaluation, and management of chILD, focusing on infants < 2 years of age[3**]. Further, specific chILD diagnoses are now included in the International Classification of Diseases 9th edition[4**].
Table 1.
Classification of childhood ILD
| Disorders more prevalent in infancy | |
| Category | Specific Diagnoses* |
| Diffuse developmental disorders | Acinar dysplasia Congenital alveolar dysplasia Alveolar-capillary dysplasia with pulmonary vein misalignment |
| Lung growth abnormalities | Pulmonary hypoplasia Chronic neonatal lung disease (BPD) Associated chromosomal disorders (Trisomy 21, others) Associated with congenital heart disease |
| Specific conditions of uncertain etiology | Neuroendocrine cell hyperplasia of infancy (NEHI) Pulmonary interstitial glycogenosis (PIG) |
| Surfactant dysfunction | Mutations in SFTPB, SFTPC, ABCA3, NKX2.1/TTF1 Histology consistent with surfactant dysfunction disorder but without recognized genetic etiology |
| Disorders not specific to infancy | |
| Disorders of normal host (immune-competent) | Infectious/post-infectious Aspiration Related to environmental agents (hypersensitivity pneumonitis, toxic inhalation) Eosinophilic pneumonia |
| Disorders related to systemic disease processes | Immune-related disorders (SLE, polymyositis/dermatomyositis, systemic sclerosis) Storage diseases Sarcoidosis Langerhans cell histiocytosis Malignant infiltrates |
| Disorders of immune-compromised host | Opportunistic infections Transplantation/rejection syndromes |
| Disease masquerading as ILDs | Arterial hypertensive vasculopathy Veno-occlusive disease Lymphatic disorders |
| Unclassified | Conditions that do not clearly fit into specific category (i.e. end-stage disease, inadequate or non-diagnostic biopsy specimen) |
Abbreviations: BPD=bronchopulmonary dysplasia; ILD=interstitial lung disease; SLE=systemic lupus erythematosis;
Includes examples of specific diagnoses classified within each category.
Adapted from [1] Deutsch et al, AJRCCM 2007.
Epidemiology
Overall, ILD is rare in children. Studies have estimated a prevalence of 3.6 cases per million in the United Kingdom and Ireland[5] and 1.32 cases per million in Germany[6]. In France, beginning in 2006, a National Reference Center for Rare Lung Diseases (RespiRare) was created to centralize data collection, and over 200 cases of ILD were identified over 3 years, though a specific diagnosis could not be established for ~25% of cases [7*]. Retrospective studies in North America suggest that increased clinician awareness of childhood ILD and improved use of diagnostic tools has resulted in greater case ascertainment[2,8**], but the prevalence of these disorders may still be under-estimated.
Diagnostic approach
Children with ILD typically manifest non-specific respiratory signs and symptoms, including tachypnea, hypoxemia, crackles, cough, and poor growth. Because these symptoms overlap those seen in many more common conditions, the first step in diagnostic evaluation is to exclude more common causes of diffuse lung disease (i.e. cystic fibrosis, immunodeficiency, congenital heart disease, pulmonary infection, primary ciliary dyskinesia, and recurrent aspiration) (Table 2). After excluding or treating these more common causes of lung disease, the term “chILD syndrome”[9] is then used to refer to children who meet 3 out 4 of the following criteria,: (1) respiratory symptoms (e.g. cough, rapid and or difficult breathing, or exercise intolerance; (2) respiratory signs (e.g. resting tachypnea, adventitious sounds, retractions, digital clubbing, failure to thrive, or respiratory failure; (3) hypoxemia; and (4) diffuse parenchymal abnormalities on chest imaging.
Table 2.
Initial Diagnostic Approach for ILD: Exclude more common conditions
| Possible diagnoses to exclude before evaluating for childhood ILD* |
Diagnostic Approaches |
|---|---|
| Infection | Appropriate cultures Consider bronchoscopy and bronchoalveolar lavage |
| Cystic fibrosis | Sweat chloride |
| Immunodeficiency (primary vs. secondary)** | Complete blood count and differential, HIV, immunoglobulins, vaccine response, others as indicated |
| Recurrent aspiration | Barium swallow study |
| Congenital heart disease/pulmonary hypertension | Echocardiogram, cardiac catheterization (select cases) |
Note that identification of these diagnoses does not completely preclude diagnosis of ILD. If respiratory symptoms persist despite treatment of the identified abnormalities or severity is out proportion to the identified causes, additional ILD evaluations may be further considered.
Certain forms of ILD also occur in children with immunodeficiency and immune dysfunction.
The recently published ATS clinical guideline describes the primary diagnostic tools used for evaluation of childhood ILD, emphasize (1) bronchoscopy with bronchoalveolar lavage (BAL), (2) chest CT, (3) genetic testing, and (4) lung biopsy[3**]. Not all tests are needed in all cases. Generally, the evaluation proceeds from least to most invasive procedures, although the sequence depends on the context, acuity and severity of the patient’s condition. Pulmonary function testing (PFT) and assessment of oxygenation with sleep and exercise (or feeding in infants) are used to characterize the degree and nature of physiologic impairment. Further, screening for pulmonary hypertension may influence the pace of diagnostic evaluations, alter treatment, and impact prognosis, as pulmonary hypertension associated with ILD predicts higher mortality[2,10]. The primary diagnostic modalities used in childhood ILD are reviewed below and summarized in Table 3.
Table 3.
Summary of key methods for evaluation of children with suspected ILD
| Test | Pros | Cons | Comments |
|---|---|---|---|
| Flexible bronchoscopy with bronchoalveolar lavage | Evaluate anatomy of airways and sample alveolar cellular composition Obtain specimen for microbiological evaluation Cytologic studies may support specific diagnoses (i.e. pulmonary hemorrhage, PAP) |
Though minimally invasive, general anesthesia required in children Potential risks: hypoxemia, bronchospasm Rarely provides definitive diagnosis of ILD |
Infection, aspiration, or airway anatomical abnormalities may be co-morbid conditions |
| Chest CT | Characterizes nature, extent, and distribution of diseased lung Provides findings that are specific for some types of ILD and thus may provide definitive diagnosis in some cases May guide lung selection of lung biopsy sites if needed |
May require sedation or anesthesia for raised volumes and to reduce motion artifact in young children (CV-HRCT) Involves radiation exposure |
Technique critical, with coordination between radiologist and anesthesiologist for infants/young children Protocols developed for infants and children limit radiation exposure |
| Genetic testing | Can provide definitive diagnosis for known single-gene disorders Non-invasive May lead to avoidance of unnecessary diagnostic procedures Potentially provides prognostic information |
Relative cost Known limitations of current sequencing technologies |
Genetic basis not known for many forms of childhood ILD |
| Lung biopsy | Gold-standard for diagnosis of many forms of ILD* | Invasive Small number of cases remain unclassifiable even after lung biopsy |
VATS is preferred due to lower rates of morbidity compared to open lung biopsy Tissue processing critical |
Genetic testing is considered a gold-standard for certain forms of childhood ILD and generally obviates the need for lung biopsy if positive
Definition of abbreviations: CV-HRCT = controlled ventilation high resolution chest CT, ILD=interstitial lung disease, PAP = pulmonary alveolar proteinosis
Bronchoscopy with bronchoalveolar lavage (BAL)
This is a common, invasive procedure that is performed to evaluate children with suspected ILD. In addition to enabling evaluation of airway anatomy and physiology, airway and alveolar samples are obtained for cytology and microbiologic diagnosis. Bronchoscopy is relatively safe, widely available, and may help diagnose infection, aspiration, hemorrhage, or pulmonary alveolar proteinosis (PAP).
Imaging studies
Chest CT is very useful for defining the extent and pattern of disease with resolution that is superior to plain chest radiographs. Common findings in chILD may include ground glass opacification, consolidation, and septal thickening. Findings may be suggestive or even specific for some types of ILD, including surfactant dysfunction disorders, bronchiolitis obliterans and Neuorendocrine cell Hyperplasia of Infancy (NEHI)[3**,11–14] and therefore may reduce need for lung biopsy. In cases where lung biopsy is required, CT imaging will guide the choice of biopsy sites. In infants, anesthesia or controlled ventilation techniques are often needed to decrease motion artifact and atelectasis that may obscure detection of lung pathology[15,16]. Imaging protocols designed specifically for young children at experienced centers significantly reduce radiation exposure[17].
Genetic tests
The availability of clinical genetic testing now allows for non-invasive definitive diagnosis in some cases. The currently known genetic causes of chILD include abnormalities in the genes encoding surfactant protein B (SFTPB), surfactant protein C (SFTPC), ATP-binding cassette transporter A-3 (ABCA3), GM-CSF receptors α and β (CSFRA and CSFRB), and thyroid transcription factor-1 (NKX2.1/TTF1)[3**]. The choice of specific genetic tests should be guided by the family history and clinical context. A specific diagnosis provides clinically useful information for the great majority of cases as it informs management, prognosis and genetic counseling. Currently, only a subset of types of childhood ILD has a defined genetic basis. However, it is likely that additional disease-associated genes will be identified in the future.
Lung Biopsy
In the absence of genetic diagnosis, lung biopsy remains the gold standard for diagnosis of many forms of chILD. To optimize diagnostic yield, standardized protocols have been developed[18], that require timely and effective communication between the clinician, radiologist, and surgeon to select proper biopsy site(s) and process tissue, including fixation in glutaraldehyde for electron microscopy.
Approach to classification of childhood ILD
Definitive diagnosis can be achieved in most cases using the approaches described above. The published classification system (Table 1) provides a useful framework for organizing the large spectrum of ILD diagnoses and has been validated in several studies. However, despite invasive evaluations, a portion of ILD cases (11% in one study)[1] remain unclassifiable, attributable in part to insufficient tissue sampling. Further, the current classification has some conceptual and practical limitations, and recent studies have suggested modifications to better incorporate cases diagnosed using genetic and/or radiographic criteria [8**] and the breadth of diseases seen in older children[19*].
As reviewed below, there several forms of ILD where considerable progress in clinical description and understanding of disease pathogenesis has recently unfolded.
ILD due to defects in NKX2.1/TTF-1
TTF-1 (encoded by the gene NKX2.1) is a transcription factor and early marker of epithelial differentiation in the lung. TTF-1 is critical for lung development and surfactant homeostasis, regulating expression of molecules such as surfactant proteins B and C, and the ATP-binding cassette transporter 3 (ABCA3). Mutations or deletions of one NKX2.1 allele can result in neurologic abnormalities including chorea, hypothyroidism, and neonatal respiratory distress syndrome (RDS) that have been clinically characterized as the brain-thyroid-lung syndrome (MIM 610978)[20,21]. A recent study found highly variable clinical, radiographic, and histologic phenotypes in children with NKX2.1 mutations or deletions. Some children presented with respiratory failure in the newborn period while others manifested chronic ILD later in life[22**]. Genetic testing for mutations and deletions in NKX2.1 is recommended in infants with chILD syndrome and evidence of thyroid dysfunction or neurologic deficits, though lung disease can also occur in the absence of brain or thyroid abnormalities. Although no specific molecular-based therapy is currently available, identification of this mutation in a child with ILD can obviate the need for additional invasive testing and prompt aggressive infection prevention measures, as recurrent pulmonary infections have been reported in children with NKX2.1 haploinsufficiency[20,21,22**].
ILD due to ABCA3 mutations
Recessive mutations in the gene encoding ABCA-3 have been associated with neonatal respiratory failure in full-term infants (attributed to defects in surfactant metabolism) [23–25] as well as ILD in older children and adults[12,26–28]. In addition, there are several prior reports of patients with a clinical presentation consistent with ABCA3 deficiency, with identification of only a single mutation in ABCA3. Another case report demonstrated ABCA3 deficiency by histologic and ultrastructural analysis, yet no mutations were detected by exon-based sequencing[29]. A recent publication establishes one potential explanation for such cases, as a novel intronic variant was identified in a full-term infant with neonatal respiratory failure and radiographic evidence of ILD, but for whom initial conventional sequencing only identified a single ABCA3 mutation[30*]. In addition, 2 cases have now been published demonstrating ILD due to gene deletions (one in ABCA3 and one in SFTPC)[31*]. These cases highlight the importance of understanding the limitations of specific genetic testing protocols, as traditional single-gene exon-based sequencing will not detect deleterious mutations in non-coding regions, deletions, or duplications within the candidate gene. Thus, the clinician must re-assess the clinical suspicion for ILD and pursue additional evaluation if the radiographic or histologic features support a specific diagnosis.
While homozygous or compound heterozygous mutations in ABCA3 are a cause of lethal neonatal failure in term infants, Wambach et al. found that a having a single ABCA3 mutation significantly increases risk for RDS in term or late pre-term infants[32**], without ILD. This study estimated a carrier allele frequency of ~1 in 3100 individuals of European-descent and ~1 in 18,000 individuals of African-descent, making these frequencies similar to cystic fibrosis. This study demonstrates how insights from studying rare conditions can contribute to understanding pathogenesis of more common conditions.
ILD due to surfactant protein C (SFTPC) gene mutations
While SFTPB mutations are almost exclusively associated with lethal neonatal lung disease, SFTPC mutations can be associated with ILD presenting in older children and adults[33–36]. The clinical presentation and natural history of lung disease in children with SFTPC mutations is highly variable, though systematic long-term data are lacking. A recent publication by Avital et al describes the outcomes of 5 children with SFTPC mutations who are now young adults. The authors suggest these children may have benefited from early hydroxychloroquine treatment[37*], though clinical trials are needed to formally test treatment regimens, particularly in light of the known variability in natural history.
The pathogenesis of SFTPC mutations is thought to be due to cytotoxic accumulation of misfolded surfactant protein-C (SP-C) proprotein and endoplasmic reticulum (ER) stress in at least some cases[38–42]. A recent in vitro study showed that the small molecule 4-pheynlbutryic acid (PBA) restores proper trafficking of one form of mutant SP-C[43*]. While further pre-clinical and clinical trials are needed, this study emphasizes the potential importance of understanding genotype-specific disease pathogenesis in developing patient-specific targeted therapies.
Neuroendocrine cell hyperplasia of infancy (NEHI)
Infants with NEHI typically present during early infancy with indolent onset of persistent tachypnea, retractions, and hypoxemia[44]. Lung biopsies have shown a characteristic excess of neuroendocrine cells in the setting of otherwise strikingly normal histology[45,46]. Although lung biopsy has been the traditional gold-standard for diagnosis, NEHI is associated with a highly specific chest CT pattern such that many patients are now diagnosed without lung biopsy (Figure 1). In one study, the sensitivity of chest CT in diagnosing NEHI was 78% compared to lung biopsy, and the specificity was 100%[14].
Figure 1.
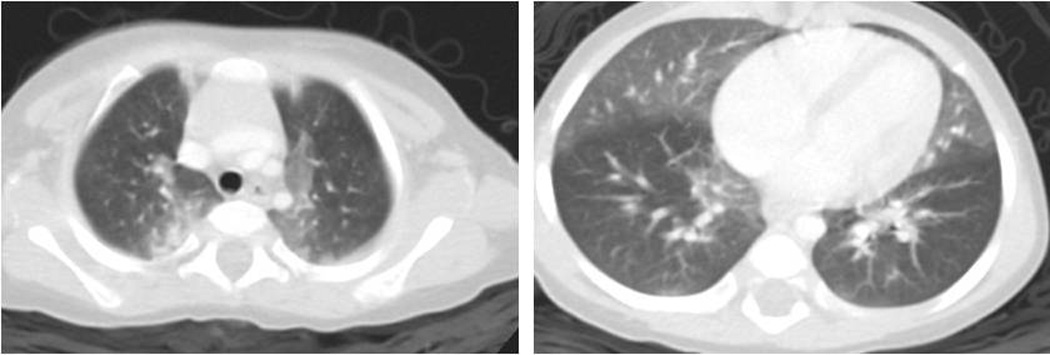
High resolution computed tomography (CT) image showing geographic ground-glass attenuation in bilateral perihilar regions, right middle lobe, and lingula in a pattern considered characteristic for neuroendocrine cell hyperplasia of infancy (NEHI). Note that the absence of bronchiectasis, architectural distortion or other significant findings is required for radiographic diagnosis of NEHI. Air-trapping is commonly demonstrated through comparison of inspiratory and expiratory images (not shown).
Other non-invasive means of evaluating children with NEHI include infant pulmonary function testing (iPFT). A retrospective single center study demonstrated significant obstruction and air-trapping in NEHI patients[47*]. There is some evidence that physiologic obstruction may correlate with extent of neuroendocrine cell prevalence[46] and with future abnormalities in physiologic parameters[47*]. Additional prospective and longitudinal studies will be needed to better define the diagnostic accuracy and prognostic value of iPFTs in NEHI. iPFT requires sedation, experienced personnel, and specialized equipment that is not available at all pediatric pulmonary centers.
Despite needing supplemental oxygen for many years and exhibiting significant respiratory distress during infancy and early childhood, current experience indicates that NEHI patients gradually improve over time, though long-term data are lacking. Recent longitudinal studies from Finland and Brazil examined small cohorts (n=9 and 12, respectively) of infants followed for 3–10 years after initial diagnosis[48*,49]. The majority of infants in both studies received corticosteroids, but did not show clinical response and continued to exhibit physiologic evidence of airflow obstruction during the follow-up period. This is consistent with prior reports and the overall lack of inflammation observed in NEHI lung biopsies, and emphasizes how establishing a diagnosis of NEHI can mitigate use of unnecessary therapies [44,45].
Reported familial cases suggest that there may be a genetic basis for NEHI. Using a candidate gene approach, Young et al. report a novel heterozygous mutation in NKX2.1 in a subject with classic NEHI presentation and biopsy, and this mutation segregated with lung disease in 5 other members of the family. However, mutations in NKX2.1 were not found in 8 other unrelated individuals with sporadic and familial NEHI, suggesting that NKX2.1 mutations are not the predominant cause of NEHI[50*]. Future studies using whole genome or exome sequencing may reveal additional genetic causes and may provide insight into the underlying pathogenesis of this disease.
Management and treatment
The pathogenesis of many forms of ILD remains poorly understood and treatment approaches remain largely empirical. Indeed, there have been no controlled trials of any therapeutic intervention in chILD. Management is largely supportive, including supplemental oxygen and ventilatory support, nutritional support, proper immunizations, and avoidance of harmful environmental exposures. While rheumatologic disorders generally respond well to immunosuppressive medications, there is no clear evidence of efficacy of systemic corticosteroids or hydroxychloroquine in most other forms of childhood ILD. Treatment decisions are thus highly individual, carefully considering potential treatment associated side effects. Lung transplantation is an option for children with end-stage lung disease[51*]. Genetic counseling and family support are also important components of care.
Conclusion
Childhood ILD is a group of heterogeneous disorders which are associated with significant morbidity and sometimes mortality. Recently published guidelines describing systematic diagnostic approaches and establishment of standardized nomenclature are improving clinical care and promoting advances in mechanistic understanding of disease pathogenesis. An increasing proportion of cases are now diagnosed without lung biopsy through use of chest CT imaging patterns and genetic testing. Obtaining a specific diagnosis often has important implications in patient management and prognosis, as well as for genetic counseling. Ongoing research to reveal genetic and molecular defects will ultimately lead to specific therapies in this diverse group of rare respiratory diseases. Moreover, these rare disorders continue to provide insight into pathogenesis of more common forms of lung disease.
KEY POINTS.
Childhood ILD manifests non-specific respiratory signs and symptoms including tachypnea, hypoxemia, crackles, cough, and poor growth, and systematic evaluation for more common diseases presenting similarly should be undertaken prior to consideration of ILD.
ILD in children younger than 2 years of age is typically distinct from ILD presenting in older children and adults.
Specific forms of ILD (i.e. NEHI, and disorders of surfactant metabolism) may be diagnosed without lung biopsy.
Clinical guidelines for evaluating and classifying ILD in children < 2 years of age have been developed. Application of this classification system has been validated in recent studies.
Acknowledgments
Funding: NIH HL119503 (LRY)
Abbreviations
- ABCA3
ATP-binding cassette member A-3
- BAL
bronchoalveolar lavage
- chILD
childhood interstitial lung disease
- CSFRA and CSFRB
genes encoding the GM-CSF receptors α and β, respectively
- CT
computed tomography
- ILD
interstitial lung disease
- iPFT
infant pulmonary function testing
- NEHI
neuroendocrine cell hyperplasia of infancy
- NKX2.1/TTF1
gene encoding the thyroid transcription factor-1
- PAP
pulmonary alveolar proteinosis
- PFT
pulmonary function testing
- RDS
respiratory distress syndrome
- SFTPB
gene encoding surfactant protein B
- SFTPC
gene encoding surfactant protein C
Footnotes
The authors report no conflicts of interest.
References
- 1.Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 2007;176(11):1120–1128. doi: 10.1164/rccm.200703-393OC. Epub 2007/09/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das S, Langston C, Fan LL. Interstitial lung disease in children. Curr Opin Pediatr. 2011;23(3):325–331. doi: 10.1097/MOP.0b013e3283464a37. Epub 2011/05/17. [DOI] [PubMed] [Google Scholar]
- 3. Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, Castile RG, et al. An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 2013;188(3):376–394. doi: 10.1164/rccm.201305-0923ST. This is the first clinical practice guideline focused specifically to children with ILD, it represents the synthesis of many discoveries and multidisciplinary efforts which have transformed the understanding and approach to childhood ILD over the past decade. The guideline provides useful algorithms and technical considerations for diagnostic modalities including genetic testing, chest CT scans, infant pulmonary function testing, and lung biopsy in young children undergoing evaluation for suspected ILD.
- 4. Popler J, Lesnick B, Dishop MK, Deterding RR. New coding in the International Classification of Diseases, Ninth Revision, for children's interstitial lung disease. Chest. 2012;142(3):774–780. doi: 10.1378/chest.12-0492. Epub 2012/09/06. This article summarizes new ICD-9 codes which have been developed recognizing the specific forms of ILD distinct in children. Implementation is expected to improve epidemiologic and other studies aimed at understanding and improving the clinical care of children with these rare disorders.
- 5.Dinwiddie R, Sharief N, Crawford O. Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and Ireland. Pediatr Pulmonol. 2002;34(1):23–29. doi: 10.1002/ppul.10125. Epub 2002/07/12. [DOI] [PubMed] [Google Scholar]
- 6.Griese M, Haug M, Brasch F, Freihorst A, Lohse P, von Kries R, et al. Incidence and classification of pediatric diffuse parenchymal lung diseases in Germany. Orphanet journal of rare diseases. 2009;4:26. doi: 10.1186/1750-1172-4-26. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nathan N, Taam RA, Epaud R, Delacourt C, Deschildre A, Reix P, et al. A national internet-linked based database for pediatric interstitial lung diseases: the French network. Orphanet journal of rare diseases. 2012;7:40. doi: 10.1186/1750-1172-7-40. Epub 2012/06/19. This study reports the scope and rapid ascertainment of childhood ILD in France after implementation of a national registry effort.
- 8. Soares JJ, Deutsch GH, Moore PE, Fazili MF, Austin ED, Brown RF, et al. Childhood interstitial lung diseases: an 18-year retrospective analysis. Pediatrics. 2013;132(4):684–691. doi: 10.1542/peds.2013-1780. Epub 2013/10/02. This study demonstrates that cases of newly described forms of childhood interstitial lung diseases likely occur at all children’s hospitals. With advances in genetic testing and recognition of imaging patterns, a significant portion of cases are identifiable with noninvasive evaluations.
- 9.Fan LL, Deterding RR, Langston C. Pediatric interstitial lung disease revisited. Pediatr Pulmonol. 2004;38(5):369–378. doi: 10.1002/ppul.20114. Epub 2004/09/18. [DOI] [PubMed] [Google Scholar]
- 10.Fan LL, Kozinetz CA. Factors influencing survival in children with chronic interstitial lung disease. Am J Respir Crit Care Med. 1997;156(3 Pt 1):939–942. doi: 10.1164/ajrccm.156.3.9703051. Epub 1997/10/06. [DOI] [PubMed] [Google Scholar]
- 11.Copley SJ, Coren M, Nicholson AG, Rubens MB, Bush A, Hansell DM. Diagnostic accuracy of thin-section CT and chest radiography of pediatric interstitial lung disease. AJR Am J Roentgenol. 2000;174(2):549–554. doi: 10.2214/ajr.174.2.1740549. Epub 2000/02/05. [DOI] [PubMed] [Google Scholar]
- 12.Doan ML, Guillerman RP, Dishop MK, Nogee LM, Langston C, Mallory GB, et al. Clinical, radiological and pathological features of ABCA3 mutations in children. Thorax. 2008;63(4):366–373. doi: 10.1136/thx.2007.083766. Epub 2007/11/21. [DOI] [PubMed] [Google Scholar]
- 13.Lynch DA, Hay T, Newell JD, Jr, Divgi VD, Fan LL. Pediatric diffuse lung disease: diagnosis and classification using high-resolution CT. AJR Am J Roentgenol. 1999;173(3):713–718. doi: 10.2214/ajr.173.3.10470910. Epub 1999/09/02. [DOI] [PubMed] [Google Scholar]
- 14.Brody AS, Guillerman RP, Hay TC, Wagner BD, Young LR, Deutsch GH, et al. Neuroendocrine cell hyperplasia of infancy: diagnosis with high-resolution CT. AJR Am J Roentgenol. 2010;194(1):238–244. doi: 10.2214/AJR.09.3385. Epub 2009/12/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long FR, Castile RG, Brody AS, Hogan MJ, Flucke RL, Filbrun DA, et al. Lungs in infants and young children: improved thin-section CT with a noninvasive controlled-ventilation technique--initial experience. Radiology. 1999;212(2):588–593. doi: 10.1148/radiology.212.2.r99au06588. Epub 1999/08/03. [DOI] [PubMed] [Google Scholar]
- 16.Long FR, Castile RG. Technique and clinical applications of full-inflation and end-exhalation controlled-ventilation chest CT in infants and young children. Pediatr Radiol. 2001;31(6):413–422. doi: 10.1007/s002470100462. Epub 2001/07/05. [DOI] [PubMed] [Google Scholar]
- 17.Frush DP. Pediatric CT: practical approach to diminish the radiation dose. Pediatr Radiol. 2002;32(10):714–717. doi: 10.1007/s00247-002-0797-1. discussion 51-4. Epub 2002/09/24. [DOI] [PubMed] [Google Scholar]
- 18.Langston C, Patterson K, Dishop MK, Askin F, Baker P, Chou P, et al. A protocol for the handling of tissue obtained by operative lung biopsy: recommendations of the chILD pathology co-operative group. Pediatr Dev Pathol. 2006;9(3):173–180. doi: 10.2350/06-03-0065.1. Epub 2006/09/02. [DOI] [PubMed] [Google Scholar]
- 19. Rice A, Tran-Dang MA, Bush A, Nicholson AG. Diffuse lung disease in infancy and childhood: expanding the chILD classification. Histopathology. 2013;63(6):743–755. doi: 10.1111/his.12185. Epub 2013/10/15. This study reports the histopathologic findings of a large number of diffuse lung disease cases in the United Kingdom. The authors suggest need for modifications to the current chILD classification system to better incorporate the scope of entities seen in older children.
- 20.Krude H, Schutz B, Biebermann H, von Moers A, Schnabel D, Neitzel H, et al. Choreoathetosis, hypothyroidism, and pulmonary alterations due to human NKX2-1 haploinsufficiency. J Clin Invest. 2002;109(4):475–480. doi: 10.1172/JCI14341. Epub 2002/02/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carre A, Szinnai G, Castanet M, Sura-Trueba S, Tron E, Broutin-L'Hermite I, et al. Five new TTF1/NKX2.1 mutations in brain-lung-thyroid syndrome: rescue by PAX8 synergism in one case. Hum Mol Genet. 2009;18(12):2266–2276. doi: 10.1093/hmg/ddp162. Epub 2009/04/02. [DOI] [PubMed] [Google Scholar]
- 22. Hamvas A, Deterding RR, Wert SE, White FV, Dishop MK, Alfano DN, et al. Heterogeneous Pulmonary Phenotypes Associated With Mutations in the Thyroid Transcription Factor Gene NKX2-1. Chest. 2013;144(3):794–804. doi: 10.1378/chest.12-2502. Epub 2013/02/23. This article provides detailed clinical, radiographic, and lung histologic findings in 21 cases of ILD due to NKX2.1/TTF1 mutations and deletions. The study expands the known phenotypes associated with this disorder.
- 23.Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350(13):1296–1303. doi: 10.1056/NEJMoa032178. Epub 2004/03/27. [DOI] [PubMed] [Google Scholar]
- 24.Garmany TH, Moxley MA, White FV, Dean M, Hull WM, Whitsett JA, et al. Surfactant composition and function in patients with ABCA3 mutations. Pediatr Res. 2006;59(6):801–805. doi: 10.1203/01.pdr.0000219311.14291.df. Epub 2006/04/28. [DOI] [PubMed] [Google Scholar]
- 25.Brasch F, Schimanski S, Muhlfeld C, Barlage S, Langmann T, Aslanidis C, et al. Alteration of the pulmonary surfactant system in full-term infants with hereditary ABCA3 deficiency. Am J Respir Crit Care Med. 2006;174(5):571–580. doi: 10.1164/rccm.200509-1535OC. Epub 2006/05/27. [DOI] [PubMed] [Google Scholar]
- 26.Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. Am J Respir Crit Care Med. 2005;172(8):1026–1031. doi: 10.1164/rccm.200503-504OC. Epub 2005/06/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young LR, Nogee LM, Barnett B, Panos RJ, Colby TV, Deutsch GH. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest. 2008;134(1):192–195. doi: 10.1378/chest.07-2652. Epub 2008/07/17. [DOI] [PubMed] [Google Scholar]
- 28.Flamein F, Riffault L, Muselet-Charlier C, Pernelle J, Feldmann D, Jonard L, et al. Molecular and cellular characteristics of ABCA3 mutations associated with diffuse parenchymal lung diseases in children. Hum Mol Genet. 2012;21(4):765–775. doi: 10.1093/hmg/ddr508. Epub 2011/11/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gower WA, Wert SE, Ginsberg JS, Golan A, Whitsett JA, Nogee LM. Fatal familial lung disease caused by ABCA3 deficiency without identified ABCA3 mutations. J Pediatr. 2010;157(1):62–68. doi: 10.1016/j.jpeds.2010.01.010. Epub 2010/03/23. [DOI] [PubMed] [Google Scholar]
- 30. Agrawal A, Hamvas A, Cole FS, Wambach JA, Wegner D, Coghill C, et al. An intronic ABCA3 mutation that is responsible for respiratory disease. Pediatr Res. 2012;71(6):633–637. doi: 10.1038/pr.2012.21. Epub 2012/02/18. This study reports a case in which ILD was strongly suspected but only one ABCA3 mutation was initially identified in coding regions. This case illustrates the potential limitations of current clinical sequencing technology and the need for appropriate clinical suspicion to pursue additional testing in selected cases.
- 31. Henderson LB, Melton K, Wert S, Couriel J, Bush A, Ashworth M, et al. Large ABCA3 and SFTPC Deletions Resulting in Lung Disease. Annals of the American Thoracic Society. 2013 doi: 10.1513/AnnalsATS.201306-170OC. Epub 2013/09/13. This study reports two cases in which large deletions were identified in children with suspected ILD, but for whom clinical genetic testing was unrevealing.
- 32. Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, et al. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012;130(6):e1575–e1582. doi: 10.1542/peds.2012-0918. Epub 2012/11/21. This study demonstrates that single ABCA3 mutations are common in the population and contribute to the risk of a common respiratory problem in infants. This is a prime example of how findings from a rare disease can lead to mechanistic understanding of more common diseases.
- 33.Nogee LM, Dunbar AE, 3rd, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–579. doi: 10.1056/NEJM200102223440805. Epub 2001/02/24. [DOI] [PubMed] [Google Scholar]
- 34.Thomas AQ, Lane K, Phillips J, 3rd, Prince M, Markin C, Speer M, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–1328. doi: 10.1164/rccm.200112-123OC. Epub 2002/05/07. [DOI] [PubMed] [Google Scholar]
- 35.Cameron HS, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, et al. A common mutation in the surfactant protein C gene associated with lung disease. J Pediatr. 2005;146(3):370–375. doi: 10.1016/j.jpeds.2004.10.028. Epub 2005/03/10. [DOI] [PubMed] [Google Scholar]
- 36.van Moorsel CH, van Oosterhout MF, Barlo NP, de Jong PA, van der Vis JJ, Ruven HJ, et al. Surfactant protein C mutations are the basis of a significant portion of adult familial pulmonary fibrosis in a dutch cohort. Am J Respir Crit Care Med. 2010;182(11):1419–1425. doi: 10.1164/rccm.200906-0953OC. Epub 2010/07/27. [DOI] [PubMed] [Google Scholar]
- 37. Avital A, Hevroni A, Godfrey S, Cohen S, Maayan C, Nusair S, et al. Natural history of five children with surfactant protein C mutations and interstitial lung disease. Pediatr Pulmonol. 2013 doi: 10.1002/ppul.22971. Epub 2013/12/19. This study reports features and long-term follow-up for five patients with SFTPC mutations.
- 38.Kabore AF, Wang WJ, Russo SJ, Beers MF. Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues. J Cell Sci. 2001;114(Pt 2):293–302. doi: 10.1242/jcs.114.2.293. Epub 2001/01/10. [DOI] [PubMed] [Google Scholar]
- 39.Wang WJ, Mulugeta S, Russo SJ, Beers MF. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J Cell Sci. 2003;116(Pt 4):683–692. doi: 10.1242/jcs.00267. Epub 2003/01/23. [DOI] [PubMed] [Google Scholar]
- 40.Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am J Respir Cell Mol Biol. 2005;32(6):521–530. doi: 10.1165/rcmb.2005-0009OC. Epub 2005/03/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. Epub 2008/04/09. [DOI] [PubMed] [Google Scholar]
- 42.Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, Keranen H, et al. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci U S A. 2012;109(7):2325–2329. doi: 10.1073/pnas.1114740109. Epub 2012/02/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stewart GA, Ridsdale R, Martin EP, Na CL, Xu Y, Mandapaka K, et al. 4-Phenylbutyric acid treatment rescues trafficking and processing of a mutant surfactant protein-C. Am J Respir Cell Mol Biol. 2012;47(3):324–331. doi: 10.1165/rcmb.2012-0003OC. Epub 2012/03/31. This study utilized in vitro cell culture systems to study the trafficking surfactant protein C mutations associated with lung disease. The findings suggest that 4-Phenylbutyric acid may warrant further study for treatment of ILD associated with certain SFTPC mutations. The results also emphasize the need for detailed understanding of mutation-specific disease biology for personalized medicine.
- 44.Deterding RR, Fan LL, Morton R, Hay TC, Langston C. Persistent tachypnea of infancy (PTI)--a new entity. Pediatr Pulmonol. 2001;(Suppl 23):72–73. Epub 2002/03/12. [PubMed] [Google Scholar]
- 45.Deterding RR, Pye C, Fan LL, Langston C. Persistent tachypnea of infancy is associated with neuroendocrine cell hyperplasia. Pediatr Pulmonol. 2005;40(2):157–165. doi: 10.1002/ppul.20243. Epub 2005/06/21. [DOI] [PubMed] [Google Scholar]
- 46.Young LR, Brody AS, Inge TH, Acton JD, Bokulic RE, Langston C, et al. Neuroendocrine cell distribution and frequency distinguish neuroendocrine cell hyperplasia of infancy from other pulmonary disorders. Chest. 2011;139(5):1060–1071. doi: 10.1378/chest.10-1304. Epub 2010/10/05. [DOI] [PubMed] [Google Scholar]
- 47. Kerby GS, Wagner BD, Popler J, Hay TC, Kopecky C, Wilcox SL, et al. Abnormal infant pulmonary function in young children with neuroendocrine cell hyperplasia of infancy. Pediatr Pulmonol. 2013;48(10):1008–1015. doi: 10.1002/ppul.22718. Epub 2012/11/22. This article reports the infant PFT findings from the largest number of NEHI cases to date, and demonstrates that high-quality infant PFTs are feasible in this population. The physiologic abnormalities observed in this disease are intriguing, and further study is warranted to define the diagnostic specificity and prognostic value of the findings.
- 48.Gomes VC, Silva MC, Maia JHF, Daltro P, Ramos SG, Brody AS, et al. Diagnostic criteria and follow-up in neuroendocrine cell hyperplasia of infancy: a case series. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia. 2013;39(5):569–578. doi: 10.1590/S1806-37132013000500007. Epub 2013/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lukkarinen H, Pelkonen A, Lohi J, Malmstrom K, Malmberg LP, Kajosaari M, et al. Neuroendocrine cell hyperplasia of infancy: a prospective follow-up of nine children. Archives of disease in childhood. 2013;98(2):141–144. doi: 10.1136/archdischild-2012-302115. Epub 2012/11/20. This study reports the features and clinical course of 9 children with NEHI, providing important information about the longitudinal course.
- 50. Young LR, Deutsch GH, Bokulic RE, Brody AS, Nogee LM. A Mutation in TTF1/NKX2.1 Is Associated With Familial Neuroendocrine Cell Hyperplasia of Infancy. Chest. 2013;144(4):1199–1206. doi: 10.1378/chest.13-0811. Epub 2013/06/22. This study reports the first identified genetic cause for NEHI, and that NEHI can be caused by genetic mechanisms. However, as NKX2.1/TTF1 mutations were not identified in other familial and sporadic NEHI cases, the findings suggest that it is not the predominant cause.
- 51. Rama JA, Fan LL, Faro A, Elidemir O, Morales DL, Heinle JS, et al. Lung transplantation for childhood diffuse lung disease. Pediatr Pulmonol. 2013;48(5):490–496. doi: 10.1002/ppul.22634. Epub 2012/09/06. This article reports the experience in lung transplantation of childhood diffuse lung diseases. It highlights that lung transplantation is an option for severe or progressive forms of childhood ILD.