

Abstract
Cytotoxic chemotherapeutic drugs, especially when used in combination, are widely employed to treat a variety of cancers in patients but often lead to serious symptoms that negatively affect physical functioning and quality of life. There is compelling evidence that implicates cytotoxic chemotherapy-induced inflammation in the etiology of these symptoms. Because IL-1β plays a central role as an initiator cytokine in immune responses, we compared doxorubicin, a drug known to induce IL-1β production, with ten other commonly prescribed chemotherapeutic drugs in their ability to lead to processing and secretion of IL-1β by primary mouse macrophages. Seven of them (melphalan, cisplatin, vincristine, etoposide, paclitaxel, methotrexate, and cytarabine) caused the production of IL-1β in cells pretreated with lipopolysaccharide. When delivered in combination with doxorubicin, one of the drugs, vincristine, was also capable of synergistically activating the NLRP3-dependent inflammasome and increasing expression of IL-1β, IL-6, and CXCL1. The absence of TNF-α and IL-1 signaling caused a partial reduction in the production of mature IL-1β. Three small-molecule inhibitors known to suppress activity of kinases situated upstream of mitogen-activated kinases (MAPKs) inhibited the expression of IL-1β, IL-6, and CXCL1 when doxorubicin and vincristine were used singly or together, so specific kinase inhibitors may be useful in reducing inflammation in patients receiving chemotherapy.
Keywords: inflammasome, cytokines, nilotinib, ponatinib, sorafenib
Introduction
Cytotoxic chemotherapy in cancer patients often results in a myriad of undesirable symptoms that affect physical functioning and quality of life.1-5 These symptoms, including fatigue, decreased appetite, disturbed sleep, cognitive difficulties, changes in body composition, and depression, may also discourage patients from adhering to treatment.6 There is compelling evidence that implicates cytotoxic chemotherapy-induced inflammation in the etiology of these symptoms (for 2 recent reviews see refs. 7 and 8). In this regard cancer treatment related symptoms have been likened to sickness behavior, a normal physiological response to harmful stimuli triggered by the production of the pro-inflammatory cytokine IL-1β.7 Cancer chemotherapeutic drugs are cytotoxic by nature, resulting in the release of intracellular contents that trigger the production of inflammatory cytokines by immune cells.9-13 In addition, the drugs frequently damage immune cells directly, causing these cells to synthesize cytokines.14-16 Studies in both human patients and animal models have demonstrated an association between inflammatory cytokines and cancer treatment-related symptoms.17-21 In severe cases, tumor lysis syndrome occurs when chemotherapeutic agents lead to rapid and massive lysis of malignant cells, releasing an overwhelming amount of inflammatory signals into the systemic circulation, resulting in significant metabolic damage that can be life-threatening.22
Combination chemotherapy became an established practice when increased numbers of patients with lymphomas or leukemias were treated successfully by regimens that consisted of multiple drugs. Before combination chemotherapy became widely used, most remissions previously were short-lived after single-agent therapies and were limited to few tumor types. More recently, simultaneous delivery of multiple drugs such as doxorubicin and vincristine23-26 has been adopted as an established practice because of several advantages, including decreasing the chance that the tumor becomes resistant to treatment and targeting multiple pathways at the same time to maximize tumor killing.27-29 However, the simultaneous administration of different medications may increase the likelihood that toxicity will occur.23
Many cytotoxic chemotherapeutic drugs such as cisplatin, doxorubicin, etoposide, and 5-fluorouracil (5-FU) activate the mitogen-activated protein kinases cascade,14,30-32 which plays a central role in the production of inflammatory cytokines that may lead to sickness behavior. ZAK, an upstream MAPK, is required for the activation of JNK and p38 MAPK by doxorubicin in epithelial cells and macrophages.33,34 IL-1β and IL-18 are first produced as a biologically inactive precursor that requires processing by the inflammasome, a multi-protein complex consisting of caspase 1 or 11 (caspase 4 and 5 in humans), a member of the NLRP family such as NLRP3, and often adaptor molecules such as ASC.35,36 Various mechanisms for the activation of the inflammasomes have been proposed.37-39 Bacterial pore-forming toxins, bacterial and viral pathogens, asbestos, silica, ATP, double-stranded RNA, uric acid crystals, and translation inhibitors (such as emetine and ribotoxic stressors including doxorubicin) stimulate IL-1β processing via the NLRP3-inflammasome. Of cancer chemotherapeutic agents, gemcitabin and 5-FU have been shown to activate the inflammasome in myeloid-derived suppressor cells;40 doxorubicin has been shown to do the same in macrophages.34,41
Although doxorubicin is able to activate the NLRP3-dependent inflammasome,34 it is unknown whether other chemotherapeutic drugs also share a common ability to induce the processing of IL-1β. It is also unclear whether cytotoxic chemotherapeutic agents, which are more commonly administered together, have additive or synergistic effects on inflammatory cytokine production by immune cells. In this study we screened 11 cancer chemotherapeutic agents from distinct drug classes for their ability to stimulate the processing and secretion of IL-1β by mouse bone marrow-derived macrophages (BMDM). We found that like doxorubicin, seven other drugs (melphalan, cisplatin, vincristine, etoposide, paclitaxel, methotrexate, and cytarabine) also stimulated the production of IL-1β. Because doxorubicin and vincristine are commonly used together in combination chemotherapy, we determine whether these two drugs have additive or synergistic affects on inflammatory cytokine production. When compared with treatment by each drug singly, the combination of doxorubicin and vincristine synergistically activated NLRP3-mediated processing of IL-1β and synergistically blocked protein synthesis. Moreover, these drugs in combination also induced a synergistic increase in the expression of IL-1β, IL-6, and CXCL1 at both the RNA and protein level. The expression was partially reduced in cells lacking the IL-1 and TNF-α receptors and in the presence of anakinra, an IL-1 receptor antagonist. In contrast, the expression was almost completely inhibited by small-molecule kinase inhibitors nilotinib, ponatinib, and sorafenib. Taken together, these data demonstrate that doxorubicin and vincristine synergistically increase both the expression of IL-1β, IL-6, and CXCL1 and the processing and release of IL-1β and that small molecule kinase inhibitors may prove useful in reducing possible inflammatory side effects of chemotherapeutic drugs.
Results
Mechanistically distinct cytotoxic chemotherapeutic agents activate the NLRP3-dependent processing of IL-1β
Doxorubicin activates the NLRP3-inflammasome to promote the processing of pro-ILβ to mature IL-1β.41 To determine whether mechanistically distinct cytotoxic chemotherapeutic agents share a common ability to promote the processing and release of IL-1β, we incubated LPS-primed BMDM with clinically relevant concentrations (corresponding to peak plasma levels) of azacitidine (0.75 µg/mL), cisplatin (17 µM), cytarabine (2 µM), etoposide (33 µM), fludarabine (1.54 µg/mL), melphalan (2.8 µg/mL), methotrexate (2.3 µM), paclitaxel (1 µM), vincristine (0.4 µM), 5-FU (0.5 mM), or doxorubicin (5 µM). Like doxorubicin, (in decreasing ability) melphalan, cisplatin, vincristine, etoposide, paclitaxel, methotrexate, and cytarabine caused the release of mature IL-1β into the culture medium, albeit to a lesser extent (Fig. 1). In contrast, azacitidine, fludarabine, and 5-FU did not induce IL-1β processing and release.
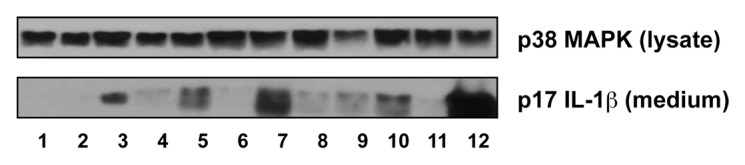
Figure 1. Mechanistically distinct chemotherapeutic drugs caused the release of mature IL-1β by BMDM. After serum deprivation for 0.5 h, BMDM were pre-treated with 50 ng/mL LPS for 4 h and then washed away. The cells were then treated with the indicated chemotherapeutic drugs for 18 h. LPS-primed BMDM were treated with medium alone (lane 1), azacitidine (lane 2), cisplatin (lane 3), cytarabine (lane 4), etoposide (lane 5), fludarabine (lane 6), melphalan (lane 7), methotrexate (lane 8), paclitaxel (lane 9), vincristine (lane 10), 5-FU (lane 11), or doxorubicin (lane 12). Western blots of cell lysates and medium samples were then processed using antibodies against total p38 MAPK in the cell lysates (as loading control) or IL-1β in the medium.
Doxorubicin and vincristine synergistically induce IL-1β production
In this experiment we determined whether cytotoxic chemotherapeutic agents, that are commonly administered together, have additive or synergistic effects on inflammatory signaling. When LPS-primed cells were exposed simultaneously to doxorubicin and vincristine, levels of IL-1β released into the culture medium were significantly increased compared with cells treated singly (Fig. 2A). The production of mature IL-1β by these agents, singly or in combination, was absent in NLRP3-deficient BMDM (Fig. 2B), suggesting a central role for the NLRP3-inflammasome in the conversion of pro-IL-1β to mature IL-1β. The increased release of IL-1β that occurred following co-treatment of BMDM with doxorubicin and vincristine may result from released IL-1β and/or TNF-α acting in an autocrine or paracrine fashion, thereby augmenting the inflammatory effects of the drugs. To examine this possibility, we cultured BMDM from WT and mice lacking both TNFR1 and IL-1R1 (Fig. 2C). Mutant BMDM treated with doxorubicin alone or doxorubicin and vincristine released significantly lower levels of IL-1β into the medium than treated WT BMDM (lanes 2, 4, 6, and 8). Nonetheless, the synergistic effect of doxorubicin and vincristine on the production of IL-1β was still observed in cells lacking TNFR1 and IL-1R1 (lanes 6, 7, and 8).
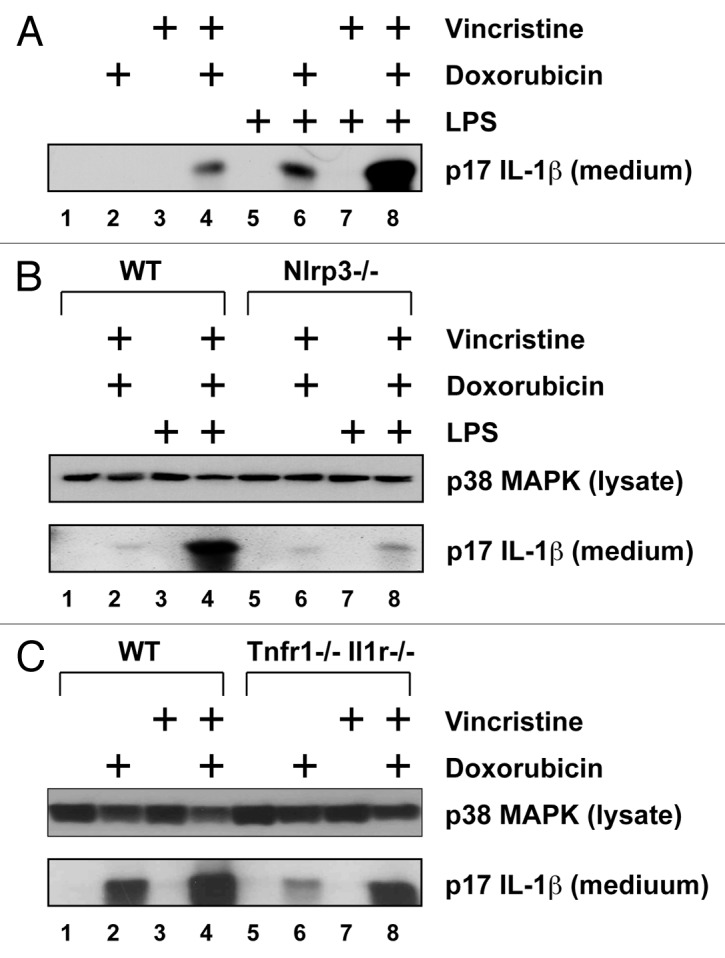
Figure 2. Doxorubicin and vincristine synergistically induce IL-1β production. BMDM from WT or mutant mice were pre-treated LPS for 4 h and then with doxorubicin, vincristine, or both for 18 h. Western blots of cell lysates and medium samples were then processed using antibodies against total p38 MAPK in the cell lysates (as loading control) or IL-1β in the medium. (A) LPS-primed or unprimed BMDM were treated with doxorubicin, vincristine, or both as indicated. (B) LPS-primed or unprimed BMDM from WT or Nlrp3−/− mice were treated with doxorubicin and vincristine as indicated. (C) LPS-primed BMDM from WT or Tnfr1−/− Il1r1−/− mice were treated with doxorubicin, vincristine, or both as indicated.
Doxorubicin and vincristine synergistically increase the RNA levels of IL-1β, IL-6, and CXCL1
In addition to augmenting the processing of pro-IL-1β to mature IL-1β, vincristine and doxorubicin may also increase the level of IL-1β in the medium by elevating the expression of pro-IL-1β. To examine the effects of doxorubicin and vincristine on the expression of RNA encoding pro-IL-1β and other inflammatory cytokines, the RNA of treated BMDM was extracted for real-time PCR analysis (Fig. 3). Combining doxorubicin and vincristine induced a significantly higher level of expression of RNA encoding IL-1β, IL-6, and CXCL-1 (Gro-α or KC), compared with addition of doxorubicin or vincristine alone (P < 0.001). Increased expression of TNF-α and CCL2 (MCP-1) was not observed in any treatments (data not shown). Of interest, cells co-treated with doxorubicin and vincristine caused the accumulation of mature IL-1β in the medium even in the absence of LPS priming (lane 4 in Fig. 2A). This finding is consistent with doxorubicin and vincristine being able to enhance the expression of IL-1β resulting in the accumulation of intracellular pro-IL-1β.

Figure 3. Doxorubicin and vincristine synergistically increased gene expression. RNA was extracted from LPS-unprimed BMDM after 12 h of exposure to doxorubicin, vincristine, or both in the presence or absence of anakinra. The expression of IL-1β, IL-6, and CXCL1 was quantitated using real-time RT-PCR.
Anakinra partially blocks the expression of IL-6 and CXCL1
We sought to determine whether the synergistic increase in the expression of IL-1β, IL-6, and CXCL-1 could be due to the paracrine and/or autocrine binding of released IL-1β to the IL-1R receptor. This was achieved using anakinra, a competitive inhibitor of IL-1β signaling.42 Because increased expression of TNF-α was not detected after treatment with doxorubicin, vincristine, or both, we did not use an inhibitor of the TNF-α pathway. In the presence of 10 µg/mL anakinra, the expression of IL-1β caused by the co-incubation with doxorubicin and vincristine was increased by 22%, while the expression of IL-6 and CXCL1 was decreased by 63% and 32%, respectively (P < 0.001) (Fig. 3). These results suggest that synthesis and release of IL-1β augment the expression of IL-6 and CXCL1 following exposure to doxorubicin and vincristine.
Doxorubicin and vincristine inhibit protein translation
We previously reported that a variety of agents that activate the NLRP3-inflammasome, including doxorubicin, also inhibit protein synthesis.41,43 To determine whether the synergistic effect of doxorubicin and vincristine on IL-1β secretion was due to a synergistic inhibition of protein synthesis, we measured the incorporation of [3H]-leucine into BMDM after treatment with doxorubicin, vincristine, or both. After 1.5 h, the level of leucine incorporation was similar in all treatments (Fig. 4). At 3 h, leucine incorporation was 90% of control levels following exposure to vincristine, whereas leucine incorporation by doxorubicin or doxorubicin plus vincristine was reduced to approximately 60%. At 6 h, vincristine began to enhance the inhibitory effect of doxorubicin, resulting in greater inhibition of protein synthesis in the presence of both drugs than in the presence of either drug added singly. When both doxorubicin and vincristine were present for 12 h, leucine incorporation was reduced to 12%, compared with 40% and 70% in response to single treatments with doxorubicin and vincristine, respectively. These data demonstrate that vincristine augmented the doxorubicin-induced inhibition of translation, but only modestly inhibited translation when added alone.
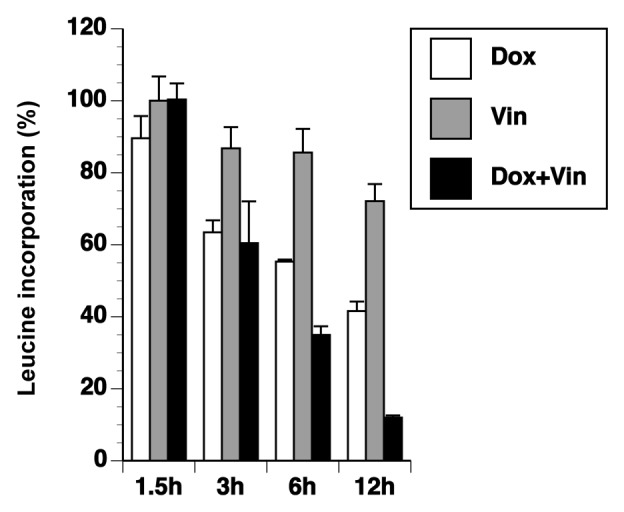
Figure 4. Doxorubicin and vincristine affects incorporation of leucine. Cells were exposed to doxorubicin, vincristine, or both for various times as indicated. [3H]-leucine was added for the final 30 min. The level of radioactive incorporation was quantitated using liquid scintillation.
Small-molecule kinase inhibitors block the expression of IL-1, IL-6, and CXCL1
Because anakinra only partially inhibited the inflammatory gene expression following doxorubicin and vincristine, we searched for more effective inhibitors that may have potential therapeutic value in blocking the inflammatory response to cytotoxic chemotherapeutic agents and resulting side effects. By targeting BCR-ABL, nilotinib44 and ponatinib45 are two small-molecular kinase inhibitors that are used in the treatment of chronic myelogenous leukemia. Sorafenib46 inhibits vascular endothelial growth factor receptor and platelet-derived growth factor receptor and is clinically used to treat renal cell carcinoma, hepatocellular carcinoma, and thyroid carcinoma. We have previously shown that all three of these tyrosine kinase inhibitors block the expression of inflammatory genes by BMDM following exposed to doxorubicin.34 In the presence of these inhibitors, the expression of RNA encoding IL-1β, IL-6, and CXCL-1 by doxorubicin, vincristine, or both was blocked (Fig. 5A). Media was collected from these cultures for measurement of IL-1β, IL-6, and CXCL-1. Consistent with the expression of RNA, the protein levels of IL-1β, IL-6, and CXCL-1 were highly elevated in the combined presence of doxorubicin and vincristine, compared with either compound alone, and these levels were substantially reduced in the presence of nilotinib, ponatinib, or sorafenib (Fig. 5B).

Figure 5. Small-molecule kinase inhibitors block the expression of IL-1β, IL-6, and CXCL1. BMDM were treated with doxorubicin, vincristine, or both for 12 h in the presence or absence of nilotinib, ponatinib, or sorafenib as indicated. (A) RNA was extracted from the cells and processed for real-time RT-PCR. (B) Cytokine levels in the medium were quantitated using a multiplex assay.
Both doxorubicin and vincristine activate the phosphorylation of JNK and p38 MAPK
ZAK is a MAP3K whose activation has been tied to expression of inflammatory cytokines.32 Because nilotinib, ponatinib, and sorafenib each block ZAK-mediated expression of genes by doxorubicin,34 we employed BMDM isolated from two separate ZAK-deficient mice to determine whether ZAK played a role in MAPK activation following treatment with doxorubicin, vincristine, or both (Fig. 6). In WT cells, doxorubicin and vincristine individually caused phosphorylation of both subunits of JNK, although the increase by doxorubicin was weaker (lanes 2 and 3). When doxorubicin and vincristine were added simultaneously, the level of JNK phosphorylation was similar to that when vincristine was added alone (lanes 3 and 4). Doxorubicin or vincristine, added singly or in combination, induced similar levels of phosphorylation of p38 MAPK (lanes 2, 3, and 4). Compared with WT cells, the phosphorylation of JNK by doxorubicin in Zak−/− cells was undetected (lanes 6 and 10), whereas the phosphorylation of JNK by vincristine (lanes 7 and 11) or by both drugs (lanes 8 and 12) was present but reduced in amount in the ZAK-deficient cells. The phosphorylation of p38 MAPK in Zak−/− cells after treatment with doxorubicin (lanes 6 and 10), vincristine (lanes 7 and 11), or both (lanes 8 and 12) was similar to the level in untreated cells (lanes 5 and 9). These data demonstrate that, like doxorubicin, vincristine activates p38 MAPK in a ZAK-dependent manner, but that activation of JNK is only partly ZAK-dependent.
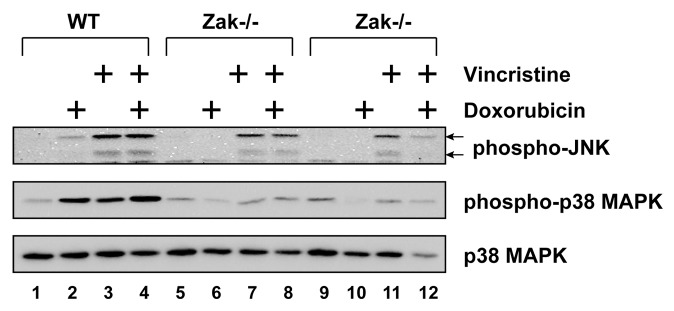
Figure 6. Doxorubicin and vincristine activated p38 MAPK through ZAK. BMDM from WT or Zak−/− mice were treated with doxorubicin, vincristine, or both for 12 h. Western blots of cell lysates were processed using antibodies against phosphorylated JNK, phosphorylated p38 MAPK, or total p38 MAPK.
Cisplatin and vincristine display additive, not synergistic, effects
Among the chemotherapeutic drugs tested in Figure 1A, cisplatin was also able to activate the processing of IL-1β. To test whether cisplatin, a synthetic platinum-containing compound which causes DNA crosslinking, is also capable of synergistically activating the expression of inflammatory genes and processing of IL-1β, we treated BMDM with cisplatin and vincristine, singly and in combination. Compared with vincristine, cisplatin was a weak activator of the expression of IL-1β, IL-6, and CXCL-1 (Fig. 7A) and of the processing of IL-1β (Fig. 7B). When cells were incubated in both cisplatin and vincristine, the level of IL-1β expression was similar to the level caused by vincristine alone (Fig. 7A). We also observed an additive, but not synergistic, increase in the amount of mature IL-1β secreted into the medium (Fig. 7B).

Figure 7. Cisplatin and vincristine fail to show a synergistic effect in gene expression or IL-1β processing. (A) RNA was extracted from BMDM exposed to cisplatin, vincristine, or both for 12 h. Gene expression was quantitated using real-time RT-PCR. (B) LPS-primed BMDM were treated with cisplatin, vincristine, or both. Levels of IL-1β in the medium were detected using western blotting.
Discussion
Among a panel of 11 chemotherapeutic drugs that have different mechanisms of action, doxorubicin was the most potent in the ability to induce IL-1β processing and secretion by LPS-primed BMDM (Fig. 1). Seven other drugs were also able to induce release of mature IL-1β. In contrast, azacitidine, fludarabine, and 5-FU, which are all nucleoside analogs, did not stimulate IL-1β processing. Therefore, in addition to pathogens, plant and bacterial toxins, antibiotics, ATP, and crystals, cytotoxic chemotherapeutic drugs (excluding some nucleoside analogs) may now be added as a new class of agents that can activate IL-1β processing.43,47 It is known that, compared with lymphocytes, macrophages have much lower intracellular concentrations of the active metabolites of many nucleoside analogs,48 In addition, macrophages are known to be resistant to physiological concentrations of 5-FU49 and fludarabine,50 so the inability of certain nucleoside analogs to induce IL-1β production may be due to poor uptake of these drugs by macrophages. Interestingly, IL-1β was detected after exposure of LPS-pretreated BMDM to cytarabine, which is another nucleoside analog, suggesting that not all nucleoside analogs behave identically.
It is unknown which characteristics are shared by melphalan, cisplatin, vincristine, etoposide, paclitaxel, methotrexate, cytarabine, and doxorubicin that contribute to their ability to induce the production of mature IL-1β. The ability to inhibit protein synthesis (Fig. 4) may underlie this effect. Our finding that doxorubicin and vincristine synergistically enhance IL-1β production and inhibit protein synthesis is consistent with this idea. These data demonstrated for the first time that two agents can synergistically activate the NLRP3-dependent inflammasome in LPS-primed BMDM (Fig. 2). In contrast, when vincristine and cisplatin were used together, the effect on IL-1β processing was additive and not synergistic (Fig. 7). It is clear that chemotherapeutic drugs differ in their ability to augment the processing of IL-1β and the expression of inflammatory mediators. Additional experiments are needed to determine systematically which drugs, especially when used in combination, can induce an inflammatory response.
Doxorubicin and vincristine are commonly used together in the treatment of Hodgkin lymphoma, B-cell lymphomas, lung cancer, Wilms tumor, multiple myeloma, thymic carcinoma, and Kaposi sarcoma, raising the concern that patients undergoing such therapy may express elevated levels of inflammatory cytokines, which in turn may lead to increased cancer treatment related symptoms. The advantages of combination chemotherapy need to be balanced with consideration of serious side effects. Because the inhibition of protein synthesis appears to be mechanistically linked to the production of IL-1β, assays that measure protein synthesis in cytokine-producing cells may be effective in predicting the ability of drugs to activate the inflammasome and cause inflammation-induced symptoms. Ultimately, using drug combinations that enhance tumor killing without enhancing side effects would be clearly be optimal.
Doxorubicin-treated BMDM from Tnfr1−/− Il1r1−/− mice expressed lower amounts of IL-1β than WT BMDM (Fig. 2C), and anakinra was partially able to block the increase in gene expression following co-treatment with doxorubicin and vincristine (Fig. 3). These results suggest that IL-1β may act in an autocrine or paracrine fashion to increase the production of IL-1β, perhaps by activating NFκB, a transcription factor important in the expression of many pro-inflammatory genes.51,52 Doxorubicin and vincristine each activate JNK and p38 MAPK (Fig. 6). Should secreted IL-1β result in activation of NFκB in these cells, the participation of both MAPK and NFκB pathways may prove to be responsible for the observed synergistic increase in gene expression.
Unlike anakinra, the three small-molecule kinase inhibitors (nilotinib, ponatinib, and sorafenib) were able to suppress the expression of IL-1β, IL-6, and CXCL1 nearly to baseline levels (Fig. 5), suggesting that these drugs may potentially be useful in lowering the level of inflammation in patients. These three kinase inhibitors have been identified as putative ZAK inhibitors that block doxorubicin-mediated increase in the phosphorylation of MAPKs in keratinocytes and BMDM33,34 and in the expression of inflammatory proteins in BMDM.34 Indeed, because BMDM deficient in ZAK showed decreased phosphorylation of p38 MAPK (Fig. 6), ZAK may regulate the pro-inflammatory effects of doxorubicin and vincristine, at least in part. Additional experiments are needed to determine whether ZAK may play a role in the pro-inflammatory effects of other chemotherapeutic drugs and whether these inhibitors may be beneficial in countering side effects. Interestingly, nilotinib has been shown to reduce the cardiotoxicity due to doxorubicin in rats53 and may even contribute to reversing multi-drug resistance by sensitizing cancer cells to killing by doxorubicin54 or vincristine.55 Therefore, these studies provide additional incentives to introduce ZAK inhibitors such as nilotinib with chemotherapeutic drugs such as doxorubicin and vincristine to increase tumor killing and reduce systemic toxicity and other side effects.
Reducing IL-1β levels may lead to a reduction of the undesirable side effects of some chemotherapeutic drugs. However, IL-1β may also promote the eradication of the tumor that the drugs are targeting, perhaps due to changes in the tumor microenvironment and in the immune responses of the body. Additional research, especially employing in vivo models, needs to be performed to study any effects on tumor responses and animal survival.
Materials and Methods
Chemicals and antibodies
Azacitidine (Sigma), cisplatin (Teva), cytarabine (Sigma), etoposide (Teva), fludarabine (Sagent), melphalan (Sigma), methotrexate (Hospira), paclitaxel (Hospira), vincristine (Hospira), 5-FU (Teva), doxorubicin (APP Pharmaceuticals), and anakinra (Amgen) were stored according to manufacturer’s recommendation. Lipopolysaccharide (LPS) from E. coli serotype 0111:B4 was purchased from Enzo Life Sciences. Nilotinib and sorafenib (LC Laboratories) and ponatinib (Tocris Bioscience) were dissolved in DMSO and stored at −80 °C. Insulin was purchased from Sigma. Trichloroacetic acid (TCA) was purchased from Fisher Scientific. Antibody against IL-1β (ab9722) was purchased from Abcam, phospho-JNK (9251) and phospho-p38 MAPK (9211) from Cell Signaling Technology, and p38 MAPK (sc-535) from Santa Cruz Biotechnology.
Mice
All animal procedures were approved by the Institutional Animal Care and Use Committee at Oregon Health and Science University and Massachusetts General Hospital. C57BL/6J mice, mice lacking NLRP3, and mice lacking both the TNF-α type 1 receptor and IL-1 type 1 receptor (B6;129S-Tnfrsf1atm1Imx Il1r1tm1Imx/J) (10–12 wk of age) were purchased from the Jackson Laboratory. Generation of mice lacking ZAK is described in a separate manuscript under preparation.
Isolation and treatment of bone marrow-derived macrophages (BMDM)
Mice, 8–10 wk of age, were used throughout the experiments. Bone marrow cells from the femurs and tibias of wild-type C57BL/6 (WT), Nlrp3−/−, Tnfr1−/− Il1r1−/−, and Zak−/− mice were flushed with and cultured in α-Minimum Essential Medium (Corning Cellgro), supplied with 10% fetal bovine serum (Hyclone), 50 μg/mL gentamicin (Life Technologies), and 100 ng/mL recombinant mouse colony-stimulating factor 1 (R&D Systems) for 72 h on non-tissue culture-treated 10-cm petri dishes. BMDM were passaged and cultured for an additional 72 h. Cells were plated onto 12-well or 24-well tissue culture plates and cultured for 24 h before initiating experimental treatment. Nilotinib, ponatinib, sorafenib (all at 1 μM), or anakinra (10 μg/mL) was added 30 min before the addition of doxorubicin, vincristine, or both.
Immunoblotting
BMDM were lysed in 2× electrophoresis sample buffer. Proteins were separated on a denaturing polyacrylamide gel in the presence of sodium dodecyl sulfate and transferred onto polyvinylidene difluoride membranes according to standard laboratory procedures. Proteins from BMDM media supernatants were first precipitated using TCA plus 200 μg insulin carrier protein and separated by 13% SDS-PAGE gels. Membranes were incubated with the indicated antibodies and the corresponding horseradish peroxidase-conjugated secondary antibodies. Signals were detected by using enhanced chemiluminescence. Experiments were repeated at least three times and representative experiments are shown.
Measurement of protein synthesis by incorporation of [3H]-leucine
BMDM were cultured in 24-well tissue culture plates. Treatments were performed in leucine-free/serum-free Dulbecco modified Eagle medium (BioWhittaker), for the indicated times. Prior to harvesting, the cells were pulse-labeled with 1 μCi [3H]-leucine (Perkin Elmer) in leucine-free medium for times specified in the figure legends. An equal volume of 10% TCA was added to terminate incorporation. Wells were washed in water, and 88% formic acid was added to solubilize the TCA-insoluble proteins. The samples were counted in a liquid scintillation counter. In each experiment, triplicate wells were used per experimental point.
Real-time RT-PCR
Total RNA from frozen tissues was isolated using TRIzol (Invitrogen) following the manufacturer’s instructions. RNA was treated with DNase I (Invitrogen) and reverse-transcribed with SuperScript II and oligo dT primer (Invitrogen). Real-time PCR was performed using SYBR Green reagents on a ViiA 7 Real-Time PCR System (Applied Biosystems); fold induction was calculated with the absolute quantification method using levels of glyceraldehyde phosphate dehydrogenase (GAPDH) for normalization. The nucleotide sequences of the primers used in this study have been previously published.56 Experiments were repeated at least three times, and representative data are shown. Statistical analyses were performed using t test.
Measurement of inflammatory cytokines from culture supernatant
Levels of IL-1β, IL-6, and CXCL1 were measured using bead-based immunoassays (EMD Millipore). Data were collected and analyzed using the Luminex-100 system Version IS (Luminex). A four- or five-parameter regression formula was used to calculate the sample concentrations from the standard curves.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dakshina M. Jandhyala for providing the Zak−/− mice. These studies were supported by grants AI105933.5 (to B.E.M.) and 1R01NR013171–01A1 (to L.J.W.) from the National Institutes of Health.
Glossary
Abbreviations:
- 5-FU
5-fluorouracil
- BMDM
bone marrow-derived macrophages
- GAPDH
glyceraldehyde phosphate dehydrogenase
- LPS
lipopolysaccharide
- MAPK
mitogen activated protein kinase
- TCA
trichloroacetic acid
References
- 1.Schwartz AL, Nail LM, Chen S, Meek P, Barsevick AM, King ME, Jones LS. Fatigue patterns observed in patients receiving chemotherapy and radiotherapy. Cancer Invest. 2000;18:11–9. doi: 10.3109/07357900009023057. [DOI] [PubMed] [Google Scholar]
- 2.Glaus A, Crow R, Hammond S. A qualitative study to explore the concept of fatigue/tiredness in cancer patients and in healthy individuals. Support Care Cancer. 1996;4:82–96. doi: 10.1007/BF01845757. [DOI] [PubMed] [Google Scholar]
- 3.Irvine D, Vincent L, Graydon JE, Bubela N, Thompson L. The prevalence and correlates of fatigue in patients receiving treatment with chemotherapy and radiotherapy. A comparison with the fatigue experienced by healthy individuals. Cancer Nurs. 1994;17:367–78. doi: 10.1097/00002820-199410000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Jacobsen PB, Hann DM, Azzarello LM, Horton J, Balducci L, Lyman GH. Fatigue in women receiving adjuvant chemotherapy for breast cancer: characteristics, course, and correlates. J Pain Symptom Manage. 1999;18:233–42. doi: 10.1016/S0885-3924(99)00082-2. [DOI] [PubMed] [Google Scholar]
- 5.Nail LM, Winningham ML. Fatigue and weakness in cancer patients: the symptoms experience. Semin Oncol Nurs. 1995;11:272–8. doi: 10.1016/S0749-2081(05)80008-7. [DOI] [PubMed] [Google Scholar]
- 6.Dodd MJ, Miaskowski C, Lee KA. Occurrence of symptom clusters. J Natl Cancer Inst Monogr. 2004:76–8. doi: 10.1093/jncimonographs/lgh008. [DOI] [PubMed] [Google Scholar]
- 7.Wood LJ, Weymann K. Inflammation and neural signaling: etiologic mechanisms of the cancer treatment-related symptom cluster. Curr Opin Support Palliat Care. 2013;7:54–9. doi: 10.1097/SPC.0b013e32835dabe3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bower JE, Lamkin DM. Inflammation and cancer-related fatigue: mechanisms, contributing factors, and treatment implications. Brain Behav Immun. 2013;30(Suppl):S48–57. doi: 10.1016/j.bbi.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jassar AS, Suzuki E, Kapoor V, Sun J, Silverberg MB, Cheung L, Burdick MD, Strieter RM, Ching LM, Kaiser LR, et al. Activation of tumor-associated macrophages by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid induces an effective CD8+ T-cell-mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res. 2005;65:11752–61. doi: 10.1158/0008-5472.CAN-05-1658. [DOI] [PubMed] [Google Scholar]
- 10.Kriegler AB, Bernardo D, Verschoor SM. Protection of murine bone marrow by dexamethasone during cytotoxic chemotherapy. Blood. 1994;83:65–71. [PubMed] [Google Scholar]
- 11.Logan RM, Stringer AM, Bowen JM, Yeoh AS, Gibson RJ, Sonis ST, Keefe DM. The role of pro-inflammatory cytokines in cancer treatment-induced alimentary tract mucositis: pathobiology, animal models and cytotoxic drugs. Cancer Treat Rev. 2007;33:448–60. doi: 10.1016/j.ctrv.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Tannenbaum CS, Wicker N, Armstrong D, Tubbs R, Finke J, Bukowski RM, Hamilton TA. Cytokine and chemokine expression in tumors of mice receiving systemic therapy with IL-12. J Immunol. 1996;156:693–9. [PubMed] [Google Scholar]
- 13.Wood LJ, Nail LM, Perrin NA, Elsea CR, Fischer A, Druker BJ. The cancer chemotherapy drug etoposide (VP-16) induces proinflammatory cytokine production and sickness behavior-like symptoms in a mouse model of cancer chemotherapy-related symptoms. Biol Res Nurs. 2006;8:157–69. doi: 10.1177/1099800406290932. [DOI] [PubMed] [Google Scholar]
- 14.Elsea CR, Roberts DA, Druker BJ, Wood LJ. Inhibition of p38 MAPK suppresses inflammatory cytokine induction by etoposide, 5-fluorouracil, and doxorubicin without affecting tumoricidal activity. PLoS One. 2008;3:e2355. doi: 10.1371/journal.pone.0002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White CM, Martin BK, Lee LF, Haskill JS, Ting JP. Effects of paclitaxel on cytokine synthesis by unprimed human monocytes, T lymphocytes, and breast cancer cells. Cancer Immunol Immunother. 1998;46:104–12. doi: 10.1007/s002620050468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sodhi A, Pai K. Increased production of interleukin-1 and tumor necrosis factor by human monocytes treated in vitro with cisplatin or other biological response modifiers. Immunol Lett. 1992;34:183–8. doi: 10.1016/0165-2478(92)90211-6. [DOI] [PubMed] [Google Scholar]
- 17.Schubert C, Hong S, Natarajan L, Mills PJ, Dimsdale JE. The association between fatigue and inflammatory marker levels in cancer patients: a quantitative review. Brain Behav Immun. 2007;21:413–27. doi: 10.1016/j.bbi.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Cheung YT, Lim SR, Ho HK, Chan A. Cytokines as mediators of chemotherapy-associated cognitive changes: current evidence, limitations and directions for future research. PLoS One. 2013;8:e81234. doi: 10.1371/journal.pone.0081234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang XS, Williams LA, Krishnan S, Liao Z, Liu P, Mao L, Shi Q, Mobley GM, Woodruff JF, Cleeland CS. Serum sTNF-R1, IL-6, and the development of fatigue in patients with gastrointestinal cancer undergoing chemoradiation therapy. Brain Behav Immun. 2012;26:699–705. doi: 10.1016/j.bbi.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith LB, Leo MC, Anderson C, Wright TJ, Weymann KB, Wood LJ. The role of IL-1β and TNF-α signaling in the genesis of cancer treatment related symptoms (CTRS): a study using cytokine receptor-deficient mice. Brain Behav Immun. 2014;38:66–76. doi: 10.1016/j.bbi.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganz PA, Bower JE, Kwan L, Castellon SA, Silverman DH, Geist C, Breen EC, Irwin MR, Cole SW. Does tumor necrosis factor-alpha (TNF-α) play a role in post-chemotherapy cerebral dysfunction? Brain Behav Immun. 2013;30(Suppl):S99–108. doi: 10.1016/j.bbi.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vodopivec DM, Rubio JE, Fornoni A, Lenz O. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:468452. doi: 10.1155/2012/468452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delbaldo C, Michiels S, Syz N, Soria JC, Le Chevalier T, Pignon JP. Benefits of adding a drug to a single-agent or a 2-agent chemotherapy regimen in advanced non-small-cell lung cancer: a meta-analysis. JAMA. 2004;292:470–84. doi: 10.1001/jama.292.4.470. [DOI] [PubMed] [Google Scholar]
- 24.Ehrhardt H, Schrembs D, Moritz C, Wachter F, Haldar S, Graubner U, Nathrath M, Jeremias I. Optimized anti-tumor effects of anthracyclines plus Vinca alkaloids using a novel, mechanism-based application schedule. Blood. 2011;118:6123–31. doi: 10.1182/blood-2010-02-269811. [DOI] [PubMed] [Google Scholar]
- 25.Zeller WJ, Berger M, Schmähl D. Synergistic action of vincristine and adriamycin in the treatment of experimental rat leukemia L5222. Cancer Res. 1979;39:1071–3. [PubMed] [Google Scholar]
- 26.Zimmermann GR, Lehár J, Keith CT. Multi-target therapeutics: when the whole is greater than the sum of the parts. Drug Discov Today. 2007;12:34–42. doi: 10.1016/j.drudis.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Dy GK, Adjei AA. Systemic cancer therapy: evolution over the last 60 years. Cancer. 2008;113(Suppl):1857–87. doi: 10.1002/cncr.23651. [DOI] [PubMed] [Google Scholar]
- 28.Ramaswamy S. Rational design of cancer-drug combinations. N Engl J Med. 2007;357:299–300. doi: 10.1056/NEJMcibr072593. [DOI] [PubMed] [Google Scholar]
- 29.Frei E., 3rd Curative cancer chemotherapy. Cancer Res. 1985;45:6523–37. [PubMed] [Google Scholar]
- 30.Brozovic A, Osmak M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007;251:1–16. doi: 10.1016/j.canlet.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Fan M, Chambers TC. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist Updat. 2001;4:253–67. doi: 10.1054/drup.2001.0214. [DOI] [PubMed] [Google Scholar]
- 32.Olson JM, Hallahan AR. p38 MAP kinase: a convergence point in cancer therapy. Trends Mol Med. 2004;10:125–9. doi: 10.1016/j.molmed.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Sauter KA, Magun EA, Iordanov MS, Magun BE. ZAK is required for doxorubicin, a novel ribotoxic stressor, to induce SAPK activation and apoptosis in HaCaT cells. Cancer Biol Ther. 2010;10:258–66. doi: 10.4161/cbt.10.3.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong J, Smith LB, Magun EA, Engstrom T, Kelley-Howard K, Jandhyala DM, Thorpe CM, Magun BE, Wood LJ. Small molecule kinase inhibitors block the ZAK-dependent inflammatory effects of doxorubicin. Cancer Biol Ther. 2013;14:56–63. doi: 10.4161/cbt.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pétrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 36.Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity. 2006;24:317–27. doi: 10.1016/j.immuni.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Tsuchiya K, Hara H. The inflammasome and its regulation. Crit Rev Immunol. 2014;34:41–80. doi: 10.1615/CritRevImmunol.2013008686. [DOI] [PubMed] [Google Scholar]
- 38.Vladimer GI, Marty-Roix R, Ghosh S, Weng D, Lien E. Inflammasomes and host defenses against bacterial infections. Curr Opin Microbiol. 2013;16:23–31. doi: 10.1016/j.mib.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruchard M, Mignot G, Derangère V, Chalmin F, Chevriaux A, Végran F, Boireau W, Simon B, Ryffel B, Connat JL, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 41.Sauter KA, Wood LJ, Wong J, Iordanov M, Magun BE. Doxorubicin and daunorubicin induce processing and release of interleukin-1β through activation of the NLRP3 inflammasome. Cancer Biol Ther. 2011;11:1008–16. doi: 10.4161/cbt.11.12.15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arend WP. Interleukin 1 receptor antagonist. A new member of the interleukin 1 family. J Clin Invest. 1991;88:1445–51. doi: 10.1172/JCI115453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vyleta ML, Wong J, Magun BE. Suppression of ribosomal function triggers innate immune signaling through activation of the NLRP3 inflammasome. PLoS One. 2012;7:e36044. doi: 10.1371/journal.pone.0036044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94:1765–9. doi: 10.1038/sj.bjc.6603170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–12. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ranieri G, Gadaleta-Caldarola G, Goffredo V, Patruno R, Mangia A, Rizzo A, Sciorsci RL, Gadaleta CD. Sorafenib (BAY 43-9006) in hepatocellular carcinoma patients: from discovery to clinical development. Curr Med Chem. 2012;19:938–44. doi: 10.2174/092986712799320736. [DOI] [PubMed] [Google Scholar]
- 47.Sutterwala FS, Ogura Y, Flavell RA. The inflammasome in pathogen recognition and inflammation. J Leukoc Biol. 2007;82:259–64. doi: 10.1189/jlb.1206755. [DOI] [PubMed] [Google Scholar]
- 48.Gavegnano C, Detorio MA, Bassit L, Hurwitz SJ, North TW, Schinazi RF. Cellular pharmacology and potency of HIV-1 nucleoside analogs in primary human macrophages. Antimicrob Agents Chemother. 2013;57:1262–9. doi: 10.1128/AAC.02012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vĕtvicka V, Bilej M, Kincade PW. Resistance of macrophages to 5-fluorouracil treatment. Immunopharmacology. 1990;19:131–8. doi: 10.1016/0162-3109(90)90048-J. [DOI] [PubMed] [Google Scholar]
- 50.Magnani M, Balestra E, Fraternale A, Aquaro S, Paiardini M, Cervasi B, Casabianca A, Garaci E, Perno CF. Drug-loaded red blood cell-mediated clearance of HIV-1 macrophage reservoir by selective inhibition of STAT1 expression. J Leukoc Biol. 2003;74:764–71. doi: 10.1189/jlb.0403156. [DOI] [PubMed] [Google Scholar]
- 51.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 52.Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol. 2006;290:L622–45. doi: 10.1152/ajplung.00477.2005. [DOI] [PubMed] [Google Scholar]
- 53.Zhou ZY, Wan LL, Yang QJ, Han YL, Li Y, Yu Q, Guo C, Li X. Evaluation of the pharmacokinetics and cardiotoxicity of doxorubicin in rat receiving nilotinib. Toxicol Appl Pharmacol. 2013;272:238–44. doi: 10.1016/j.taap.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 54.Villar VH, Vögler O, Martínez-Serra J, Ramos R, Calabuig-Fariñas S, Gutiérrez A, Barceló F, Martín-Broto J, Alemany R. Nilotinib counteracts P-glycoprotein-mediated multidrug resistance and synergizes the antitumoral effect of doxorubicin in soft tissue sarcomas. PLoS One. 2012;7:e37735. doi: 10.1371/journal.pone.0037735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen T, Kuang YH, Ashby CR, Lei Y, Chen A, Zhou Y, Chen X, Tiwari AK, Hopper-Borge E, Ouyang J, et al. Imatinib and nilotinib reverse multidrug resistance in cancer cells by inhibiting the efflux activity of the MRP7 (ABCC10) PLoS One. 2009;4:e7520. doi: 10.1371/journal.pone.0007520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Korcheva V, Wong J, Corless C, Iordanov M, Magun B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am J Pathol. 2005;166:323–39. doi: 10.1016/S0002-9440(10)62256-0. [DOI] [PMC free article] [PubMed] [Google Scholar]