

Abstract
Recent studies suggest that arylhydrocarbon receptor (AhR) may be a target for a number of diseases. Natural product malassezin is a AhR agonist with an interesting 2,3′-diindolylmethane skeleton. We have prepared a series of analogues of natural product malassezin using our recently developed method and tested the activity of these analogues against AhR in a cell-based assay. We found that a methyl substituent at 1′-N can significantly increase the activity and the 2-formyl group is not critical for some diindolylmethanes.
Keywords: AhR, diindolylmethane, agonist, indole, transcription factor
The arylhrydrocarbon receptor (AhR) is a basic helix-loop-helix transcription factor that is well conserved across many species.1 AhR readily binds to various endogenous and xenobiotic polyaromatic heterocycles.2 It is perhaps best known for its role in conferring the toxicity of environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD or dioxin) 1 (Figure 1).3 AhR binds to TCDD with remarkably high affinity and upon binding, the ligand-receptor complex travels to the nucleus where AhR then dimerizes with the aryl hydrocarbon receptor translocator (ARNT).4 It is this active dimer that functions to promote or repress the transcription of a multitude of different genes, most notably CYP1A1.5 AhR activation is highly regulated by a negative feedback mechanism to prevent continuous signaling.6 It is believed that prolonged AhR signaling is very unfavorable.7 Therefore, it has been proposed that the toxicity of dioxin is due to its exceptionally long metabolic stability (half-life of 7–10 years in human),3 resulting in a continuously activated AhR.
Figure 1.

Representative Synthetic and Natural AhR Agonists
The physiological role of AhR has been a long sought problem.2–3 Recent data showed that AhR was involved in many important biological processes such as immune cell differentiation,8 intestinal function,9 and development of prostate.10 It has been postulated that AhR may be a potential target for the treatment of benign prostatic hyperplasia,11 reduction of undesired immune responses during organ transplantation,12 inflammation disorders,13 and certain types of cancer.14 Peterson and Safe reported that synthetic AhR agonist 6-methyl-1,3,8-trichlorodibenzofuran (MCDF) 2 blocked vascular endothelial growth factor in prostate and conferred protection against prostate cancer in vivo.15 Safe also showed that a variety of substituted 3,3′-diindolylmethanes (DIMs) 3, which are also AhR agonists, inhibit tumor growth in rat models.16 Natural products indolo[3,2-b]carbazole (ICZ) 4, 6-formylindolo[3,2-b]carbazole (FICZ) 5, and malassezin 6 have been demonstrated to be agonists of AhR. Both ICZ and FICZ are categorized as indolocarbazoles,17 while malassezin18 is a formylated 2,3′-diindolylmethane.19 Recent studies suggested that FICZ had anti-asthmatic effects by inhibiting Th2 cytokine production in a mouse model.20 Herein we report our efforts towards the development of selective AhR modulators by preparing an assortment of 2,3′-diindolylmethanes.
We have recently reported a platinum-catalyzed indole annulation/arylation cascade reaction for the synthesis of diindolylmethane 9 from propargylic ether 7 (Scheme 1) and demonstrated its utility in the total synthesis of natural product malassezin.21 Based on this method, we prepared a collection of malassezin analogues 10 and 11 following the sequence in Scheme 1. After the formation of 9 using the previously established indole annulation/arylation protocol, we cleaved the Boc-group under thermal conditions to produce diindolylmethanes 10, which were then treated with POCl3 and DMF to give the formylated malassezin analogues 11.18,21
Scheme 1.

Synthesis of 2,3′-Diindolylmethanes
We next turned our attention to the investigation of the bioactivity of malassezin analogues. We employed the well-documented ethoxyresorufin-O-deethylase (EROD) assay to determine the EC50 values of our compounds towards AhR activation in HepG2 cells.22 This assay measures the induction of cytochrome P450-1A1 (CYP1A1), which is a major outcome of AhR activation.23 CYP1A1 selectively converts 7-ethoxyresorufin to a fluorescent product resorufin.
Malassezin was served as the positive control (Table 1). The EC50 values of our malassezin analogues varied quite dramatically depending on their respective substitution pattern. For the halogenated 2,3′-diindolylmethanes 11a–c, 5′-chlorination resulted in a compound (11a) more potent than the positive control, while 7′-chlorination gave a slightly weaker agonist (11b). Compound 11c, with a methoxy substitution on the 5′-position, behaved similarly to malassezin. Interestingly, compound 11d with an N-methyl group on the 1′-position was surprisingly potent; its EC50 value was about five times lower than that of malassezin.
Table 1.
AhR Activity of Diindolylmethanes
| Compounds | EC50 (μM) | |
|---|---|---|
| 6 |
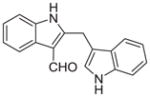
|
0.27 ± 0.1 |
| 11a |
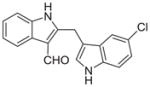
|
0.093 ± 0.15 |
| 11b |
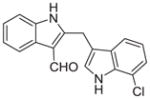
|
0.32 ± 0.1 |
| 11c |
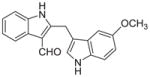
|
0.23 ± 0.1 |
| 11d |
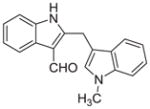
|
0.055 ± 0.03 |
| 6′ |
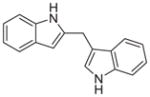
|
0.215 ± 0.46 |
| 10a |
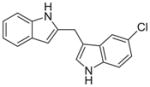
|
> 6.7 |
| 10c |
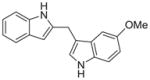
|
0.094 ± 0.27 |
| 10d |
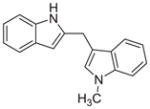
|
0.086 ± 0.04 |
We then investigated the importance of the formyl group in malassezin analogues for AhR activation. In the case of compound pair 10a/11a, the formyl group contributed significantly to the potency of 11a. In contrast, other non-formylated compounds including 6′, 10c, and 10d have similar potency as their formylated counterparts (6, 11c, and 11d).
Up to 12 fold increase of signal over DMSO treatment was observed for many of the malassezing analogues. Even at 10 nM concentration, compounds 11a and 11d could induce 5 and 6 fold increase of signals, respectively (Figure 2).
Figure 2.

Fold increase of signals (Y-axis) for compounds 6, 11a, 11d over DMSO negative control at 10nM.
In summary, we have synthesized a collection of novel analogues of the natural product malassezin and investigated their bioactivity in an EROD assay. It was found that some of these compounds were more potent than the parent malassezin and may become lead compounds for further diseases-relevant studies.
Supplementary Material
Acknowledgments
We thank the NIH (R01GM088285) and the University of Wisconsin for financial support. We also thank the University of Wisconsin-Madison Small Molecule Screening and Synthesis Facility (SMSSF) for their assistance with the EROD assay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Hahn ME. Chem Biol Interact. 2002;141:131. doi: 10.1016/s0009-2797(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 2.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Toxicol Sci. 2011;124:1. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bock KW. Biol Chem. 2013;394:729. doi: 10.1515/hsz-2012-0340. [DOI] [PubMed] [Google Scholar]
- 4.a) Knutson JC, Poland A. Cell. 1982;30:225. doi: 10.1016/0092-8674(82)90028-9. [DOI] [PubMed] [Google Scholar]; b) Poland A, Knutson JC. Annu Rev Pharmacol Toxicol. 1982;22:517. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt JV, Bradfield CA. Annu rev Cell Dev Biol. 1996;12:55. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 6.Chang CY, Puga A. Mol Cell Biol. 1998;18:525. doi: 10.1128/mcb.18.1.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell KA, Elferink CJ. Biochem Pharmacol. 2009;77:947. doi: 10.1016/j.bcp.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Nature. 2008;453:65. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]; b) Stevens EA, Mezrich JD, Bradfield CA. Immunology. 2009;127:299. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Kiss EA, Vonarbourg C, Kopfmann S, Hobeika E, Finke D, Esser C, Diefenbach A. Science. 2011;334:1561. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]; b) Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF, Wilhelm C, Veldhoen M. Cell. 2011;147:629. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]
- 10.Vezina CM, Lin TM, Peterson RE. Biochem Pharmacol. 2009;77:566. doi: 10.1016/j.bcp.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta V, Vezina CM. Differentiation. 2011;82:211. doi: 10.1016/j.diff.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Voorhis M, Fechner JH, Zhang X, Mezrich JD. Transplantation. 2013;95:983. doi: 10.1097/TP.0b013e31827a3d1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Beamer CA, Shepherd DM. Semin Immunopathol. 2013;35:693. doi: 10.1007/s00281-013-0391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Busbee PB, Rouse M, Nagarkatti M, Nagarkatti PS. Nutrition Rev. 2013;71:353. doi: 10.1111/nure.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Safe S, Lee SO, Jin UH. Toxicol Sci. 2013;135:1. doi: 10.1093/toxsci/kft128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fritz WA, Lin TM, Safe S, Moore RW, Peterson RE. Biochem Pharmacol. 2009;77:1151. doi: 10.1016/j.bcp.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDougal A, Gupta MS, Morrow D, Ramamoorthy K, Lee JE, Safe SH. Breast Cancer Res Treat. 2001;66:147. doi: 10.1023/a:1010608000074. [DOI] [PubMed] [Google Scholar]
- 17.a) Knölker HJ, Reddy KR. Chem Rev. 2002;102:4303. doi: 10.1021/cr020059j. [DOI] [PubMed] [Google Scholar]; b) Schmidt AW, Reddy KR, Knölker HJ. Chem Rev. 2012;112:3193. doi: 10.1021/cr200447s. [DOI] [PubMed] [Google Scholar]; c) Wincent E, Amini N, Luecke S, Glatt H, Bergman J, Crescenzi C, Rannug A, Rannug U. J Biol Chem. 2009;284:2690. doi: 10.1074/jbc.M808321200. [DOI] [PubMed] [Google Scholar]
- 18.a) Wille G, Mayser P, Thoma W, Monsees T, Baumgart A, Schmitz HJ, Schrenk D, Polborn K, Steglich W. Bioorg Med Chem. 2001;9:955. doi: 10.1016/s0968-0896(00)00319-9. [DOI] [PubMed] [Google Scholar]; b) Kramer HJ, Podobinska M, Bartsch A, Battmann A, Thoma W, Bernd A, Kummer W, Irlinger B, Steglich W, Mayser P. Chembiochem. 2005;6:860. doi: 10.1002/cbic.200400247. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen LP, Bradfield CA. Chem Res Toxicol. 2008;21:102. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong KT, Hwang SJ, Oh GS, Park JH. Int Immunopharmacol. 2012;13:377. doi: 10.1016/j.intimp.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Shu D, Winston-McPherson GN, Song W, Tang W. Org Lett. 2013;15:4162. doi: 10.1021/ol4018408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Donato MT, Gomezlechon MJ, Castell JV. Anal Biochem. 1993;213:29. doi: 10.1006/abio.1993.1381. [DOI] [PubMed] [Google Scholar]; b) van Tonder JJ. Thesis. University of Pretoria Z.A; 2011. [Google Scholar]
- 23.Whyte JJ, Jung RE, Schmitt CJ, Tillitt DE. Crit Rev Toxicol. 2000;30:347. doi: 10.1080/10408440091159239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.