

Abstract
The cell surface receptor low-density lipoprotein receptor-related protein 5 (LRP5) is a key regulator of bone mass and bone strength. Heterozygous missense mutations in LRP5 cause autosomal dominant high bone mass (HBM) in humans by reducing binding to LRP5 by endogenous inhibitors, such as sclerostin (SOST). Mice heterozygous for a knockin allele (Lrp5p.A214V) that is orthologous to a human HBM-causing mutation have increased bone mass and strength. Osteogenesis Imperfecta (OI) is a skeletal fragility disorder predominantly caused by mutations that affect type I collagen. We tested whether the LRP5 pathway can be used to improve bone properties in animal models of OI. First, we mated Lrp5+/p.A214V mice to Col1a2+/p.G610C mice, which model human type IV OI. We found that Col1a2+/p.G610C;Lrp5+/p.A214V offspring had significantly increased bone mass and strength compared to Col1a2+/p.G610C;Lrp5+/+ littermates. The improved bone properties were not due to altered mRNA expression of type I collagen or its chaperones, nor were they due to changes in mutant type I collagen secretion. Second, we treated Col1a2+/p.G610C mice with a monoclonal antibody that inhibits sclerostin activity (Scl-Ab). We found that antibody treated mice had significantly increased bone mass and strength compared to vehicle treated littermates. These findings indicate increasing bone formation, even without altering bone collagen composition, may benefit patients with OI.
Introduction
Osteogenesis Imperfecta (OI) is a genetic disorder in which skeletal fragility is a hallmark feature (1). Most patients with OI have mutations in genes encoding type I collagen, COL1A1 and COL1A2, or in genes encoding proteins that participate in the assembly, modification, and/or secretion of type I collagen (2). Bisphosphonates, which inhibit osteoclast function, are currently used “off label” with various degrees of success in patients with OI (3-5). The rationale for using bisphosphonates in OI is that inhibiting osteoclast-mediated degradation of abnormal bone enable osteoblasts to effect an increase in bone mass and, as a consequence, an increase in bone strength. Bisphosphonates have been reported to increase bone mineral density and reduce fracture rates in patients with OI (3, 5). However, the effectiveness of bisphosphonate therapy in patients with OI as well as the optimal treatment regimen remain controversial (6, 7). Also of concern are the long-term side effects of bisphosphonates, since these agents may ultimately increase skeletal fragility by facilitating microdamage accumulation and potentially increasing the risk of “atypical” (e.g. subtrochanteric) and non-united fractures (8, 9).
The cell surface receptor low-density lipoprotein receptor-related protein 5 (LRP5) regulates bone mass and bone strength in humans. Loss-of-function mutations in LRP5 cause the Osteoporosis-Pseudoglioma syndrome, an autosomal recessive disorder presenting with very low bone density and fractures (10). Specific missense mutations in LRP5 cause an autosomal dominant phenotype characterized by high bone mass (HBM) and increased bone strength (11, 12). In vitro and in vivo studies suggest that LRP5 functions in the Wnt signaling pathway and that the HBM-causing missense mutations make LRP5 resistant to its endogenous inhibitors Dickkopf1 (DKK1) and sclerostin (SOST) (12-15).
The HBM phenotype has been recapitulated in knockin mice that have Lrp5 missense mutations orthologous to those found in human patients (16). These Lrp5 knockin mice (e.g., Lrp5+/p.A214V) have improved bone mass and bone strength compared to wild-type mice. Dual fluorochrome labeling and quantitative histomorphometry of bone in the knockin mice indicate that the principal effect of the HBM-causing alleles is increased bone formation rather than decreased bone resorption (16). These data indicate that increasing LRP5 signaling could be an effective strategy for improving bone mass and bone strength in humans.
One strategy for increasing LRP5 signaling derives from the identification of mutations that compromise the expression or function of the LRP5 inhibitor SOST/sclerostin in individuals with van Buchem Disease or Sclerosteosis, respectively (17-20). These findings suggested that reducing sclerostin activity would increase bone mass, and led to the development of monoclonal antibodies that inhibit sclerostin activity. These antibodies improved bone properties in wild-type mice, rats, monkeys and humans, and are effective in animal models of disuse-induced and ovariectomy-induced bone loss (21-26).
Here we describe two proof-of-principle experiments, which show that increasing bone anabolism via the LRP5 pathway significantly improves bone mass and bone strength in the Col1a2+/p.G610C mouse model of OI. Col1a2+/p.G610C mice have a missense mutation in the α2 chain of type I collagen which is identical to that found in a large kindred affected with a moderate form of OI (27). The Col1a2+/p.G610C mice have lower bone density and bone strength than their wild-type littermates (27). In the first experiment we crossed Lrp5+/p.A214V mice with Col1a2+/p.G610C mice and determined the effect of the LRP5 HBM allele on bone properties in the offspring. In the second experiment, we administered a sclerostin inhibiting antibody (Scl-Ab) (26) or vehicle alone to wild-type and to Col1a2+/p.G610C mice and determined the effect on bone properties.
Materials and Methods
Mouse strains and genotyping
Col1a2+/p.G610C mice have been previously described (27) and were obtained on a fixed C57BL/6 background from The Jackson Laboratory (Bar Harbor, ME). Lrp5+/p.A214V mice (16) have been maintained on a fixed 129/SvJ background. Tail snip DNA was recovered for PCR genotyping using the HotSHOT method (28). Genotyping was performed as previously described (16, 27).
Mouse care and handling
For the experiments involving genetic crosses, male Col1a2+/p.G610C mice were mated with female Lrp5+/p.A214V mice to generate offspring with the following 4 genotypes: wild-type (Col1a2+/+;Lrp5+/+), OI (Col1a2+/p.G610C;Lrp5+/+), OI with HBM (Col1a2+/p.G610C;Lrp5+/p.A214V), and HBM (Col1a2+/+;Lrp5+/p.A214V). Offspring were tail clipped for DNA extraction and given ear tags for identification before 10 days-of-age, weaned before 28 days-of-age, and then group housed as same-sex littermates. When 11-weeks-old, each mouse was given a single intraperitoneal (IP) injection of calcein green (Sigma-Aldrich, St. Louis, MO; 10 mg/kg), followed 4 days later by an IP dose of alizarin complexone (Sigma-Aldrich, St. Louis, MO; 20 mg/kg). When 12-weeks-old, each mouse was anesthetized with IP ketamine/xylazine and measures of bone mineral density (BMD) and bone mineral content (BMC) were obtained with the Piximus II dual energy x-ray absorptiometer (GE Lunar, Madison, WI, USA). While still anesthetized, the mouse was euthanized and blood was collected by cardiac puncture. Both femurs were then removed. The left femur were fixed in 10% formalin and the right femur was dissected free of soft tissue, wrapped in sterile phosphate buffered saline (PBS)-soaked gauze (Gibco-Life Technologies, Grand Island, NY), and stored frozen at -20°C.
For the experiments involving treatment with Scl-Ab, male Col1a2+/p.G610C mice were mated with wild-type 129/SvJ females. Tail clipping, ear-tagging, weaning, and group housing of offspring were performed as described earlier. BMD and BMC measures were obtained when the mice were 6-weeks-old. Mice were then randomized based on sex and Col1a2 genotype to receive twice-weekly subcutaneous injections of either Scl-AbIII (25 mg/kg in PBS) or PBS for a total of 6 weeks. Scl-AbIII (Amgen, Inc., Thousand Oaks, CA) is a ratized monoclonal antibody against sclerostin (26); antibody stocks were stored frozen, thawed and diluted to 5.56 mg/mL in PBS prior to use. When treatment was initiated, each mouse received a single IP dose of demeclocycline HCl (Sigma-Aldrich, St. Louis, MO: 75 mg/kg). Mice were weighed every three doses and the dose of Scl-Ab was adjusted accordingly. When 11-weeks-old, each mouse was given a single IP injection of calcein green, followed 4 days later by an IP dose of alizarin complexone, as described earlier. When 12-weeks-old, bone mineral density (BMD) and bone mineral content (BMC) were measured and, while still anesthetized, mice were euthanized and blood was collected by cardiac puncture. Both femurs were then removed and processed as described earlier. No significant differences were detected between the wild-type (Col1a2+/+;Lrp5+/+) and OI (Col1a2+/p.G610C;Lrp5+/+) groups compared to their genetically identical counterparts that received six weeks of vehicle (PBS) treatment and thus the data from the PBS treated and non-treated animals were combined for the analysis of the genetic cross experiments.
Assessment of bone properties
MicroCT (μCT) measurements of the left femur (midshaft cortical bone and distal trabecular bone) were performed as previously described (16). After μCT measurements were obtained, the left femur was embedded in methyl methacrylate, sectioned, and imaged for quantitative histomorphometry as previously described (16, 29). Calcein and alizarin complexone labeling was used for quantitative histomorphometry in the genetic experiment and demeclocycline, calcein and alizarin complexone labeling was used in the Scl-Ab experiment. Osteoblast and osteoclast covered surfaces were measured in distal femur trabecular bone from MacNeal/von Kassa- and Trap- stained sections as described previously (16).
The right femur was used for biomechanical testing in a three-point bending assay as previously described (16). Briefly, the frozen femur was brought slowly to room temperature (∼1.5 h) in a saline bath, positioned with the posterior side down across the two bottom supports of the three-point bending apparatus and mounted in a Bose Electroforce 3200 electromagnet test machine (Bose, Eden Praire, MN), which has a force resolution of 0.01 N. Each femur was loaded to failure in monotonic compression using a crosshead speed of 0.2 mm s-1, during which force and displacement measurements were collected every 0.005 seconds.
Measurement of serum biomarkers for collagen synthesis and degradation
Cardiac puncture was performed in order to obtain the maximum amount of blood. Following cardiac puncture, blood was then immediately put into Amber serum separator tubes (BD, Franklin Lakes, NJ) and centrifuged at 1723 × g for 15 min at 4°C. After centrifugation, the serum was stored at -20°C until further use. Serum procollagen type 1 amino-terminal propeptide (P1NP) and C-terminal telopeptide of type I collagen (CTX) were measured in duplicate with commercially available ELISA assays following the manufacturer's instructions (Immunodiagnostic Systems, Scottsdale, AZ).
Bone Collagen Analysis
Femurs from male 12-week-old Col1a2+/+;Lrp5+/+, Col1a2+/p.G610C;Lrp5+/+, and Col1a2+/p.G610C;Lrp5+/p.A214V mice were recovered following euthanasia. Mid-shaft bone was decalcified overnight in 0.1M HCl at 4°C, minced and heat denatured at 90°C for 10 min in SDS sample buffer. Equal amounts of demineralized tissue were separated by 6% SDS-PAGE.
Individual α-chains were cut from the gel and subjected to in-gel trypsin digestion (30, 31). Electrospray MS was performed on the tryptic peptides using an LTQ XL ion-trap mass spectrometer equipped with in-line liquid chromatography (LC) (Thermo Scientific, Waltham, MA) using a C4 5 μm capillary column (300 μm × 150 mm; Higgins Analytical RS-15M3-W045) eluted at 4.5 μl min. Proteome Discoverer search software (Thermo Scientific) was used for peptide identification using the NCBI protein database.
RNA-seq Analysis
Messenger RNA (mRNA) expression in the tibiae of male 12-week-old Col1a2p.G610C/+;Lrp5+/+ (n=3 mice, 6 tibiae) and Col1a2p.G610C/+;Lrp5p.A214V/+ (n=3 mice, 6 tibiae) mice were compared as previously described (32). Briefly, tibiae from each mouse were extracted within 10 minutes of euthanasia and cleaned of soft tissues. Epiphyses were excised, bone marrow was removed through centrifugation for 1 min at >15,000 × g, and the remaining diaphyseal bone was immediately frozen in liquid nitrogen. Each frozen tibia was homogenized and total RNA was extracted. mRNA was enriched and reverse transcribed with random hexamers. The resultant cDNA constructs were amplified and washed to remove primer-dimers. Individual mRNA libraries were pooled (n=8 per lane) and sequenced on an Illumina HiSeq 2000 machine (Illumina Inc., San Diego, CA) to obtain 50 basepair paired-end reads.
Raw sequence data were mapped to the mouse genome (mm9) using RUM (33). Normalized numbers of reads uniquely mapping to exons were used when quantifying gene expression. Differential expression was calculated with an EdgeR (p<0.05), where the results were corrected for multiple hypothesis testing (false discovery rate<0.05) (34). The significant differences were filtered with leave-one-out cross validation to eliminate outlier effects due to a single sample or animal. Transcripts of skeletal muscle, bone marrow and blood origin, which could potentially contaminate bone RNA-seq libraries, were also excluded in the analyses. A complete list of these transcripts and additional details of the library preparation and data analysis protocols are provided elsewhere (32).
Statistical Analyses
Bone density and strength properties were tested for normality using the Shapiro-Wilk W statistic and no significant departures or outliers were detected for males or females on any parameter (35). Therefore, two-way mixed model analysis of variance (ANOVA) with genotype and treatment as factors was applied with least-significant difference (LSD) post-hoc testing used to compare groups and assess genotype and treatment effects (36). The sample sizes of 7-17 animals per group for each gender provided 80% power (α=0.05, β=0.2) for detecting mean differences of 30% (moderate effect sizes of 0.8 or larger) at 12 weeks of age (version 7.0, nQuery Advisor, Statistical Solutions, Saugus, MA). All data, except where indicated, are presented as mean ± 1 SD with two-tailed p < 0.05 considered statistically significant. Statistical analysis was performed using SPSS software (version 19.0, SPSS Inc./IBM, Chicago, IL).
Results
As anticipated from previously published work (27), offspring with the OI mutation alone (i.e., Col1a2+/p.G610C;Lrp5+/+) had lower BMD, cortical thickness and bone strength than their wild-type, same sex littermates (Figure 1). Offspring with the Lrp5 HBM allele alone (i.e., Col1a2+/+;Lrp5+/p.A214V) had higher BMD, trabecular bone volume/total volume BV/TV, cortical thickness, and bone strength than their wild-type, same sex littermates (Figure 1). Importantly, we found that offspring with the OI and Lrp5 HBM alleles (i.e., Col1a2+/p.G610C;Lrp5+/p.A214V) had measures of bone mass, volume, and strength that were significantly higher than their littermates with OI alone and either comparable to or higher than their wild-type littermates (Figure 1).
Figure 1.
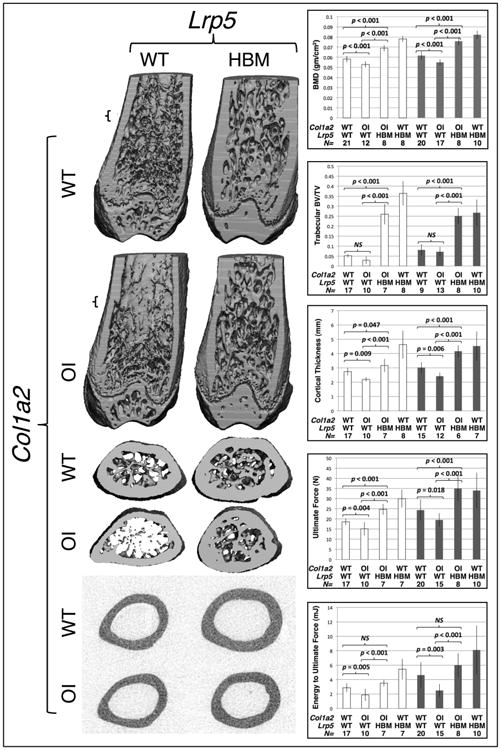
The Lrp5p.A214V allele improves bone properties in wild-type and in OI mice. (left panels) 3-D μCT reconstructions of the distal and midshaft femurs from male 12-week-old wild-type (Col1a2+/+;Lrp5+/+), OI (Col1a2+/p.G610C;Lrp5+/+), OI with HBM (Col1a2+/p.G610C;Lrp5+/p.A214V), and HBM (Col1a2+/+;Lrp5+/p.A214V) mice. Small brackets in the coronal section of the distal femurs indicate the location of the transverse sections depicted immediately below. (right panels) Graphs depicting mean (± SD) measures of bone mineral density (BMD), distal femur trabecular bone volume/total volume (BV/TV), midshaft femur cortical thickness, ultimate force to failure, and energy to ultimate force in female (open bars) and male (shaded bars) 12-week-old wild-type, OI, OI with HBM, and HBM mice. Genotypes, wild-type (WT) or heterozygous knockin (OI or HBM, with respect to the Lrp5 and Col1a2) are indicated, as is the number (N) of animals with each genotype that were studied. Brackets indicate comparisons and p values between OI and wild-type mice, between OI with HBM and OI mice, and between OI with HBM and wild-type mice. NS – not significant.
To determine the mechanism responsible for the improved bone properties in the OI with HBM mice, we performed quantitative histomorphometry after dual fluorochrome labeling, collagen mass spectroscopy, and RNA-seq on mouse bone; we also measured serum markers of bone formation and degradation (Figure 2). We found that the OI with HBM mice (Col1a2+/p.G610C;Lrp5+/p.A214V) had a significantly increased bone formation rate (BFR) compared to the OI mice (Col1a2+/p.G610C;Lrp5+/+) (Figure 2) at both the cortical and trabecular bone surfaces. MS/BS was also increased at the trabecular bone surface. We found no difference between osteoblast surface or osteoclast surface in trabecular bone between the OI with HBM mice (Col1a2+/p.G610C;Lrp5+/p.A214V) compared to the OI mice (Col1a2+/p.G610C;Lrp5+/+) (Figure 2). We did not find any significant differences in the levels of serum P1NP or CTX, except between male mice with OI compared to wild-type mice (Figure 2). Yields of collagen type I chains extracted from equal starting weights of demineralized bone tissue for the mass spectroscopic analyses suggested a higher concentration of collagen in the matrix of the OI with HBM bone, but no significant difference in ratio of wild-type to mutant α2(I) chains between OI with HBM and OI bone (Figure 2). We obtained averages of ∼13 million RNA-seq reads per bone library, with ∼99% reads mapping to the mouse genome, ∼87% uniquely aligning, and ∼14% being PCR duplicates. Only 9 genes had statistically significant differences in gene expression between the OI with HBM tibiae and OI tibiae (Figure 2); none of the 9 genes have a known role in type I collagen expression, trafficking, secretion, or assembly. Also, since RNA-seq provides expression data at single nucleotide resolution, we measured the relative expression of the wild-type and mutant Col1a2 alleles in bone, and found no difference between OI with HBM mice and OI mice (data not shown).
Figure 2.
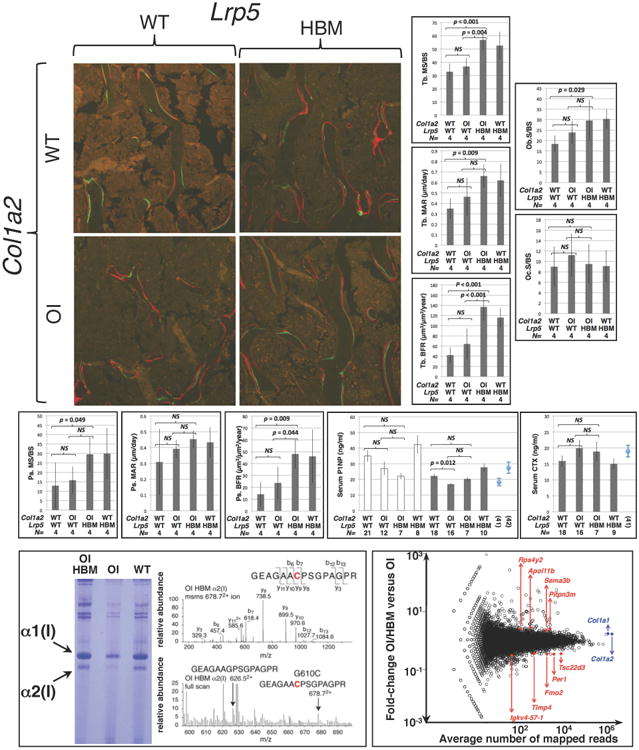
The Lrp5p.A214V allele increases bone formation and does not alter mutant collagen deposition in Col1a2+/p.G610C mice. (top panels) Fluorescence micrographs of dual fluorochrome (calcein green and alizarin complexone) labeled trabecular femur bone from male 12-week-old wild-type (Col1a2+/+;Lrp5+/+), OI (Col1a2+/p.G610C;Lrp5+/+), OI with HBM (Col1a2+/p.G610C;Lrp5+/p.A214V), and HBM (Col1a2+/+;Lrp5+/p.A214V) mice. (top graphs) Graphs depicting mean (± SD) trabecular mineralizing surface/total bone surface (MS/BS), trabecular mineral apposition rate (MAR), and trabecular bone formation rates (BFR) in male (shaded bars) 12-week-old mice, and osteoblast surface and osteoclast surface in male (shaded bars) 12-week-old mice. (middle graphs) Graphs depicting mean (± SD) periosteal mineralizing surface/total bone surface (MS/BS), periosteal mineral apposition rate (MAR), and periosteal bone formation rates (BFR) in male (shaded bars) 12-week-old mice, and serum P1NP and CTX (mean ± SE) in female (open bars) and male (shaded bars) 12-week-old mice. CTX data for female mice are similar to those of males, but not shown. P1NP and CTX graphs show values (mean ± SE) from wild-type mice reported from other papers (41, 42). Genotypes, wild-type (WT) or heterozygous knockin (OI or HBM, with respect to the Lrp5 and Col1a2) are indicated, as is the number (N) of animals with each genotype that were studied. Brackets indicate comparisons and p values between OI and wild-type mice, between OI with HBM and OI mice, and between OI with HBM and wild-type mice. NS – not significant. (bottom left panel) Coomassie blue stained SDS-PAGE gel containing α1(I) and α2(I) polypeptide chains (arrows) recovered from OI with HBM, OI, and WT bone. Representative mass spectroscopy data for the OI with HBM bone show the mutant α2(I) polypeptide is present and comparable in abundance to wild-type α2(I) polypeptide, which was also observed in OI bone (data not shown). (bottom right panel) Graph depicting mean fold-changes in gene expression between OI with HBM bone and OI bone (y-axis) and the average number of mapped reads/gene (x-axis). Circles represent individual genes. The 9 genes whose expression differed significantly between the two genotypes are indicated and shaded red. The Col1a1 and Col1a2 genes, whose expression did not differ significantly between the two genotypes, are indicated and shaded blue.
With respect to the sclerostin antibody experiments in wild-type and OI (Col1a2+/p.G610C) mice, no complications occurred during the 6 weeks the mice received either vehicle or antibody. At the time these mice were randomized into the vehicle or antibody treatment cohorts, their bone parameters were indistinguishable from their sex and genotype matched littermates (data not shown). But as anticipated from previously published work (37), wild-type mice that received 6 weeks of antibody had BMD, BV/TV, and bone strength measures that were significantly higher than the wild-type mice that received only vehicle (Figure 3). We also observed significant increases in BMD, BV/TV, and bone strength in the Col1a2+/p.G610C mice that received 6 weeks of antibody compared to those that received only vehicle (Figure 3). Importantly, the Col1a2+/p.G610C mice that received 6 weeks of antibody attained bone parameters that were comparable to or greater than those of vehicle treated wild-type mice (Figure 3).
Figure 3.
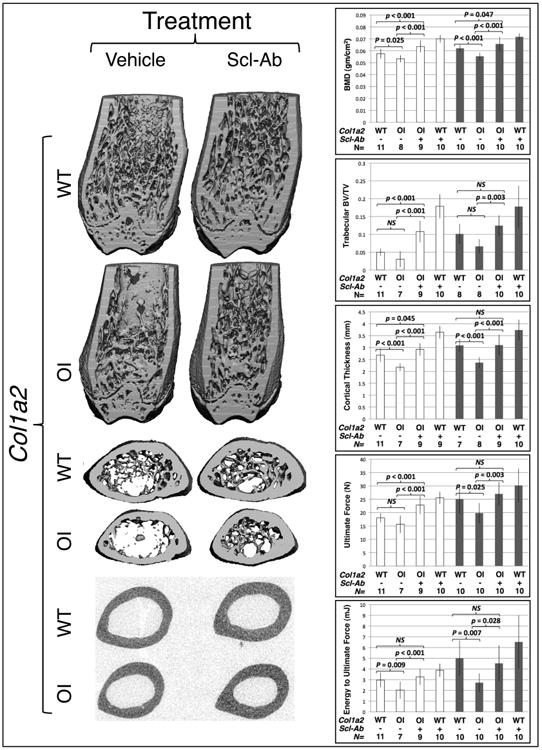
Sclerostin inhibiting antibody treatment improves bone properties in wild-type and in OI mice. (left panels) 3-D μCT reconstructions of the distal and midshaft femurs from male 12-week-old wild-type (Col1a2+/+) and OI (Col1a2+/p.G610C) mice that received 6 weeks of vehicle or Scl-Ab. (right panels) Graphs depicting mean (± SD) measures of bone mineral density (BMD), distal femur trabecular bone volume/total volume (BV/TV), midshaft femur cortical thickness, ultimate force to failure, and energy to ultimate force in female (open bars) and male (shaded bars) 12-week-old wild-type and OI mice that received 6 weeks of vehicle or Scl-Ab. Col1a2 genotypes, wild-type (WT) or heterozygous knockin (OI) are indicated, as is the number (N) of animals with each genotype that were studied. Brackets indicate comparisons and p values between vehicle treated OI and vehicle treated wild-type mice, between Scl-Ab treated OI and vehicle treated OI mice, and between Scl-Ab treated OI and vehicle treated wild-type mice. NS – not significant.
Quantitative histomorphometry revealed a trend towards increased periosteal mineralizing surface (MS/BS) in Col1a2+/p.G610C mice that received antibody versus vehicle alone, as well as significant increases in all measures of bone formation in trabecular bone (Figure 4). There was increased osteoblast surface, but not osteoclast surface present in trabecular bone (Figure 4). Treatment with antibody did not appear to affect levels of serum P1NP or CTX in mice with OI (Figure 4).
Figure 4.
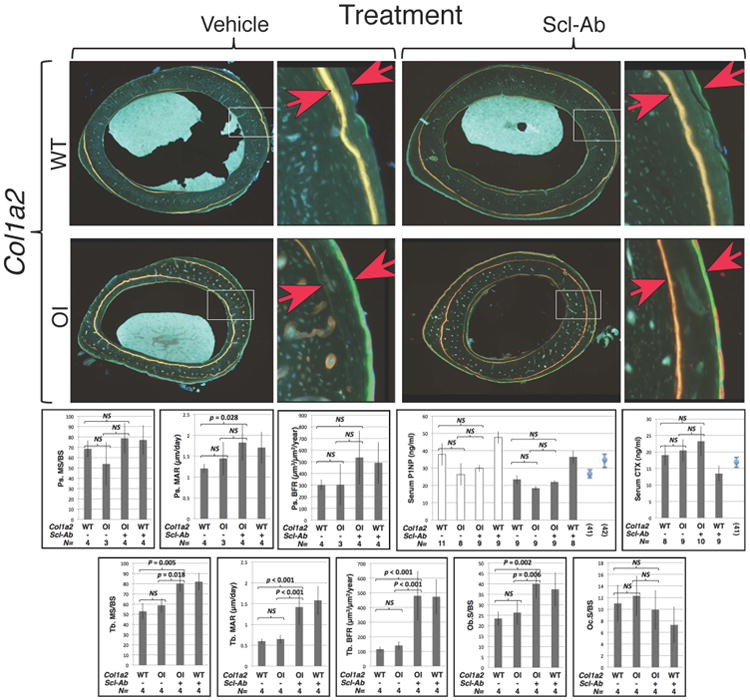
Sclerostin inhibiting antibody therapy increases bone formation in wild-type and in OI mice. (top panels) Fluorescence micrographs of dual fluorochrome labeled midshaft femur bone from male 12-week-old wild-type and OI (Col1a2+/p.G610C) mice that received 6 weeks of vehicle or Scl-Ab. Higher magnification images represent the areas indicated with white rectangles. The red arrows indicate the fluorochrome (democlocycline and calcein green/alizarin complexone) labels. Images of dual flurochrome labeled trabecular bone were similar to those seen in Figure 2 (data not shown). (middle graphs) Graphs depicting mean (± SD) periosteal mineralizing surface/total bone surface (MS/BS), periosteal mineral apposition rate (MAR), and periosteal bone formation rates (BFR) in male (shaded bars) 12-week-old mice, and serum P1NP and CTX (mean ± SE) in female (open bars) and male (shaded bars) 12-week-old mice. CTX data for female mice are similar to those of males but not shown. P1NP and CTX graphs show values (mean ± SE) from wild-type mice reported from other papers (41, 42). (bottom graphs) Graphs depicting mean (± SD) trabecular mineralizing surface/total bone surface (MS/BS), trabecular mineral apposition rate (MAR), and trabecular bone formation rates (BFR) in male (shaded bars) 12-week-old mice, and osteoblast surface and osteoclast surface in male (shaded bars) 12-week-old mice.Col1a2 genotypes, wild-type (WT) or heterozygous knockin (OI) are indicated, as is the number (N) of animals with each genotype that were studied. Brackets indicate comparisons and p values between vehicle treated OI and vehicle treated wild-type mice, between Scl-Ab treated OI and vehicle treated OI mice, and between Scl-Ab treated OI and vehicle treated wild-type mice. NS – not significant.
Discussion
Our data indicate that enhancing LRP5-mediated signaling either using an Lrp5 HBM allele or 6 weeks of treatment with sclerostin antibody significantly increases bone mass, volume, and strength in the Col1a2+/p.G610C mouse model of OI (Figures 1 and 3). These LRP5-mediated improvements in bone properties are most likely due to increased bone anabolism, as indicated by the increased BV/TV and the higher rates of bone formation in trabecular bone (Figures 1, 2, 3 and 4).
The improvement in bone properties is most likely the result of increasing bone quantity, rather than improving the quality of bone. Enhancing LRP5 signaling did not alter the ratio of wild-type to mutant α2(I) polypeptides that were deposited in bone matrix, as measured by mass spectroscopy of pepsin digested α2(I) chains (Figure 2). Furthermore, RNA-seq data did not identify Lrp5 HBM-associated changes in the expression of genes encoding type I collagen, its chaperones, other bone matrix proteins, or proteins involved in the misfolded protein response pathways in the OI mice (Figure 2). We cannot exclude modest changes in mRNA expression being responsible for the improved bone properties in the OI with HBM mice, since our RNA-seq method is not sensitive for changes in gene expression that are less than 1.7-fold (32).
The improvements in bone properties observed in the genetic cross were greater than those observed in the sclerostin antibody study. However, we gave mice antibody from 6 to 12 weeks-of-age, whereas the Lrp5 HBM allele was present from conception. Therefore, it is possible that earlier initiation of antibody therapy would result in even greater improvements in bone properties. In fact, earlier administration of antibody may improve bone properties to a greater extent than the Lrp5 HBM allele, since sclerostin inhibits LRP5 and LRP6 (38, 39), and both receptors have anabolic effects on bone (40).
Sinder et al. (2013) recently administered Scl-Ab to a different mouse model of OI, the Brtl mouse (37). A 2-week course of Scl-Ab increased BFR and bone strength significantly, the latter measured by 4-point bending (37). Consistent with our finding that LRP5 signaling increased the quantity of bone in the Col1a2+/p.G610C mice, but not its quality, Sinder et al. (2013) found no effect of Scl-Ab on bone quality in the Brtl mice using nano-indentation.
Enhanced LRP5 signaling did not cause consistent changes in the serum markers of collagen formation and turnover between mouse genotypes or treatment groups (Figures 2 and 4). This may be because our cohort sizes were underpowered to detect small changes. The P1NP and CTX values we obtained were similar to those reported by other investigators using these same assays (6, 41, 42). The Sinder et al. (2013) study detected a significant change in a serum marker of osteoblast activity, Osteocalcin, but not a marker of osteoclast activity, TRACP5b, following 2 weeks of antibody-mediated sclerostin inhibition (37). This is consistent with our finding of increased osteoblast surface, but no change in osteoclast surface following six weeks of antibody therapy. Whether Osteocalcin and TRACP5b are better indicators of changes in bone homeostasis that occur following sclerostin inhibition, or whether there is a greater change in serum markers after a short treatment period (i.e., 2-weeks) compared to a longer period (6 weeks), remain to be determined.
Mouse models have been generated with mutations encompassing the spectrum of clinical severity and types of OI that occur in human patients (43-50). Importantly, our studies and those of Sinder et al. (2013) were performed in mice that secrete mutant collagen into the extracellular matrix. At least one mouse model of OI has a mutation that causes excessive intracellular retention of abnormal collagen and endoplasmic reticulum stress (46). Intracellular retention of abnormal collagen has also been reported in one human family segregating severe OI (51). Therefore, enhancing LRP5 signaling in mice, and in humans, having mutations that affect the intracellular trafficking of type I collagen could worsen the skeletal phenotype rather than improve it.
We conclude that enhancing LRP5-mediated signaling is an effective strategy for improving bone properties in at least one mouse model of OI, Col1a2+/p.G610C. This improvement is more likely the result of increased bone production, rather than improved bone quality. In contrast to bisphosphonate therapy, which is anti-catabolic and does not significantly increase bone mass in adults with OI, drugs targeting the anabolic LRP5 pathway could be used to improve bone mass and bone strength in adult patients with OI. We obtained a significant increase in bone strength after 6 weeks of therapy using sclerostin inhibitory antibody. Earlier and longer administration of sclerostin antibody may cause even greater benefit. Although we did not directly compare antibody to bisphosphonate in the Col1a2+/p.G610C mice, alendronate did not cause a significant increase in bone strength after 6 weeks of therapy in the Brtl mice (52). It remains important to study additional animal models of OI in order to determine how fragility phenotypes caused by other pathologic mechanisms respond to anabolic therapy. Such studies will help identify patients with OI who are most likely to benefit from strategies that promote bone anabolism via LRP5 signaling.
Acknowledgments
The authors thank the other members of their laboratories for technical support, Melissa Cummings and Animal Resources at Children's Hospital for assistance with DEXA analysis and mouse care. Scl-AbIII was provided by Amgen Inc. and UCB Pharma. This work was supported by the following grants: Osteogenesis Imperfecta Foundation Michael Geisman Fellowship (CMJ), Pediatric Endocrinology Society Research Fellowship (CMJ), NIAMS/NIH AR37694 and AR37318 (DRE), NICHD/NIH HD070394 (DRE), NIAMS/NIH R01AR053237 (AGR), NIAMS/NIH R21AR062326 (MLW) and Howard Hughes Medical Institute (MLW).
Footnotes
Disclosures: All authors state that they have no conflicts of interest.
At that time the studies were performed, none of the authors reported any financial or other conflicts of interest.
Author Contribution Statement: CMJ preformed the mouse studies and oversaw all experiments. LAB and HJR contributed to genotyping, mouse care and injections. UMA performed the RNAseq studies. MAS performed the ELISA assays. MAW and DE performed the bone collagen analysis. DZ performed the statistical analysis. LED and AGR performed the microCT, 3-point bending and quantitative histomorphometry studies. CMJ, AGR and MLW designed experiments, and provided reagents and financial support. CMJ and MLW prepared the first draft of the manuscript. All co-authors contributed detailed methods and results, revised the manuscript and approved the submitted version.
References
- 1.Cundy T. Recent advances in osteogenesis imperfecta. Calcif Tissue Int. 2012 Jun;90(6):439–49. doi: 10.1007/s00223-012-9588-3. Epub 2012/03/28. eng. [DOI] [PubMed] [Google Scholar]
- 2.Byers PH, Pyott SM. Recessively inherited forms of osteogenesis imperfecta. Annu Rev Genet. 2012;46:475–97. doi: 10.1146/annurev-genet-110711-155608. Epub 2012/11/14. eng. [DOI] [PubMed] [Google Scholar]
- 3.Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998 Oct 1;339(14):947–52. doi: 10.1056/NEJM199810013391402. [DOI] [PubMed] [Google Scholar]
- 4.Falk MJ, Heeger S, Lynch KA, DeCaro KR, Bohach D, Gibson KS, et al. Intravenous bisphosphonate therapy in children with osteogenesis imperfecta. Pediatrics. 2003 Mar;111(3):573–8. doi: 10.1542/peds.111.3.573. Epub 2003/03/04. eng. [DOI] [PubMed] [Google Scholar]
- 5.Alharbi M, Pinto G, Finidori G, Souberbielle JC, Guillou F, Gaubicher S, et al. Pamidronate treatment of children with moderate-to-severe osteogenesis imperfecta: a note of caution. Horm Res. 2009 Jan;71(1):38–44. doi: 10.1159/000173740. Epub 2008/11/29. eng. [DOI] [PubMed] [Google Scholar]
- 6.Bradbury LA, Barlow S, Geoghegan F, Hannon RA, Stuckey SL, Wass JA, et al. Risedronate in adults with osteogenesis imperfecta type I: increased bone mineral density and decreased bone turnover, but high fracture rate persists. Osteoporos Int. 2012 Jan;23(1):285–94. doi: 10.1007/s00198-011-1658-2. Epub 2011/07/09. eng. [DOI] [PubMed] [Google Scholar]
- 7.Phillipi CA, Remmington T, Steiner RD. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Database Syst Rev. 2008;(4):CD005088. doi: 10.1002/14651858.CD005088.pub2. Epub 2008/10/10. eng. [DOI] [PubMed] [Google Scholar]
- 8.Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, et al. Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res. 2010 Nov;25(11):2267–94. doi: 10.1002/jbmr.253. Epub 2010/09/16. eng. [DOI] [PubMed] [Google Scholar]
- 9.Sellmeyer DE. Atypical fractures as a potential complication of long-term bisphosphonate therapy. JAMA. 2010 Oct 6;304(13):1480–4. doi: 10.1001/jama.2010.1360. Epub 2010/10/07. eng. [DOI] [PubMed] [Google Scholar]
- 10.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001 Nov 16;107(4):513–23. doi: 10.1016/s0092-8674(01)00571-2. Epub 2001/11/24. eng. [DOI] [PubMed] [Google Scholar]
- 11.Little RD, Carulli JP, Del Mastro RG, Dupuis J, Osborne M, Folz C, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am J Hum Genet. 2002 Jan;70(1):11–9. doi: 10.1086/338450. Epub 2001/12/13. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002 May 16;346(20):1513–21. doi: 10.1056/NEJMoa013444. Epub 2002/05/17. eng. [DOI] [PubMed] [Google Scholar]
- 13.Semenov MV, He X. LRP5 mutations linked to high bone mass diseases cause reduced LRP5 binding and inhibition by SOST. J Biol Chem. 2006 Dec 15;281(50):38276–84. doi: 10.1074/jbc.M609509200. Epub 2006/10/21. eng. [DOI] [PubMed] [Google Scholar]
- 14.Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, et al. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcif Tissue Int. 2008 Jun;82(6):445–53. doi: 10.1007/s00223-008-9130-9. Epub 2008/06/04. eng. [DOI] [PubMed] [Google Scholar]
- 15.Ellies DL, Viviano B, McCarthy J, Rey JP, Itasaki N, Saunders S, et al. Bone density ligand, Sclerostin, directly interacts with LRP5 but not LRP5G171V to modulate Wnt activity. J Bone Miner Res. 2006 Nov;21(11):1738–49. doi: 10.1359/jbmr.060810. Epub 2006/09/28. eng. [DOI] [PubMed] [Google Scholar]
- 16.Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011 Jun;17(6):684–91. doi: 10.1038/nm.2388. Epub 2011/05/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001 Mar;68(3):577–89. doi: 10.1086/318811. Epub 2001/02/17. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balemans W, Ebeling M, Patel N, Van Hul E, Olson P, Dioszegi M, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001 Mar 1;10(5):537–43. doi: 10.1093/hmg/10.5.537. Epub 2001/02/22. eng. [DOI] [PubMed] [Google Scholar]
- 19.Staehling-Hampton K, Proll S, Paeper BW, Zhao L, Charmley P, Brown A, et al. A 52-kb deletion in the SOST-MEOX1 intergenic region on 17q12-q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002 Jun 15;110(2):144–52. doi: 10.1002/ajmg.10401. Epub 2002/07/13. eng. [DOI] [PubMed] [Google Scholar]
- 20.Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002 Feb;39(2):91–7. doi: 10.1136/jmg.39.2.91. Epub 2002/02/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agholme F, Li X, Isaksson H, Ke HZ, Aspenberg P. Sclerostin antibody treatment enhances metaphyseal bone healing in rats. J Bone Miner Res. 2010 Nov;25(11):2412–8. doi: 10.1002/jbmr.135. Epub 2010/05/26. eng. [DOI] [PubMed] [Google Scholar]
- 22.Ominsky MS, Li C, Li X, Tan HL, Lee E, Barrero M, et al. Inhibition of sclerostin by monoclonal antibody enhances bone healing and improves bone density and strength of nonfractured bones. J Bone Miner Res. 2011 May;26(5):1012–21. doi: 10.1002/jbmr.307. Epub 2011/05/05. eng. [DOI] [PubMed] [Google Scholar]
- 23.Ominsky MS, Vlasseros F, Jolette J, Smith SY, Stouch B, Doellgast G, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010 May;25(5):948–59. doi: 10.1002/jbmr.14. Epub 2010/03/05. eng. [DOI] [PubMed] [Google Scholar]
- 24.Padhi D, Jang G, Stouch B, Fang L, Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011 Jan;26(1):19–26. doi: 10.1002/jbmr.173. Epub 2010/07/02. eng. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Warmington KS, Niu QT, Asuncion FJ, Barrero M, Grisanti M, et al. Inhibition of sclerostin by monoclonal antibody increases bone formation, bone mass, and bone strength in aged male rats. J Bone Miner Res. 2010 Dec;25(12):2647–56. doi: 10.1002/jbmr.182. Epub 2010/07/20. eng. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Ominsky MS, Warmington KS, Morony S, Gong J, Cao J, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009 Apr;24(4):578–88. doi: 10.1359/jbmr.081206. Epub 2008/12/04. eng. [DOI] [PubMed] [Google Scholar]
- 27.Daley E, Streeten EA, Sorkin JD, Kuznetsova N, Shapses SA, Carleton SM, et al. Variable bone fragility associated with an Amish COL1A2 variant and a knock-in mouse model. J Bone Miner Res. 2010 Feb;25(2):247–61. doi: 10.1359/jbmr.090720. Epub 2009/07/15. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000 Jul;29(1):52–4. doi: 10.2144/00291bm09. Epub 2000/07/25. eng. [DOI] [PubMed] [Google Scholar]
- 29.Sawakami K, Robling AG, Ai M, Pitner ND, Liu D, Warden SJ, et al. The Wnt co-receptor LRP5 is essential for skeletal mechanotransduction but not for the anabolic bone response to parathyroid hormone treatment. J Biol Chem. 2006 Aug 18;281(33):23698–711. doi: 10.1074/jbc.M601000200. Epub 2006/06/23. eng. [DOI] [PubMed] [Google Scholar]
- 30.Hanna SL, Sherman NE, Kinter MT, Goldberg JB. Comparison of proteins expressed by Pseudomonas aeruginosa strains representing initial and chronic isolates from a cystic fibrosis patient: an analysis by 2-D gel electrophoresis and capillary column liquid chromatography-tandem mass spectrometry. Microbiology. 2000 Oct;146(Pt 10):2495–508. doi: 10.1099/00221287-146-10-2495. Epub 2000/10/06. eng. [DOI] [PubMed] [Google Scholar]
- 31.Eyre DR, Weis MA, Wu JJ. Advances in collagen cross-link analysis. Methods. 2008 May;45(1):65–74. doi: 10.1016/j.ymeth.2008.01.002. Epub 2008/04/30. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ayturk UM, Jacobsen CM, Christodoulou DC, Gorham J, Seidman JG, Seidman CE, et al. An RNA-seq protocol to identify mRNA expression changes in mouse diaphyseal bone: Applications in mice with bone property altering Lrp5 mutations. J Bone Miner Res. 2013 Apr 2; doi: 10.1002/jbmr.1946. Epub 2013/04/05. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant GR, Farkas MH, Pizarro AD, Lahens NF, Schug J, Brunk BP, et al. Comparative analysis of RNA-Seq alignment algorithms and the RNA-Seq unified mapper (RUM) Bioinformatics. 2011 Sep 15;27(18):2518–28. doi: 10.1093/bioinformatics/btr427. Epub 2011/07/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan 1;26(1):139–40. doi: 10.1093/bioinformatics/btp616. Epub 2009/11/17. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shapiro SS, Wilk WB. An analysis of variance test for normality. Biometrika. 1965;52:591–611. [Google Scholar]
- 36.Sokal RR. Biometry: The principles and practice of statistics in biological research. 3. New York: W. H. Freeman; 1995. [Google Scholar]
- 37.Sinder BP, Eddy MM, Ominsky MS, Caird MS, Marini JC, Kozloff KM. Sclerostin antibody improves skeletal parameters in a Brtl/+ mouse model of osteogenesis imperfecta. J Bone Miner Res. 2013 Jan;28(1):73–80. doi: 10.1002/jbmr.1717. Epub 2012/07/28. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005 May 20;280(20):19883–7. doi: 10.1074/jbc.M413274200. Epub 2005/03/22. eng. [DOI] [PubMed] [Google Scholar]
- 39.Semenov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005 Jul 22;280(29):26770–5. doi: 10.1074/jbc.M504308200. Epub 2005/05/24. eng. [DOI] [PubMed] [Google Scholar]
- 40.Holmen SL, Giambernardi TA, Zylstra CR, Buckner-Berghuis BD, Resau JH, Hess JF, et al. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res. 2004 Dec;19(12):2033–40. doi: 10.1359/JBMR.040907. Epub 2004/11/13. eng. [DOI] [PubMed] [Google Scholar]
- 41.Motyl KJ, Dick-de-Paula I, Maloney AE, Lotinun S, Bornstein S, de Paula FJ, et al. Trabecular bone loss after administration of the second-generation antipsychotic risperidone is independent of weight gain. Bone. 2012 Feb;50(2):490–8. doi: 10.1016/j.bone.2011.08.005. Epub 2011/08/23. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shahnazari M, Wronski T, Chu V, Williams A, Leeper A, Stolina M, et al. Early response of bone marrow osteoprogenitors to skeletal unloading and sclerostin antibody. Calcif Tissue Int. 2012 Jul;91(1):50–8. doi: 10.1007/s00223-012-9610-9. Epub 2012/05/31. eng. [DOI] [PubMed] [Google Scholar]
- 43.Bogan R, Riddle RC, Li Z, Kumar S, Nandal A, Faugere MC, et al. A mouse model for human osteogenesis imperfecta type VI. J Bone Miner Res. 2013 Feb 14; doi: 10.1002/jbmr.1892. Epub 2013/02/16. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vranka JA, Pokidysheva E, Hayashi L, Zientek K, Mizuno K, Ishikawa Y, et al. Prolyl 3-hydroxylase 1 null mice display abnormalities in fibrillar collagen-rich tissues such as tendons, skin, and bones. J Biol Chem. 2010 May 28;285(22):17253–62. doi: 10.1074/jbc.M110.102228. Epub 2010/04/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006 Oct 20;127(2):291–304. doi: 10.1016/j.cell.2006.08.039. Epub 2006/10/24. eng. [DOI] [PubMed] [Google Scholar]
- 46.Lisse TS, Thiele F, Fuchs H, Hans W, Przemeck GK, Abe K, et al. ER stress-mediated apoptosis in a new mouse model of osteogenesis imperfecta. PLoS Genet. 2008 Feb;4(2):e7. doi: 10.1371/journal.pgen.0040007. Epub 2008/02/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aubin I, Adams CP, Opsahl S, Septier D, Bishop CE, Auge N, et al. A deletion in the gene encoding sphingomyelin phosphodiesterase 3 (Smpd3) results in osteogenesis and dentinogenesis imperfecta in the mouse. Nat Genet. 2005 Aug;37(8):803–5. doi: 10.1038/ng1603. Epub 2005/07/19. eng. [DOI] [PubMed] [Google Scholar]
- 48.Forlino A, Porter FD, Lee EJ, Westphal H, Marini JC. Use of the Cre/lox recombination system to develop a non-lethal knock-in murine model for osteogenesis imperfecta with an alpha1(I) G349C substitution. Variability in phenotype in BrtlIV mice. J Biol Chem. 1999 Dec 31;274(53):37923–31. doi: 10.1074/jbc.274.53.37923. Epub 1999/12/23. eng. [DOI] [PubMed] [Google Scholar]
- 49.Chipman SD, Sweet HO, McBride DJ, Jr, Davisson MT, Marks SC, Jr, Shuldiner AR, et al. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1993 Mar 1;90(5):1701–5. doi: 10.1073/pnas.90.5.1701. Epub 1993/03/01. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartung S, Jaenisch R, Breindl M. Retrovirus insertion inactivates mouse alpha 1(I) collagen gene by blocking initiation of transcription. Nature. 1986 Mar 27 2;Apr 27 2;320(6060):365–7. doi: 10.1038/320365a0. Epub 1986/03/02. eng. [DOI] [PubMed] [Google Scholar]
- 51.Lindahl K, Barnes AM, Fratzl-Zelman N, Whyte MP, Hefferan TE, Makareeva E, et al. COL1 C-propeptide cleavage site mutations cause high bone mass osteogenesis imperfecta. Hum Mutat. 2011 Jun;32(6):598–609. doi: 10.1002/humu.21475. Epub 2011/02/24. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Uveges TE, Kozloff KM, Ty JM, Ledgard F, Raggio CL, Gronowicz G, et al. Alendronate treatment of the brtl osteogenesis imperfecta mouse improves femoral geometry and load response before fracture but decreases predicted material properties and has detrimental effects on osteoblasts and bone formation. J Bone Miner Res. 2009 May;24(5):849–59. doi: 10.1359/JBMR.081238. Epub 2008/12/31. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]