

Abstract
Crohn’s disease (CD) and ulcerative colitis (UC) are the two main forms of inflammatory bowel disease (IBD) with both overlapping and distinct clinical, pathological and biomolecular features. It has been suggested that pediatric IBD is a distinct disease entity, with probably different disease subtypes.The aim of this study is to review and summarize the evolution of the current concept of pediatric IBD. The results of this review reinforce the idea that pediatric CD and UC may be further classified in various clinicopathologic entities. For clinicians and pathologists convenience, practical algorithms for the distinction of the various subphenotypes of pediatric IBD are also provided.
Keywords: Pediatric inflammatory bowel disease, Crohn’s disease, Ulcerative disease, Histopathology, Molecular biology
Core tip: The review contains the most recent data of the literature and suggests a clinical- pathological heterogeneity of the inflammatory bowel disease (IBD) in childhood. It provides diagrams that display the main anatomo-clinical and biomolecular correlations and that may be encountered in IBD children. These diagnostic patterns and correlations may be useful in clinical practice for pediatric IBD.
INTRODUCTION
Inflammatory bowel disease (IBD) represents a group of idiopathic, chronic, inflammatory intestinal conditions in which complex interactions among genetic, immune, and environmental factors are involved. Crohn’s disease (CD) and ulcerative colitis (UC) are the two most common forms of IBD with both overlapping and distinct clinical, pathological and biomolecular features. Traditionally, UC is defined as a disease involving the colonic mucosa in a diffuse, continuous manner, always affecting the rectum. In contrast, CD may involve any part of the gastrointestinal tract and frequently shows discontinuous or segmental involvement. However, IBD may be better considered as a syndrome of complex disorders with a significant heterogeneity in disease presentation and course[1-5]. It has also been suggested that pediatric IBD is characterized by distinct phenotypic differences including disease type, disease location, disease behaviour, gender preponderance and genetically attributable risk, compared to adult-onset IBD[6-11]. The incidence of IBD in childhood is rising worldwide[12,13]. Rates are highest in North America and Europe, with rapid increases noted in developing nations adopting a Westernized lifestyle. Childhood and adolescent IBD accounts for nearly 30% of total cases[14-17]. Pediatric IBD demonstrates a pattern with CD predominating over UC. In particular, the incidence of CD has risen markedly, while a rather stable incidence of pediatric onset UC has been reported[18]. A recent study based on the Swedish population found that the incidence of CD was 9.2 per 100000 per person years. The incidence of UC in children over the same time period was 2.8 per 100000 per person years[19]. Similar rates have been reported by other studies[20-24].
Recent population based studies have demonstrated a significant male excess in incidence of pediatric CD[25]. Pediatric CD more often involves the ileocolonic/colonic regions, whereas adult CD does not demonstrate a high proportion of colonic disease[7]. Furthermore, a variety of phenotypic characteristics have been described in pediatric UC thanks to increasing diagnostic accuracy[26,27]. Reflecting these trends, IBD classification has been changed from the Vienna statement[28], through the Montreal classification[29] to the recent pediatric Paris classification[30].
Although the precise etiology of IBD remains elusive, both animals and human studies point towards a strong genetic susceptibility. Genome wide association studies (GWAS) identified over 160 susceptibility loci/genes that are significantly associated with IBD[31-33]. However, newer genomics technologies are now beginning to complement GWAS findings and add to our understanding of the molecular genetic universe of IBD. Genetic studies have provided detailed appreciation of the molecular architecture of IBD, and, in particular, the areas of overlap between CD and UC (such as Th17 pathways) and the pathways which are disease-specific. Moreover, the genes implicated in childhood-onset and adult-onset IBD often overlap, suggesting similar contributory genetic predisposition and pathophysiological pathways. For CD, gene discoveries have focused on defective processing of intracellular bacteria, autophagy, and innate immunity. For UC, the focus has been on barrier function.
A further addition to the complexity of understanding disease mechanisms is that a susceptibility allele often requires other genetic and non-genetic cues to manifest the disease[34]. The variable concordance rate in monozygotic twins of 27%-50% in CD compared with 15%-19% in UC suggests that non-genetic factors may have an even more important role in UC than in CD[35,36].
In the present review, we summarize current knowledge concerning the correlation between clinicopathologic features and genetic profiles of pediatric IBD in order to offer practical algorithms for the categorization of the vast majority in this group of lesions.
PARIS CLASSIFICATION
The issue of subclassification of IBD by phenotype has been reviewed in recent years. The World Congress of Gastroenterology in Vienna, in 1998, considered age of onset (A), disease location (L), and disease behaviour (B) as predominant phenotypic elements[28]. The Montreal revision of the Vienna classification did not changed the three predominant parameters of age at diagnosis, location, and behaviour, but modification within each of these categories was made[29]. However, the criteria of Montreal Classification has inherent limitations with respect to classification of pediatric IBD. In the pediatric Paris classification[30] growth failure in the patient at any time was added as G1 vs G0 (never growth failure) to classic phenotypic elements (age at diagnosis, disease location, and disease behaviour). The comparisons between the Montreal and Paris classifications for CD and UC are shown in Tables 1 and 2, respectively.
Table 1.
Montreal and Paris classifications for Crohn’s disease
| Montreal classification | Paris classification | |||
| Age at diagnosis | A1 | < 17 yr | A1a | < 10 yr |
| A1b | 10-16 yr | |||
| A2 | 17-40 yr | A2 | 17-40 yr | |
| A3 | > 40 yr | A3 | > 40 yr | |
| Location | L1 | Ileal disease | L1 | Distal 1/3 ileum ± limited cecal disease colonic disease ileocolonic disease isolated |
| L2 | Colonic disease | L2 | upper disease | |
| L3 | Ileocolonic disease | L3 | ||
| L4 | Isolated upper disease | L4 | ||
| L4a | Upper disease proximal to ligament of Treitz | |||
| L4b | Upper disease distal to ligament of Treitz and proximal to distal 1/3 ileum | |||
| Behavior | B1 | Non-stricturing, non-penetrating | B1 | Non-stricturing, non-penetrating |
| B2 | Stricturing | B2 | Stricturing | |
| B3 | Penetrating | B3 | Penetrating | |
| B2B3 | both stricturing and penetrating | |||
| P | Perianal disease modifier | P | Perianal disease modifier | |
| Growth | Not applicable | G0 | No evidence of growth delay | |
| G1 | Growth delay | |||
Table 2.
Montreal and Paris classifications for ulcerative colitis
| Montreal classification | Paris classification | |||
| Extent | E1 | Ulcerative proctitis | E1 | Ulcerative proctitis |
| E2 | Leftsided colitis distal to splenic flexure | E2 | Leftsided colitis distal to splenic flexure | |
| E3 | Extensive colitis distal to hepatic flexure | E3 | Extensive colitis proximal to splenic flexure | |
| Severity | S0 | Clinical remission | E4 | Pancolitis, proximal to hepatic flexure |
| S1 | Mild ulcerative colitis | S0 | Never severe | |
| S2 | Moderate ulcerative colitis | S1 | Ever severe | |
| S3 | Severe ulcerative colitis | |||
IBD UNCLASSIFIEDAND “INDETERMINATE” COLITIS
The term “indeterminate colitis” was originally coined for IBD resections with features of both UC and CD, usually in the setting of severe acute or “fulminant” colitis[37]. Over the years the term has been adopted by clinicians to describe patients in whom a diagnosis of UC or CD cannot be made based on standard clinical testing, including colonoscopy, imaging, laboratory tests and biopsy[38]. However, this term has been used incorrectly with considerable confusion among clinicians and pathologists. Recently, it has been recommended that the term “indeterminate colitis” be reserved only for patients for whom a surgical specimen is available and the term “colonic IBD type unclassified” (IBD-U) for patients with no surgical specimen available and for whom endoscopy is inconclusive and histological changes do not fit with either CD or UC[29]. It remains controversial whether IBD-U constitutes a problem of classification or an IBD subtype distinct from CD and UC. Some authors believe that IBD-U is not a third form of IBD with specific diagnostic criteria, being a provisional diagnosis of exclusion used until a diagnosis of UC or CD is made with certainty[39,40]. Instead, other authors consider IBD-U as a distinct phenotype of IBD for the following reasons: (1) A recent meta-analysis showed that IBD-U is more common in children accounting for 12.7% of all cases of IBD vs 6% in adults[41]; (2) Children with IBD-U have a disease that rapidly progresses to pancolitis[42,43]; (3) Although many patients with IBD-U will be reclassified as having CD or UC on long-term follow-up evaluation, a significant proportion of them will still carry the diagnosis of indeterminate colitis[44]; (4) Epidemiologic data have shown that clinical course and prognosis of IBD-U could be worse compared with UC, especially concerning outcome of surgery with greater risk of pouchitis[45]; and (5) As will be discussed in the next section, IBD-U is diagnosed in a large subgroup of patients at a very early age (0-2 years, infantile IBD)[30-46].
AGE OF ONSET
An important modification recommended by the Paris classification[30] includes age at onset as A1a (0 to < 10 years) and A1b (10 to < 17 years). In both the Montreal[29] and Paris[30] Classification systems, A2 and A3 account for age of diagnosis at 17-40 years, and > 40 years, respectively.
Although rare, IBD may occur before the age of 2 years. Therefore, the Paris classification[30] suggested the possibility of distinguishing a separate group of children diagnosed with IBD at a very early age (0-2 years, infantile IBD). This subgroup is characterized by a high rate of consanguinity, more severe disease course, association with primary immunodeficiency and resistance to immunosuppressive treatment[45,46]. Abscess formation, anal fissures and enterocutaneous or rectovaginal fistulae complicate the disease and frequently require partial or total colectomy[30,47].
The suspicion of a monogenetic cause of these early onset forms was recently confirmed by the discovery of mutations in the genes coding for one of the two IL10 receptors causing impaired IL10 signalling[48-52].
The region encoding IL10 was originally identified by a German GWAS in UC, and the association documented with non-coding variants upstream of the IL10 gene[53]. Subsequently, an international GWAS meta-analysis showed that this region is associated with CD[54]. Through a genetic-linkage analysis and candidate-gene sequencing on samples from two unrelated consanguineous families with children affected by early-onset IBD, Glocker et al[55] identified three distinct homozygous mutations in genes IL10RA and ILRB. These genes encode the IL10R1 and IL10R2 proteins, respectively, which form a heterotetramer to make up the IL10 receptor. Functional experiments have shown that the IL10RA and ILRB gene mutations abrogate IL10 signalling and lead to severe intestinal inflammation. Loss of function mutation in IL10 and IL10R was also identified in 66 patients with very early onset IBD. In this study, it has been found that in 5 patients with IL10R deficiency, the allogenic hematopoietic stem cell transplantation induced sustained clinical remission with a median follow-up time of 2 years[49]. Recently, Moran et al[50] have identified a novel homozygous, splice-site point mutation in IL10RA in an infantile-onset IBD Caucasian female. The patient was also affected by significant arthritis and folliculitis. The mutation caused a premature stop codon (P206X) and IL10 insensitivity. Moreover, 188 children with early-onset IBD and 188 healthy subjects have been studied. In the discovery cohort, five IL10RA polymorphisms associated with UC have been found[50]. These studies show the role of immune pathways in early onset IBD pathogenesis. Indeed, IL10 is an anti-inflammatory cytokine secreted by a variety of cell types and is critical for maintaining immune homeostasis in the gastrointestinal (GI) tract. IL10 restricts excessive immune response[56]. In particular, IL10 limits the secretion of proinflammatory cytokines, such as tumour necrosis factor α (TNF-α) and IL12[57]. Moreover, the assembly of IL10R1 results in the activation of the receptor-associated Janus tyrosine kinases, JAK1 and Tyk2, leading to the phosphorylation of STAT3 (signal transducer and activator of transcription 3) and the induction of STAT3-dependent genes[58]. A severe enterocolitis has been found in mice that are deficient in either IL10 or IL10R2. These data underline the pivotal role of IL10 in the mediation of signalling that controls inflammation in the gut.
Other monogenic primary immunodeficiencies showing IBD-like gastrointestinal pathology include Wiskott-Aldrich syndrome[47], chronic granulomatous disease[47], XIAP deficiency[55,59], X-linked (IPEX) syndrome[60] and nuclear factor kB essential modulator (NEMO) deficiency[61]. It is also noteworthy that a large subgroup of these patients have IBD-U[30,42]. Therefore, we think that IBD-U is an IBD subtype distinct from CD and UC as it constitutes a histopathological substrate of a clinicopathologic entity with characteristic epidemiological (< 2 years old), clinical ( severe clinical course with pancolitis), and genetic features (i.e., mutation in interleukin-10 receptor and interleukin gene or other primary immunodeficiencies). It is, therefore, plausible that infantile IBD (< 2 years) may be incorporated into future modifications of the Paris classification. Figure 1 summarizes the main clinicopathologic characteristics of infantile IBD.
Figure 1.
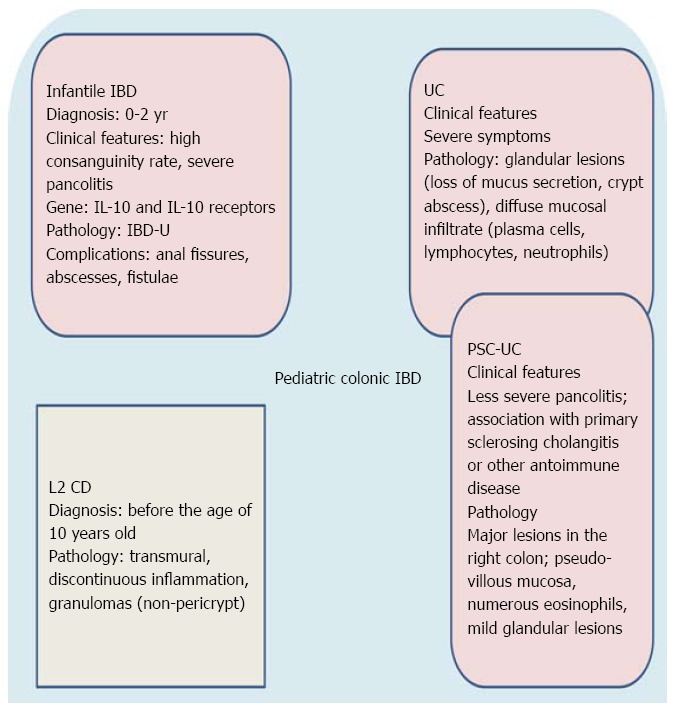
Distinct clinicopathological and biomolecular features of the pediatric colonic inflammatory bowel disease. IBD: Inflammatory bowel disease; CD: Crohn’s disease; UC: Ulcerative colitis.
CD
Disease location
According to the Paris classification[30], CD location is categorized as follows: (L1) involvement of the terminal ileum only, with limited or no cecal disease; (L2) colonic involvement only; (L3) involvement of both the terminal ileum and colon; (L4) isolated upper gastrointestinal disease. L4 is further separated into esophagogastroduodenal disease (L4A), jejuna/proximal ileal disease (L4B), or both L4A and L4B. Adolescents more often present with ileal disease (L1), whereas children have a tendency to present with isolated colonic disease (L2). Pediatric patients with L2 disease are less likely to have esophagogastroduodenal involvement or stricturing disease behaviour than patients with L1 and L3 disease[62]. These data support the existence of discrete subtypes of CD, which are, in part, defined by the predominant anatomical location of disease[7] and summarized in Table 3 and Figure 2.
Table 3.
Salient histopathological and biomolecular features in pediatric inflammatory bowel disease
| Gene | Genetic alterations | Histopathology | Clinical features | Ref. |
| IL10RA | G141R | IBD-U | Infantile IBD | [48-50] |
| IL10RB | W159X | |||
| NOD2 | R702W, G908R, L1007fsinsC | Abnormal Paneth cells, granuloma-poor | L1 CD | [72] |
| ATG16L1 | T300A | |||
| NOD2 | R702W, G908R, L1007fsinsC | L1 CD, | [81-87] | |
| Structuring (B2) behavior | [82,83,87] | |||
| Early surgery | [83] | |||
| Growth delay | [83,84] | |||
| Higher disease activity | [83,88] | |||
| NOD2 | R702W, G908R, L1007fsinsC | Diminished immunohistochemical | L1, L3 CD | [106-108] |
| Expression of alpha-defensin in small intestinal Paneth cells | ||||
| TLRs | TLR-4 Asp299gly | CD, UC | [111] | |
| IRGM | rs4958847 | Fistulizing (B3) CD | [135] | |
| ILRL1 and IL33 | Upexpression of mRNA | Extensive UC | [138-140,150] | |
| β-defensin | Low gene copy number | Diminished immunohistochemical expression of beta-defensin 2 | L2 CD | [104,105] |
| HBD2 | ||||
| NCF4 | Rs4821544 polymorphism | Perianal CD | [157] |
IBD: Inflammatory bowel disease; CD: Crohn’s disease; UC: Ulcerative colitis.
Figure 2.
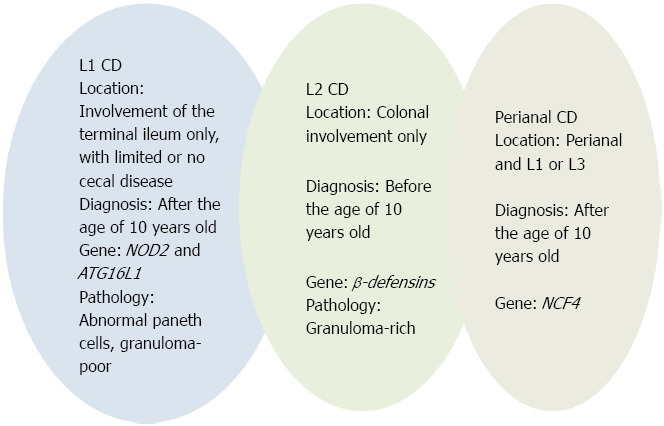
Subtypes of Crohn’s disease. CD: Crohn’s disease.
Paneth cells are protagonist in the L1CD
Histologic, immunologic and biomolecular evidence strongly suggests a key role for Paneth cells in L1 CD. Histologically, Paneth cells are abundant in the small intestine and are occasionally found in the cecum and proximal ascending colon[63]. In parallel, L1 (distal 1/3 ileum ± limited cecal disease) CD location corresponds to the normal distribution of Paneth cells.
From an immunological viewpoint, Paneth cells, by virtue of their vast repertoire of effector molecules, are multifunctional cells. They produce antimicrobial proteins such as alpha-defensins HD5 and HD6, lysozyme, secretory phospholypase A2 and the lectinRegIIIγ[64]. Wehkamp et al[65] showed that ileal (L1) but not isolated colonic (L2) CD is associated with a diminished synthesis of Paneth cell defensins. Wehkamp and Stange[66], therefore, proposed the term “Paneth’s disease” to describe a complex disease in which Paneth cells might explain the poor antimicrobial capability of (L1) CD. Paneth cells have also been shown to contain TNF-α transcripts. With ultrastructural immunogold methods, Beil et al[67] showed TNF-α in mature and immature secretory granules of Paneth cells. One of the major biological roles of TNF-α is in the host defence to bacterial, viral and parasitic infections, and Paneth cells are considered to be the major source of TNF-α in the normal bowel[67].Clinical and molecular studies implicate TNF-α as a key mediator in the initiation and propagation of CD[68]. This is evidenced by an increased amount of TNF in inflammatory cells infiltrating ileal tissue (eosinophils, mast cells, neutrophils, macrophages, fibroblasts) and a marked clinical response of TNF-α antagonists in patients with active CD[69]. Therefore, abnormal production of TNF-α protein by Paneth cell may be involved in the pathogenesis of L1 CD[69].
From a histopathological viewpoint, Paneth cell depletion occurs in the most heavily inflamed areas of the ileal mucosa[70]. Recently, Günther et al[71] showed a significant decrease in the number of Paneth cells and a high number of dying cells with shrunken eosinophilic cytoplasm at the crypt base in histological samples from terminal ileum of patients with active CD. Paneth cells showed ultrastructural signs of non-apoptotic cell death, such as organelle swelling, vacuole formation and the lack of blebbing. The data of Günther et al[71] suggest that necroptosis (a form of non-apoptotic cell death) of Paneth cells might be involved in the pathogenesis of CD.
Recently, VanDussen et al[72] have studied the correlation between Paneth cell phenotype (based on lysozyme-positive secretory granule morphology) and NOD2 and ATG16L1 genotype. They observed an inverse correlation between abnormal Paneth cells (with disordered, diminished, diffuse, or excluded granule phenotypes) and the presence of granulomas[72]. The cumulative number of NOD2 and ATG16L1 risk alleles had an additive effect on the proportion of abnormal Paneth cells. Moreover, high proportions of abnormal Paneth cells were associated with shorter time to disease recurrence after surgery. VanDussen et al[72] suggest stratifying CD based on Paneth cell phenotypes rather than the presence of granulomas. In fact, granulomas could be sparse and more likely to be undersampled, especially in biopsy specimens, whereas Paneth cell phenotypes are more easily analyzed within limited samples. Therefore, these authors concluded that histologic analysis of Paneth cell phenotypes can be used to divide patients with CD into subgroups with distinct pathognomonic and clinical features[49]. However, further correlative histopathologic and biomolecular studies are necessary to clarify the role of necroptosis and/or granule changes of Paneth cells in L1 CD characterized by abundant or few granulomas (so-called granuloma-rich or granuloma-poor CD).
Biomolecular studies of antimicrobial function in Paneth cells
Defective antimicrobial function in Paneth cells has been described by a variety of mechanisms including mutations in the innate immune receptor NOD 2, defensins, Toll-like receptors (TLRs), the autophagy protein ATG16L1, IRGM gene.
NOD2 gene
With the pivotal study of Hugot et al[73], who discovered the very first and also strongest susceptibility gene for CD in NOD2/CARD15, “IBD gene hunting” was opened. NOD2 is an intracytoplasmic member of the family of intracellular NLR (NOD like receptors) able to recognize pathogen associated molecular patterns (PAMPs) and to modulate an inflammatory response through enhanced NF-κB activation[74,75]. NOD2 is highly expressed in cells of the phagocyte system and is involved in the production of defensins in response to gut microbiota. Three single nucleotide polimorphisms (SNPs) within the NOD2/CARD15 gene (R702W, G908R, and 1007fsinsC) have been established as independent risk factors for CD in Caucasians[76,77] and they represent 82% of the mutations in NOD2[78]. Interestingly, the NOD2/CARD15 mutations are absent or very rare in Asians, Arabs, Africans, and African Americans[79]. NOD2/CARD15 variants have been associated to more severe CD, a greater need for surgery and a younger age at onset[80]. In children, a correlation with ileal (L1) localization[81-87], stricturing behavior[82,83,87], early surgery[83], growth delay[83,84] and higher disease activity[83,88] has been found. However, these results have not been confirmed by other studies[89-91]. The precise mechanistic relationship between NOD2/CARD15 and CD remains controversial. Interestingly, a study by Zelinkova et al[92] suggested that NOD2 mutations may result in perpetuation of mucosal inflammation through insufficient pathogen elimination.
The important role of NOD2 in the pathogenesis of CD has been underlined by recent studies on microRNAs (miRNAs)[93]. miRNAs are short non-coding RNAs that have emerged as key modulators of various cellular processes at the post-transcriptional level[94-96]. There are recent reports on their role in the regulation of intestinal permeability as the loss of intestinal miRNAs impairs the epithelial barrier function, and causes acute inflammation[97]. Some Authors showed that miRNA-122 regulates intestinal permeability tight junctions (TJ) by targeting occluding mRNA degradation[98-102]. Moreover, Chen et al[102] found that NOD2 is a functional target of miRNA-122.
Other mechanisms have been proposed for NOD2/CARD15 mutations in recent years, ranging from abnormal Paneth cell function with reduced defensin secretion, altered modulation of Toll-like receptor signaling and a reduced ability to trigger autophagy[103]. Several studies have shown the role of defensins in the pathogenesis of CD[104-107]. In particular, ileal (L1) and ileal colonic (L3) CD is characterized by a specific decrease in small intestinal Paneth cell human α-defensin HD-5 and-6[106-108]. In a group of pediatric CD, Perminow et al[109] studied the role of HD5 and TCF-4, a Wnt-signaling transcription factor which controls Paneth cell defensin expression. They showed a low intestinal expression of HD5 and TCF4 mRNA in ileal CD, confirming the important role of antimicrobial host defense in pediatric patients.
Toll-like receptors (TLRs) have an important role in the pathogenesis of CD. They are crucial components of innate immunity and cell surface molecules that also detect normal and pathogenic microbial agents and can trigger antimicrobial host defense responses. TLRs are abundantly expressed on the surface of monocytes, macrophages, and dendritic and epithelial cells[110]. In IBD mucosa, dendritic cells are activated and there are increased levels of TLR2 and TLR4. These TLRs mediate the recognition of bacterial lipoproteins and LPS, respectively. An association has been described between the TLR4 D299G SNP and IBD, in UC as well as CD patients[111]. This SNP is associated with impaired LPS signaling and increased susceptibility to gram-negative infections, thus supporting the role of gram-negative bacteria in the pathogenesis of IBD. Polymorphisms have been described in IBD with regard to the TLR9 gene, but their functional significance is still unclear[112].
Genes of the autophagy: A key role in IBD
The autophagy 16-like 1 (ATG16L1), immunity-related guanosine triphosphate M (IRGM), and leucine-rich repeat kinase 2 (LRRK2) genes, which regulate autophagy, have been associated with CD in GWAS[113,114].
Autophagy describes the sequestration of intracellular material such as obsolete organelles or large unfolded protein within membranes, and their trafficking to fuse with lysosome with a subsequent degradation of the contents. Autophagy is now recognized as playing a key role in innate immunity against intracellular micro-organisms.
ATG16L1 is essential for all forms of autophagy as it has a role in the clearance of intracellular bacteria[115]. It interacts with two other autophagy proteins, ATG5 and ATG12, to forms a complex essential for the process of autophagy[116]. The association at ATG16L1 with CD seems to be entirely accounted for by the T300A coding variant which maps to a highly conserved part of the gene adjacent to the coiled-coil domain. ATGL16L1 mutations cause a deficiency of the correspondent protein and disrupt the recruitment of the ATG12-ATG5. Therefore, autophagosome formation and degradation of proteins with a long half-life are severely impaired in ATG16L1-deficient cells[117,118]. The decreased autophagy impairs immune tolerance by autoantigen presentation on major histocompatibility complex class II molecules and causes immune inflammation[119].
ATG16L1 gene mutations could also impair the mechanisms that involve autophagy and apoptosis as there is an acceleration in the rate of epithelial cell apoptosis and an inhibition of inflammatory cell apoptosis in CD and UC[120,121]. Several studies have been performed on the role of ATG16L1 gene in the pathogenesis of IBD.
The T300A allele was correlated with the incidence of CD in three populations from Germany, Hungary and the Netherlands[122]. In contrast, no correlation between T300A allele and UC was detected. However, the results of other studies on the association of T300A allele with predisposition to CD and UC, are inconsistent[122], also in child-onset IBD cases[123]. Recently, a meta-analysis performed on twenty-five studies of CD, 14 of which involved cases of UC[124] showed that the T300A allele confers a susceptibility to CD and to UC. However, ATG6L1 was associated with the risk of child-onset CD, but not with child-onset UC probably because there are few studies on pediatric UC. Recently, a functional link between NOD2/CARD15, ATG16L1 and autophagy has been provided[125-128].
IRGM gene
In the Wellcome Trust Case-Control Consortium (WTCCC) GWA scan, a highly significant association between variant flanking IRGM and susceptibility to CD has been shown. The IRGM gene is located on chromosome 5q33.1, and is required during the initiation phase of autophagy, when it localizes to bacteria- containing autophagic vacuoles. IRGM and autophagy are involved in clearance of intracellular organisms such as M. tubercolusis[129,130] and the CD-associated IRGM variant is predicted to affect autophagic control of Salmonella typhimurium[131].
The IRGM risk alleles for CD are non-coding and appear to affect mRNA transcription or stability. Over the last 5 years three distinct mechanisms have been identified which might explain the impact on IRGM expression. Cooney et al[131] discovered a large copy number variant upstream of IRGM which correlated with tissue-specific expression effects. Prescott et al[132] reported a disruption of a transcription factor binding site in the IRGM promoter. Most recently, Brest et al[133] found that a synonymous coding variant of IRGM alters the binding domain of miRNA196, a family of miRNAs, hence affecting mRNA stability and gene translation. In this study, it has been shown that microRNA196 is overexpressed in CD patients and that it downregulates an IRGM protective variant but not the risk-associated allele.
Lapaquette et al[134] found that the reduced IRGM gene expression correlated with impaired clearance by macrophages of CD-associated adherent-invasive Escherichia coli. A recent Italian study[135] reported the association between CD and two risk SNPs (rs1000113 and rs4958847) for IRGM, irrespective of age, but not in UC. In addition, a trend to B3 (penetrating) disease behavior in patients with IRGM SNPs was suggested[135]. Moon et al[136] showed that IRGM SNP rs10065172 was significantly associated with CD susceptibility. They also reported a protective role of SNP rs72553867.
By suppressive subtractive hybridization (SSH) technique, Sim et al[137] studied the differential expression gene profiles in ileal biopsies from CD children. Twenty-eight genes previously reported in association with adult CD, and 47 new genes were identified. It is significant that some adult CD genes have also been found in early-onset CD. Indeed, it underlines that, in some cases, there is a common genetic pathway between pediatric and adult CD cases. Several genes reported in the study are involved in microbial pathogenesis, antigen presentation, inflammation, regulation of epithelial barrier function, vesicular transport or cell differentiation and proliferation. Recent studies have found that IL33 expression is enhanced in the inflamed colonic mucosa of IBD, especially in UC[138,139], and the IL33/IL1RL1 signalling axis has been implicated in the IBD pathogenesis[140]. Recently, new genes have been identified by GWAS in IBD[141-150]. In particular, in 2008, Kugathasan et al[141] performed the first GWAS in a cohort with pediatric disease onset, identifying two new loci on 20q13 and 21q22. This paper reported the associations with two intronic SNPs (rs2315008 and rs4809330) at the 20q13 locus. These two SNPs map to the zinc finger CCCH-type with G patch domain (ZGPAT) gene which is located in a region containing eight potential candidate genes for CD. Moreover, an association, at the 21q22 region, of CD with the intergenic SNP rs2836878 has been shown that implicated the nearby PSMG1 gene[141].
A follow-up scan on an extended cohort of 3426 childhood-onset IBD (European/North America collaboration) identified five more new loci associated with pediatric IBD[147]. These loci included 16p11 near the cytokine gene IL27, 22q12, 10q22, 2q37 and 19q13.11. The results of this study showed that IL27 is a promising candidate gene for pediatric CD. Recently, Latiano et al[149] have studied a large Italian cohort of adult and early-onset IBD to verify the role of new genes involved in the immune response and inflammation (PTGER4, HLA-BTNL2, TNFSF15, NKX2-3, ZNF365, IFNG, PTPN2, and PSMG1).
Role of colonic epithelial cells producing β-defensins in colonic (L2) CD location.
Since Paneth cells are rare in the colon, the contribution of α-defensins to antimicrobial defence in the large intestine is only limited. In contrast, β-defensins HDB1-3 are secreted by columnar and goblet cells in the colon[151,152]. A deficiency of β-defensin HDBD2 has been shown by molecular biology in L2 (colonic) CD and confirmed on the protein level by immunohistochemistry[104]. This observed colonic defect of the antimicrobial barrier caused by a diminished expression of β-defensins may allow luminal microbes to attach to and invade the mucosa triggering the inflammation[153].
CD behaviour
The Montreal classification[29] describes three behaviours for CD: nonstricturing nonpenetrating disease (B1), stricturing disease (B2), penetrating/fistulizing disease (B3). In addition, the Paris classification[30] proposes a new classification B2B3 to identify patients with both B2 and B3 phenotypes (either at the same or different times). Disease locations are the most important factor identified in determining the risk of developing either a structuring or penetrating complication (more complications with ileal, less with colonic disease)[154]. To date, there have been few pediatric studies that have evaluated the association between CD behavior and genotype with prolonged follow-up. In a study by Shaoul et al[155], pediatric CD at the end of follow-up (mean 4.9 years) was classified as inflammatory (78%), stricturing (17%) and penetrating (7%). Moreover, a role for NOD2 in CD behavior (as opposed to location) was not supported in this study. NOD2 genotype was clearly associated with ileal involvement (L1), but not as an independent risk factor for stricturing, penetrating disease or a need for surgery[155]. A recent study showed that L1 CD and stricturing disease behavior are more common in children diagnosed after 10 years of age than in younger patients[39].
Approximately 10% of newly diagnosed pediatric patients shows perianal CD at time of diagnosis[156]. The Vienna classification did not distinguish luminal and perianal fistulising disease into different categories[28]. Subsequent evidence suggested that perianal and luminal fistulae often occur completely independently of each other. According to the Montreal and Paris classifications[29,30], a separate perianal modifier was added that can coexist with any disease behaviour. Perianal CD is defined as inflammation at or near the anus, including tags, fissures, fistulae, abscesses, or stenosis. A recent study by Eglinton et al[157] suggests specific associations with perianal CD patients compared with patients without perianal involvement. These associations include younger age at diagnosis, male gender, ileal (L1 or L3) location, and complicated disease behaviour (B2 + B3). In addition, an association with the NCF4 gene was demonstrated[157]. Taken together, literature data suggest that perianal CD represents a distinct disease phenotype, summarized in Figure 2.
UC
UC is classically defined as a chronic inflammation characterized by a continuous involvement of the colonic mucosa without granulomas on biopsy, affecting the rectum and a variable extent of the colon in continuity, and characterized by a relapsing, remittent course[158]. However, several studies suggest that pediatric UC may present with atypical phenotypes, such as macroscopical rectal sparing, macroscopic skip lesions in the colon, periappendiceal inflammation, backwash ileitis, limited upper gastrointestinal inflammation, and extensive colitis with less severe and less diffuse architectural abnormalities[159,160]. Thus, diagnosis of UC in pediatric patients may be particularly difficult. The recent Paris classification[30], which examined this issue, adds a new category (E4) for pancolitis (inflammation extending proximal to the hepatic flexure) (Table 2). Moreover, a disease behavior classification of S0 or S1 was adopted, with the latter denoting the presence of severe disease at any time in patient history (Table 2). The classification of UC based on anatomical extent has clinical relevance, as it affects the choice of therapy and the mode of delivery of drugs. For example, oral therapy is the first choice for UC extending above the splenic flexure[161].
Primary sclerosing cholangitis (PSC) is a cholestatic liver disease of unknown etiology, commonly associated with IBD and characterized by inflammation, fibrosis, and stenoses of the biliary tree leading to liver cirrhosis[162]. In most patients (80%-90%) the IBD can be classified as UC. Several features of UC in PSC differ from those of a general UC population (increased frequency of pancolitis, “backwash ileitis”, rectal sparing and colorectal carcinoma), and these observations have led to the hypothesis that UC in PSC may represent a specific phenotypic entity (PSC-UC)[163]. There are a few studies on PSC-UC in pediatric patients. Ordonez et al[164] studied twenty-eight consecutive children with UC associated with PSC, celiac disease, or autoimmune hepatitis comparing them with a matched control group of 27 children with isolated UC. At diagnosis, pancolitis was seen in 18/28 UC associated with autoimmunity patients compared with 8/27 in UC. Pathological findings were also different from isolated UC: (1) major lesions predominantly located in the right colon; (2) pseudo-villous appearance of the mucosa, and strong infiltration with eosinophils; and (3) mild glandular lesions. Evolution in UC associated with autoimmune disease was less aggressive, requiring less corticosteroids/immunomodulators[164]. In conclusion, clinical, histological, and molecular analyses reveal marked differences between pediatric patients with isolated UC and those with associated autoimmune phenomena, supporting the hypothesis of a distinct autoimmune presentation of UC (Figure 1).
Goblet cells as protagonist in the UC
Mucus produced by goblet cells forms a key component of the mucosal barrier. Gel-forming mucins of intestinal mucus are arranged into a bilayer with a firm inner layer devoid of bacteria and a looser outer layer with MUC2 being major constituent of both[165]. The mucus layer has a crucial role in intestinal homeostasis, as decreased levels of goblet cells, leading to reduced mucin secretion, are a hallmark of human UC[166]. The thickness of the mucus layer in the healthy colon is between 100 and 300 μm, increasing from the ascending colon to the rectum, whereas in UC, this mucus layer is thinner, more variable and in part denuded[167-169]. Taken together, several lines of evidence strongly suggest a key role for goblet cells in UC.
Molecular biology of UC
GWAS and candidate gene association studies identified several UC susceptibility loci, including 7 that overlap with CD (e.g., IL23 pathways genes, NKX2-3 and IL10)[170]. In a recent study, three new UC specific loci (HNF4A, CDH1 and LAMB1) has been found that are involved in the regulation of barrier function[171].
An association was seen at rs6017342 SNP which maps within a recombination hotspot on chromosome 20q13 in which the 3’ untraslated region (UTR) of the HNF4A gene is located. This gene encodes the transcription factor hepatocyte nuclear factor 4 α which regulates the expression of multiple components within the adherens junction, the tight junction and desmosome[172].
Cell-cell junctions have an important role on epithelial organization and barrier function. Moreover, in the embryonic age, the HNF4A gene participates in the development of mammalian gastrointestinal tract[173]. CDH1 gene which encodes E-cadherin has been located on chromosome 16q22. E-cadherin is a transmembrane glycoprotein, a key component of the adherens junction and mediates intercellular adhesion in the intestinal epithelium[171]. It also participates in processes of epithelial restitution and repair following the damage of mucosa. Indeed, there is a significant reduced expression of CDH1 in areas of active UC[174]. Interestingly, two CDH1 variants associated with UC[171] have also been associated with colorectal cancer[175], a possible complication of UC. The recent hypothesis is that HNF4A and E-cadherin co-participate to maintain the integrity of the epithelial intestinal barrier. LAMB1 gene is located on chromosome 7q31[171] and encodes the laminin β 1 subunit, which is detected in laminins-1, -2 and-10. In UC, the expression of laminins in the intestinal basement membrane is downregulated[171].
In 2011, Anderson et al[176] identified 29 additional UC risk loci from a meta-analysis and IL1R2, IL8RA/B, IL7R, IL12B, DAP, PRDM1, JAK2, IRF5, GNA12 and LSP1 were proposed as new important candidate genes in UC pathogenesis. Several genes were associated with cytokines and cytokine receptors, key regulators of citokyne-mediated signalling pathways, innate and adaptive immune response, macrophage activation and regulation of apoptosis. Interestingly, an association with DAP gene (death-associated protein) has been found. The expression of DAP kinase increases with inflammation in UC[177] and is a negative regulator of autophagy[178]. Therefore, the association with DAP suggests a possible link between autophagy and UC.
PRDM1, IRF5 and NKX2-3 genes could have a key role for transcriptional regulation in UC pathogenesis. GNA12 which plays a fundamental role in the tight junction assembly in epithelial cells has been identified at the 7p22 locus[179]. It is the most likely UC candidate gene from those described in the meta-analysis of Anderson et al[176]. Barrier integrity is important in UC pathogenesis given previous associations to HNF4A, CDH1 and LAMB1 genes[171]. Further studies are, however, need to verify the role of the genes in pediatric UC. The salient histopathological and biomolecular features of pediatric IBD are shown in Table 3.
CONCLUSION
In conclusion, our review reinforce the idea that pediatric UC and CD may be further classified into various clinicopathologic entities, based on genotype-phenotype correlation reported in recent literature. Therapy for complex genetic diseases such as IBD is difficult. For example, treatment with ant-TNF-α monoclonal antibodies does not induce remission in the majority of CD cases. It is becoming increasingly apparent that novel strategies to define and stratify IBD patients, that are based on serum, DNA and histopathology, will be needed to progress towards improved diagnostics, prognostics and therapeuthics.
Footnotes
P- Reviewer: Singhal S, van Langenberg DR S- Editor: Zhai HH L- Editor: A E- Editor: Liu XM
References
- 1.Walfish A, Sachar D. Phenotype classification in IBD: Is there an impact on therapy? Inflamm Bowel Dis. 2007;13:1573–1575. doi: 10.1002/ibd.20232. [DOI] [PubMed] [Google Scholar]
- 2.Louis E, Van Kemseke C, Reenaers C. Necessity of phenotypic classification of inflammatory bowel disease. Best Pract Res Clin Gastroenterol. 2011;25 Suppl 1:S2–S7. doi: 10.1016/S1521-6918(11)70003-8. [DOI] [PubMed] [Google Scholar]
- 3.Targan SR, Hawkey CJ. Indeterminate colitis. In: Hawkey CJ, Bosch J, Richter JE, Garcia-Tsao G, Chan FKL, et al., editors. Textbook of Clinical Gastroenterology and Hepatology. Chichester: Wiley-Blackwell; 2012. pp. 394–398. [Google Scholar]
- 4.Sachar DB, Walfish A. Inflammatory bowel disease: one or two diseases? Curr Gastroenterol Rep. 2013;15:298. doi: 10.1007/s11894-012-0298-9. [DOI] [PubMed] [Google Scholar]
- 5.Kaser A, Zeissig S, Blumberg RS. Genes and environment: how will our concepts on the pathophysiology of IBD develop in the future? Dig Dis. 2010;28:395–405. doi: 10.1159/000320393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kelsen J, Baldassano RN. Inflammatory bowel disease: the difference between children and adults. Inflamm Bowel Dis. 2008;14 Suppl 2:S9–S11. doi: 10.1002/ibd.20560. [DOI] [PubMed] [Google Scholar]
- 7.Levine A. Pediatric inflammatory bowel disease: is it different? Dig Dis. 2009;27:212–214. doi: 10.1159/000228552. [DOI] [PubMed] [Google Scholar]
- 8.Sagiv-Friedgut K, Karban A, Weiss B, Shaoul R, Shamir R, Bujanover Y, Reif S, Boaz M, Shani I, Levine A, et al. Early-onset Crohn disease is associated with male sex and a polymorphism in the IL-6 promoter. J Pediatr Gastroenterol Nutr. 2010;50:22–26. doi: 10.1097/MPG.0b013e3181b7a6a4. [DOI] [PubMed] [Google Scholar]
- 9.Gupta N, Bostrom AG, Kirschner BS, Ferry GD, Winter HS, Baldassano RN, Gold BD, Abramson O, Smith T, Cohen SA, et al. Gender differences in presentation and course of disease in pediatric patients with Crohn disease. Pediatrics. 2007;120:e1418–e1425. doi: 10.1542/peds.2007-0905. [DOI] [PubMed] [Google Scholar]
- 10.Biank V, Broeckel U, Kugathasan S. Pediatric inflammatory bowel disease: clinical and molecular genetics. Inflamm Bowel Dis. 2007;13:1430–1438. doi: 10.1002/ibd.20213. [DOI] [PubMed] [Google Scholar]
- 11.Van Limbergen J, Russell RK, Drummond HE, Aldhous MC, Round NK, Nimmo ER, Smith L, Gillett PM, McGrogan P, Weaver LT, et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology. 2008;135:1114–1122. doi: 10.1053/j.gastro.2008.06.081. [DOI] [PubMed] [Google Scholar]
- 12.Heyman MB, Kirschner BS, Gold BD, Ferry G, Baldassano R, Cohen SA, Winter HS, Fain P, King C, Smith T, et al. Children with early-onset inflammatory bowel disease (IBD): analysis of a pediatric IBD consortium registry. J Pediatr. 2005;146:35–40. doi: 10.1016/j.jpeds.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 13.Ponder A, Long MD. A clinical review of recent findings in the epidemiology of inflammatory bowel disease. Clin Epidemiol. 2013;5:237–247. doi: 10.2147/CLEP.S33961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russel MG. Changes in the incidence of inflammatory bowel disease: what does it mean? Eur J Intern Med. 2000;11:191–196. doi: 10.1016/s0953-6205(00)00090-x. [DOI] [PubMed] [Google Scholar]
- 15.Pappa HM, Semrin G, Walker TR, Grand RJ. Pediatric inflammatory bowel disease. Curr Opin Gastroenterol. 2004;20:333–340. doi: 10.1097/00001574-200407000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Hait E, Bousvaros A, Grand R. Pediatric inflammatory bowel disease: what children can teach adults. Inflamm Bowel Dis. 2005;11:519–527. doi: 10.1097/01.mib.0000166932.66853.fd. [DOI] [PubMed] [Google Scholar]
- 17.Murch SH, Baldassano R, Buller H, Chin S, Griffiths AM, Hildebrand H, Jasinsky C, Kong T, Moore D, Orsi M. Inflammatory bowel disease: Working Group report of the second World Congress of Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2004;39 Suppl 2:S647–S654. doi: 10.1097/00005176-200406002-00011. [DOI] [PubMed] [Google Scholar]
- 18.Kugathasan S, Judd RH, Hoffmann RG, Heikenen J, Telega G, Khan F, Weisdorf-Schindele S, San Pablo W, Perrault J, Park R, et al. Epidemiologic and clinical characteristics of children with newly diagnosed inflammatory bowel disease in Wisconsin: a statewide population-based study. J Pediatr. 2003;143:525–531. doi: 10.1067/s0022-3476(03)00444-x. [DOI] [PubMed] [Google Scholar]
- 19.Malmborg P, Grahnquist L, Lindholm J, Montgomery S, Hildebrand H. Increasing incidence of paediatric inflammatory bowel disease in northern Stockholm County, 2002-2007. J Pediatr Gastroenterol Nutr. 2013;57:29–34. doi: 10.1097/MPG.0b013e31828f21b4. [DOI] [PubMed] [Google Scholar]
- 20.Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis. 2011;17:423–439. doi: 10.1002/ibd.21349. [DOI] [PubMed] [Google Scholar]
- 21.Grieci T, Bütter A. The incidence of inflammatory bowel disease in the pediatric population of Southwestern Ontario. J Pediatr Surg. 2009;44:977–980. doi: 10.1016/j.jpedsurg.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 22.Bernstein CN, Wajda A, Svenson LW, MacKenzie A, Koehoorn M, Jackson M, Fedorak R, Israel D, Blanchard JF. The epidemiology of inflammatory bowel disease in Canada: a population-based study. Am J Gastroenterol. 2006;101:1559–1568. doi: 10.1111/j.1572-0241.2006.00603.x. [DOI] [PubMed] [Google Scholar]
- 23.Perminow G, Brackmann S, Lyckander LG, Franke A, Borthne A, Rydning A, Aamodt G, Schreiber S, Vatn MH. A characterization in childhood inflammatory bowel disease, a new population-based inception cohort from South-Eastern Norway, 2005-07, showing increased incidence in Crohn’s disease. Scand J Gastroenterol. 2009;44:446–456. doi: 10.1080/00365520802647434. [DOI] [PubMed] [Google Scholar]
- 24.Lehtinen P, Ashorn M, Iltanen S, Jauhola R, Jauhonen P, Kolho KL, Auvinen A. Incidence trends of pediatric inflammatory bowel disease in Finland, 1987-2003, a nationwide study. Inflamm Bowel Dis. 2011;17:1778–1783. doi: 10.1002/ibd.21550. [DOI] [PubMed] [Google Scholar]
- 25.Sawczenko A, Sandhu BK. Presenting features of inflammatory bowel disease in Great Britain and Ireland. Arch Dis Child. 2003;88:995–1000. doi: 10.1136/adc.88.11.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yantiss RK, Odze RD. Diagnostic difficulties in inflammatory bowel disease pathology. Histopathology. 2006;48:116–132. doi: 10.1111/j.1365-2559.2005.02248.x. [DOI] [PubMed] [Google Scholar]
- 27.Magro F, Langner C, Driessen A, Ensari A, Geboes K, Mantzaris GJ, Villanacci V, Becheanu G, Borralho Nunes P, Cathomas G, Fries W, Jouret-Mourin A, Mescoli C, de Petris G, Rubio CA, Shepherd NA, Vieth M, Eliakim R; European Society of Pathology (ESP); European Crohn's and Colitis Organisation (ECCO) European consensus on the histopathology of inflammatory bowel disease. J Crohns Colitis. 2013;7:827–851. doi: 10.1016/j.crohns.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Gasche C, Scholmerich J, Brynskov J, D’Haens G, Hanauer SB, Irvine EJ, Jewell DP, Rachmilewitz D, Sachar DB, Sandborn WJ, et al. A simple classification of Crohn’s disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm Bowel Dis. 2000;6:8–15. doi: 10.1097/00054725-200002000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19 Suppl A:5A–36A. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 30.Levine A, Griffiths A, Markowitz J, Wilson DC, Turner D, Russell RK, Fell J, Ruemmele FM, Walters T, Sherlock M, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17:1314–1321. doi: 10.1002/ibd.21493. [DOI] [PubMed] [Google Scholar]
- 31.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140:1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denson LA, Long MD, McGovern DP, Kugathasan S, Wu GD, Young VB, Pizarro TT, de Zoeten EF, Stappenbeck TS, Plevy SE, et al. Challenges in IBD research: update on progress and prioritization of the CCFA’s research agenda. Inflamm Bowel Dis. 2013;19:677–682. doi: 10.1097/MIB.0b013e31828134b3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham DB, Xavier RJ. From genetics of inflammatory bowel disease towards mechanistic insights. Trends Immunol. 2013;34:371–378. doi: 10.1016/j.it.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brant SR. Update on the heritability of inflammatory bowel disease: the importance of twin studies. Inflamm Bowel Dis. 2011;17:1–5. doi: 10.1002/ibd.21385. [DOI] [PubMed] [Google Scholar]
- 36.Halfvarson J. Genetics in twins with Crohn’s disease: less pronounced than previously believed? Inflamm Bowel Dis. 2011;17:6–12. doi: 10.1002/ibd.21295. [DOI] [PubMed] [Google Scholar]
- 37.Price AB. Overlap in the spectrum of non-specific inflammatory bowel disease--’colitis indeterminate’. J Clin Pathol. 1978;31:567–577. doi: 10.1136/jcp.31.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geboes K, Colombel JF, Greenstein A, Jewell DP, Sandborn WJ, Vatn MH, Warren B, Riddell RH. Indeterminate colitis: a review of the concept--what’s in a name? Inflamm Bowel Dis. 2008;14:850–857. doi: 10.1002/ibd.20361. [DOI] [PubMed] [Google Scholar]
- 39.Martland GT, Shepherd NA. Indeterminate colitis: definition, diagnosis, implications and a plea for nosological sanity. Histopathology. 2007;50:83–96. doi: 10.1111/j.1365-2559.2006.02545.x. [DOI] [PubMed] [Google Scholar]
- 40.Feakins RM. Inflammatory bowel disease biopsies: updated British Society of Gastroenterology reporting guidelines. J Clin Pathol. 2013;66:1005–1026. doi: 10.1136/jclinpath-2013-201885. [DOI] [PubMed] [Google Scholar]
- 41.Prenzel F, Uhlig HH. Frequency of indeterminate colitis in children and adults with IBD - a metaanalysis. J Crohns Colitis. 2009;3:277–281. doi: 10.1016/j.crohns.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Carvalho RS, Abadom V, Dilworth HP, Thompson R, Oliva-Hemker M, Cuffari C. Indeterminate colitis: a significant subgroup of pediatric IBD. Inflamm Bowel Dis. 2006;12:258–262. doi: 10.1097/01.MIB.0000215093.62245.b9. [DOI] [PubMed] [Google Scholar]
- 43.Romano C, Famiani A, Gallizzi R, Comito D, Ferrau’ V, Rossi P. Indeterminate colitis: a distinctive clinical pattern of inflammatory bowel disease in children. Pediatrics. 2008;122:e1278–e1281. doi: 10.1542/peds.2008-2306. [DOI] [PubMed] [Google Scholar]
- 44.Malaty HM, Mehta S, Abraham B, Garnett EA, Ferry GD. The natural course of inflammatory bowel disease-indeterminate from childhood to adulthood: within a 25 year period. Clin Exp Gastroenterol. 2013;6:115–121. doi: 10.2147/CEG.S44700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruel J, Ruane D, Mehandru S, Gower-Rousseau C, Colombel JF. IBD across the age spectrum-is it the same disease? Nat Rev Gastroenterol Hepatol. 2014;11:88–98. doi: 10.1038/nrgastro.2013.240. [DOI] [PubMed] [Google Scholar]
- 46.Ruemmele FM, El Khoury MG, Talbotec C, Maurage C, Mougenot JF, Schmitz J, Goulet O. Characteristics of inflammatory bowel disease with onset during the first year of life. J Pediatr Gastroenterol Nutr. 2006;43:603–609. doi: 10.1097/01.mpg.0000237938.12674.e3. [DOI] [PubMed] [Google Scholar]
- 47.Cannioto Z, Berti I, Martelossi S, Bruno I, Giurici N, Crovella S, Ventura A. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur J Pediatr. 2009;168:149–155. doi: 10.1007/s00431-008-0721-2. [DOI] [PubMed] [Google Scholar]
- 48.Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schäffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kotlarz D, Beier R, Murugan D, Diestelhorst J, Jensen O, Boztug K, Pfeifer D, Kreipe H, Pfister ED, Baumann U, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 50.Moran CJ, Walters TD, Guo CH, Kugathasan S, Klein C, Turner D, Wolters VM, Bandsma RH, Mouzaki M, Zachos M, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–123. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Begue B, Verdier J, Rieux-Laucat F, Goulet O, Morali A, Canioni D, Hugot JP, Daussy C, Verkarre V, Pigneur B, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106:1544–1555. doi: 10.1038/ajg.2011.112. [DOI] [PubMed] [Google Scholar]
- 52.Shah N, Kammermeier J, Elawad M, Glocker EO. Interleukin-10 and interleukin-10-receptor defects in inflammatory bowel disease. Curr Allergy Asthma Rep. 2012;12:373–379. doi: 10.1007/s11882-012-0286-z. [DOI] [PubMed] [Google Scholar]
- 53.Franke A, Balschun T, Karlsen TH, Sventoraityte J, Nikolaus S, Mayr G, Domingues FS, Albrecht M, Nothnagel M, Ellinghaus D, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–1323. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 54.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glocker E, Grimbacher B. Inflammatory bowel disease: is it a primary immunodeficiency? Cell Mol Life Sci. 2012;69:41–48. doi: 10.1007/s00018-011-0837-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 57.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- 58.Williams L, Bradley L, Smith A, Foxwell B. Signal transducer and activator of transcription 3 is the dominant mediator of the anti-inflammatory effects of IL-10 in human macrophages. J Immunol. 2004;172:567–576. doi: 10.4049/jimmunol.172.1.567. [DOI] [PubMed] [Google Scholar]
- 59.Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. doi: 10.1097/GIM.0b013e3182088158. [DOI] [PubMed] [Google Scholar]
- 60.Agarwal S, Mayer L. Diagnosis and treatment of gastrointestinal disorders in patients with primary immunodeficiency. Clin Gastroenterol Hepatol. 2013;11:1050–1063. doi: 10.1016/j.cgh.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Orange JS, Geha RS. Finding NEMO: genetic disorders of NF-[kappa]B activation. J Clin Invest. 2003;112:983–985. doi: 10.1172/JCI19960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de Bie CI, Paerregaard A, Kolacek S, Ruemmele FM, Koletzko S, Fell JM, Escher JC. Disease phenotype at diagnosis in pediatric Crohn’s disease: 5-year analyses of the EUROKIDS Registry. Inflamm Bowel Dis. 2013;19:378–385. doi: 10.1002/ibd.23008. [DOI] [PubMed] [Google Scholar]
- 63.Stappenbeck TS. Paneth cell development, differentiation, and function: new molecular cues. Gastroenterology. 2009;137:30–33. doi: 10.1053/j.gastro.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 64.Ouellette AJ. Paneth cell alpha-defensin synthesis and function. Curr Top Microbiol Immunol. 2006;306:1–25. doi: 10.1007/3-540-29916-5_1. [DOI] [PubMed] [Google Scholar]
- 65.Wehkamp J, Wang G, Kübler I, Nuding S, Gregorieff A, Schnabel A, Kays RJ, Fellermann K, Burk O, Schwab M, et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J Immunol. 2007;179:3109–3118. doi: 10.4049/jimmunol.179.5.3109. [DOI] [PubMed] [Google Scholar]
- 66.Wehkamp J, Stange EF. Paneth’s disease. J Crohns Colitis. 2010;4:523–531. doi: 10.1016/j.crohns.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 67.Beil WJ, Weller PF, Peppercorn MA, Galli SJ, Dvorak AM. Ultrastructural immunogold localization of subcellular sites of TNF-alpha in colonic Crohn‘s disease. J Leukoc Biol. 1995;58:284–298. doi: 10.1002/jlb.58.3.284. [DOI] [PubMed] [Google Scholar]
- 68.Beisner J, Stange EF, Wehkamp J. Paneth cell function--implications in pediatric Crohn disease. Gut Microbes. 2011;2:47–51. doi: 10.4161/gmic.2.1.14649. [DOI] [PubMed] [Google Scholar]
- 69.Keshav S. Paneth cells: leukocyte-like mediators of innate immunity in the intestine. J Leukoc Biol. 2006;80:500–508. doi: 10.1189/jlb.1005556. [DOI] [PubMed] [Google Scholar]
- 70.Kelly P, Feakins R, Domizio P, Murphy J, Bevins C, Wilson J, McPhail G, Poulsom R, Dhaliwal W. Paneth cell granule depletion in the human small intestine under infective and nutritional stress. Clin Exp Immunol. 2004;135:303–309. doi: 10.1111/j.1365-2249.2004.02374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–339. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology. 2014;146:200–209. doi: 10.1053/j.gastro.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 74.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 75.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 76.Ahmad T, Armuzzi A, Bunce M, Mulcahy-Hawes K, Marshall SE, Orchard TR, Crawshaw J, Large O, de Silva A, Cook JT, et al. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology. 2002;122:854–866. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 77.Rigoli L, Romano C, Caruso RA, Lo Presti MA, Di Bella C, Procopio V, Lo Giudice G, Amorini M, Costantino G, Sergi MD, et al. Clinical significance of NOD2/CARD15 and Toll-like receptor 4 gene single nucleotide polymorphisms in inflammatory bowel disease. World J Gastroenterol. 2008;14:4454–4461. doi: 10.3748/wjg.14.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Parkes M. The genetics universe of Crohn’s disease and ulcerative colitis. Dig Dis. 2012;30 Suppl 1:78–81. doi: 10.1159/000341130. [DOI] [PubMed] [Google Scholar]
- 79.Chamaillard M, Iacob R, Desreumaux P, Colombel JF. Advances and perspectives in the genetics of inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2006;4:143–151. doi: 10.1016/j.cgh.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 80.Weersma RK, Stokkers PC, van Bodegraven AA, van Hogezand RA, Verspaget HW, de Jong DJ, van der Woude CJ, Oldenburg B, Linskens RK, Festen EA, et al. Molecular prediction of disease risk and severity in a large Dutch Crohn’s disease cohort. Gut. 2009;58:388–395. doi: 10.1136/gut.2007.144865. [DOI] [PubMed] [Google Scholar]
- 81.Wine E, Reif SS, Leshinsky-Silver E, Weiss B, Shaoul RR, Shamir R, Wasserman D, Lerner A, Boaz M, Levine A. Pediatric Crohn’s disease and growth retardation: the role of genotype, phenotype, and disease severity. Pediatrics. 2004;114:1281–1286. doi: 10.1542/peds.2004-0417. [DOI] [PubMed] [Google Scholar]
- 82.Kugathasan S, Collins N, Maresso K, Hoffmann RG, Stephens M, Werlin SL, Rudolph C, Broeckel U. CARD15 gene mutations and risk for early surgery in pediatric-onset Crohn’s disease. Clin Gastroenterol Hepatol. 2004;2:1003–1009. doi: 10.1016/s1542-3565(04)00452-5. [DOI] [PubMed] [Google Scholar]
- 83.Russell RK, Drummond HE, Nimmo EE, Anderson N, Smith L, Wilson DC, Gillett PM, McGrogan P, Hassan K, Weaver LT, et al. Genotype-phenotype analysis in childhood-onset Crohn’s disease: NOD2/CARD15 variants consistently predict phenotypic characteristics of severe disease. Inflamm Bowel Dis. 2005;11:955–964. doi: 10.1097/01.mib.0000183423.38037.f3. [DOI] [PubMed] [Google Scholar]
- 84.Tomer G, Ceballos C, Concepcion E, Benkov KJ. NOD2/CARD15 variants are associated with lower weight at diagnosis in children with Crohn’s disease. Am J Gastroenterol. 2003;98:2479–2484. doi: 10.1111/j.1572-0241.2003.08673.x. [DOI] [PubMed] [Google Scholar]
- 85.Cucchiara S, Latiano A, Palmieri O, Staiano AM, D’Incà R, Guariso G, Vieni G, Rutigliano V, Borrelli O, Valvano MR, et al. Role of CARD15, DLG5 and OCTN genes polymorphisms in children with inflammatory bowel diseases. World J Gastroenterol. 2007;13:1221–1229. doi: 10.3748/wjg.v13.i8.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Levine A, Kugathasan S, Annese V, Biank V, Leshinsky-Silver E, Davidovich O, Kimmel G, Shamir R, Palmieri O, Karban A, et al. Pediatric onset Crohn’s colitis is characterized by genotype-dependent age-related susceptibility. Inflamm Bowel Dis. 2007;13:1509–1515. doi: 10.1002/ibd.20244. [DOI] [PubMed] [Google Scholar]
- 87.Ferraris A, Knafelz D, Torres B, Fortina P, Castro M, Dallapiccola B. Analysis of CARD15 gene variants in Italian pediatric patients with inflammatory bowel diseases. J Pediatr. 2005;147:272–273. doi: 10.1016/j.jpeds.2005.03.039. [DOI] [PubMed] [Google Scholar]
- 88.Roesler J, Thürigen A, Sun L, Koch R, Winkler U, Laass MW, Gahr M, Rösen-Wolff A, Henker J. Influence of CARD15 mutations on disease activity and response to therapy in 65 pediatric Crohn patients from Saxony, Germany. J Pediatr Gastroenterol Nutr. 2005;41:27–32. doi: 10.1097/01.mpg.0000165017.00562.27. [DOI] [PubMed] [Google Scholar]
- 89.Shaoul R, Karban A, Weiss B, Reif S, Wasserman D, Pacht A, Eliakim R, Wardi J, Shirin H, Wine E, et al. NOD2/CARD15 mutations and presence of granulomas in pediatric and adult Crohn’s disease. Inflamm Bowel Dis. 2004;10:709–714. doi: 10.1097/00054725-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 90.Weiss B, Shamir R, Bujanover Y, Waterman M, Hartman C, Fradkin A, Berkowitz D, Weintraub I, Eliakim R, Karban A. NOD2/CARD15 mutation analysis and genotype-phenotype correlation in Jewish pediatric patients compared with adults with Crohn’s disease. J Pediatr. 2004;145:208–212. doi: 10.1016/j.jpeds.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 91.Ideström M, Rubio C, Granath F, Finkel Y, Hugot JP. CARD15 mutations are rare in Swedish pediatric Crohn disease. J Pediatr Gastroenterol Nutr. 2005;40:456–460. doi: 10.1097/01.mpg.0000150423.38210.2e. [DOI] [PubMed] [Google Scholar]
- 92.Zelinkova Z, van Beelen AJ, de Kort F, Moerland PD, Ver Loren van Themaat E, te Velde AA, van Deventer SJ, de Jong EC, Hommes DW. Muramyl dipeptide-induced differential gene expression in NOD2 mutant and wild-type Crohn’s disease patient-derived dendritic cells. Inflamm Bowel Dis. 2008;14:186–194. doi: 10.1002/ibd.20308. [DOI] [PubMed] [Google Scholar]
- 93.Iborra M, Bernuzzi F, Correale C, Vetrano S, Fiorino G, Beltrán B, Marabita F, Locati M, Spinelli A, Nos P, et al. Identification of serum and tissue micro-RNA expression profiles in different stages of inflammatory bowel disease. Clin Exp Immunol. 2013;173:250–258. doi: 10.1111/cei.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 95.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 96.Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18:997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 97.McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, Kaestner KH. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology. 2010;139:1654–1664, 1664.e1. doi: 10.1053/j.gastro.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 99.Boutz DR, Collins PJ, Suresh U, Lu M, Ramírez CM, Fernández-Hernando C, Huang Y, Abreu Rde S, Le SY, Shapiro BA, et al. Two-tiered approach identifies a network of cancer and liver disease-related genes regulated by miR-122. J Biol Chem. 2011;286:18066–18078. doi: 10.1074/jbc.M110.196451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology. 2011;141:1323–1333. doi: 10.1053/j.gastro.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanaan Z, Rai SN, Eichenberger MR, Barnes C, Dworkin AM, Weller C, Cohen E, Roberts H, Keskey B, Petras RE, et al. Differential microRNA expression tracks neoplastic progression in inflammatory bowel disease-associated colorectal cancer. Hum Mutat. 2012;33:551–560. doi: 10.1002/humu.22021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen Y, Wang C, Liu Y, Tang L, Zheng M, Xu C, Song J, Meng X. miR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn’s disease. Biochem Biophys Res Commun. 2013;438:133–139. doi: 10.1016/j.bbrc.2013.07.040. [DOI] [PubMed] [Google Scholar]
- 103.Noomen CG, Hommes DW, Fidder HH. Update on genetics in inflammatory disease. Best Pract Res Clin Gastroenterol. 2009;23:233–243. doi: 10.1016/j.bpg.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 104.Wehkamp J, Fellermann K, Herrlinger KR, Baxmann S, Schmidt K, Schwind B, Duchrow M, Wohlschläger C, Feller AC, Stange EF. Human beta-defensin 2 but not beta-defensin 1 is expressed preferentially in colonic mucosa of inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2002;14:745–752. doi: 10.1097/00042737-200207000-00006. [DOI] [PubMed] [Google Scholar]
- 105.Fellermann K, Stange DE, Schaeffeler E, Schmalzl H, Wehkamp J, Bevins CL, Reinisch W, Teml A, Schwab M, Lichter P, et al. A chromosome 8 gene-cluster polymorphism with low human beta-defensin 2 gene copy number predisposes to Crohn disease of the colon. Am J Hum Genet. 2006;79:439–448. doi: 10.1086/505915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc Natl Acad Sci USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zilbauer M, Jenke A, Wenzel G, Goedde D, Postberg J, Phillips AD, Lucas M, Noble-Jamieson G, Torrente F, Salvestrini C, et al. Intestinal alpha-defensin expression in pediatric inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:2076–2086. doi: 10.1002/ibd.21577. [DOI] [PubMed] [Google Scholar]
- 109.Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, Vatn MH, Wehkamp J. Defective paneth cell-mediated host defense in pediatric ileal Crohn’s disease. Am J Gastroenterol. 2010;105:452–459. doi: 10.1038/ajg.2009.643. [DOI] [PubMed] [Google Scholar]
- 110.Cook DN, Pisetsky DS, Schwartz DA. Toll-like receptors in the pathogenesis of human disease. Nat Immunol. 2004;5:975–979. doi: 10.1038/ni1116. [DOI] [PubMed] [Google Scholar]
- 111.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Devière J, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 113.Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39:830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ishihara S, Aziz MM, Yuki T, Kazumori H, Kinoshita Y. Inflammatory bowel disease: review from the aspect of genetics. J Gastroenterol. 2009;44:1097–1108. doi: 10.1007/s00535-009-0141-8. [DOI] [PubMed] [Google Scholar]
- 117.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 118.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Münz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–449. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 120.Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulation of epithelial apoptosis and barrier repair in active Crohn's disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302. doi: 10.1136/gut.2003.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 122.Büning C, Durmus T, Molnar T, de Jong DJ, Drenth JP, Fiedler T, Gentz E, Todorov T, Haas V, Buhner S, et al. A study in three European IBD cohorts confirms that the ATG16L1 c.898A& gt; G (p.Thr300Ala) variant is a susceptibility factor for Crohn’s disease. J Crohns Colitis. 2007;1:70–76. doi: 10.1016/j.crohns.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 123.Zhang HF, Qiu LX, Chen Y, Zhu WL, Mao C, Zhu LG, Zheng MH, Wang Y, Lei L, Shi J. ATG16L1 T300A polymorphism and Crohn’s disease susceptibility: evidence from 13,022 cases and 17,532 controls. Hum Genet. 2009;125:627–631. doi: 10.1007/s00439-009-0660-7. [DOI] [PubMed] [Google Scholar]
- 124.Cheng JF, Ning YJ, Zhang W, Lu ZH, Lin L. T300A polymorphism of ATG16L1 and susceptibility to inflammatory bowel diseases: a meta-analysis. World J Gastroenterol. 2010;16:1258–1266. doi: 10.3748/wjg.v16.i10.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Okazaki T, Wang MH, Rawsthorne P, Sargent M, Datta LW, Shugart YY, Bernstein CN, Brant SR. Contributions of IBD5, IL23R, ATG16L1, and NOD2 to Crohn’s disease risk in a population-based case-control study: evidence of gene-gene interactions. Inflamm Bowel Dis. 2008;14:1528–1541. doi: 10.1002/ibd.20512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- 127.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhães JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 128.Kuballa P, Huett A, Rioux JD, Daly MJ, Xavier RJ. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PLoS One. 2008;3:e3391. doi: 10.1371/journal.pone.0003391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, Ponpuak M, Master S, Pilli M, White E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol. 2010;12:1154–1165. doi: 10.1038/ncb2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 131.Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90–97. doi: 10.1038/nm.2069. [DOI] [PubMed] [Google Scholar]
- 132.Prescott NJ, Dominy KM, Kubo M, Lewis CM, Fisher SA, Redon R, Huang N, Stranger BE, Blaszczyk K, Hudspith B, et al. Independent and population-specific association of risk variants at the IRGM locus with Crohn’s disease. Hum Mol Genet. 2010;19:1828–1839. doi: 10.1093/hmg/ddq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hébuterne X, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet. 2011;43:242–245. doi: 10.1038/ng.762. [DOI] [PubMed] [Google Scholar]
- 134.Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010;12:99–113. doi: 10.1111/j.1462-5822.2009.01381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Latiano A, Palmieri O, Cucchiara S, Castro M, D’Incà R, Guariso G, Dallapiccola B, Valvano MR, Latiano T, Andriulli A, et al. Polymorphism of the IRGM gene might predispose to fistulizing behavior in Crohn’s disease. Am J Gastroenterol. 2009;104:110–116. doi: 10.1038/ajg.2008.3. [DOI] [PubMed] [Google Scholar]
- 136.Moon CM, Shin DJ, Kim SW, Son NH, Park A, Park B, Jung ES, Kim ES, Hong SP, Kim TI, et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm Bowel Dis. 2013;19:106–114. doi: 10.1002/ibd.22972. [DOI] [PubMed] [Google Scholar]
- 137.Sim WH, Wagner J, Cameron DJ, Catto-Smith AG, Bishop RF, Kirkwood CD. Expression profile of genes involved in pathogenesis of pediatric Crohn’s disease. J Gastroenterol Hepatol. 2012;27:1083–1093. doi: 10.1111/j.1440-1746.2011.06973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Seidelin JB, Bjerrum JT, Coskun M, Widjaya B, Vainer B, Nielsen OH. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett. 2010;128:80–85. doi: 10.1016/j.imlet.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 139.Sponheim J, Pollheimer J, Olsen T, Balogh J, Hammarström C, Loos T, Kasprzycka M, Sørensen DR, Nilsen HR, Küchler AM, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177:2804–2815. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Beltrán CJ, Núñez LE, Díaz-Jiménez D, Farfan N, Candia E, Heine C, López F, González MJ, Quera R, Hermoso MA. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1097–1107. doi: 10.1002/ibd.21175. [DOI] [PubMed] [Google Scholar]
- 141.Kugathasan S, Baldassano RN, Bradfield JP, Sleiman PM, Imielinski M, Guthery SL, Cucchiara S, Kim CE, Frackelton EC, Annaiah K, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet. 2008;40:1211–1215. doi: 10.1038/ng.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Funke B, Autschbach F, Kim S, Lasitschka F, Strauch U, Rogler G, Gdynia G, Li L, Gretz N, Macher-Goeppinger S, et al. Functional characterisation of decoy receptor 3 in Crohn’s disease. Gut. 2009;58:483–491. doi: 10.1136/gut.2008.148908. [DOI] [PubMed] [Google Scholar]
- 143.Amre DK, Mack DR, Morgan K, Fujiwara M, Israel D, Deslandres C, Seidman EG, Lambrette P, Costea I, Krupoves A, et al. Investigation of reported associations between the 20q13 and 21q22 loci and pediatric-onset Crohn’s disease in Canadian children. Am J Gastroenterol. 2009;104:2824–2828. doi: 10.1038/ajg.2009.430. [DOI] [PubMed] [Google Scholar]
- 144.Amre DK, Mack DR, Morgan K, Israel D, Deslandres C, Seidman EG, Lambrette P, Costea I, Krupoves A, Fegury H, et al. Association between genome-wide association studies reported SNPs and pediatric-onset Crohn’s disease in Canadian children. Hum Genet. 2010;128:131–135. doi: 10.1007/s00439-010-0835-2. [DOI] [PubMed] [Google Scholar]
- 145.Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O’Neil NJ, Petalcorin MI, Martin JS, Collis SJ, Cantor SB, Auclair M, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–271. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bai C, Connolly B, Metzker ML, Hilliard CA, Liu X, Sandig V, Soderman A, Galloway SM, Liu Q, Austin CP, et al. Overexpression of M68/DcR3 in human gastrointestinal tract tumors independent of gene amplification and its location in a four-gene cluster. Proc Natl Acad Sci USA. 2000;97:1230–1235. doi: 10.1073/pnas.97.3.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, Kugathasan S, Bradfield JP, Walters TD, Sleiman P, Kim CE, Muise A, Wang K, Glessner JT, Saeed S, Zhang H, Frackelton EC, Hou C, Flory JH, Otieno G, Chiavacci RM, Grundmeier R, Castro M, Latiano A, Dallapiccola B, Stempak J, Abrams DJ, Taylor K, McGovern D; Western Regional Alliance for Pediatric IBD, Silber G, Wrobel I, Quiros A; International IBD Genetics Consortium, Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmuda MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ; NIDDK IBD Genetics Consortium, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E; Belgian-French IBD Consortium; Wellcome Trust Case Control Consortium, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwillam R, Tremelling M, Delukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ, Heyman MB, Ferry GD, Kirschner B, Lee J, Essers J, Grand R, Stephens M, Levine A, Piccoli D, Van Limbergen J, Cucchiara S, Monos DS, Guthery SL, Denson L, Wilson DC, Grant SF, Daly M, Silverberg MS, Satsangi J, Hakonarson H. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009;41:1335–1340. doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Steinman L. A brief history of T(H)17, the first major revision in the T(H)1/T(H)2 hypothesis of T cell-mediated tissue damage. Nat Med. 2007;13:139–145. doi: 10.1038/nm1551. [DOI] [PubMed] [Google Scholar]
- 149.Latiano A, Palmieri O, Latiano T, Corritore G, Bossa F, Martino G, Biscaglia G, Scimeca D, Valvano MR, Pastore M, et al. Investigation of multiple susceptibility loci for inflammatory bowel disease in an Italian cohort of patients. PLoS One. 2011;6:e22688. doi: 10.1371/journal.pone.0022688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Latiano A, Palmieri O, Pastorelli L, Vecchi M, Pizarro TT, Bossa F, Merla G, Augello B, Latiano T, Corritore G, et al. Associations between genetic polymorphisms in IL-33, IL1R1 and risk for inflammatory bowel disease. PLoS One. 2013;8:e62144. doi: 10.1371/journal.pone.0062144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhao C, Wang I, Lehrer RI. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett. 1996;396:319–322. doi: 10.1016/0014-5793(96)01123-4. [DOI] [PubMed] [Google Scholar]
- 152.Frye M, Bargon J, Lembcke B, Wagner TO, Gropp R. Differential expression of human alpha- and beta-defensins mRNA in gastrointestinal epithelia. Eur J Clin Invest. 2000;30:695–701. doi: 10.1046/j.1365-2362.2000.00696.x. [DOI] [PubMed] [Google Scholar]
- 153.Gersemann M, Wehkamp J, Fellermann K, Stange EF. Crohn‘s disease--defect in innate defence. World J Gastroenterol. 2008;14:5499–5503. doi: 10.3748/wjg.14.5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gupta N, Bostrom AG, Kirschner BS, Ferry GD, Gold BD, Cohen SA, Winter HS, Baldassano RN, Abramson O, Smith T, et al. Incidence of stricturing and penetrating complications of Crohn’s disease diagnosed in pediatric patients. Inflamm Bowel Dis. 2010;16:638–644. doi: 10.1002/ibd.21099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shaoul R, Karban A, Reif S, Weiss B, Shamir R, Tamir A, Davidovich O, Halevi J, Silver EL, Levine A. Disease behavior in children with Crohn’s disease: the effect of disease duration, ethnicity, genotype, and phenotype. Dig Dis Sci. 2009;54:142–150. doi: 10.1007/s10620-008-0326-7. [DOI] [PubMed] [Google Scholar]
- 156.de Zoeten EF, Pasternak BA, Mattei P, Kramer RE, Kader HA. Diagnosis and treatment of perianal Crohn disease: NASPGHAN clinical report and consensus statement. J Pediatr Gastroenterol Nutr. 2013;57:401–412. doi: 10.1097/MPG.0b013e3182a025ee. [DOI] [PubMed] [Google Scholar]
- 157.Eglinton TW, Roberts R, Pearson J, Barclay M, Merriman TR, Frizelle FA, Gearry RB. Clinical and genetic risk factors for perianal Crohn’s disease in a population-based cohort. Am J Gastroenterol. 2012;107:589–596. doi: 10.1038/ajg.2011.437. [DOI] [PubMed] [Google Scholar]
- 158.Stange EF, Travis SP, Vermeire S, Reinisch W, Geboes K, Barakauskiene A, Feakins R, Fléjou JF, Herfarth H, Hommes DW, et al. European evidence-based Consensus on the diagnosis and management of ulcerative colitis: Definitions and diagnosis. J Crohns Colitis. 2008;2:1–23. doi: 10.1016/j.crohns.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 159.Levine A, de Bie CI, Turner D, Cucchiara S, Sladek M, Murphy MS, Escher JC. Atypical disease phenotypes in pediatric ulcerative colitis: 5-year analyses of the EUROKIDS Registry. Inflamm Bowel Dis. 2013;19:370–377. doi: 10.1002/ibd.23013. [DOI] [PubMed] [Google Scholar]
- 160.Aloi M, D’Arcangelo G, Pofi F, Vassallo F, Rizzo V, Nuti F, Di Nardo G, Pierdomenico M, Viola F, Cucchiara S. Presenting features and disease course of pediatric ulcerative colitis. J Crohns Colitis. 2013;7:e509–e515. doi: 10.1016/j.crohns.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 161.Aloi M, Nuti F, Stronati L, Cucchiara S. Advances in the medical management of paediatric IBD. Nat Rev Gastroenterol Hepatol. 2014;11:99–108. doi: 10.1038/nrgastro.2013.158. [DOI] [PubMed] [Google Scholar]
- 162.Joo M, Abreu-e-Lima P, Farraye F, Smith T, Swaroop P, Gardner L, Lauwers GY, Odze RD. Pathologic features of ulcerative colitis in patients with primary sclerosing cholangitis: a case-control study. Am J Surg Pathol. 2009;33:854–862. doi: 10.1097/PAS.0b013e318196d018. [DOI] [PubMed] [Google Scholar]
- 163.Loftus EV, Harewood GC, Loftus CG, Tremaine WJ, Harmsen WS, Zinsmeister AR, Jewell DA, Sandborn WJ. PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut. 2005;54:91–96. doi: 10.1136/gut.2004.046615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ordonez F, Lacaille F, Canioni D, Talbotec C, Fournet JC, Cerf-Bensussan N, Goulet O, Schmitz J, Ruemmele FM. Pediatric ulcerative colitis associated with autoimmune diseases: a distinct form of inflammatory bowel disease? Inflamm Bowel Dis. 2012;18:1809–1817. doi: 10.1002/ibd.22864. [DOI] [PubMed] [Google Scholar]
- 165.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 167.Gersemann M, Stange EF, Wehkamp J. From intestinal stem cells to inflammatory bowel diseases. World J Gastroenterol. 2011;17:3198–3203. doi: 10.3748/wjg.v17.i27.3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gersemann M, Becker S, Kübler I, Koslowski M, Wang G, Herrlinger KR, Griger J, Fritz P, Fellermann K, Schwab M, et al. Differences in goblet cell differentiation between Crohn’s disease and ulcerative colitis. Differentiation. 2009;77:84–94. doi: 10.1016/j.diff.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 169.Strugala V, Dettmar PW, Pearson JP. Thickness and continuity of the adherent colonic mucus barrier in active and quiescent ulcerative colitis and Crohn’s disease. Int J Clin Pract. 2008;62:762–769. doi: 10.1111/j.1742-1241.2007.01665.x. [DOI] [PubMed] [Google Scholar]
- 170.Doecke JD, Simms LA, Zhao ZZ, Huang N, Hanigan K, Krishnaprasad K, Roberts RL, Andrews JM, Mahy G, Bampton P, et al. Genetic susceptibility in IBD: overlap between ulcerative colitis and Crohn’s disease. Inflamm Bowel Dis. 2013;19:240–245. doi: 10.1097/MIB.0b013e3182810041. [DOI] [PubMed] [Google Scholar]
- 171.Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Battle MA, Konopka G, Parviz F, Gaggl AL, Yang C, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc Natl Acad Sci USA. 2006;103:8419–8424. doi: 10.1073/pnas.0600246103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Garrison WD, Battle MA, Yang C, Kaestner KH, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology. 2006;130:1207–1220. doi: 10.1053/j.gastro.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Karayiannakis AJ, Syrigos KN, Efstathiou J, Valizadeh A, Noda M, Playford RJ, Kmiot W, Pignatelli M. Expression of catenins and E-cadherin during epithelial restitution in inflammatory bowel disease. J Pathol. 1998;185:413–418. doi: 10.1002/(SICI)1096-9896(199808)185:4<413::AID-PATH125>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 175.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, Chandler I, Vijayakrishnan J, Sullivan K, Penegar S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–1435. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kuester D, Guenther T, Biesold S, Hartmann A, Bataille F, Ruemmele P, Peters B, Meyer F, Schubert D, Bohr UR, et al. Aberrant methylation of DAPK in long-standing ulcerative colitis and ulcerative colitis-associated carcinoma. Pathol Res Pract. 2010;206:616–624. doi: 10.1016/j.prp.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 178.Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol. 2010;20:1093–1098. doi: 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 179.Sabath E, Negoro H, Beaudry S, Paniagua M, Angelow S, Shah J, Grammatikakis N, Yu AS, Denker BM. Galpha12 regulates protein interactions within the MDCK cell tight junction and inhibits tight-junction assembly. J Cell Sci. 2008;121:814–824. doi: 10.1242/jcs.014878. [DOI] [PubMed] [Google Scholar]