

SUMMARY
The human kinome includes between 500 and 600 known kinases and open reading frames (ORFs) that play key roles in regulating many cellular processes. Past studies adopting loss-of-function approaches have identified some kinases whose activities are required for hepatitis C virus (HCV) life cycle. Here, by screening a retroviral cDNA library of 192 active human kinases, we found that three of them, namely cyclin-dependent kinases regulatory subunit 1 (CKS1B), mitogen-activated protein kinase kinase 5 (MAP2K5) and protein kinase C and casein kinase substrate in neurons 1 (PACSIN1), potently suppressed HCV infection. The expression of these kinases did not induce the production of type I interferon (IFN) and interferon-stimulated genes (ISGs); instead, they inhibited HCV at postentry stages. Specifically, CKS1B and MAP2K5 significantly inhibited viral RNA replication. PACSIN1, by contrast, inhibited HCV infection by decreasing the level of HCV p7. Altogether, the identification of human protein kinases that exert an anti-HCV activity highlighted the potential of combating HCV infection by activating specific kinase-mediated pathways, offering an alternative strategy of treatment.
Keywords: hepatitis C virus, human kinase, viral hepatitis
INTRODUCTION
Hepatitis C virus affects approximately 170 million people worldwide and is a major cause of liver diseases. The World Health Organization estimates that the virus infects 3% of the global population and that 3–4 million new infections occur annually [1,2]. HCV has the ability to establish persistent infections, and some of prevalent HCV genotypes respond to treatment poorly [3–7]. Standard treatment of chronic HCV infection is a combination therapy of polyethylene glycol-conjugated interferon-alpha and ribavirin [8]. Although this combination therapy elicits a strong inhibitory effect on HCV replication, patients often have to endure severe side effects, such as flu-like symptoms, depression and anaemia, that often result in poor patient compliance [9,10]. Two HCV protease inhibitors, Merck’s boceprevir and Vertex’s telaprevir, were approved in May 2011 by the Food and Drug Administration (FDA) to treat chronic HCV infection [11]. Resistance to these drugs, however, has emerged rapidly in clinics. A successful treatment for HCV infection is expected to involve a combination therapy with multiple inhibitors of different targets.
Hepatitis C virus infection has been linked to host kinases. Casein kinases I and II and choline kinase alpha (CHKA) [12,13], AKT, p70s6K, MEK, MKKI and CKII [14] have been reported to be important for HCV replication. The casein kinase II (CKII), in particular, has been implicated as a kinase that phosphorylates the viral protein NS5A [15]. As kinases are the promising targets for drugs, full kinome or partial kinome has been screened for those which are required for HCV infection through siRNA or chemical inhibitor screenings [16–22]. One recent study by Reiss and colleagues described a screening of siRNA library targeting all known and predicted human kinases and found phosphatidylinositol–4 kinase III alpha (PI4KIIIα) to be important in HCV entry and replication [16]. This kinase was previously reported by another group, in which they used a siRNA library targeting 140 cellular membrane-trafficking genes and systematically evaluated their impact on the production of infectious HCV and HCV subgenomic replicon replication [23]. Here, we report the identification of three kinases whose activities suppress HCV infection through expressing a cDNA library of 192 human kinases.
MATERIALS AND METHODS
Full details are available in supplemental materials
Cell line
The human kidney epithelial cell line Lenti-X 293T was purchased from Clontech. The human liver cell line Huh7.5.1 line was provided by Dr. Francis Chisari (Scripps Research Institute) [24]. The HCV genotype 1b-replicon-containing cell line (2–3+) and the cured cell line (2–3c) were generous gifts from Dr. Stanley Lemon (University of Texas Medical Branch, Galveston, TX, USA) [25]. All cell lines were maintained in DMEM supplemented with 5% penicillin and streptomycin, 1% NEAA and 10% foetal bovine serum (FBS) (Gemini Bio-Products, West Sacramento, CA, USA).
Plasmids
The kinase library consisting of 192 human myristoylated kinases was purchased from Addgene. These kinases were shipped as glycerol stock in two 96-well plates, referred to as plates 1 and 2 (Table S1). These kinases contain kinase-related open reading frames (ORFs) cloned into a retroviral vector which adds a myristoylation sequence and flag-epitope tag to each ORF. The plates were stored at −80 °C.
Virus packaging
To perform primary screening, retroviral packaging was assembled by transfecting a pool of eight kinase DNAs, MLV-gag-pol and VSV-G packaged in 293T Lenti-X cell line. The kinases were pooled in groups of eight in the primary screening to make the screening manageable. The plasmids were transfected using 1 μg/μL of polyethylenimine (PEI), and after 48 h, the supernatant containing the packaged virus was collected and stored at −80 °C. In the secondary screening, the same method was applied, except that the kinases in each pool were individually packaged. The virus collected from packaging was used to infect Huh 7.5.1 cells (MOI 1≃3), incubated with the cells in the presence of 4 μg/mL polybrene at 37 °C for 3–6 h prior to the removal of virus.
Immunostaining
Huh 7.5.1 cells were seeded at approximately 4 × 104 cells/well in a 24-well plate covered with collagen-coated coverslips and maintained in complete DMEM at 37 °C and 5% CO2. Next day, the kinase plasmids were transfected into Huh 7.5.1 by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and subsequently subjected to JFH1-AM2 virus infection for 2 h. Forty eight hours postinfection, samples were washed three times in 1X PBS (Invitrogen) for 3 min. Cells were then fixed in 2% paraformaldehyde (PFA) for 10 min and permeabilized with 0.2% Triton-X 100 for 10 min. Then, the cells were incubated for 1 h in a humid chamber with primary antibodies: mouse α-Flag (1:1000), human α-HCV E2 (1:200) and/or mouse α-NS5A (1:2000). Cells were next washed in 1X PBS three times for 10 min each, and secondary antibodies fluorescently conjugated to Alexa Fluor 568, Alexa Fluor 488 and/or FITC were used and incubated for another 1 h in a humid chamber. The coverslips with samples were washed with 1X PBS three times for 10 min each, and Draq 5 (Cell Signaling, Danvers, MA, USA) was used to stain the nucleus (1:5000) during the second wash. The coverslips were mounted with homemade mounting solution, and images were captured using confocal microscopy (Zeiss Meta 510) using a 40× objective lens.
Statistical analysis
Bar graphs were plotted to show mean ± standard deviation (SD) of at least three independent experiments. Statistical analyses were performed using Prism 5 (GraphPad Software, La Jolla, CA, USA). A P value of <0.05 in the Student’s t-test was considered statistically significant.
RESULTS
Identification of kinases that suppress hepatitis C virus infection
The Addgene 192 kinases can be categorized according to cellular localization, biological processes and molecular functions (Fig. S1). Primary screening was performed in pools of eight kinases in each group. For example, virus pool group 1 had kinases A1 through H1, and virus pool group 2 had kinases A2 through H2. Knocking down HCV entry factor claudin-1 by shRNA (shRNA Cl-1) was included as a positive control. Following the expression of kinases, cell culture-grown HCV-carrying luciferase (HCVcc-luc) was added to the cells at a low multiplicity of infection (MOI) of 0.1 to allow spreading of the infection. The results showed that virus pool 1 (A1-H1) from plate 1 and virus pool 7 (A7-H7) from plate 2 had significant regulatory effects on HCVcc infection (Fig. S2a,b). To rule out the possible contribution of altered cell viability due to active kinase activities, the cellular ATP levels in kinase-expressing cells, which reflect the cell viability and metabolism, were measured using a Cell Titer Glo kit. As shown in Fig. S2c,d, short-term expression of these active kinases in Huh 7.5.1 cells did not significantly alter cell viability and metabolism.
Next, a secondary screening was conducted to locate the individual kinase from virus pool 1 (A1-H1) from plate 1 and virus pool 7 (A7-H7) from plate 2. To this end, virus expressing individual kinase from these two pools was packaged and used in the study. It was found that three kinases CKS1B (A1), MAP2K5 (C1) and PACSIN1 (H1) from plate 1 consistently inhibited HCVcc infection, whereas the expression of individual kinase from pool 7 of plate 2 failed to consistently enhance HCVcc infection and hence omitted from subsequent evaluations (Fig. 1).
Fig. 1.

Secondary screen identified three kinases that inhibited HCVcc infection. (a,b) Kinases in group 1 of plate 1 were individually expressed in Huh 7.5.1 cells to determine their effects upon HCVcc-luc infection. (c,d) Kinases in group 7 of plate 2 were individually expressed in Huh7.5.1 cells to determine their individual effects upon HCVcc-luc infection. Values are expressed as mean ± SD (n = 3) *P < 0.05, ****P < 0.0001.
CKS1B, MAP2K5 and PACSIN1 inhibited HCV, but not Dengue virus (DENV) infection
In order to verify the observed inhibitory effects on HCVcc infection in the above study, kinase-expressing Huh7.5.1 cells were infected with wild-type JFH1 virus (HCVcc) and then immunostained for viral core and E2 proteins. As shown in Fig. 2a, the expression of CKS1B (A1), MAP2K5 (C1) and PACSIN1 (H1) significantly reduced HCVcc infection in terms of both the number of infected cells and the size of the foci. The intracellular viral RNA also decreased accordingly (Fig. S3). By contrast, expression of the kinase STK17B (B1) or CSNK1A1L (negative controls) had no effect. Interestingly, none of the three kinases exhibited any inhibitory effect upon DENV2 infection (Fig. 2b and Fig. S4).
Fig. 2.

CKS1B, MAP2K5 and PACSIN1 inhibited HCV, but not DENV2. (a) Approximately 4 × 104 cells were seeded on collagen-coated glass coverslips in 24-well plates, and the cells were then infected by JFH1-based HCVcc (MOI ~0.3). Forty eight hours postinfection, cells were stained for HCV E2 (red), HCV core (green) and nuclei (blue) and imaged under confocal microscope. (b) Huh7.5.1 were first infected by retroviruses expressing individual kinases for 24 h followed by infection with DENV2 (Thailand 16681 strain) (MOI = 1). Infected cells were stained for the presence of viral protein prM (2H2 antibody) (red). Nuclei were stained by Draq 5 (blue), and images were taken by using a Zeiss Meta LSM 510 confocal microscope. The images are representative of at least three independent experiments.
Expression and cellular distribution of CKS1B, MAP2K5 and PACSIN1
Computational analysis based on existing microarray data suggests that CKS1B is expressed in liver tissue or cell lines, whereas MAP2K5 expression level is low (Fig. S5). Subsequently, we performed confocal microscopy to examine the localization pattern of the activated kinases. Because there are no commercial antibodies available that give satisfactory staining of endogenous kinases, individual kinase genes were subcloned into the retroviral vector pQCXIP in which a Flag tag was inserted to the N-terminus of each gene. All constructs allowed expressions of corresponding kinases as demonstrated by Western blotting (Fig. 3a). When examined by confocal microscopy, CKS1B displayed a predominantly nuclear localization pattern with a low level of cytoplasmic distribution. STK17B exhibited an exclusive nuclear distribution. Both MAP2K5 and PACSIN1 localized to the cytoplasm (Fig. 3b). Interestingly, co-staining using the lipophilic dye Bodipy 493/503 revealed that MAP2K5 surrounded lipid droplets, the important cellular organelle for HCV assembly, in a manner very similar to HCV core protein (Fig. 3c,d).
Fig. 3.

Subcellular locations of CKS1B, MAP2K5 and PACSIN1. (a) Expression of Flag-tagged kinases in 293T cells. Each kinase was subcloned into retroviral vector pQCXIP that contains a Flag tag at the N-terminus of each gene. 0.5 μg plasmid DNA was transfected into 293T cells. Forty eight hours post-transfection, total cell lysates were prepared and resolved on a 15% SDS-PAGE followed by immunoblotting using the anti-Flag (top panel) and anti-β-actin antibody (bottom panel). (b) Cellular distribution of kinases. Flag-tagged kinases were expressed in Lenti-X 293T cells and immunostained with anti-Flag antibody. Images were taken under a 40× objective using the Carl Zeiss Meta LSM510 laser scanning confocal microscope. Nuclei were stained by Draq 5 (blue). The result is a representation of at least three independent experiments. (c) Localization of kinases with respect to lipid droplets. Flag-tagged CKS1B, MAP2K5 and PACSIN1 plasmids were transfected into Huh7.5.1 cells. Cells were fixed and stained for each kinase (red) and then lipid droplets (green). For reference, HCV-infected Huh7.5.1 cells were stained for core and lipid droplet. The data represent two independent experiments. (d) MAP2K5 surrounded lipid droplets. 3D scanning of MAP2K5-expressing Huh7.5.1 cells was performed. Diagonally projected images (XY, XZ and YZ) are shown here. Consistent with what has been reported, HCV core surrounded lipid droplets where the virus assembly occurs. Similarly, MAP2K5 appeared to cover the lipid droplets although this does not necessarily mean a direct interaction between them. The data represent two independent experiments.
CKS1B and MAP2K5 inhibited hepatitis C virus replication
To determine at what stage of the viral life cycle that the screened kinases are inhibiting, lentiviral particles bearing HCV envelope proteins (HCVpp) were packaged for the assessment of HCV entry. As shown in Fig. 4a, the presence of active kinases did not exhibit any effect on HCV entry.
Fig. 4.

Kinase expressions inhibited HCV replication, but not entry. (a) Identified kinases did not inhibit HCV entry. The kinases were transiently expressed in Huh7.5.1 cells and subsequently infected with HCVpp, VSV-Gpp or HCVcc-luc. The% infection from the mock group (parental cells) was set to 100. Results are presented as mean ± SD (n = 3). (b) CKS1B and MAP2K5 suppressed HCV RNA replication. HCV replicon cells (2–3+) were retrovirally transduced to express individual kinases. Total RNA was isolated and analysed by real-time RT-PCR. The positive control treatment was IFN-α. (c) Replicon cells were transfected with the indicated amount of plasmid DNA shown above panel B, to achieve a dose-dependent effect. In both b and c, viral RNA copies were normalized to the values obtained by measuring a housekeeping gene (RPS 11). Results are presented as mean ± SD (n = 2). * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
Subsequently, individual kinases were expressed via retroviral transduction into a HCV full-length replicon cell line named replicon (2–3+), which harbours a full-length genotype 1b HCV genome that actively replicates without secreting infectious virus. Such a system permits direct assessment of the effect of a kinase on stages of viral RNA (vRNA) replication. As shown in Fig. 4a, addition of recombinant IFN-α inhibited HCV replication by nearly 100-fold. Expressions of CKS1B and MAP2K5 also significantly suppressed vRNA levels, although the inhibition exerted by MAP2K5 was less pronounced. Notably, PAC-SIN1 did not seem to inhibit HCV RNA replication. The two kinases that showed inhibition on viral replication (CKS1B and MAP2K5) were further evaluated in a dose-dependent experiment for their inhibitory effects. As shown in Fig. 4c, there was an inverse correlation between the amount of kinase plasmid that was transfected into replicon cells and the detected vRNA copies. This result corroborated the finding that CKS1B and MAP2K5 inhibit HCV RNA replication.
CKS1B, MAP2K5 and PACSIN1 did not induce the production of IFNs
Kinase-mediated activation of cellular pathways may result in the production of type I interferon, which is known to potently inhibit HCV replication. To explore this possibility, luciferase reporter assays were performed where the interferon response gene (ISG) promoter-driven (IRSE-Luc) and the IFN-beta promoter-driven (p125-Luc) luciferase reporter constructs were co-transfected into Huh7.5.1 cells with each kinase construct. As shown in Fig. 5, whereas MAVS, an important signalling molecule downstream of the RIG-I-mediated antiviral pathway [26], strongly activated both ISRE-luc and p125-luc, none of the kinases activated the transcription of reporter genes, suggesting that signalling from these kinases is not likely to directly induce interferon or ISG production.
Fig. 5.

Kinase expressions did not induce I interferon (IFN) production. (a,b) Expression of kinases did not induce transcription from IFN or interferon-stimulated genes (ISG) promoters. Huh 7.5.1 cells were preseeded in a 24-well plate. The next day, 0.1 μg of reporter construct was co-transfected with 0.2 μg of indicated kinase expression plasmid using Lipofectamine 2000 reagent. Forty eight hours post-transfection, cells were lysed and luciferase activity was determined. pISRE-Luc contains five copies of ISRE sites from the ISG54 promoter upstream of the luciferase gene, and p125 contains the IFN-β promoter. (c) Kinase expression did not suppress new castle disease virus (NDV) infection. Huh7.5.1 cells that express individual kinases were seeded on glass coverslips (24-well plate) and infected by NDV-GFP virus using the protocol described in Supplementary materials. Sixteen hours postinfection, the cells were fixed and imaged for infection (demonstrated as the expression of GFP). Fluorescence was measured by using J software and calculating corrected total cell fluorescence (CTCF). Data shown here are the representatives of three independent experiments.
To validate the above observations, an IFN bioassay was performed using a published protocol that is based on the detection of a new castle disease virus (NDV) that expresses green fluorescent protein (GFP) [27]. In this bioassay, the presence of type I IFN suppresses the NDV-GFP infections, resulting in reduction in the GFP signal. As shown in Fig. 5c, whereas the addition of recombinant human IFN-α significantly decreased the GFP signal, none of the kinases had any effect on NDV-GFP infection. This result confirmed that overexpression of kinases in Huh7.5.1 cells did not lead to the production of type I interferon.
CKS1B, MAP2K5 and PACSIN1 did not alter cell cycle
The kinase CKS1B belongs to the highly conserved cyclin kinase subunit 1 (CKS1) protein family, which interacts with cyclin-dependent kinases (CDKs) and controls cell cycle progression [28,29]. HCV replication, at least in cell culture, is known to be affected by cell cycle [25]. Thus, it was necessary to investigate the effect of these kinases on cell cycle progression, to rule out the possibility of cell cycle alteration. Because it was previously observed that short-term expression of these active kinases in Huh 7.5.1 cells did not significantly alter cell viability/metabolism, it was not anticipated that the expression of these kinases would significantly alter the cell cycle progression. Consistent with this, the activated kinases did not show any alteration in the cell cycle progression (Fig. S6).
PACSIN1 expression decreased the level of hepatitis C virus p7
Because PACSIN1 did not show any significant effect on viral replication nor did it co-localize with lipid droplets in previous experiments, we co-transfected the PACSIN1 plasmid with the viral protein plasmids in 293T cells and attempted to determine whether that alters the viral protein expression. All ten HCV proteins were co-transfected with PACSIN1 initially, although only p7 and NS4B are shown in Fig. 6. The presence of PACSIN1 decreased the expression of HCV p7 in a dose-dependent manner. NS4B is included in the figure to show that this effect was specific to p7.
Fig. 6.
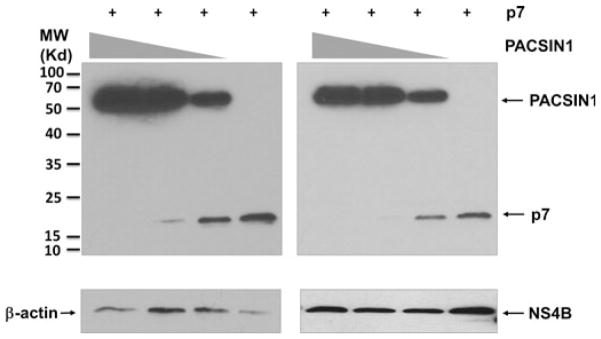
PACSIN1 decreased the level of p7. PACSIN1 and p7 were co-transfected in 293T cells, followed by Western blotting. As PACSIN1 expression decreased (0.5, 0.3 and 0.2 μg), p7 viral protein expression increased. NS4B is shown as a control, and β-actin was a loading control. The Western blot on the left is a longer exposure to illustrate the trend in p7 viral protein expression.
DISCUSSION
The completion of the human genome sequence has led to the identification of approximately 500–600 known kinases and ORFs, which are potentially important in regulating many cellular processes [30]. Interestingly, HCV infections, in particular, have been linked to host kinases. The objective of this study was to screen for novel human kinases that suppress HCV infection by expressing active kinases in cells. This approach is different from the screens in the past, in which kinases were either knocked down by siRNA or blocked by chemical inhibitors (loss-of-function approach). We found that CKS1B, MAP2K5 and PACSIN1 potently inhibited HCV infection and their mechanisms of inhibition were also investigated. The limitation of the study is that we have yet to confirm that the observed inhibitory effects were due to the active kinase activities. Because there were no known substrates for the three kinases that can be used in in vitro kinase assay, an alternative approach to confirm the kinase activation would have been to utilize kinase inhibitors, but to our knowledge there are no available inhibitors of CKS1B and PACSIN1, while MAP2K5 inhibitor is rather nonspecific.
CKS1B is a 79-amino-acid cyclin-dependent kinase that normally localizes in the cytoplasm. CKS1B protein binds to the catalytic subunit of the CDKs and presumably regulates cell cycle [31]. MAP2K5 belongs to the MAP kinase kinase family. It localizes in the cytoplasm also and specifically interacts with and activates MAPK7/ERK5. Interestingly, inhibition of MEK/ERK signalling reportedly up-regulated HCV replication and production [32]. PACSIN1, protein kinase C and casein kinase substrate in neurons 1, is a 50-kd protein that localizes in cytoplasm. It is highly expressed in brain and to a lower level in heart, pancreas and liver [33]. Functional characterization revealed that these three kinases did not inhibit HCV entry. However, CKS1B and MAP2K5 both suppressed viral RNA replication albeit to varied degrees. Confocal microscopy studies showed that MAP2K5 localized to the cytoplasm and surrounded lipid droplets, suggesting that MAP2K5 may have an additional effect on the viral assembly. Future study is necessary to address this possibility.
We initially proposed three mechanisms by which the identified kinases inhibit HCV replication: (i) production of IFN, (ii) altered cell cycle and (iii) hyperphosphorylation of viral protein NS5A. This may be especially true for CKS1B given its nuclear localization. However, assays measuring interferon production and cell cycle analysis did not show any significant changes in the presence of activated kinases. Therefore, CKS1B might indirectly affect HCV replication via an unknown mechanism. It has been reported that hyperphosphorylation of NS5A resulted in decreased HCV replication [34]. To explore the potential effect of kinase activation on the phosphorylation status of HCV NS5A, Huh 7.5.1 cells were infected by vaccinia virus expressing T7 polymerase (MOI 3) for 1 h followed by transfection of HCVcc-JFH1 construct and individual kinase expression plasmids. Immunoblotting of NS5A using the 9E10 antibody failed to detect any change to the previously reported hyper- and hypophosphorylated NS5A (p58 and p56) [14] in the presence of kinases (data not shown). Therefore, the possibility of the activated kinases directly phosphorylating NS5A phosphorylation appears low; it remains possible, however, that kinases can act upon a cellular target or targets that are important for HCV replication.
The observation that the overexpression of PACSIN1 decreased p7 viral protein is striking. Currently, specific functions to HCV proteins have been well documented except for the role of the small membrane protein p7. While p7 is essential for infection, it is not necessary for RNA replication [35,36]. In vitro studies have reported that p7 oligomerizes and functions to conduct ions across artificial membrane systems [35,37–39]. Steinmann et al. [35] reported that p7 is critical for virus assembly and release, but not essential for its infectivity. Thus, while the basic structural features of p7 are becoming gradually known, the exact mechanism of p7 in viral assembly and release, and the conditions that lead to the assembly of a functional channel, still remains to be better understood. PACSIN1 belongs to the PACSIN family of three cytoplasmic phosphoproteins that play a role in vesicle formation and transport. Notably, all three PACSIN isoforms including PACSIN1 were shown to bind to endocytic proteins and inhibit endocytosis [40]. Thus, PACSIN1 may down-regulate p7 through a yet-to-be-identified mechanism and hence inhibit HCV assembly and/or release.
In summary, we identified three host kinases that inhibit HCV infection at different stages of the viral life cycle through an unbiased screen of 192 human kinases. While the mechanisms by which these kinases inhibit HCV infection warrant further investigation, compounds that activate the three kinases may be developed as novel therapeutic options for chronic HCV infection.
Supplementary Material
Acknowledgments
We thank Drs. T. Wakita, C. Rice, F. Chisari, S. Lemon and F. Cosset for providing cell lines and reagents. We thank Dr. Chao Qiu for critical reading of this manuscript. This manuscript is dedicated to the memory of Nun Wang (1986–2013), a friend and an outstanding scientist whose scientific aspiration and perseverance has been instrumental to this work.
FUNDING
This work was supported by NIH R01DK088787 grant (to T.W.).
Abbreviations
- CKS1B
CDC28 protein kinase regulatory subunit 1B
- HCV
hepatitis C virus
- HCVcc
cell culture-grown HCV
- HCVpp
lentiviral particles pseudotyped with HCV envelope proteins
- MAP2K5
mitogen-activated protein kinase kinase 5
- MOI
multiplicity of infection
- PACSIN1
protein kinase C and casein kinase substrate in neurons 1
- VSVpp
HIV particles pseudotyped with vesicular stomatitis virus envelope protein G
Footnotes
CONFLICT OF INTEREST
The authors have no competing interest.
Additional Supporting Information may be found in the online version of this article:
Figure S1: Categorizations of 192 human kinases.
Figure S2: Initial screening of the myristoylated kinase library.
Figure S3: CKS1B, MAP2K5 and PACSIN1 inhibited HCV.
Figure S4: Huh7.5.1 were first infected by retrovirus expressing individual kinase for 24 h followed by infection with DENV2 (Thailand 16681strain) (MOI = 0.1).
Figure S5: Gene expression profiles of CKS1B and MAP2K5.
Figure S6: Kinase expression did not alter cell cycle.
References
- 1.Brown RS. Hepatitis C and liver transplantation. Nature. 2005;436(7053):973–978. doi: 10.1038/nature04083. Epub 2005/08/19. [DOI] [PubMed] [Google Scholar]
- 2.Major ME, Feinstone SM. The molecular virology of hepatitis C. Hepatology. 1997;25(6):1527–1538. doi: 10.1002/hep.510250637. Epub 1997/06/01. [DOI] [PubMed] [Google Scholar]
- 3.Sharma SD. Hepatitis C virus: molecular biology & current therapeutic options. Indian J Med Res. 2010;131:17–34. Epub 2010/02/20. [PubMed] [Google Scholar]
- 4.Seeff LB. Natural history of chronic hepatitis C. Hepatology. 2002;36(5 Suppl 1):S35–S46. doi: 10.1053/jhep.2002.36806. Epub 2002/10/31. [DOI] [PubMed] [Google Scholar]
- 5.Strader DB, Wright T, Thomas DL, Seeff LB. Diagnosis, management, and treatment of hepatitis C. Hepatology. 2004;39(4):1147–1171. doi: 10.1002/hep.20119. Epub 2004/04/02. [DOI] [PubMed] [Google Scholar]
- 6.Boehm JS, Zhao JJ, Yao J, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. doi: 10.1016/j.cell.2007.03.052. Epub 2007/06/19. [DOI] [PubMed] [Google Scholar]
- 7.Rual JF, Hirozane-Kishikawa T, Hao T, et al. Human ORFeome version 1. 1: a platform for reverse proteomics. Genome Res. 2004;14(10B):2128–2135. doi: 10.1101/gr.2973604. Epub 2004/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaefer M, Capuron L, Friebe A, et al. Hepatitis C infection, antiviral treatment and mental health: a European expert consensus statement. J Hepatol. 2012;57(6):1379–1390. doi: 10.1016/j.jhep.2012.07.037. [DOI] [PubMed] [Google Scholar]
- 9.Butt AA, Wagener M, Shakil AO, Ahmad J. Reasons for non-treatment of hepatitis C in veterans in care. J Viral Hepatitis. 2005;12(1):81–85. doi: 10.1111/j.1365-2893.2005.00547.x. Epub 2005/01/19. [DOI] [PubMed] [Google Scholar]
- 10.Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–965. doi: 10.1016/s0140-6736(01)06102-5. Epub 2001/10/05. [DOI] [PubMed] [Google Scholar]
- 11.Assis DN, Lim JK. New pharmacotherapy for hepatitis C. Clin Pharmacol Ther. 2012;92(3):294–305. doi: 10.1038/clpt.2012.103. [DOI] [PubMed] [Google Scholar]
- 12.Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300(5622):1145–1148. doi: 10.1126/science.1082604. Epub 2003/04/19. [DOI] [PubMed] [Google Scholar]
- 13.Tellinghuisen TL, Foss KL, Treadaway J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008;4(3):e1000032. doi: 10.1371/journal.ppat.1000032. Epub 2008/03/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu D, Lemm JA, O’Boyle DR, 2nd, et al. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J Gen Virol. 2011;92(Pt 11):2502–2511. doi: 10.1099/vir.0.034801-0. Epub 2011/07/29. [DOI] [PubMed] [Google Scholar]
- 15.Quintavalle M, Sambucini S, Summa V, et al. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J Biol Chem. 2007;282 (8):5536–5544. doi: 10.1074/jbc.M610486200. Epub 2006/12/15. [DOI] [PubMed] [Google Scholar]
- 16.Reiss S, Rebhan I, Backes P, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9(1):32–45. doi: 10.1016/j.chom.2010.12.002. Epub 2011/01/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tai AW, Benita Y, Peng LF, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5(3):298–307. doi: 10.1016/j.chom.2009.02.001. Epub 2009/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaillancourt FH, Pilote L, Cartier M, et al. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology. 2009;387(1):5–10. doi: 10.1016/j.virol.2009.02.039. Epub 2009/03/24. [DOI] [PubMed] [Google Scholar]
- 19.Borawski J, Troke P, Puyang X, et al. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol. 2009;83 (19):10058–10074. doi: 10.1128/JVI.02418-08. Epub 2009/07/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coller KE, Berger KL, Heaton NS, Cooper JD, Yoon R, Randall G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009;5 (12):e1000702. doi: 10.1371/journal.ppat.1000702. Epub 2009/12/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q, Brass AL, Ng A, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci USA. 2009;106(38):16410–16415. doi: 10.1073/pnas.0907439106. Epub 2009/09/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141(5):799–811. doi: 10.1016/j.cell.2010.03.050. Epub 2010/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger KL, Cooper JD, Heaton NS, et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106(18):7577–7582. doi: 10.1073/pnas.0902693106. Epub 2009/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102(26):9294–9299. doi: 10.1073/pnas.0503596102. Epub 2005/06/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scholle F, Li K, Bodola F, Ikeda M, Luxon BA, Lemon SM. Virus-host cell interactions during hepatitis C virus RNA replication: impact of polyprotein expression on the cellular transcriptome and cell cycle association with viral RNA synthesis. J Virol. 2004;78(3):1513– 1524. doi: 10.1128/JVI.78.3.1513-1524.2004. Epub 2004/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NFkappaB and IRF 3. Cell. 2005;122 (5):669–682. doi: 10.1016/j.cell.2005.08.012. Epub 2005/08/30. [DOI] [PubMed] [Google Scholar]
- 27.Biswas N, Liu S, Ronni T, et al. The ubiquitin-like protein PLIC-1 or ubiquilin 1 inhibits TLR3-Trif signaling. PLoS One. 2011;6(6):e21153. doi: 10.1371/journal.pone.0021153. Epub 2011/06/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhan F, Colla S, Wu X, et al. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2- and p27Kip1-dependent and -independent mechanisms. Blood. 2007;109 (11):4995–5001. doi: 10.1182/blood-2006-07-038703. Epub 2007/02/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin-Ezquerra G, Salgado R, Toll A, et al. CDC28 protein kinase regulatory subunit 1B (CKS1B) expression and genetic status analysis in oral squamous cell carcinoma. Histol Histopathol. 2011;26(1):71–77. doi: 10.14670/HH-26.71. Epub 2010/12/01. [DOI] [PubMed] [Google Scholar]
- 30.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298 (5600):1912–1934. doi: 10.1126/science.1075762. Epub 2002/12/10. [DOI] [PubMed] [Google Scholar]
- 31.Westbrook L, Manuvakhova M, Kern FG, Estes NR, 2nd, Ramanathan HN, Thottassery JV. Cks1 regulates cdk1 expression: a novel role during mitotic entry in breast cancer cells. Cancer Res. 2007;67(23):11393–11401. doi: 10.1158/0008-5472.CAN-06-4173. Epub 2007/12/07. [DOI] [PubMed] [Google Scholar]
- 32.Ndjomou J, Park IW, Liu Y, Mayo LD, He JJ. Up-regulation of hepatitis C virus replication and production by inhibition of MEK/ERK signaling. PLoS One. 2009;4(10):e7498. doi: 10.1371/journal.pone.0007498. Epub 2009/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez-Otano I, Lujan R, Tavalin SJ, et al. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat Neurosci. 2006;9(5):611–621. doi: 10.1038/nn1680. Epub 2006/04/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans MJ, Rice CM, Goff SP. Phosphorylation of hepatitis C virus non-structural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci USA. 2004;101(35):13038–13043. doi: 10.1073/pnas.0405152101. Epub 2004/08/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007;3(7):e103. doi: 10.1371/journal.ppat.0030103. Epub 2007/07/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Griffin SD, Harvey R, Clarke DS, Barclay WS, Harris M, Rowlands DJ. A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J Gen Virol. 2004;85 (Pt 2):451–461. doi: 10.1099/vir.0.19634-0. Epub 2004/02/11. [DOI] [PubMed] [Google Scholar]
- 37.Griffin SD, Beales LP, Clarke DS, et al. The p7 protein of hepatitis C virus forms an ion channel that is blocked by the antiviral drug, Amantadine. FEBS Lett. 2003;535 (1–3):34–38. doi: 10.1016/s0014-5793(02)03851-6. Epub 2003/02/01. [DOI] [PubMed] [Google Scholar]
- 38.Haqshenas G, Mackenzie JM, Dong X, Gowans EJ. Hepatitis C virus p7 protein is localized in the endoplasmic reticulum when it is encoded by a replication-competent genome. J Gen Virol. 2007;88(Pt 1):134–142. doi: 10.1099/vir.0.82049-0. Epub 2006/12/16. [DOI] [PubMed] [Google Scholar]
- 39.Montserret R, Saint N, Vanbelle C, et al. NMR structure and ion channel activity of the p7 protein from hepatitis C virus. J Biol Chem. 2010;285(41):31446–31461. doi: 10.1074/jbc.M110.122895. Epub 2010/07/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Modregger J, Ritter B, Witter B, Paulsson M, Plomann M. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci. 2000;113(Pt 24):4511–4521. doi: 10.1242/jcs.113.24.4511. Epub 2000/11/18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.