

Abstract
Purpose
A recent experiment indicated that a loss of function mutation in the murine Katnal1 gene resulted in male factor infertility due to premature exfoliation of spermatids. This study investigated the relevance of this gene to infertility in humans.
Methods
Multiple methods of genetic analysis were employed to investigate whether mutations in human KATNAL1 have a causative role in male infertility. This was a genetic association study, which included DNA samples from 105 men with non-obstructive azoospermia (NOA) and 242 anonymous sperm donor controls. 28 commercially available TaqMan SNP assays were used to haplotype samples from both groups and genetically tag regions of interest across the entire gene. AmpliSeq primers were then designed for identified regions so that targeted next-generation sequencing (NGS) could be used to identify causative variants.
Results
Four SNPs in the 3’UTR demonstrated a putative association with NOA. The AmpliSeq primers designed for the 3’UTR provided 83 % coverage of the 7,202 basepairs within the regions of interest. Variant sites were analyzed against genetic models to identify sequence polymorphisms which associated with NOA. No variants met standard criteria for significance when tested between the groups.
Conclusions
This study suggests a lack of association of KATNAL1 gene sequence variants and azoospermia in humans.
Keywords: KATNAL1, Azoospermia, Genetic association, Next generation sequencing, Tagging SNP
Introduction
Male factor infertility can stem from numerous pre-, post-, and testicular etiological factors, among which are genetic mutations [1, 2]. Chromosomal abnormalities such as Y-chromosome microdeletions and aneuploidy are the most common genetic abnormalities associated with male infertility, responsible for around 5 % of cases in males and 15 % in azoospermic males [3]. Single gene and polygenic mutations can be causative of various infertility phenotypes as well, and genetic association studies using knockout models and ENU mutagenesis screens, as well as microarrays and hybridization arrays for whole genome analyses, have been conducted in attempts to identify responsible genes [4–6]. The advent of targeted next generation sequencing has furthered these attempts by allowing for large scale studies to be conducted at base pair resolution for both mutations and their surrounding sequences.
The relevance of associated SNPs and genes involved in male specific infertility can be difficult to discern since more than 2,300 genes have been predicted to be involved in spermatogenesis alone [7]. Investigating genes based on specific phenotypes, however, can help narrow the number of candidate genes. For example, the human katanin p60 subunit A-like 1 (KATNAL1) and katanin p80 subunit B-like 1 (KATNBL1) genes have been identified in murine models as causative of male-specific infertility due to two different loss-of-function point mutations. Katanin is a member of the AAA ATPase super family that uses energy from nucleotide hydrolysis to sever and disassemble microtubules through the catalytic p60 subunit and centrosome-targeting regulatory p80 subunit [8–11]. In Katnbl1 mutant mice, a guanine to thymine mutation in exon 9 caused oligoasthenoteratozoospermia due to decreased sperm production, motility, and abnormal sperm morphology [12], while in Katnal1 mutant mice, Sertoli cell microtubules were disrupted causing the premature exfoliation of spermatids due to a thymine to guanine mutation in exon 7 resulting in the absence of mature sperm [13].
Spermatogenesis maturation arrest in humans is most typically seen during meiosis, indicating that mutations in genes essential for meiosis are potentially causative of infertility [14]. Katanin has a conserved functional role in meiosis, with mutations in functional homologs (MEI-1 and MEI-2) revealing roles in meiotic spindle function and regulation of microtubules in C. elegans [15–17], and flagellar central apparatus assembly and microtubule severing in Chlamydomonas [18]. Based on the murine mutation and the conserved functional role of Katanin, the KATNAL1 gene was investigated for further associations with infertility by using a combination of genetic association and next generation sequencing. Specifically, this study aimed to identify tagging SNPs associated with azoospermia across KATNAL1 through a genetic association study and then used a targeted next generation sequencing approach to further investigate regions of interest for novel or functional polymorphisms associated with this phenotype.
Materials and methods
Ethics
This study was done under IRB approval. The samples were selected from an existing repository consisting of individuals seeking fertility treatment.
Population
Samples were identified from a cohort of patients undergoing IVF treatment in Northern New Jersey. Cases consisted of men with non-obstructive azoospermia (NOA) and severe oligospermia, while controls consisted of men with normal sperm concentration. Cases (n = 105) had at least one semen specimen submitted for analysis that revealed azoospermia without a known cause. Samples were excluded from this cohort if they had: undescended testicles, vasectomy/vasovasostomy, cancer treatments (chemotherapy, radiation or radical orchiectomy), Y-chromosome microdeletions, aneuploidy (including the sex chromosomes), retrograde ejaculation, congenital bilateral absence of the vas deferens, bilateral vas deferens occlusion as a result of inguinal hernia repair, etc. The control group (n = 242) consisted of anonymous sperm donor samples.
SNP selection
HapMap was used to identify 59 SNPs present in KATNAL1. 28 of these SNPs were selected based on haplo-block correlation and distribution across KATNAL1. Only tagging SNPs that had primers commercially available through Life Technologies (Life Technologies [LTI], Carlsbad, CA, USA) were considered for inclusion. The selected SNPs (see Fig. 1) captured 86 % of the targeted alleles, with a mean r-squared value of 0.999. The 28 SNPs also did not have high marker-to-marker linkage disequilibrium with their neighboring selected SNPs, with r-squared values ranging from 0–0.477 [19]. After the initial results were viewed, four SNPs were excluded from further analysis; SNPs rs12866391 and rs7327725 were excluded due to complete homozygosity, and rs7328946 and rs9550539 were excluded for violating Hardy-Weinberg equilibrium across both the case and control sample groups. Samples were then filtered to include only those with ≥ 90 % call rates before statistical analysis was performed.
Fig. 1.
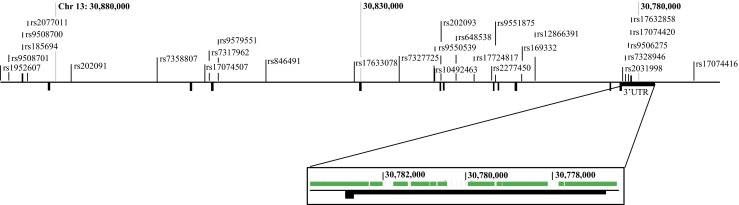
The locations of the 28 tagging SNPs used in the genetic association study are depicted spanning the KATNAL1 gene. The magnification of the 3’UTR shows the regions that were covered by the AmpliSeq design (green) for next generation sequencing
Genotyping through allelic discrimination
Genotyping was done on the QuantStudio™12 K Flex Real-Time PCR System (LTI). 347 samples were normalized to 50 ng/uL. Three samples did not have enough starting gDNA, so whole genome amplification with the REPLI-g Mini Kit (QIAGEN Inc, Germantown, MD, USA) was performed prior to use. 2.5 uL of TaqMan® OpenArray® Genotyping Master Mix (LTI) and 2.5 uL of each gDNA sample were premixed in a 384 well plate and loaded onto the genotyping plates using the QuantStudio™ 12 K Flex OpenArray® AccuFill™ System. The samples were plated in duplicate, and results were analyzed in TaqMan Genotyperv1.2 (LTI). Samples that were called unamplified or undetermined were rerun using duplex real time PCR reactions followed by allelic discrimination on the ABI PRISM® 7900 HT Sequence Detection System using SDS 2.3 software (LTI). A no template control (NTC) consisting of water was included in each run.
SNP statistical analysis
Association analysis was performed in Golden Helix SNP & Variation Suite (SVS) v7.x software (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com). Additive, Dominant, Recessive, and Basic Allele associations were performed, as well as Principal component analysis.
Next generation sequencing data acquisition
Sample library preparation was done according to the procedure specified in the Ion AmpliSeq Library Preparation User Guide (LTI). Custom AmpliSeq primers were designed using Ion AmpliSeq Designer to cover the region stretching from the 3’UTR to the last exon in KATNAL1 (Fig. 1). Forty-nine amplicons 200 bp in length were designed to cover the 7.2 kilobase region (chromosome 13: 30,776,500-30,783,702) (hg19). The primer design gave 83.48 % coverage, excluding 1,190 base pairs due to repetitive sequences and low GC content (between 0–19.685). Input DNA consisted of 2 different DNA pools each at 20 ng, with sequence targets amplified for 21 cycles using the 2X primer pool protocol, followed by adapter and barcode ligation. Molar concentrations for each amplicon were obtained using the Bioanalyzer and Agilent High-Sensitivity DNA microfluidic chips (Agilent Technologies), and the samples were then normalized to 22 pmol/L. Template preparation was done according to the Ion One Touch protocol (LTI), which used the Ion PGMTM Template OT2 200 Kit for template preparation and the Ion PGMTM Sequencing 200 Kit v2.0 for sequencing. 318 Chips were used, with 12–24 samples barcoded per chip (with each sample having 2 barcodes for each primer pool).
NGS data analysis
Fastq files [20] for all of the barcoded samples were obtained from the Ion Torrent Server. Ion Reporter Software 1.4 was used to analyze the variants. The reference file was generated from the .BED file that the Ion AmpliSeq primers were designed from, and the variant caller parameters were the same as for the built-in CHPv2 workflow. The output vcf files contained the SNP and INDELs annotated for each sample.
Results
Association analysis using the additive model with the NOA samples (n = 90) compared against the control samples (n = 226) was performed for twenty-four SNPs to explore possible associations with NOA (overall call rate 97.06 %). The statistical results are listed in Table 1. As indicated, SNP rs17074420 had a significant uncorrected p-value, p = 0.004, while rs17074416, rs17632858, and rs9506275, showed putative associations at p = 0.025, p = 0.068, and p = 0.060, respectively. While none of the p-values survived Bonferroni correction, the corrected p-value for rs17074420 showed a putative association at p = 0.098. Dominant and basic allele models yielded similar results, while the recessive model did not have any significant associations (results not shown).
Table 1.
Genetic Association Values
| Marker | Corr/Trend P | BonferroniCorrected P | Corr/Trend Full Scan Permutation* P | Odds Ratio ((Dd) vs. (dd))** |
|---|---|---|---|---|
| rs17074416 | 0.025 | 0.589 | 0.351 | 1.392 |
| rs17632858 | 0.068 | 1 | 0.666 | 1.445 |
| rs17074420 | 0.004 | 0.098 | 0.070 | 2.546 |
| rs9506275 | 0.060 | 1 | 0.611 | 0.686 |
| rs2031998 | 0.884 | 1 | 1 | 0.914 |
| rs169332 | 0.386 | 1 | 1 | 1.319 |
| rs9551875 | 0.764 | 1 | 1 | 1.004 |
| rs2277450 | 0.368 | 1 | 1 | 1.301 |
| rs17724817 | 0.585 | 1 | 1 | 0.999 |
| rs648538 | 0.050 | 1 | 0.571 | 0.797 |
| rs202093 | 0.310 | 1 | 0.997 | 1.380 |
| rs10492463 | 0.749 | 1 | 1 | 0.835 |
| rs17633078 | 0.109 | 1 | 0.821 | 1.228 |
| rs846491 | 0.196 | 1 | 0.959 | 0.705 |
| rs9579551 | 0.286 | 1 | 0.993 | 1.426 |
| rs7317962 | 0.634 | 1 | 1 | 0.563 |
| rs17074507 | 0.947 | 1 | 1 | 0.773 |
| rs7358807 | 0.312 | 1 | 0.998 | 1.398 |
| rs202091 | 0.397 | 1 | 1 | 0.734 |
| rs2077011 | 0.600 | 1 | 1 | 1.301 |
| rs9508700 | 0.093 | 1 | 0.765 | 0.671 |
| rs185694 | 0.564 | 1 | 1 | 0.791 |
| rs9508701 | 0.468 | 1 | 1 | 0.806 |
| rs1952607 | 0.646 | 1 | 1 | 1.038 |
* Permutation analysis done at 10,000 permutations
** dd = Major Allele
In order to ensure that results were not affected by population stratification, a PCA was performed for the twenty-four SNPs across all samples. While there was no difference observed in the distribution of cases and controls in the PCA space (p > 0.05), there was not enough power to definitively rule out PCA without examining additional SNPs. However the observed results suggest that population stratification was not a strong confounding factor.
Since rs17074420 was located in the 3’UTR of KATNAL1, the entire 3’UTR (7.2 kilobases, chromosome 13: 30,776,500-30,783,702) was selected for further investigation via next generation sequencing. Samples carrying rs17074420 were roughly matched at a 2:1 ratio with randomly selected samples that did not carry the SNP, yielding an overall sample size of 194 samples (105 cases and 89 controls). Ion Reporter Software 1.4 was used to identify variant sites.
From the entire sequenced 3’UTR region, four SNP sites had both genotyping data from the original association study done by qPCR and allelic discrimination, and from the second phase by next-generation sequencing, allowing for comparisons to be made between the genotype calls assigned to each sample. The similarity between the two methods ranged from 90.2 % to 94.8 %, which is within the expected range identified for cross-platform replication of next-generation sequencing data [21–23].
A total of 60 SNPs were identified in the 3’UTR region (42 variants with dbSNP identification numbers [24] and 18 variants that were novel). Most variants were covered at an average depth of ~500 reads. All of the novel variants were observed only in the heterozygous state, with seventeen of these variants appearing only once. Because of this, Fisher exact tests (2-tailed, 95 % CI) indicated that these results were insignificant (Fisher p-value = >0.9 for variants present in only one sample, and 0.585 or 0.876 when present in more than one sample). A Fisher exact test was also done comparing the total number of minor alleles observed between the cases and controls for these novel variants, but the result was also insignificant (Fisher p-value = 1.00). Table 2 shows a summary of these novel SNPs and their locations in KATNAL1. None of the regions where the SNPs were located were known to have roles in regulation.
Table 2.
Novel variants detected in the 3’UTR region
| Chr 13 Position | No. Cases (Het) | No. Controls (Het) | Ref Allele | Alt Allele |
|---|---|---|---|---|
| 30,776,977 | 1 | 2 | A | G |
| 30,777,467 | 0 | 1 | C | A |
| 30,777,656-30,777,666 | 0 | 1 | AAGAGGGAAAC | - |
| 30,778,287 | 1 | 0 | A | C |
| 30,778,292 | 0 | 1 | C | T |
| 30,779,639 | 0 | 1 | T | C |
| 30,779,734 | 0 | 1 | C | A |
| 30,779,755 | 2 | 0 | G | C |
| 30,779,960 | 0 | 1 | A | ATCGTGGTTGCC |
| 30,780,749 | 0 | 1 | A | C |
| 30,780,773 | 1 | 0 | T | C |
| 30,781,443 | 1 | 0 | T | C |
| 30,781,636-30,781-642 | 1 | 0 | AATGTAA | -- |
| 30,781,682 | 0 | 1 | A | C |
| 30,781,712 | 1 | 0 | T | A |
| 30,782,331 | 1 | 0 | A | G |
| 30,783,284 | 1 | 0 | A | G |
| 30,783,564 | 1 | 0 | C | T |
The 42 variants that did have dbSNP identification numbers had been previously reported as untranslated or intron variants, and had minor allele frequencies available from the 1000 genome project. Fisher p-values were calculated for these variants as well, however none of the variants were found to be significant after Bonferroni correction, with all values driven to p = 1.00 (Table 3). Therefore no variants met the standard criteria for significance when tested between the cases and controls.
Table 3.
Known Variants Detected in the 3’UTR Region
| - | Cases | Controls | ||||
|---|---|---|---|---|---|---|
| rs# | Het Carrier | Homo Affected | Het Carrier | Homo Affected | Fisher P-Values | Bonferroni Corrected P |
| rs182105027 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs11839654 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs147850458 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs186665940 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs76781079 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs145250193 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs148949899 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs76940165 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs113998014 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs188065499 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs55836900 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs9508679 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs140253752 | 0 | 0 | 1 | 0 | 0.918 | 1 |
| rs61946916 | 0 | 7 | 1 | 3 | 0.337 | 1 |
| rs182453378 | 1 | 0 | 0 | 0 | >0.999 | 1 |
| rs114243171 | 1 | 0 | 1 | 0 | >0.999 | 1 |
| rs61435763 | 1 | 0 | 2 | 0 | 0.876 | 1 |
| rs17689385 | 1 | 0 | 3 | 0 | 0.504 | 1 |
| rs117247354 | 1 | 0 | 3 | 0 | 0.504 | 1 |
| rs78699770 | 2 | 0 | 0 | 0 | 0.585 | 1 |
| rs114829192 | 2 | 0 | 0 | 0 | 0.585 | 1 |
| rs77772670 | 2 | 0 | 0 | 0 | 0.585 | 1 |
| rs9579542 | 2 | 0 | 1 | 0 | >0.999 | 1 |
| rs144510457 | 2 | 0 | 2 | 0 | >0.999 | 1 |
| rs117991635 | 2 | 0 | 2 | 0 | >0.999 | 1 |
| rs75680479 | 5 | 0 | 9 | 0 | 0.257 | 1 |
| rs45507700 | 6 | 0 | 7 | 0 | 0.758 | 1 |
| rs9508678 | 9 | 1 | 7 | 0 | 0.719 | 1 |
| rs11620056 | 16 | 4 | 19 | 0 | 0.944 | 1 |
| rs1023104 | 17 | 1 | 22 | 0 | 0.372 | 1 |
| rs12428633 | 17 | 9 | 28 | 10 | 0.019 | 1 |
| rs9508676 | 21 | 0 | 16 | 0 | 0.873 | 1 |
| rs12430537 | 21 | 1 | 15 | 1 | 0.779 | 1 |
| rs17074420 | 21 | 1 | 20 | 1 | 0.783 | 1 |
| rs45484093 | 23 | 1 | 14 | 1 | 0.445 | 1 |
| rs9551868 | 25 | 7 | 33 | 2 | 0.885 | 1 |
| rs2031998 | 27 | 7 | 34 | 2 | 0.749 | 1 |
| rs3825533 | 28 | 5 | 34 | 3 | 0.345 | 1 |
| rs7328946 | 28 | 9 | 37 | 5 | 0.360 | 1 |
| rs112146305 | 33 | 0 | 24 | 0 | 0.637 | 1 |
| rs17632858 | 44 | 7 | 37 | 2 | 0.360 | 1 |
| rs9506275 | 52 | 18 | 45 | 15 | >0.999 | 1 |
Discussion
The lack of associated infertility phenotypes found in KATNAL1 suggests that the genotype-phenotype correlation observed for this gene in murine models may not apply to humans. While many spermatogenesis genes and processes are conserved between mice and humans [25–27], studies have shown that functional orthologs are not always representative of their corresponding human genes [28, 29], and even if they are, the expected phenotype severity can vary between species [14]. It is also possible however that there could have been a novel variant in this region that was not picked up by the association study that could have been causative of the NOA phenotype. Since sequencing was not done across the entire KATNAL1 gene, it is possible that such a variant was missed. The leucine to valine [UUA to GUA] change reported in exon 7 of murine Katnal1 [13] corresponds to position 30805474 on human chromosome 13. In terms of reported variants, the USC Genome Browser reports no SNPs at this position, and no significant association was observed when utilizing the TaqMan SNPs in the association study.
With the exception of mutations that cause very specific phenotypes, such as globozoospermia [30, 31], very few genes have been definitively linked to infertility. The numerous candidate genes identified as causative of these specific phenotypes suggest genetic heterogeneity, indicating that male factor infertility could be caused by patient-specific mutations in different genes or can result from various genetic interactions. Genomic instability may also be causative of infertility [4], with studies showing that men with good semen quality (normal and motile sperm) and concentration had dose–response decreased mortality [32] and comorbidity rates [33] than infertile men. The lack of association of KATNAL1 with the NOA phenotype may also reflect the relatively small sample size (n = 105 NOA patients) utilized in this study.
In summary, 28 tagging SNPs that spanned KATNAL1 were originally analyzed for their potential association with the NOA phenotype. Only 4 of those SNPs demonstrated a putative association with the NOA phenotype and all of them were located in the 3’UTR. Since the 3’UTR is important for various regulatory processes, including transcript cleavage, stability, and polyadenylation, as well as translational activation, mRNA localization, and binding sites for microRNAs and regulatory proteins [34, 35], the entire region was sequenced in attempts to identify SNPs that could affect functional variants located in KATNAL1 or in neighboring genes. The lack of any significant variants identified in the NOA patients in both the 3’UTR and across KATNAL1 indicates that this gene is not likely to be responsible for the NOA phenotype in male infertility patients.
Ethical Standards
The manuscript does not contain clinical studies or patient data.
Footnotes
Capsule Investigation of the KATNAL1 gene for an association with the human phenotype non-obstructive azoospermia using genetic association and next generation sequencing yielded no significant findings.
References
- 1.De Kretser DM, Baker HW. Infertility in men: recent advances and continuing controversies. J Clin Endocrinol Metab. 1999;84:3443–3450. doi: 10.1210/jcem.84.10.6101. [DOI] [PubMed] [Google Scholar]
- 2.McLachlan RI, Rajpert-De Meyts E, Hoei-Hansen CE, et al. Histological evaluation of the human testis–approaches to optimizing the clinical value of the assessment: mini review. Hum Reprod. 2007;22:2–16. doi: 10.1093/humrep/del279. [DOI] [PubMed] [Google Scholar]
- 3.O'Flynn O'Brien KL, Varghese AC, Agarwal A. The genetic causes of male factor infertility: a review. Fertil Steril. 2010;93:1–12. doi: 10.1016/j.fertnstert.2009.10.045. [DOI] [PubMed] [Google Scholar]
- 4.Aston KI, Carrell DT. Emerging evidence for the role of genomic instability in male factor infertility. Syst Biol Reprod Med. 2012;58:71–80. doi: 10.3109/19396368.2011.635751. [DOI] [PubMed] [Google Scholar]
- 5.Waclawska A, Kurpisz M. Key functional genes of spermatogenesis identified by microarray analysis. Syst Biol Reprod Med. 2012;58:229–235. doi: 10.3109/19396368.2012.693148. [DOI] [PubMed] [Google Scholar]
- 6.Aston KI, Carrell DT. Genome-wide study of single-nucleotide polymorphisms associated with azoospermia and severe oligozoospermia. J Androl. 2009;30:711–725. doi: 10.2164/jandrol.109.007971. [DOI] [PubMed] [Google Scholar]
- 7.Schultz N, Hamra FK, Garbers DL. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci U S A. 2003;100:12201–12206. doi: 10.1073/pnas.1635054100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McNally FJ, Vale RD. Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell. 1993;75:419–429. doi: 10.1016/0092-8674(93)90377-3. [DOI] [PubMed] [Google Scholar]
- 9.Hartman JJ, Mahr J, McNally K, et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell. 1998;93:277–287. doi: 10.1016/s0092-8674(00)81578-0. [DOI] [PubMed] [Google Scholar]
- 10.Hartman JJ, Vale RD. Microtubule disassembly by ATP-dependent oligomerization of the AAA enzyme katanin. Science. 1999;286:782–785. doi: 10.1126/science.286.5440.782. [DOI] [PubMed] [Google Scholar]
- 11.McNally KP, Bazirgan OA, McNally FJ. Two domains of p80 katanin regulate microtubule severing and spindle pole targeting by p60 katanin. J Cell Sci. 2000;113(Pt 9):1623–1633. doi: 10.1242/jcs.113.9.1623. [DOI] [PubMed] [Google Scholar]
- 12.O'Donnell L, Rhodes D, Smith SJ, et al. An essential role for katanin p80 and microtubule severing in male gamete production. PLoS Genet. 2012;8:e1002698. doi: 10.1371/journal.pgen.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith LB, Milne L, Nelson N, et al. KATNAL1 regulation of sertoli cell microtubule dynamics is essential for spermiogenesis and male fertility. PLoS Genet. 2012;8:e1002697. doi: 10.1371/journal.pgen.1002697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Massart A, Lissens W, Tournaye H, et al. Genetic causes of spermatogenic failure. Asian J Androl. 2012;14:40–48. doi: 10.1038/aja.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srayko M, Buster DW, Bazirgan OA, et al. MEI-1/MEI-2 katanin-like microtubule severing activity is required for Caenorhabditis elegans meiosis. Genes Dev. 2000;14:1072–1084. [PMC free article] [PubMed] [Google Scholar]
- 16.McNally KP, McNally FJ. The spindle assembly function of Caenorhabditis elegans katanin does not require microtubule-severing activity. Mol Biol Cell. 2011;22:1550–1560. doi: 10.1091/mbc.E10-12-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mains PE, Kemphues KJ, Sprunger SA, et al. Mutations affecting the meiotic and mitotic divisions of the early Caenorhabditis elegans embryo. Genetics. 1990;126:593–605. doi: 10.1093/genetics/126.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dymek EE, Smith EF. PF19 encodes the p60 catalytic subunit of katanin and is required for assembly of the flagellar central apparatus in Chlamydomonas. J Cell Sci. 2012;125:3357–3366. doi: 10.1242/jcs.096941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson AD, Handsaker RE, Pulit SL, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cock PJ, Fields CJ, Goto N, et al. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010;38:1767–1771. doi: 10.1093/nar/gkp1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam HY, Clark MJ, Chen R, et al. Performance comparison of whole-genome sequencing platforms. Nat Biotechnol. 2012;30:78–82. doi: 10.1038/nbt.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robasky K, Lewis NE. Church GM. Nat Rev Genet: The role of replicates for error mitigation in next-generation sequencing; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ratan A, Miller W, Guillory J, et al. Comparison of sequencing platforms for single nucleotide variant calls in a human sample. PLoS One. 2013;8:e55089. doi: 10.1371/journal.pone.0055089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9:677–679. [PubMed] [Google Scholar]
- 25.Borg CL, Wolski KM, Gibbs GM, et al. Phenotyping male infertility in the mouse: how to get the most out of a 'non-performer'. Hum Reprod Update. 2010;16:205–224. doi: 10.1093/humupd/dmp032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamowski S, Aston KI, Carrell DT. The use of transgenic mouse models in the study of male infertility. Syst Biol Reprod Med. 2010;56:260–273. doi: 10.3109/19396368.2010.485244. [DOI] [PubMed] [Google Scholar]
- 27.Yan W. Male infertility caused by spermiogenic defects: lessons from gene knockouts. Mol Cell Endocrinol. 2009;306:24–32. doi: 10.1016/j.mce.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seok J, Warren HS, Cuenca AG, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stouffs K, Vandermaelen D, Tournaye H, et al. Mutation analysis of three genes in patients with maturation arrest of spermatogenesis and couples with recurrent miscarriages. Reprod Biomed Online. 2011;22:65–71. doi: 10.1016/j.rbmo.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Dam AH, Koscinski I, Kremer JA, et al. Homozygous mutation in SPATA16 is associated with male infertility in human globozoospermia. Am J Hum Genet. 2007;81:813–820. doi: 10.1086/521314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu G, Shi QW, Lu GX. A newly discovered mutation in PICK1 in a human with globozoospermia. Asian J Androl. 2010;12:556–560. doi: 10.1038/aja.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jensen TK, Jacobsen R, Christensen K, et al. Good semen quality and life expectancy: a cohort study of 43,277 men. Am J Epidemiol. 2009;170:559–565. doi: 10.1093/aje/kwp168. [DOI] [PubMed] [Google Scholar]
- 33.Salonia A, Matloob R, Gallina A, et al. Are infertile men less healthy than fertile men? Results of a prospective case–control survey. Eur Urol. 2009;56:1025–1031. doi: 10.1016/j.eururo.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 34.Cannell IG, Kong YW, Bushell M. How do microRNAs regulate gene expression? Biochem Soc Trans. 2008;36:1224–1231. doi: 10.1042/BST0361224. [DOI] [PubMed] [Google Scholar]
- 35.Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]