

Abstract
NMDA receptors are important elements in pain signaling in the spinal cord dorsal horn. They are heterotetramers, typically composed of two GluN1 and two of four GluN2 subunits: GluN2A-2D. Mice lacking some of the GluN2 subunits show deficits in pain transmission yet functional synaptic localization of these receptor subtypes in the dorsal horn has not been fully resolved. In this study, we have investigated the composition of synaptic NMDA receptors expressed in monosynaptic and polysynaptic pathways from peripheral sensory fibers to lamina I neurons in rats. We focused on substance P receptor-expressing (NK1R+) projection neurons, critical for expression of hyperalgesia and allodynia. EAB-318 and (R)-CPP, GluN2A/B antagonists, blocked both monosynaptic and polysynaptic NMDA EPSCs initiated by primary afferent activation by ∼90%. Physiological measurements exploiting the voltage dependence of monosynaptic EPSCs similarly indicated dominant expression of GluN2A/B types of synaptic NMDA receptors. In addition, at synapses between C fibers and NK1R+ neurons, NMDA receptor activation initiated a secondary, depolarizing current. Ifenprodil, a GluN2B antagonist, caused modest suppression of monosynaptic NMDA EPSC amplitudes, but had a widely variable, sometimes powerful, effect on polysynaptic responses following primary afferent stimulation when inhibitory inputs were blocked to mimic neuropathic pain. We conclude that GluN2B subunits are moderately expressed at primary afferent synapses on lamina I NK1R+ neurons, but play more important roles for polysynaptic NMDA EPSCs driven by primary afferents following disinhibition, supporting the view that the analgesic effect of the GluN2B antagonist on neuropathic pain is at least in part, within the spinal cord.
Keywords: disinhibition, ifenprodil, NK1 receptor, polysynaptic, spinal cord
Introduction
Mechanical allodynia, a condition in which pain is evoked by a light touch, can develop following inflammation and nerve injury (Zeilhofer and Zeilhofer, 2008; Sandkühler, 2009). Molecular and cellular changes in the spinal cord dorsal horn contribute to development and maintenance of this condition. Some important elements of these changes and the underlying circuitry in the dorsal horn have already been identified. For example, some of the projection neurons essential to the development of mechanical allodynia in lamina I are those expressing the receptor for substance P, the NK1 receptor-expressing (NK1R+) neurons (Nichols et al., 1999). In addition, loss of inhibition in the dorsal horn accompanies inflammation and nerve injury (Moore et al., 2002; Coull et al., 2005; Scholz et al., 2005; Zeilhofer, 2005) and may be responsible for the expression of mechanical allodynia (Yaksh, 1989; Sivilotti and Woolf, 1994). Under control conditions, rat NK1R+ lamina I neurons receive excitatory synaptic drive from high threshold primary afferent fibers including Aδ and C fibers (Torsney and MacDermott, 2006). However, with disinhibition, a new and powerful polysynaptic drive becomes apparent, initiated by stimulation of A fibers, including the low threshold Aβ fibers (Baba et al., 2003; Torsney and MacDermott, 2006). All of the polysynaptic excitatory drive following disinhibition in the multineuronal pathways between A fibers and the NK1R+ lamina I neurons (Torsney and MacDermott, 2006), as well as lamina II neurons (Baba et al., 2003), is blocked by the NMDA receptor antagonist, APV. If NMDA receptors in these polysynaptic pathways are molecularly and pharmacologically distinguishable from those at primary afferent synapses, it might be possible to develop selective compounds to specifically reduce polysynaptic pathways that contribute to chronic and persistent pain while maintaining acute, physiological pain signaling.
NMDA receptors generally consist of two obligatory GluN1 subunits and two of four GluN2 subunits: GluN2A, GluN2B, GluN2C, and GluN2D (Nakanishi, 1992). Earlier functional studies show that direct synaptic inputs onto lamina I and II inhibitory and excitatory neurons are primarily mediated by NMDA receptors with GluN2A or GluN2A and GluN2B subunits (Bardoni et al., 1998; Momiyama, 2000; Shiokawa et al., 2010). Here we test the hypothesis that the polysynaptic responses between primary afferents and NK1R+ neurons may be sensitive to selective GluN2B NMDA receptor antagonist. The possibility that the GluN2B subunit has a special role in the low and high threshold polysynaptic pathways to NK1R+ neurons is suggested by observations that GluN2B-specific antagonists are able to alleviate allodynia and hyperalgesia. Intrathecal administration of GluN2B antagonists blocks or decreases certain forms of chronic pain including carrageenan-induced mechanical hyperalgesia (Taniguchi et al., 1997) and nerve injury-induced mechanical allodynia (Boyce et al., 1999; Chizh et al., 2001a). Here we use whole-cell patch-clamp recording, fluorescence imaging, and receptor pharmacology to investigate the identity of synaptic NMDA receptors expressed at primary afferent synapses onto lamina I dorsal horn neurons and in polysynaptic pathways revealed by disinhibition.
Materials and Methods
Transverse slice preparation.
All experiments were conducted with the approval of the Columbia University Institutional Animal Care and Use Committee and in accord with the Guide for the Care and Use of Laboratory Animals. Postnatal day 14–20 Sprague Dawley rats of either sex were anesthetized with isoflurane and then decapitated. Their thoracic and lumbar spines along with rib cages were taken out from the bodies with ventral side facing up and placed on an ice-surrounded dish filled with ice-cold oxygenated high Mg2+ Krebs solution (95% O2/5% CO2 saturated Krebs solution containing the following (in mm): 125 NaCl or 250 sucrose, 2.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 25 glucose, 6 MgCl2, and 1.5 CaCl2, pH 7.4) plus 1 mm kynurenic acid. Spinal cords were exposed by carefully removing the ventral vertebrae with bone cutting scissors, and then gently lifted with a pair of forceps at their rostral side while dura mater and spinal nerves were carefully cut. Dorsal roots and DRG were preserved and kept intact and remained attached to the spinal cords. After spinal cords were taken out from the vertebrae, the remaining dura mater and arachnoid membranes were removed with forceps and all ventral roots were cut close to the cord. The spinal cords were then placed, with dorsal side facing down, into a small dish (2.5 cm diameter, 2 cm height) with a piece of filter paper at the bottom. The dish was quickly filled with 2% preheated low melting point agarose (Invitrogen Life Technologies) and placed on ice immediately to make the agarose solidify. These solidified agar blocks with embedded spinal cords were taken out from the small dish and placed in the slicing chamber of a Leica VT1200S vibrating blade microtome. The piece of filter paper was then removed and transverse slices (350–450 μm) with attached dorsal roots were cut using the microtome. During the cutting, a pair of forceps was always used to lift the dorsal roots to prevent them being cut. Slices were subsequently transferred into oxygenated high Mg2+ Krebs solution (no sucrose included) in an incubation chamber at 36°C for 1 h before recording at room temperature.
Recording from pre-identified NK1R+ and NK1R− lamina I neurons.
The labeling of substance P receptor (NK1R)-expressing dorsal horn neurons with fluorescent dye has been described previously (Labrakakis and MacDermott, 2003; Tong and MacDermott, 2006; Tong et al., 2008). In brief, spinal cord slices were transferred from the incubation chamber and then incubated in tetramethylrhodamine-conjugated substance P (TMR-SP; 20–40 nm) containing high Mg2+ Krebs solution for 20–30 min at room temperature. After slices were transferred back to the incubation chamber and unbound TMR-SP was washed away for at least 20 min, slices were transferred to a submersion style chamber for recording. Lamina I NK1R+ neurons were identified as neurons showing strong labeling with TMR-SP within lamina I. Our operational definition of lamina I was the region of the dorsal horn filled with fluorescently labeled dendrites and somata as shown in Figure 1A. NK1R− neurons were neurons in lamina I that did not show any somal fluorescence. According to earlier studies, 80% of the projection neurons in lamina I are NK1R+ (Marshall et al., 1996; Todd et al., 2000; Al-Khater and Todd, 2009). Although both projection and excitatory interneurons in lamina I express NK1R (Todd et al., 1998) and constitute 45% of the lamina I neurons based on immunocytochemical staining, projection neurons are larger and show high-intensity immunofluorescence while interneurons are smaller and show only weak NK1R immunofluorescence (Cheunsuang and Morris, 2000; Al Ghamdi et al., 2009). Our labeling technique may not be as sensitive a detector of NK1R+ neurons as immunocytochemical staining and we observed a smaller percentage of lamina I NK1R+ neurons, suggesting that the majority of labeled neurons in our study were those strongly expressing NK1R and were likely to be mainly projection neurons.
Figure 1.

NMDA receptors are expressed at primary afferent synapses. A, An example showing the identification of NK1R+ neurons within lamina I of superficial dorsal horn. Left, After TMR-SP incubation and washout, NK1R+ neurons were labeled and could be identified with fluorescence microscopy. Lamina I was distinguished by its fluorescent area due to labeled cell bodies and dendrites while lamina II virtually lacked fluorescence. Right, Shows low magnification of the same slice. B, Top, Representative C fiber-evoked monosynaptic currents recorded from an NK1R+ neuron using intracellular 10 mm EGTA solution at −70 mV and +50 mV holding potentials. Bottom, Superfusion of SR95531 (10 μm), strychnine (1 μm), and NBQX (5 μm) eliminated the inhibitory inputs and AMPA EPSCs to reveal the NMDA EPSCs. The stimulating intensity was 500 μA and two broken lines indicate that monosynaptic NMDA EPSCs reach their peaks 10 ms after AMPA EPSC reach their peaks. C, In the presence of SR95531, strychnine and NBQX, complete blockade of the NMDA EPSCs by d-APV (30 μm) confirmed that NMDA and AMPA EPSCs are the major EPSCs following a single primary afferent stimulus. The data in B and C are from two different neurons.
Recording solutions.
Intracellular solution was composed of the following (in mm): 120 Cs-methylsulfonate, 10 Na-methylsulfonate, 10 TEA · Cl, 10 EGTA, 1 CaCl2, 10 HEPES, 5 QX-314 · Cl, and 2 Mg2+-ATP, pH adjusted to 7.2 with CsOH, osmolarity adjusted to 290 with sucrose. For some monosynaptic NMDA EPSC recordings, internal solution contained 130 mm Cs-methylsulfonate without TEA · Cl. We found no difference in the conductance ratio (defined below) of monosynaptic NMDA EPSCs (0.15 ± 0.03, n = 8 vs 0.20 ± 0.06, n = 12, p = 0.53) and δΕPSC rise time (defined below; 1.48 ± 0.26 ms, n = 8 vs 1.06 ± 0.17 ms, n = 12, p = 0.17) recorded from NK1R+ neurons using these two internal solutions. We therefore combined these two sets of data for further analysis. For some experiments in which intracellular Ca2+ was strongly buffered to very low concentrations, BAPTA intracellular solution was used. It contained the following (in mm): 50 Cs-methylsulfonate, 10 Na-methylsulfonate, 40 BAPTA · Cs, 4 CaCl2, 10 HEPES, 5 QX-314 · Cl or 5 QX-222 · Cl, 2 Mg2+-ATP, and 10 TEA · Cl, pH adjusted to 7.2 with CsOH, osmolarity ∼310. Junction potentials were measured and corrected in the bath before Gigaohm seal was formed for each cell.
Monosynaptic EPSC recording and analysis.
To study synaptic NMDA EPSCs from pre-identified NK1R+ or NK1R− neurons within lamina I, neurons were held at –70 mV under voltage-clamp control most of the time. Dorsal roots were stimulated using a glass suction electrode with stimuli of variable intensities and 0.1 ms duration. Monosynaptic responses were identified based on the absence of synaptic failures and low variability of latency at high-frequency stimulation (10 Hz for Aδ fibers and 1 Hz for C fibers). At these frequencies, polysynaptic responses tend to fail and markedly change their latency while monosynaptic responses maintain a constant latency. Stimulus intensity was gradually increased until the maximal response was identified. The intensity was then further increased to either 100 μA for Aδ fiber or 300 μA for C fiber activations (constant current output); 500 μA was sometimes used to ensure full activation of C fibers. Because monosynaptic EPSCs mediated by Aβ fibers only occur in 5% of the lamina I NK1R+ neurons, most of the cells were not tested with Aβ fiber intensity (20 μA; Nakatsuka et al., 2000; Torsney and MacDermott, 2006). Data were acquired and analyzed using Axopatch 200 amplifier, Digidata 1322A, and pClamp 9.2 software (Axon Instruments). Sampling rate was 10 kHz and data were filtered at 5 kHz.
One approach we used to investigate the composition of functional NMDA receptors at synapses between primary afferents and dorsal horn neurons was to compare their current–voltage (I–V) relationships at these synapses with those of GluN2A/B or GluN2C/D type NMDA receptors. SR95531 (10 μm), strychnine (1 μm), and NBQX (5 μm) were included to block GABAA, glycine, and non-NMDA receptors, respectively. The membrane potential was stepped to different values (−90 mV to +50 mV using 20 mV increment) for 350 ms before giving a stimulus to the dorsal root. To isolate the synaptic current, membrane currents recorded in the absence of dorsal root stimulation at each membrane potential were subtracted. Monosynaptic currents were averaged over two to three evoked EPSCs. The peak amplitude of NMDA EPSCs was measured at 10 ms following the first peak of AMPA EPSCs to ensure minimal contamination of polysynaptic NMDA receptors (McBain and Mayer, 1994).
Different NMDA receptor compositions show different I–V relationships (Kuner and Schoepfer, 1996). To compare the I–V relationships of the monosynaptic NMDA EPSCs with those of heterologously expressed known NMDA receptor subtypes, conductance ratio was used in a manner similar to our earlier study (Tong et al., 2008, Shiokawa et al., 2010). It was calculated based on the equation:
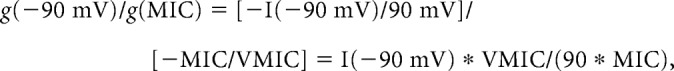 |
g(−90 mV) and g(MIC) represent the NMDA EPSC peak conductance at −90 mV holding potential and the conductance of the maximal inward current (MIC), respectively. To obtain the MIC and the membrane potential for the MIC (VMIC), a second degree polynomial equation was used to fit the three largest inward current values at negative membrane potentials within each NMDA EPSC I–V curve. MIC and VMIC were then calculated from the fitting equation. Conductance ratio has been shown to be a useful way to estimate NMDA receptor subunit composition (Tong et al., 2008; Shiokawa et al., 2010). Extracellular Mg2+ concentration was kept at 100 μm so that that the difference between the I–V relationships of GluN2A/B type and GluN2C/D type NMDA receptors is larger compared with that in 1 mm Mg2+ solution, thus making it easier to identify the subtypes of functional NMDA receptors.
In some cells, monosynaptic NMDA EPSCs showed different EPSC rise times at different membrane potentials. To analyze the variation of NMDA EPSC rise times of individual cells, we first measured the time from the stimulus artifact to 50% of the peak monosynaptic NMDA EPSC (tv, where v indicates the holding membrane potential) at each membrane potential for each cell, instead of directly measuring the 20–80% or 30–70% rise time. Because changing the membrane potentials of postsynaptic neurons does not change conduction of action potentials in primary afferent fibers or presynaptic neurotransmitter release (Billups et al., 2002), the variations of NMDA EPSC rise time are reflected in the variations of value tv. Measuring tv largely reduces measuring errors due to the small amplitudes of some EPSCs recorded close to −90 mV or near 0 mV, potentials at which NMDA receptors are mainly blocked by Mg2+ or have a small driving force for ion flow.
We therefore defined a value representing the delay of EPSCs at negative membrane potentials as
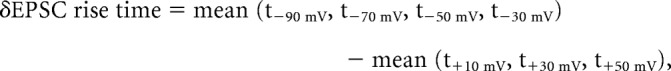 |
as illustrated in Figure 4A, bottom.
Figure 4.

Slowly rising NMDA EPSCs distort the calculation of conductance ratios. A, Top, Representative traces of monosynaptic NMDA EPSCs recorded from an NK1R+ neuron using internal EGTA solution. The inset of the NMDA I–V relationship indicates GluN2C/D-like NMDA receptors at the synapses. Middle, Shows NMDA EPSCs normalized at their peaks from the same cell. The slower EPSC rise times and faster decay times at some negative membrane potentials indicate some currents other than NMDA EPSCs were activated. Bottom, Illustrates the methodology of calculating the δEPSC rise time (see Material and Method). The broken line represents the peak of NMDA EPSCs. B, Top, An example of NMDA EPSCs recorded from an NK1R− neuron using EGTA as the internal solution. Bottom, Normalized NMDA EPSCs showed similar rise times. The inset I–V relationship of the EPSCs indicates GluN2A/B-like NMDA receptors at the synapses. C, Top, An example of NMDA EPSCs recorded with 40 mm BAPTA intracellular solution. Bottom, Normalized NMDA EPSCs with BAPTA showed similar rise times. The inset I–V relationship of the EPSCs indicates GluN2A/B-like NMDA receptors at the synapses. D, Individual recordings from NK1R+ and NK1R− neurons under EGTA and BAPTA conditions were plotted according to their g(−90 mV)/g(MIC) conductance ratio and δEPSC rise time. There was a greater percentage of NK1R+ neurons showing δEPSC < 0.7 ms (38% vs 10%, p < 0.05 for χ2 test) in the BAPTA intracellular solution in comparison with EGTA intracellular solution. We also recorded EPSCs from NK1R− neurons using BAPTA in the recording pipette. As expected, EPSCs from these neurons still showed small δEPSCs (0.17 ± 0.21 ms, n = 11) as well as conductance ratios close to those of receptors with GluN2A/B subunits (0.08 ± 0.01, n = 11). The dotted line represents δEPSC = 0.7 ms.
Large δEPSC rise times represent substantial slowing of NMDA EPSCs at negative membrane potentials compared with those at positive membrane potentials while small δEPSCs indicate similar EPSC rise times at negative and positive membrane potentials.
Polysynaptic EPSC recording and analysis.
Polysynaptic NMDA responses following primary afferent fiber stimulation were routinely elicited every 45–120 s in the presence of SR95531 (10 μm), strychnine (1 μm), and NBQX (5 μm) in 100 μm Mg2+ Krebs solution. With this time interval between stimuli, synaptic depression was minimal.
Most of the neurons tested for polysynaptic responses were recorded at −70 mV to minimize voltage-gated channel activation while some were recorded alternately at both +50 mV and −70 mV. To test the effects of different antagonists on polysynaptic responses, instead of amplitude, the area under the polysynaptic EPSC curves before, during, and after antagonist application were measured from the stimulus artifact through 1000 ms later. For some cells that were recorded at both +50 and −70 mV membrane potentials, the EPSC areas were calculated from stimulus artifact to 500 ms later.
In some experiments, we stimulated the dorsal roots with a range of intensities, including 20, 100, and 500 μA to test for Aβ, Aδ, and C fiber-driven polysynaptic activity. Neurons showing polysynaptic responses at a stimulation intensity of 20 μA were considered to receive Aβ fiber-driven polysynaptic inputs. Some of these neurons also showed additional polysynaptic responses when the intensity was increased to 100 μA or 500 μA. These neurons were considered to receive Aβ/Aδ or Aβ/C fiber inputs, respectively. Neurons showing no response at 20 μA, but a polysynaptic response at 100 μA, were considered to receive inputs driven by Aδ fibers. Neurons showing an additional response when the stimulation intensity was increased to 500 μA were considered to receive Aδ/C fiber inputs. Some neurons did not show any polysynaptic responses until the stimulation intensity reached 500 μA. These neurons were considered to receive only C fiber polysynaptic inputs. In 40 NK1R+ neurons showing polysynaptic responses, 18 contained initial monosynaptic responses. In 31 NK1R− neurons tested, 7 contained initial monosynaptic responses. For these cells, we calculated the polysynaptic EPSC area from 300 ms following the monosynaptic EPSC peak through 800 ms or 1000 ms later to exclude the possible inclusion of monosynaptic NMDA EPSCs, which usually have a decay time constant faster than 100 ms (Vicini et al., 1998). To justify that 300 ms is a reasonable time to start measuring polysynaptic responses with minimal inclusion of monosynaptic responses in our preparation, we analyzed monosynaptic EPSCs with no obvious contamination of polysynaptic EPSCs at −70 mV holding potential. With single exponential decay fitting, we observed that the decay time constant for these neurons was 83 ± 11 ms (n = 10). Assuming synaptic delay from primary afferents to dorsal horn neurons to be 5 ms following stimulus artifact, monosynaptic NMDA EPSCs peak 10 ms after AMPA EPSCs and monosynaptic NMDA EPSCs follow a single exponential decay, the remaining current of NMDA EPSCs at 300 ms would be 100% * exp(−(300 − 5 − 10)/83) = 3%. In our data, the remaining current at 300 ms was 12 ± 2% (n = 10), possibly due to contamination of polysynaptic responses. Because these experiments were performed in 0.1 mm Mg2+ Krebs solution, the polysynaptic responses persisted in the presence of NBQX, in contrast to our earlier study (Torsney and MacDermott, 2006).
Statistics.
Data were expressed as mean ± SE, with n referring to the number of cells tested. Unpaired t test was used to compare the drug effects, conductance ratios, and δEPSC rise times between two populations. Paired t test was used to test the drug effects on conductance ratios and δEPSC rise time. We also used one sample t test to test the significance of drug-depressing effects. χ2 test was used to compare the percentage change of cells showing high δEPSC rise time under different intracellular solutions.
Materials.
d-APV, SR 95531 hydrobromide, QX-222 · Cl, and (R)-CPP were purchased from Tocris Cookson. QX-314 · Cl was purchased from Sigma-Aldrich or Alomone Labs. Strychnine was obtained from Sigma-Aldrich. Low melting point agarose was purchased from Invitrogen. TMR-SP was synthesized and purchased from AnaSpec and EAB-318 was provided by Wyeth Neuroscience. EAB-318 has an IC50 of 20, 80, and 3500 nm for NMDA receptors with GluN2A, GluN2B, and GluN2C, respectively (Sun et al., 2004).
Based on the previous observations that (R)-CPP has a Ki of 41, 270, 630, and 2000 nm for GluN2A, GluN2B, GluN2C, and GluN2D (Feng et al., 2004) with Hill coefficients for inhibition ranging from 0.6 to 1.4 (Porter et al., 1992; Sandberg et al., 1994; Laube et al., 1997) and that (R)-CPP has a slow dissociation rate of ∼1.1/s (Benveniste and Mayer, 1991), a brief increase of glutamate concentration in the synaptic cleft (assuming 1 mm, 1.2 ms decay time constant) during synaptic transmission (Clements et al., 1992) would be unable to facilitate a dissociation of (R)-CPP from NMDA receptors. Under this condition and assuming a hill coefficient of 0.9, (R)-CPP concentration of 200 nm should block ∼75% of the GluN2A-containing receptors and ∼30% of GluN2B-containing receptors. At an (R)-CPP concentration of 3 μm, it should block 97, 83, 68, and 40% of GluN2A, GluN2B, GluN2C, and GluN2D-containing receptors, respectively.
Results
Monosynaptic NMDA receptor-mediated EPSCs (NMDA EPSCs) evoked by primary afferent activation
To compare the NMDA receptors responsible for polysynaptic excitation of the NK1R+ neurons in lamina I to those at primary afferent synapses, we first determined NMDA receptor subtypes at the primary afferent, monosynaptic inputs onto NK1R+ neurons and, for comparison, NK1R− neurons. This was investigated using both pharmacology and ratio conductance (see Materials and Methods; Tong et al., 2008; Shiokawa et al., 2010). Neurons in these two populations in lamina I were visually identified then monosynaptic EPSCs were evoked and recorded using whole-cell patch-clamp (Fig. 1A,B, top). Upon superfusion of SR95531(10 μm) + strychnine (1 μm) + NBQX (5 μm) to block GABAA, glycine and AMPA receptors, respectively, NMDA receptor-mediated EPSCs were revealed (Fig. 1B, bottom). The NMDA receptor identity of these EPSCs was confirmed by observing a complete and reversible blockade in response to the nonselective NMDA receptor antagonist, d-APV (30 μm), as shown in Figure 1C (98.7 ± 0.3% blockade, n = 4). Because NMDA EPSCs reach their peaks ∼10 ms after AMPA EPSCs reach their peaks at the same synapses (McBain and Mayer, 1994), monosynaptic NMDA EPSC peaks were measured 10 ms after the monosynaptic AMPA EPSC peak (Fig. 1B).
Pharmacology of monosynaptic NMDA EPSCs
Our first approach to testing synaptic NMDA receptor subunit composition was to use subunit-selective antagonists on identified NK1R+ and NK1R− neurons. To remove Mg2+ blockade from the receptors, the cells were held at + 50 mV membrane potential while stimulating the dorsal roots. As shown in Figure 2, A and B, EAB-318 (200 nm), an antagonist for NMDA receptors with GluN2A/B subunit composition, depressed the peak amplitude of NMDA EPSCs by 88 ± 2% (n = 17, p < 0.05 for one sample t test) for NK1R+ neurons and 85 ± 7% (n = 4, p < 0.05 for one sample t test) for NK1R− neurons. This indicates that both NK1R+ and NK1R− neurons express mainly NMDA receptors with GluN2A/B subunits at primary afferent synapses. (R)-CPP (3 μm), another strong GluN2A/B antagonist, reduced the peak monosynaptic NMDA current by 95 ± 1% for NK1R+ neurons (n = 4, p < 0.05 for one sample t test). At this concentration, (R)-CPP blocks NMDA receptors with GluN2D subunits by only 40% (see Materials and Methods; Feng et al., 2004). Thus, consistent with our EAB-318 data, NMDA receptors at these synapses are predominately formed with GluN2A/B subunits.
Figure 2.

NMDA receptors with GluN2A subunits are the major NMDA receptors expressed at synapses between primary afferent fibers and lamina I neurons while those with GluN2B subunits are the minor forms. A, Representative traces showing the inhibiting effects of EAB-318 (200 nm) and (R)-CPP (200 nm) on monosynaptic NMDA EPSCs recorded from an NK1R+ neuron. Cells were held at + 50 mV. B, The summary of the monosynaptic NMDA receptor pharmacology. EAB-318 (200 nm) and (R)-CPP (3 μm), GluN2A/B-preferring antagonists, largely depressed the NMDA EPSC amplitude while ifenprodil (3 μm), an GluN2B-preferring antagonist, had moderate blocking effect for both NK1R+ and NK1R− neurons. (R)-CPP (200 nm), a GluN2A-preferring antagonist at lower concentration, significantly depressed the NMDA EPSC amplitude for both NK1R+ and NK1R− neurons. Asterisks indicate significantly different from control.
To specifically test for the contribution of GluN2B containing NMDA receptors at these synapses, ifenprodil (3 μm), a selective GluN2B antagonist, was used. At this concentration, ifenprodil blocks >90% of GluN1/GluN2B diheteromers, but blocks <20% of GluN1/GluN2A/GluN2B triheteromers and has virtually no effect on GluN1/GluN2A diheteromers (Hatton and Paoletti, 2005). Although ifenprodil has been reported to reduce high voltage-activated P/Q type calcium channels and affect synaptic transmission at high concentration, it has no significant effect on synaptic transmission at 5 μm (Church and Fletcher, 1995; Delaney et al., 2012). Ifenprodil was superfused onto the slices for 20–25 min. During ifenprodil application, dorsal horn neurons were continuously stimulated at 0.022–0.067 Hz to allow for activity-dependent blockade (Kew et al., 1996; Chizh et al., 2001a). Ifenprodil produced a modest inhibition of NMDA EPSCs for NK1R+ neurons (26 ± 4% inhibition, n = 12, p < 0.01 for one sample t test) and a significantly stronger inhibition for NK1R− neurons (42 ± 8% inhibition, n = 8, p < 0.01 for one sample t test, p < 0.05 when comparing the blocking effects of ifenprodil on the two neuronal populations using one tail unpaired t test) as shown in Figure 2B. Together with the strong blocking action of EAB-318, these data suggest that GluN2A is the major GluN2 subunit for NMDA receptors expressed at these synapses with GluN2B subunit as the minor form. There are fewer GluN2B-containing NMDA receptors expressed by NK1R+ neurons at primary afferent synapses compared withNK1R− neurons in lamina I (Fig. 2) and at focally stimulated synapses on interneurons in lamina II (Shiokawa et al., 2010). To further confirm that GluN2A is the dominant form at these synapses, (R)-CPP was also tested at a lower concentration (200 nm), which would be expected to block 75% of GluN2A type NMDA receptors and 30% of GluN2B NMDA receptors (see Materials and Methods). Under these conditions, peak NMDA EPSC amplitude was decreased by 64 ± 7% (n = 7, p < 0.05 for one sample t test) for NK1R+ neurons and 53 ± 15% (n = 4, p < 0.05 for one sample t test) for NK1R− neurons (Fig. 2B). This is consistent with the idea that NMDA receptors with GluN2A subunits are the major component contributing to monosynaptic NMDA EPSCs for both lamina I neuronal types.
NMDA receptors are also expressed at or near presynaptic terminals of primary afferent fibers within the superficial dorsal horn. While exogenous application of NMDA acts to depress presynaptic glutamate release (Bardoni et al., 2004; Zeng et al., 2006), endogenous glutamate may act on these presynaptic NMDA receptors to enhance release (Thomson et al., 2006). Thus, it cannot be ruled out that inhibition by EAB-318 and (R)-CPP of NMDA EPSCs was due to drug action on presynaptic NMDA receptors. To test this possibility, we blocked postsynaptic NMDA receptors by including an intracellular NMDA receptor antagonist, MK-801 (1 mm), in the patch pipette while recording from dorsal horn neurons (Berretta and Jones, 1996; Zeng et al., 2006). This inclusion effectively eliminated postsynaptic NMDA currents without affecting postsynaptic AMPA and presynaptic NMDA receptors (data not shown). d-APV (30 μm), a strong NMDA receptor antagonist relatively nonselective for NMDA receptors of different compositions, only slightly depressed the peak amplitude of the remaining AMPA receptor-mediated EPSCs by 13 ± 4% (n = 4, p = 0.06 for one sample t test), suggesting that most of the inhibition of NMDA EPSCs by subunit-selective antagonists was due to postsynaptic, not presynaptic inhibition.
Conductance ratio method for estimating NMDA receptor subunit composition
We independently determined synaptic NMDA receptor subunit composition using the conductance ratio technique elaborated in our earlier papers (Tong et al., 2008; Shiokawa et al., 2010). In brief, we use the subunit-dependent differences in voltage dependence of NMDA receptor-mediated conductance reported by Kuner and Schoepfer (1996) to estimate the relative contribution of GluN2A/B and GluN2C/D subunits to NMDA currents. The current–voltage (I–V) relationships of NMDA currents, both those activated by agonist application (Tong et al., 2008) and by synaptic activation (Shiokawa et al., 2010), were obtained and their conductance ratios, g(−90 mV)/g(MIC), calculated. The conductance ratio is the ratio of the conductance of NMDA receptors at −90 mV relative to the conductance of the current recorded at the membrane potential where the NMDA inward current was maximal. These measurements are illustrated in Figure 3B. Based on earlier studies using a heterologous expression system and recording in bath with 100 μm Mg2+, NMDA receptors containing GluN1/GluN2A or GluN1/GluN2B show g(−90 mV)/g(MIC) conductance ratios of 0.06–0.07. NMDA receptors containing GluN1/GluN2C or GluN1/GluN2D have ratios of 0.38–0.40 (Kuner and Schoepfer, 1996). Thus, smaller conductance ratios indicate expression of a higher percentage of GluN2A/B-containing, Mg2+-sensitive NMDA receptors. Using bath application of NMDA, we observed that NMDA receptors with high Mg2+ sensitivity (GluN2A/B) and low Mg2+ sensitivity (GluN2C/D) were expressed by NK1R+ projection neurons within the superficial dorsal horn lamina I (Tong et al., 2008). We also observed that in lamina II, all focally evoked synaptic NMDA currents were mediated by GluN2A/B only (Shiokawa et al., 2010). In the present study, synaptic NMDA receptors were selectively activated by endogenous synaptically released glutamate from primary afferent central terminals. NMDA EPSCs evoked by dorsal root stimulation were recorded at eight different holding potentials from −90 mV to +50 mV at 20 mV increments (Fig. 3A). The current–voltage relationship of the monosynaptic NMDA EPSCs for each cell was obtained by plotting the peak amplitudes of NMDA EPSCs as a function of the holding potential as shown in Figure 3B.
Figure 3.

NK1R+ neurons show higher mean conductance ratio of monosynaptic NMDA EPSCs compared with NMDA EPSCs from NK1R− neurons. A, Representative traces of monosynaptic NMDA EPSCs recorded from an NK1R− neuron at different membrane potentials (−90 mV to + 50 mV at 20 mV increment) using internal EGTA solution. B, I–V relationship of the NMDA EPSC traces from A. MIC, VMIC, g(−90 mV), and g(MIC) are illustrated (see Material and Method). C, Summary of the conductance ratios for NK1R+ and NK1R− neurons. Dots represent the conductance ratios of individual neurons. Both types of cells were patched using internal EGTA (10 mm) solution. The broken lines represent the conductance ratios of pure GluN2A/B type and GluN2C/D type NMDA receptors, respectively.
The conductance ratio for each cell, g(−90 mV)/g(MIC), was calculated from its current voltage (I–V) relationship (see Material and Methods). Figure 3B illustrates how the g(−90 mV) and g(MIC) were determined. Overall, the average conductance ratios of the monosynaptic NMDA EPSCs for NK1R+ and NK1R− neurons were 0.18 ± 0.04 (n = 20) and 0.08 ± 0.01 (n = 10), respectively (p < 0.05; Fig. 3C). The conductance ratio obtained from NK1R− neurons suggests that GluN2A/B type NMDA receptors are the major NMDA receptors at the primary afferent synapses.
The slowly rising currents recorded from NK1R+ neurons
While acquiring sets of NMDA EPSCs recorded as a function of membrane potential, it became apparent that NMDA EPSCs from some neurons had temporally different waveforms. This was seen as slower rise times and more rapid decay times at some of the negative holding potentials between −30 mV and −90 mV. As an example of this, Figure 4A, top, shows monosynaptic NMDA EPSC traces recorded from an NK1R+ neuron at different membrane potentials and the inset shows the current–voltage relationship. This NK1R+ neuron has a calculated conductance ratio of 0.25, a value that falls between the conductance ratios of pure GluN2A/B and GluN2C/D type NMDA receptors. However, it is visually apparent that at least two of the synaptic currents have different time courses compared with other currents in the series as shown in Figure 4A, top and middle. If these altered NMDA EPSC time courses are caused by the activation of a secondary current, such as Ca2+ or Na+-activated currents, it raises the possibility that the synaptic current amplitudes are not a pure reflection of NMDA currents. This in turn suggests that the calculated conductance ratios based on amplitude measurements may be compromised by these additional currents. In addition, these secondary currents at negative membrane potentials are expected to contribute to the total impact of excitatory drive at the synapse under study.
To identify neurons having variable EPSC rise times at negative membrane potentials, we calculated δEPSC rise time, an index that tracks the variability of NMDA EPSC rise times at negative membrane potentials compared with EPSC rise times at positive membrane potentials (see Material and Methods; Fig. 4A, bottom). Smaller δEPSC rise times indicate that rise times are similar for NMDA EPSCs at both negative and positive membrane potentials, while those with higher δEPSC rise times indicate variable NMDA EPSC rise times at negative membrane potentials.
NMDA receptors normally have a constant EPSC rise time at different membrane potentials (Billups et al., 2002). Accordingly, NMDA EPSCs recorded from NK1R− neurons (Fig. 4B) showed limited δEPSC rise times (0.27 ± 0.28 ms, n = 10), suggesting that there was no substantial variability in δEPSC rise times in NK1R− neurons. These NMDA EPSCs also had low conductance ratios (0.08 ± 0.01, n = 10), indicating that GluN2A/B type NMDA receptors are the major monosynaptic NMDA receptors expressed at the synapses between primary afferents and lamina I, NK1R− neurons, consistent with our earlier pharmacological results.
The strongly altered rise times of the NMDA EPSCs recorded from some of the NK1R+ neurons at negative membrane potentials suggested that additional currents were activated during EPSCs. Some channels, for example, Ca2+-activated potassium channels, K(Ca), may be activated by the elevated intracellular Ca2+ associated with neuronal activity and responsible for the generation of slowly rising currents at negative membrane potentials (Vergara et al., 1998; Alpert and Alford, 2013). To prevent activation of putative Ca2+-dependent currents, we used 40 mm BAPTA intracellular solution to rapidly chelate intracellular Ca2+. Figure 4C is an example of such a recording. The mean δEPSC rise time recorded from NK1R+ neurons with BAPTA (0.85 ± 0.13 ms, n = 51) was smaller than that recorded in EGTA (1.23 ± 0.15 ms, n = 20, p < 0.05) but was still much higher than that of NK1R− neurons using EGTA (0.27 ± 0.28 ms, n = 10), suggesting that the variability in rise time was reduced but not eliminated. Overall, fewer NK1R+ neurons had slowed and variable NMDA EPSC rise times when BAPTA was added to the recording solution compared with EGTA (62% vs 90% δEPSC > 0.7 ms, p < 0.05 for χ2 test).
To determine whether there was a relationship between the variable NMDA EPSC rise times and conductance ratio, we plotted conductance ratio as a function of δEPSC rise time (Fig. 4D). Figure 4D shows that when little or no rise time variability is present (δEPSC < 0.7 ms), conductance ratio is consistent with a predominantly GluN2A/B receptor subunit composition regardless of neuron population tested or Ca2+ chelator used. Conversely, when larger NMDA EPSC rise time variability is present (δEPSC > 0.7 ms), the conductance ratios tend to be widely variable for NK1R+ neurons and cannot be used to distinguish NMDA receptor subunits. This is apparent because some conductance ratio values are much greater than that predicted for even a pure GluN2C/D synapse (Fig. 4D). This suggests that conductance ratios are most informative when δEPSC is minimal. When δEPSC rise time is large, this suggests the presence of secondary currents that appear to be activated following NMDA receptor activation in a subset of the NK1R+ neurons.
The slowly rising monosynaptic NMDA EPSCs recorded from NK1R+ neurons were only observed at negative membrane potentials. Thus, it is possible that the currents were triggered by Ca2+ or Na+ influx through NMDA receptors. If this was the case, the δEPSC rise time should be reduced with decreased NMDA EPSC amplitude. To test this, EAB-318 (200 nm) was used to depress NMDA EPSC amplitude. Figure 5A shows an example of control NMDA EPSC traces (top) and their normalized forms (bottom) from an NK1R+ neuron. After EAB-318 application, the NMDA EPSCs were strongly blocked at all membrane potentials by ∼90% (Fig. 5B, top). When the remaining currents were normalized at their peak times, they had developed similar rise times (Fig. 5 compare A,B, bottom), suggesting the elimination of the secondary currents. As summarized in Figure 5C, EAB-318 largely reduced the δEPSC from 1.1 ± 0.3 to 0.2 ± 0.2 ms (n = 7, p < 0.05 for paired t test). We conclude that the synaptic inward currents recorded from NK1R+ neurons at negative membrane potentials were initiated by GluN2A/B type NMDA receptor activation, and that when altered kinetics occurred, it indicated activation of secondary current. After reduction of the NMDA EPSC amplitudes by EAB-318, the resulting EPSCs had slightly lower conductance ratios, changing from 0.23 ± 0.04 to 0.18 ± 0.02 (n = 7) ranging from 0.11 to 0.26 but still higher than pure GluN2A/B as shown in Figure 5C. This raises the possibility that the ∼10% of the current remaining following EAB-318 blockade was mediated by synaptic NMDA receptors, which included the GluN2D type in these neurons.
Figure 5.

The slowly rising component of some NMDA EPSCs were triggered by NMDA receptor activation. A, Top, Representative monosynaptic NMDA EPSCs recorded from an NK1R+ neuron upon C fiber activation. Bottom, Shows the normalized NMDA EPSCs of the same traces. B, Top, Indicates NMDA EPSCs were largely blocked by GluN2A/B-preferring antagonist EAB-318 (200 nm). Bottom, Shows normalized EPSCs from the same traces as with diminished amplitudes. Under this condition, NMDA EPSCs had similar EPSC rise times across all membrane potentials. The normalized traces are subjected to rolling averaging over 1 ms. C, Conductance ratios and δEPSC rise time changes from individual neurons are plotted. EAB-318 significantly lowered the δEPSC rise time of NMDA EPSCs from NK1R+ neurons, indicating decreased contribution from the current causing the slow rise of NMDA EPSCs.
To test if the slowly rising EPSCs were due to poor space clamp of the patched neurons, we calculated the reversal potentials of the NMDA EPSCs. The NMDA EPSCs recorded from NK1R+ and NK1R− neurons had average reversal potentials of 3.1 ± 0.8 mV (n = 71) and 1.3 ± 1.4 mV (n = 21), respectively, both close to the reversal potential of NMDA receptors ∼ 0 mV. This indicates that poor space clamp is not likely to be a major contributor to rise time variability of NMDA EPSCs.
Identification of synapses containing altered synaptic current rise times
The NMDA EPSCs recorded from NK1R+ neurons included those elicited by Aδ and C fiber activation. To identify which fiber type triggers synaptic currents with large δEPSC rise times, we classified the recorded monosynaptic NMDA EPSCs based on the thresholds of the fibers (see Material and Methods). NMDA EPSCs obtained following C fiber activation had significantly larger δEPSC rise times and conductance ratios than those obtained from Aδ fibers as shown in Figure 6 (δEPSC rise times: C fiber: 1.1 ± 0.1 ms, n = 56; Aδ fiber: 0.5 ± 0.2 ms, n = 14, p < 0.05 for unpaired t test; conductance ratios: C fiber: 0.22 ± 0.03, n = 56; Aδ fiber: 0.12 ± 0.01, n = 14, p < 0.05 for unpaired t test). These data suggest that the large δEPSC rise times are generated between primary C fibers and NK1R+ dorsal horn neurons.
Figure 6.

Monosynaptic NMDA ESPCs with slow rise times were only evoked between primary afferent C fibers and NK1R+ neurons. The same data as in Figure 4D, but classified according to Aδ and C fiber inputs. Aδ fiber-evoked NMDA EPSCs showed smaller δNMDA EPSC rise times and conductance ratios close to those of GluN2A/B type NMDA receptors, independent of the internal solution used. C fiber-evoked NMDA EPSCs showed a wide range of conductance ratios.
Polysynaptic NMDA receptors on NK1R+ and NK1R− lamina I neurons
We have investigated the NMDA receptors expressed within polysynaptic pathways from primary afferent fibers to lamina I dorsal horn neurons while synaptic inhibition was blocked. Polysynaptic NMDA responses were routinely elicited every 45–120 s in the presence of SR95531, strychnine and NBQX and in 100 μm Mg2+, and the area under the current curves were measured for analysis. When the interval between stimuli was reduced to 15 s, the responses showed progressive depression, consistent with the polysynaptic nature of the responses. Figure 7, A and B, are examples showing that both EAB-318 (200 nm) and (R)-CPP (3 μm) reversibly blocked the polysynaptic responses recorded from two NK1R+ neurons held at −70 mV. On average, EAB-318 (200 nm) and (R)-CPP (3 μm) blocked the polysynaptic responses recorded from NK1R+ neurons by 98 ± 1% (n = 6, p < 0.01 for one sample t test) and 99.8 ± 0.2% (n = 3, p < 0.01 for one sample t test), respectively, while they blocked those recorded from NK1R− neurons by 98.5 ± 0.6% (n = 6, p < 0.01) and 99.9 ± 0.1% (n = 4, p < 0.01), respectively (Fig. 7C). (R)-CPP (200 nm), a GluN2A-preferring antagonist at lower concentration, blocked the polysynaptic responses by 90 ± 5% (n = 4, p < 0.01) and 92 ± 4% (n = 3) for NK1R+ and NK1R− neurons, respectively. All these data suggest that GluN2A/B type NMDA receptors are dominant in mediating the polysynaptic responses from primary afferents to both lamina I NK1R+ and NK1R− neurons.
Figure 7.

NMDA receptors with GluN2A/B subunits are the major NMDA receptors expressed in polysynaptic pathways from primary afferent fibers to lamina I neurons. Representative traces showing the inhibiting effects of (A) EAB-318 (200 nm) and (B) (R)-CPP (3 μm) on polysynaptic NMDA EPSCs recorded from NK1R+ neurons following primary afferent activation in the presence of SR95531(10 μm), strychnine (1 μm), and NBQX (5 μm). Cells were held at −70 mV and stimulated at 0.033 Hz. C, Summary of the drug effects on the polysynaptic NMDA responses from NK1R+ and NK1R− neurons, respectively. Both EAB-318 and (R)-CPP (3 μm), GluN2A/B-preferring antagonists, largely depressed the NMDA EPSC amplitude while ifenprodil (3 μm), an GluN2B-preferring antagonist, had a wide range of blocking effects on the EPSCs from both NK1R+ and NK1R− neurons.
To determine the role of NMDA receptors with GluN2B subunits in the polysynaptic pathways, ifenprodil (3 μm) was tested on the evoked polysynaptic responses while dorsal horn neurons were continuously stimulated at low frequencies. Interestingly, ifenprodil depressed the polysynaptic responses to a highly variable degree as shown in Figure 7C. Figure 8A is an example showing the depressing effect of ifenprodil on the polysynaptic responses. The average inhibition by ifenprodil of polysynaptic activity recorded from NK1R+ neurons was 48 ± 5% (n = 25) and from NK1R− neurons was 48 ± 4% (n = 21). The average inhibition of polysynaptic activity from NK1+ neurons was greater than the effect of ifenprodil on monosynaptic inputs (26 ± 4% inhibition, p < 0.01) while no significant difference was observed for NK1R− neurons. However, the most prominent aspect of ifenprodil action on polysynaptic input to lamina I was its wide variability.
Figure 8.

Ifenprodil showed a wide range of blocking effects on the polysynaptic NMDA responses from primary afferents to individual dorsal horn neuron tested. A, An example showing the depressing effect of ifenprodil on a polysynaptic response recorded from an NK1R− neuron upon 100 μA electrical stimulation of dorsal root at a rate of 0.0167 Hz in the presence of SR95531(10 μm), strychnine (1 μm), and NBQX (5 μm). B, Summary of the effects of ifenprodil on pure Aβ, Aδ, or C fiber-evoked polysynaptic NMDA EPSCs recorded from NK1R+ and NK1R− neurons. NK1R+ neurons receiving Aβ fiber-driven polysynaptic input included neurons receiving only Aβ fiber-driven input (15/38, 39%), plus those receiving Aβ/Aδ (n = 2/38, 5%) and Aβ/C inputs (n = 4/38, 11%). Data from all of these neurons were pooled together to analyze the depressing effect of ifenprodil on Aβ fiber-evoked polysynaptic responses. NK1R− neurons receiving Aβ (n = 11/29, 38%) and Aβ/Aδ (n = 2/29, 7%; no Aβ/ C inputs) were also pooled together. The effect of ifenprodil on Aδ fiber-driven polysynaptic activity was obtained by combining the data from neurons receiving Aδ (8/38, 21% for NK1R+ and 13/29, 45% for NK1R− neurons) and Aδ/C fibers (4/38, 11% for NK1R+ and 2/29, 7% for NK1R− neurons). Only four pure C fiber-mediated polysynaptic EPSCs were obtained from NK1R+ neurons (11%) and one from NK1R− neurons (3%).
To test if the variable degree of depression by ifenprodil was associated with polysynaptic pathways driven by specific subtypes of primary afferent fibers, we classified the neurons under study into three groups (Aβ, Aδ, and C) according to the appropriate stimulation paradigm. We only tested the effect of ifenprodil on the polysynaptic responses evoked at the lowest intensity of primary afferent stimulation able to evoke polysynaptic activity. Ifenprodil depressed the Aβ-evoked NMDA polysynaptic EPSCs recorded from NK1R+ and NK1R− neurons by 45 ± 6% (n = 14) and 42 ± 5% (n = 8), respectively. Aδ fiber-evoked polysynaptic responses recorded from NK1R+ and NK1R− neurons were depressed by 36 ± 8% (n = 7) and 50 ± 6% (n = 12), respectively, upon application of ifenprodil. Very few neurons received only C fiber-driven polysynaptic inputs. In these instances, ifenprodil largely depressed the C fiber-evoked response recorded from NK1R+ neurons by 78 ± 8% (n = 4) and an NK1R− neuron by 75%, respectively (n = 1). Upon pooling the data from both NK1R+ and NK1R− neurons, it became clear that ifenprodil showed stronger mean inhibiting effects on C fiber-induced polysynaptic responses than those evoked by Aβ or Aδ fibers. In addition, in all cases of fiber type, the effect of ifenprodil was highly variable.
Discussion
We have shown that NMDA receptors with GluN2A/B subunits are the dominant synaptic NMDA receptors expressed within both monosynaptic and polysynaptic pathways from primary afferent inputs onto lamina I neurons. The widely variable effects of GluN2B antagonists on polysynaptic pathways leading to NK1R+ output neurons is most likely due to the impact of partial pharmacological block distributed within a multineuronal pathway as described below. These data suggest that selective targeting of GluN2B containing NMDA receptors may be able to depress polysynaptic activity following certain types of injury or inflammation without having the unwanted side effects of a general NMDA receptor antagonist or a selective GluN2A antagonist.
Identification of monosynaptic NMDA receptors expressed by dorsal horn neurons
In our experiments, the identification of GluN2A/B subunit containing NMDA receptors at monosynaptic sites is based on two major observations: the conductance ratio and the pharmacology of the NMDA EPSCs at primary afferent synapses. The g(−90 mV)/g(MIC) conductance ratios of the monosynaptic NMDA EPSCs recorded from both NK1R+ and NK1R− neurons were close to those of pure GluN2A/B type NMDA receptors, provided no additional currents altered the rising phase of the NMDA EPSCs at negative membrane potentials. Pharmacological evidence included the 85–90% blocking action of EAB-318 (200 nm) on monosynaptic NMDA EPSCs. Together with the results of (R)-CPP (3 μm), which blocked 95% of the monosynaptic NMDA response, we conclude that NMDA receptors with GluN2A/B subunits are the major synaptic NMDA receptors at all primary afferent synapses in lamina I.
Our earlier study on the functional NMDA receptors expressed by lamina I neurons showed expression of NMDA receptors with GluN2D subunits by NK1R+ neurons (Tong et al., 2008). In combination with our data showing GluN2A/B type NMDA receptors at synapses, we propose that NMDA receptors with GluN2D subunits may be expressed primarily extrasynaptically as originally shown (Momiyama, 2000, but see Hildebrand et al., 2014). Our data suggest that the contribution of GluN2D subunits at primary afferent synapses is minimal when activated following primary afferent stimulation.
In earlier studies, synaptic NMDA EPSCs recorded from lamina II neurons are dominated mainly by GluN2A subunits (Bardoni et al., 1998; Momiyama, 2000). At the primary afferent synapses onto lamina II neurons, ifenprodil (10 μm) was found to be an ineffective antagonist (Momiyama, 2000), contrasting with our data in which 3 μm ifenprodil blocked primary synaptic responses by an average of 30%. This difference could indicate that the primary afferent synapses onto lamina I and II neurons are different. However, it may be instead that in the earlier study, ifenprodil was not applied for long enough to allow this use dependent blocker to be effective (Kew et al., 1996).
NMDA receptors in polysynaptic pathways driven by primary afferent fibers
Synaptic NMDA receptors within polysynaptic pathways between primary afferent inputs and NK1R+ neurons in lamina I are important mediators of enhanced excitatory drive during disinhibition in the dorsal horn (Torsney and MacDermott, 2006). Although the exact pathway is still unclear, it is generally accepted that lamina I projection neurons, including NK1R+ neurons, receive some polysynaptic inputs initiated by deep dorsal horn neurons in lamina III/IV following low threshold primary afferent Aβ fiber activation under conditions of disinhibition (Torsney and MacDermott, 2006; Miraucourt et al., 2007). Ifenprodil had a wide range of potencies in blocking individual polysynaptic pathways ranging from 8 to 95%. This broad variability of ifenprodil blocking may be related to the requirement for each neuron in any polysynaptic pathway to come to threshold and fire action potentials to release transmitter and activate the next neuron in the pathway. A partial block of synaptic NMDA receptors under these conditions may prevent one of the neurons in the pathway from coming to threshold and, consequently, shutting down the rest of that polysynaptic pathway. Each polysynaptic pathway will be shut down in an all or none manner if any synapse in the pathway fails to bring the postsynaptic neuron to threshold. From this point of view, a partial block of a polysynaptic response suggests that the neuron under study normally receives input from multiple polysynaptic pathways.
Immunocytochemical and in situ studies demonstrate that GluN2B type NMDA receptors are expressed within the superficial dorsal horn (Yung, 1998; Boyce et al., 1999; Shibata et al., 1999; Nagy et al., 2004). The effectiveness of GluN2B antagonist in treating chronic pain suggests that these GluN2B type NMDA receptors within the dorsal horn are functional and activated under pathological conditions (Boyce et al., 1999; Chizh and Headley, 2005). However, there is little evidence of functional synaptic GluN2B NMDA receptors within the superficial dorsal horn in noninjured animals (Momiyama, 2000; Iwata et al., 2007) and some studies suggest that the effectiveness of GluN2B antagonists is due to their supraspinal effects (Chizh et al., 2001b; Nakazato et al., 2005). However, our study on polysynaptic responses driven by primary afferents shows that GluN2B type NMDA receptors are activated in the superficial dorsal horn following acute disinhibition, but play a more minor role in normal pain-related transmission, suggesting that these receptors may become more important when polysynaptic responses to NK1R+ neurons are unmasked (Moore et al., 2002; Scholz et al., 2005; Torsney and MacDermott, 2006). It is also possible that distinct nociceptive pathways such as NK1R+ neurons projecting to lateral parabrachial nucleus or periaqueductal gray (Al-Khater and Todd, 2009) have different levels of GluN2B expression, and it is this potential subpopulation distribution that is responsible for the variable blocking effects of ifenprodil.
Our studies of polysynaptic responses used pharmacological disinhibition to mimic the conditions of actual injury models. However, different injury models may involve different circuitry changes. For example, the percentage of neurons receiving Aδ fiber-evoked polysynaptic input jumps from 7 to 44% following spared nerve injury but hardly changes in sciatic nerve transection/chronic constriction injury model (Kohno et al., 2003). In a CFA inflammatory model, the number of neurons receiving monosynaptic C fiber input is unchanged while those receiving monosynaptic Aδ input are significantly increased (Torsney, 2011). Nevertheless, our data support the idea that GluN2B antagonist may be more effective in treating chronic pain conditions in which disinhibition is prominent, consistent with previous studies showing that GluN2B antagonist reduces nerve injury-induced mechanical allodynia (Boyce et al., 1999) where reduced inhibition in dorsal horn has been demonstrated (Moore et al., 2002).
The slow inward currents triggered following NMDA receptor activation
NMDA EPSCs recorded from 90% of the NK1R+ neurons using EGTA showed variable EPSC rise times (δEPSC > 0.7 ms). Although the decay time course of NMDA EPSCs is voltage dependent (D'Angelo et al., 1994), rise time is independent of membrane potential (Billups et al., 2002). Therefore we suggest that the greatly slowed NMDA EPSC rise times are due to the presence of additional current during the EPSC. Synaptic NMDA receptor activation was essential for triggering the slowly rising currents because δEPSC rise times were largely reduced upon partial block of NMDA EPSCs using EAB-318 superfusion. Fewer NK1R+ neurons had slowed NMDA EPSC rise times when BAPTA was in the recording solution. Because BAPTA has a faster Ca2+ buffering rate than EGTA, this outcome suggests there are at least two components causing the slowed rise of the EPSCs: one that can be blocked by BAPTA and is thus Ca2+ dependent, and one that is Ca2+ independent or inaccessible to intracellular BAPTA, and thus still present using BAPTA. These currents were absent in NMDA EPSCs recorded from NK1R− neurons or from Aδ fiber-induced monosynaptic responses regardless of how the intracellular Ca2+ was buffered, indicating that it is unique at synapses between C fibers and NK1R+ neurons.
The identity of the secondary current remains to be determined. NMDA EPSCs with slowed rise times were only triggered at negative membrane potentials. One candidate current is the Ca2+-activated potassium currents (K(Ca)). For example, Ca2+ entry through NMDA receptors could increasingly activate outward current. This would alter the rising inward synaptic current and cause an apparent slowing of time to peak. Similar coupling of NMDA receptor activation and K(Ca) has been reported in spinal cord ventral horn (Alpert and Alford, 2013). The slowly rising NMDA EPSCs recorded in the presence of BAPTA may also be triggered by Ca2+ influx, but at distal dendrites where BAPTA diffusion was limited. Other Ca2+-independent currents, for example, Na+-activated currents, cannot be ruled out (Kaczmarek, 2013). Further investigation of these currents may shed light on potential therapeutic agents to treat neuropathic pain because of their unique sites of generation.
Conclusions
In conclusion, we have investigated the expression of functional NMDA receptors at the monosynaptic and polysynaptic pathways in lamina I dorsal horn neurons following dorsal root stimulation. We provide strong evidence that NMDA receptors with GluN2B subunits provide a useful pharmacological target to suppress disinhibition-induced polysynaptic responses in the dorsal horn. Because disinhibition accompanies some injury and inflammation-induced chronic, non-nociceptive pain, our work supports a focus on GluN2B in controlling chronic pain.
Footnotes
This work was supported by a National Institutes of Health Grant NS029797. We thank Dr. Joachim Scholz for helpful comments on this manuscript.
The authors declare no competing financial interests. Publication costs were supported by Columbia Stem Cell Initiative.
References
- Al Ghamdi KS, Polgár E, Todd AJ. Soma size distinguishes projection neurons from neurokinin 1 receptor-expressing interneurons in lamina I of the rat lumbar spinal dorsal horn. Neuroscience. 2009;164:1794–1804. doi: 10.1016/j.neuroscience.2009.09.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Khater KM, Todd AJ. Collateral projections of neurons in laminae I, III, and IV of rat spinal cord to thalamus, periaqueductal gray matter, and lateral parabrachial area. J Comp Neurol. 2009;515:629–646. doi: 10.1002/cne.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert MH, Alford S. Synaptic NMDA receptor-dependent Ca(2)(+) entry drives membrane potential and Ca(2)(+) oscillations in spinal ventral horn neurons. PLoS One. 2013;8:e63154. doi: 10.1371/journal.pone.0063154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/S1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Magherini PC, MacDermott AB. NMDA EPSCs at glutamatergic synapses in the spinal cord dorsal horn of the postnatal rat. J Neurosci. 1998;18:6558–6567. doi: 10.1523/JNEUROSCI.18-16-06558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste M, Mayer ML. Structure-activity analysis of binding kinetics for NMDA receptor competitive antagonists: the influence of conformational restriction. Br J Pharmacol. 1991;104:207–221. doi: 10.1111/j.1476-5381.1991.tb12409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS. Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience. 1996;75:339–344. doi: 10.1016/0306-4522(96)00301-6. [DOI] [PubMed] [Google Scholar]
- Billups D, Liu YB, Birnstiel S, Slater NT. NMDA receptor-mediated currents in rat cerebellar granule and unipolar brush cells. J Neurophysiol. 2002;87:1948–1959. doi: 10.1152/jn.00599.2001. [DOI] [PubMed] [Google Scholar]
- Boyce S, Wyatt A, Webb JK, O'Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 1999;38:611–623. doi: 10.1016/S0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- Cheunsuang O, Morris R. Spinal lamina I neurons that express neurokinin 1 receptors: morphological analysis. Neuroscience. 2000;97:335–345. doi: 10.1016/S0306-4522(00)00035-X. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM. NMDA antagonists and neuropathic pain–multiple drug targets and multiple uses. Curr Pharm Des. 2005;11:2977–2994. doi: 10.2174/1381612054865082. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM, Tzschentke TM. NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharmacol Sci. 2001a;22:636–642. doi: 10.1016/S0165-6147(00)01863-0. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Reissmüller E, Schlütz H, Scheede M, Haase G, Englberger W. Supraspinal vs spinal sites of the antinociceptive action of the subtype-selective NMDA antagonist ifenprodil. Neuropharmacology. 2001b;40:212–220. doi: 10.1016/S0028-3908(00)00148-9. [DOI] [PubMed] [Google Scholar]
- Church J, Fletcher EJ. Blockade by sigma site ligands of high voltage-activated Ca2+ channels in rat and mouse cultured hippocampal pyramidal neurones. Br J Pharmacol. 1995;116:2801–2810. doi: 10.1111/j.1476-5381.1995.tb15929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Rossi P, Taglietti V. Voltage-dependent kinetics of N-methyl-D-aspartate synaptic currents in rat cerebellar granule cells. Eur J Neurosci. 1994;6:640–645. doi: 10.1111/j.1460-9568.1994.tb00309.x. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Power JM, Sah P. Ifenprodil reduces excitatory synaptic transmission by blocking presynaptic P/Q type calcium channels. J Neurophysiol. 2012;107:1571–1575. doi: 10.1152/jn.01066.2011. [DOI] [PubMed] [Google Scholar]
- Feng B, Tse HW, Skifter DA, Morley R, Jane DE, Monaghan DT. Structure-activity analysis of a novel NR2C/NR2D-preferring NMDA receptor antagonist: 1-(phenanthrene-2-carbonyl) piperazine-2,3-dicarboxylic acid. Br J Pharmacol. 2004;141:508–516. doi: 10.1038/sj.bjp.0705644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton CJ, Paoletti P. Modulation of triheteromeric NMDA receptors by N-terminal domain ligands. Neuron. 2005;46:261–274. doi: 10.1016/j.neuron.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Hildebrand ME, Pitcher GM, Harding EK, Li H, Beggs S, Salter MW. GluN2B and GluN2D NMDARs dominate synaptic responses in the adult spinal cord. Sci Rep. 2014;4:4094. doi: 10.1038/srep04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata H, Takasusuki T, Yamaguchi S, Hori Y. NMDA receptor 2B subunit-mediated synaptic transmission in the superficial dorsal horn of peripheral nerve-injured neuropathic mice. Brain Res. 2007;1135:92–101. doi: 10.1016/j.brainres.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK. Slack, slick and sodium-activated potassium channels. ISRN Neurosci. 2013;pii:354262. doi: 10.1155/2013/354262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol. 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Moore KA, Baba H, Woolf CJ. Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol. 2003;548:131–138. doi: 10.1113/jphysiol.2002.036186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrakakis C, MacDermott AB. Neurokinin receptor 1-expressing spinal cord neurons in lamina I and III/IV of postnatal rats receive inputs from capsaicin sensitive fibers. Neurosci Lett. 2003;352:121–124. doi: 10.1016/j.neulet.2003.08.042. [DOI] [PubMed] [Google Scholar]
- Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997;18:493–503. doi: 10.1016/S0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- Marshall GE, Shehab SA, Spike RC, Todd AJ. Neurokinin-1 receptors on lumbar spinothalamic neurons in the rat. Neuroscience. 1996;72:255–263. doi: 10.1016/0306-4522(95)00558-7. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- Miraucourt LS, Dallel R, Voisin DL. Glycine inhibitory dysfunction turns touch into pain through PKCgamma interneurons. PLoS One. 2007;2:e1116. doi: 10.1371/journal.pone.0001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama A. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. J Physiol. 2000;523:621–628. doi: 10.1111/j.1469-7793.2000.t01-1-00621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy GG, Watanabe M, Fukaya M, Todd AJ. Synaptic distribution of the NR1, NR2A and NR2B subunits of the N-methyl-d-aspartate receptor in the rat lumbar spinal cord revealed with an antigen-unmasking technique. Eur J Neurosci. 2004;20:3301–3312. doi: 10.1111/j.1460-9568.2004.03798.x. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Nakatsuka T, Ataka T, Kumamoto E, Tamaki T, Yoshimura M. Alteration in synaptic inputs through C-afferent fibers to substantia gelatinosa neurons of the rat spinal dorsal horn during postnatal development. Neuroscience. 2000;99:549–556. doi: 10.1016/S0306-4522(00)00224-4. [DOI] [PubMed] [Google Scholar]
- Nakazato E, Kato A, Watanabe S. Brain but not spinal NR2B receptor is responsible for the anti-allodynic effect of an NR2B subunit-selective antagonist CP-101,606 in a rat chronic constriction injury model. Pharmacology. 2005;73:8–14. doi: 10.1159/000081069. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Porter RH, Cowburn RF, Alasuzoff I, Briggs RS, Roberts PJ. Heterogeneity of NMDA receptors labelled with [3H]3-((+-)-2-carboxypiperazin-4-yl) propyl-1-phosphonic acid ([3H]CPP): receptor status in Alzheimer's disease brains. Eur J Pharmacol. 1992;225:195–201. doi: 10.1016/0922-4106(92)90020-V. [DOI] [PubMed] [Google Scholar]
- Sandberg MP, Ask AL, Fowler CJ. Characterization of [3H]CGS 19755 binding sites in the rat spinal cord. Neurochem Int. 1994;24:329–337. doi: 10.1016/0197-0186(94)90111-2. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Scholz J, Broom DC, Youn DH, Mills CD, Kohno T, Suter MR, Moore KA, Decosterd I, Coggeshall RE, Woolf CJ. Blocking caspase activity prevents transsynaptic neuronal apoptosis and the loss of inhibition in lamina II of the dorsal horn after peripheral nerve injury. J Neurosci. 2005;25:7317–7323. doi: 10.1523/JNEUROSCI.1526-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Watanabe M, Ichikawa R, Inoue Y, Koyanagi T. Different expressions of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid and N-methyl-D-aspartate receptor subunit mRNAs between visceromotor and somatomotor neurons of the rat lumbosacral spinal cord. J Comp Neurol. 1999;404:172–182. doi: 10.1002/(SICI)1096-9861(19990208)404:2<172::AID-CNE3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Shiokawa H, Kaftan EJ, MacDermott AB, Tong CK. NR2 subunits and NMDA receptors on lamina II inhibitory and excitatory interneurons of the mouse dorsal horn. Mol Pain. 2010;6:26. doi: 10.1186/1744-8069-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol. 1994;72:169–179. doi: 10.1152/jn.1994.72.1.169. [DOI] [PubMed] [Google Scholar]
- Sun L, Chiu D, Kowal D, Simon R, Smeyne M, Zukin RS, Olney J, Baudy R, Lin S. Characterization of two novel N-methyl-D-aspartate antagonists: EAA-090 (2-[8,9-dioxo-2,6-diazabicyclo [5.2.0]non-1(7)-en2-yl]ethylphosphonic acid) and EAB-318 (R-alpha-amino-5-chloro-1-(phosphonomethyl)-1H-benzimidazole-2-propanoic acid hydrochloride) J Pharmacol Exp Ther. 2004;310:563–570. doi: 10.1124/jpet.104.066092. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br J Pharmacol. 1997;122:809–812. doi: 10.1038/sj.bjp.0701445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson LM, Zeng J, Terman GW. Differential effect of glutamate transporter inhibition on EPSCs in the morphine naive and morphine tolerant neonatal spinal cord slice. Neurosci Lett. 2006;407:64–69. doi: 10.1016/j.neulet.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Spike RC, Polgár E. A quantitative study of neurons which express neurokinin-1 or somatostatin sst2a receptor in rat spinal dorsal horn. Neuroscience. 1998;85:459–473. doi: 10.1016/S0306-4522(97)00669-6. [DOI] [PubMed] [Google Scholar]
- Todd AJ, McGill MM, Shehab SA. Neurokinin 1 receptor expression by neurons in laminae I, III and IV of the rat spinal dorsal horn that project to the brainstem. Eur J Neurosci. 2000;12:689–700. doi: 10.1046/j.1460-9568.2000.00950.x. [DOI] [PubMed] [Google Scholar]
- Tong CK, MacDermott AB. Both Ca2+-permeable and -impermeable AMPA receptors contribute to primary synaptic drive onto rat dorsal horn neurons. J Physiol. 2006;575:133–144. doi: 10.1113/jphysiol.2006.110072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CK, Kaftan EJ, Macdermott AB. Functional identification of NR2 subunits contributing to NMDA receptors on substance P receptor-expressing dorsal horn neurons. Mol Pain. 2008;4:44. doi: 10.1186/1744-8069-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsney C. Inflammatory pain unmasks heterosynaptic facilitation in lamina I neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2011;31:5158–5168. doi: 10.1523/JNEUROSCI.6241-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signalling in superficial NK1 receptor expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/S0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Yung KK. Localization of glutamate receptors in dorsal horn of rat spinal cord. Neuroreport. 1998;9:1639–1644. doi: 10.1097/00001756-199805110-00069. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU. The glycinergic control of spinal pain processing. Cell Mol Life Sci. 2005;62:2027–2035. doi: 10.1007/s00018-005-5107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeilhofer HU, Zeilhofer UB. Spinal dis-inhibition in inflammatory pain. Neurosci Lett. 2008;437:170–174. doi: 10.1016/j.neulet.2008.03.056. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]