

Abstract
Oxidative stress plays a pathological role in the development of various diseases including diabetes, atherosclerosis, or cancer. Systemic oxidative stress results from an imbalance between oxidants derivatives production and antioxidants defenses. Reactive oxygen species (ROS) are generally considered to be detrimental for health. However, evidences have been provided that they can act as second messengers in adaptative responses to stress. Obesity represents a major risk factor for deleterious associated pathologies such as type 2 diabetes, liver, and coronary heart diseases. Many evidences regarding obesity-induced oxidative stress accumulated over the past few years based on established correlations of biomarkers or end-products of free-radical-mediated oxidative stress with body mass index. The hypothesis that oxidative stress plays a significant role in the development of metabolic disorders, especially insulin-resistance state, is supported by several studies where treatments reducing ROS production reverse metabolic alterations, notably through improvement of insulin sensitivity, hyperlipidemia, or hepatic steatosis. In this review, we will develop the mechanistic links between oxidative stress generated by adipose tissue in the context of obesity and its impact on metabolic complications development. We will also attempt to discuss potential therapeutic approaches targeting obesity-associated oxidative stress in order to prevent associated-metabolic complications.
1. Introduction
The prevalence of obesity over the past years has been in constant progression leading the World Health Organization to consider it as an epidemic pathology. Obesity is defined as an excessive accumulation of body fat mass to the extent that individual's health will be negatively affected. Indeed, obesity is considered as a top risk factor to develop deleterious associated pathologies as type 2 diabetes, liver, and coronary heart diseases.
White adipose tissue (WAT) constitutes the main energy supply in the body, which will ensure whole-body energy homeostasis by either storing excess energy in the form of lipids (namely, triacylglycerol—TG) or mobilizing fatty acids (FA) according to metabolic needs. However, the view of WAT, long-time considered as an inert storage depot, has considerably evolved since the identification of the adipocyte-secreted hormone leptin acting at the central level to control food intake [1]. This major discovery was followed by the characterization of many molecules secreted by adipocytes, called adipokines, which has revealed the endocrine potential of this tissue and its property to communicate with others [2]. Upon nutrient overloading, WAT will expand in considerable proportions through the ability of adipocyte to mechanistically adapt to increasing in their size. Thus, WAT can account for more than 50% of total body weight in obese subjects. This adipose plasticity is also supported by the constant renewal of adipocytes based on differentiation of adipocytes precursors present in WAT, which has been estimated to be about 10% per year [3]. Adipocyte represents the functional cell type specialized in lipid storage in WAT. The latter also regroups other cell types including precursors, immune cells, and endothelial cells. The rapid expansion of WAT in response to nutrient overload is signed by a profound remodeling of fat affecting all cellular components of WAT. This remodeling especially is characterized by an increased immune cells infiltration [4, 5]. Moreover, cellular stresses concomitant to fat overloading such as metabolic dysfunctions, inflammation, hypoxia, reticulum endoplasmic stress, and hypoxia will contribute to attracting and retaining inflammatory cells within the WAT (for review, [6]). The development of a chronic low-grade inflammatory state has been shown to play a central role in the development of metabolic complications associated obesity, since it has been linked to the development of insulin resistance, endothelial and microvascular dysfunctions [7]. Similar to other inflammatory processes such as microbial infection, WAT inflammation is intrinsically linked to oxidative stress.
Systemic oxidative stress is part of the numerous biological alterations reported during chronic obesity [8]. Evidences regarding obesity-induced oxidative stress are derived from several clinical studies, which have established correlations of biomarkers, or end-products of free radicals-mediated oxidative stress (lipid peroxidation or protein carbonylation products) with body mass index (BMI) [9, 10]. In contrast, an inverse relationship exists between body fat, visceral obesity, and antioxidant defense markers in obese individuals [11]. The hypothesis that oxidative stress is causative in the development of metabolic disorders, especially insulin-resistant state, has been supported by different studies where treatments reducing ROS production improve insulin sensitivity, hyperlipidemia, and hepatic steatosis [12–14]. Hypertrophied adipocytes have been reported as a significant source of ROS, promoting WAT dysfunction notably by altering adipokine production [12]. Furthermore, oxidative stress-associated obesity has also been shown to alter the function of many cell types or tissues (including vascular endothelial cells, myocytes, or pancreatic-β-cells) leading to consider oxidative stress as a contributor in obesity-related metabolic diseases.
In this review, we will expose mechanistic links between oxidative stress generated by adipose tissue in the context of obesity and its impact on metabolic complications development. We will notably focus on how WAT-associated oxidative stress might impact adipose tissue metabolism leading to WAT dysfunction. Dysregulated systemic metabolic parameters will be moreover reviewed as contributors and amplifiers of obesity-associated oxidative stress. Finally, we will discuss strategies of potential therapeutic approaches to lower obesity-associated oxidative stress and associated-metabolic complications.
2. White Adipose Tissue as a Significant Source of Reactive Oxygen Species Production during Obesity
Oxidative stress results from an imbalance between the production of ROS and biological systems' ability to detoxify the reactive intermediates or to repair the resulting damages, which can impact all components of the cell, including proteins, lipids, and DNA. Examples of ROS include superoxide anions (O2 •−), hydrogen peroxide (H2O2), and hydroxyl radical (OH•). Furthermore, reactive nitrogen species (RNS) might form by combination of nitric oxide (NO•) with O2 •− to form peroxynitrite (ONOO−) and act together with ROS to damage cells, causing nitrosative stress.
2.1. ROS Sources in Fat Cells
Different sources of intracellular ROS detailed below might participate in ROS generation by adipocytes (Figure 1).
Figure 1.
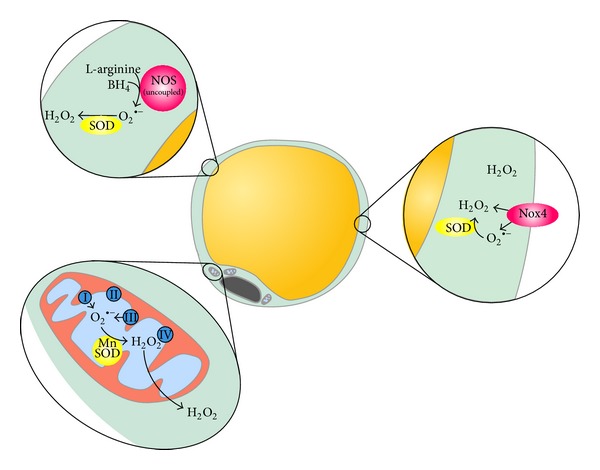
Different intracellular ROS sources participate in ROS generation by adipocytes. Mitochondria, as the core machinery for energy production through oxidative phosphorylation, is considered as a main source of O2 •−, predominantly produced at complexes I and III. Superoxide anions will be further converted to H2O2 by mitochondrial manganese superoxide dismutase (MnSOD). ROS can also be produced by NADPH oxidases enzymes, in which Nox4 is the main isoform in adipocytes. This isoform presents the particularity to primarily generate H2O2, whereas other NADPH oxidases would generate O2 •− later converted to H2O2 by endogenous SOD. Nitric oxide synthase, whose eNOS and iNOS isoforms are abundantly expressed by adipocytes, might represent another source of ROS since they can be uncoupled to produce O2 •− in the absence of sufficient amounts of substrates.
2.1.1. ROS-Derived Mitochondria
ROS can derive from mitochondria. It is currently accepted that between 0.15% and 2% of O2 consumption is incompletely metabolized and results in O2 •− production, predominantly at complexes I and III [15]. Increase in nutrient uptake into adipocytes will consequently increase mitochondrial substrate load, which by increasing electron transport chain (ETC) activity will result in enhanced O2 •− by-products [16]. However, the mitochondrial origin of ROS in adipocytes is still a matter of debate. Indeed, mitochondrial metabolic flux as a basis for increased adipocyte ROS production is controversial in regard to substrate utilization in adipocytes. Free fatty acids (FFA) entering adipocytes are rapidly and predominantly converted to fatty acyl-CoA to be ultimately stored as TGs without significant mitochondrial oxidation [17]. Moreover, excess energy derived from glucose carbons undergoing glycolysis will rather enter the lipogenic pathway than the Krebs cycle, to contribute to lipid droplets fat storage [18]. In agreement with the latter, excess of glucose or palmitate does not increase mitochondrial oxidative phosphorylation or beta-oxidation in 3T3-L1 adipocytes [19].
2.1.2. NADPH Oxidases
Various enzymes inside the cells can also produce ROS. Particularly, the family of NAPDH oxidases (NOX) is considered to be an important source of ROS generation [20]. NOX are membrane-bound enzyme complexes that transfer electrons from NADPH to oxygen. Generated O2 •− are further converted into H2O2, longer-lived membrane-permeable ROS, predominantly by superoxide dismutase (SOD). Seven isoforms of the catalytic subunit of NADPH oxidase have been identified in mammalian cells, known as Nox1 to Nox5, Duox1 and Duox2. NOX4, which does not require other activators and has sustained activity, is the major isoform expressed in adipocytes and is increased in fat cells exposed to excess glucose or palmitate [21]. In addition, silencing of NOX4 in 3T3-L1 adipocytes inhibits palmitate- and glucose-stimulated ROS generation underlying the importance of nonmitochondrial sources of ROS in adipocytes [19]. The molecular mechanism underlying activation of NOX by FFA has been linked to stimulated-diacylglycerol synthesis, which, connected to PKC activation, leads to the activation of NAPDH oxidase [22]. An intimate cross talk between NAPDH and mitochondria nevertheless exists since mitochondria constitute a target for ROS produced by NOX but also a significant source of ROS, which under certain conditions may stimulate NADPH oxidases [23]. It has been in fact demonstrated that mitochondria-targeted antioxidants break this vicious cycle, inhibiting ROS production by mitochondria and reducing NADPH oxidase activity [24].
2.1.3. eNOS Uncoupling
Another enzyme, which may be an important contributor to ROS generation, is nitric oxide synthase (NOS) since O2 •− can react avidly with vascular NO• to form ONOO−. Under certain pathological conditions, for instance, when the availability of NOS cofactors tetrahydrobiopterin (BH4) or substrate L-arginine is too low, the enzymatic activity of NOS can be uncoupled to produce O2 •− rather than NO [25, 26]. The transformation of eNOS from a protective enzyme to a contributor to oxidative stress has been well described in endothelial cells and observed in different models of cardiovascular diseases, including patients with cardiovascular risk factors [27]. In many cases, supplementation with BH4 is sufficient to correct eNOS dysfunction in animal models and patients [28, 29]. Endothelial NO synthase- (eNOS-) and inducible NO synthase- (iNOS-) dependent NO are abundant in adipocytes. iNOS expression has been shown to be increased in WAT derived from diet-induced or genetic models of obesity [30]. Similarly, both eNOS and iNOS are expressed at higher levels in WAT from obese patients compared to lean controls [31, 32]. Despite important role attributed to NO in many adipocyte metabolic functions [32], fat cell NO metabolism remains incompletely understood and contribution of eNOS uncoupling to ROS production has not been yet clearly established.
2.2. Enhanced ROS Production of Obese Adipose Tissues
2.2.1. Elevated ROS Production by Hypertrophied Adipocytes
To determine whether fat accumulation is primarily involved in increased oxidative stress-associated obesity, Furukawa et al. have analyzed lipid peroxidation and H2O2 production in adipose tissues from obese KKAy mice [14]. They have found a specific elevation of lipid peroxidation and H2O2 production in WAT from these obese rodents, but not in liver, muscles, or aorta. Increased oxidative stress has been further described in WAT of other models of obesity such as high-fat diet [33] or ob/ob mice [12]. In agreement, adipocytes isolated from mice fed with high-fat diet [34] or exposed to nutrient excess in vivo [35] display significantly elevated ROS in vitro. Whereas it appears that fat accumulation parallels ROS production, as confirmed by increased ROS production during adipocyte 3T3-L1 conversion [36], contribution of the different pathways involved in this elevation of ROS is still debated. Importance of NAPDH-derived ROS are suggested by selective induction of expression of NAPDH subunits in WAT from KKAy obese mice together with the ability of NADPH inhibitors to reduce ROS production in isolated obese adipocytes [14, 34]. However, exposition of in vitro 3T3-L1 adipocytes to high-levels of glucose or FFA has been shown to increase mitochondrial ROS production [37, 38]. Accordingly, hyperglycemia-induced ROS production in 3T3-L1 adipocytes can significantly be reduced with pharmacological agents lowering the mitochondrial membrane potential, subsequent to overexpression of uncoupling protein-1 or SOD also suggesting a contribution of mitochondrial ROS [35]. Moreover, a number of studies have provided direct evidence of mitochondrial dysfunction associated with obesity, related to increased ROS production, supporting the development of insulin resistance [39]. Also aminooxidases, as a source of H2O2, are highly expressed in adipose tissues; they are unlikely to contribute to elevation of adipocyte production since they display reduced activities in WAT from obese subjects [40].
2.2.2. Decreased Antioxidant Defenses in Obese Adipose Tissues
To prevent free radical damages, the organism has developed antioxidant defenses largely based on antioxidant enzymes able to scavenge ROS. SODs are responsible for the reduction of O2 •− to H2O2 and multiple enzymes will remove H2O2 including peroxiredoxins (PRXs), glutathione peroxidases (GPXs), and catalase (CAT). These antioxidant enzymes systems are active in fat cells isolated from rats, although their activities are lower than in liver [41]. Their activities might moreover depend on fat pad localization inasmuch as redox status differences have been reported between epididymal and inguinal fat pads from obese Zucker rats [42]. Silencing of the antioxidant enzyme glutathione-S-transferase (GSTA4) in cultured adipocytes or its invalidation in mice both results in increased ROS production and mitochondrial dysfunction [33]. These results highlight the importance of ROS scavenging processes in fat cells. Despite the presence of active antioxidant system, obese state is associated with a decrease in antioxidant defenses. For instance, a decrease in expression and activities of antioxidant enzymes such as SOD, GPX, or catalase have been reported in WAT from obese mice models [14]. Similarly, expression of the antioxidant enzyme GST-A4 in humans has been found considerably reduced in obese insulin-resistant subjects [33].
Additionally, adipose tissue represents also a preferential storage site for natural antioxidants compounds, as liposoluble vitamins (e.g., vitamins A and E) or carotenoids [43]. However, obese people present generally a relatively low total antioxidant status (TAS) characterized by lower levels of serum vitamins A, E, C, and β-carotene as well as glutathione [44, 45]. Although adipose tissue storage generally equilibrates with circulating levels of molecules [46, 47], fat can also act as sink concentrating vitamins in adipocyte lipid droplets therefore limiting their bioavailability [41, 48].
Altogether, sluggish behavior of the ROS scavenging system might contribute to exacerbating the existing high levels of ROS produced by hypertrophied adipocytes. In addition, the topology of the ROS production has also to be considered. Depending on detoxifying enzymes altered, they can modify ROS species and consequently downstream redox signaling.
3. ROS-Derived Adipocytes as Potential Instigator of Adipose Tissue Dysfunction
Numerous experimental findings favor the notion that ROS may act as second messengers triggering dedicated adaptative cellular machinery that increase the resistance of organism to stress [49]. Among ROS species, O2 •− H2O2 or NO• can act as signaling molecules. For instance, in the field of aging, longevity-promoting interventions such as caloric restriction and physical activity both increase mitochondrial metabolism [50]. This activation promotes mitochondrial formation of ROS signals, which act as mild stressors, and drive an adaptative response through the induction of specific oxidative stress-sensitive pathways resulting in lifespan extension and health promotion [49, 51, 52]. In recent years, it has therefore become apparent that low levels of ROS may be required for normal cellular functioning and intracellular signaling. However, this does not negate the involvement of chronic ROS in the progression of pathologies such as diabetes. This has led to apply the theory of hormesis, concept that describes the drug action of low-dose stimulation and high-dose inhibition [50]. In addition, ROS diffusion potential is also likely to determine metabolic effects. ROS are described as short-life time species, with limited diffusion capacities especially for O2 •− and H2O2, which mainly remain in aqueous phases [53]. OH• is nevertheless able to penetrate deeply in phospholipid bilayers membranes but its limited lifetime (around few nanoseconds) will considerably reduce its diffusion [54]. Therefore, depending on the degree of ROS generation, typology, and distribution, enhanced production of ROS by hypertrophied adipocytes is likely to affect metabolic pathways of cellular components of WAT as described below.
3.1. Oxidative Stress and Insulin Response
In adipocytes, early reports have highlighted that insulin may elicit H2O2 production in adipocytes [55]. This transient production in response to insulin involved NADPH oxidase, which produces O2 •− that spontaneously dismutates to H2O2 [56]. A number of insulin effects on adipocytes may be mimicked by H2O2 when added in physiological concentrations on fat cells [57, 58]. For instance, H2O2 enhances glucose transporters GLUT translocation and consequently glucose uptake [59] as well as lipid synthesis [55] whereas it inhibits stimulated lipolysis [60]. The rapid formation of H2O2 in insulin-treated adipocytes 3T3-L1 adipocytes causes the oxidative inhibition of protein-tyrosine phosphatases and enhances the tyrosine phosphorylation of proteins in the early insulin action cascade [61]. These data point out a novel regulatory mechanism complementing the early steps in insulin amplification signaling. Accordingly, mice invalidated for GPX1 have enhanced insulin sensitivity, effect that can be reversed by the antioxidant N-acetylcysteine [62]. This study therefore points out that subtle increase in physiological ROS production, particularly in muscle, might reflect the contribution of ROS to insulin sensitivity early in the development of insulin resistance, prior to the onset of hyperglycemia/hyperlipidemia and frank diabetes.
Whereas transient ROS produced by physiological stimuli such as insulin may be beneficial, sustained ROS generation impairs insulin response and might therefore initiate WAT dysfunction (Figure 2). Indeed, exposition of 3T3-L1 adipocytes to micromolar concentrations of H2O2, in response to glucose oxidase, alters the expression of GLUT transporters associated and reduced insulin-stimulated transport of glucose and lipogenesis [63]. Recent data confirm that chronic elevation of intracellular ROS levels in adipocytes subsequent to mitochondrial dysfunction results in insulin resistance through attenuation of insulin signaling [64]. Furthermore, in vivo evidence for the involvement of obesity-induced ROS in promoting insulin resistance has been provided using obese rodent models [12, 65]. In particular, the antioxidant MnTBAP improves insulin sensitivity in ob/ob mice that are hyperglycemic and insulin-resistant [12]. Additionally, inhibition of muscle mitochondrial ROS generation in high-fat fed rodents prevents hyperglycemia and insulin resistance [65].
Figure 2.
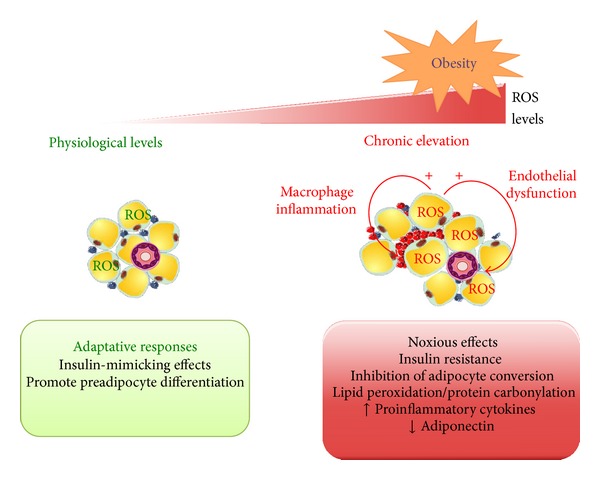
Adaptative or deleterious metabolic responses depending on ROS levels on adipose tissue metabolism. Depending on ROS intracellular levels, adipocytes will trigger different metabolic responses. Physiological levels of ROS, maintained by efficient detoxification system, are able to induce insulin-mimicking effects of H2O2 and to favor adipogenesis, which can be seen as an adaptative response in order to cope with nutrient overloading. In contrast, excessive or inappropriate redox balance will lead to considerable raise in intracellular ROS which will have detrimental effects particularly by altering insulin signaling, adipokine secretion, and adipocyte precursors conversion. Lipid peroxidation, protein carbonylation, and proinflammatory cytokines secretion are increased in obese adipocytes following exposure to high levels of ROS. Moreover, enhancement of ROS production by hypertrophied adipocytes will affect neighboring environment, namely, immune cells infiltrated in WAT or endothelial cells in close vicinity of perivascular adipose tissue. Macrophages are schematically represented as blue (M2 type) or red cells (M1) within adipocytes.
Lipid aldehydes generated following lipid peroxidation of polyunsaturated acyl chains by O2 •− have also been shown to interfere with insulin signaling. Such reactive lipid aldehydes, including trans-4-hydroxy-2-nonenal (4-HNE), a major oxidation product containing n-6 polyunsaturated acyl groups, can covalently carbonylate proteins leading to their loss of their function. Accumulating evidences suggest important roles of these end-products in the development of insulin resistance as well as other metabolic complications associated with obesity [66]. HNE are increased in the blood and muscle tissue of obese subjects compared to normal weight subjects [67]. Of interest, expression of HNE adducts of many proteins in subcutaneous abdominal depots has been found to be higher in type 2 diabetes than in similarly obese, nondiabetic controls [66]. Muscle protein HNE adducts have been directly correlated to the severity of insulin resistance in muscle tissues [68]. They can impair skeletal muscle sensitivity by inducing oxidative damage on key components of insulin signaling [69] and inhibit glucose-induced insulin secretion [70]. Similarly, deleterious effects of 4-HNE have been reported in adipocytes. Indeed, 4-HNE impair IRS function [71], decrease adiponectin secretion [72], alter protein function following carbonylation [73], and increase lipolysis [74]. Additionally, 4-HNE might link oxidative stress and chronic inflammation in adipocytes since addition of peroxidation products to 3T3-L1 adipose cells induces cyclooxygenase-2 (COX-2) expression through p38MAPK activation [75].
Finally, exposition of adipocytes to high ROS levels suppresses adiponectin expression and secretion [14, 64], an adipokine which displays insulin-sensitizing, antiatherogenic, and anti-inflammatory properties (for review, [76]). Accordingly, human serum adiponectin levels have been inversely correlated with systemic oxidative stress [14, 77]. Regulation of adiponectin production by oxidative stress has been demonstrated in vivo in obese mice treated with the NADPH inhibitor apocynin, which reduces ROS production [14]. Such treatment led to improvement of insulin resistance and restoration of adiponectin production of obese rodents. This protective role of adiponectin against oxidative stress likely involved inhibition of inducible iNOS and the suppression of the expression of the gp91phox subunit of NAPDH oxidase as demonstrated in experiments using myocardial infarction reperfusion model [78]. While similar effects were observed in endothelial cells [79], such a protective effect has not yet been evidenced in adipocytes. Systemic decreased levels of adiponectin will moreover contribute to maintain high levels of oxidative stress within WAT.
3.2. Adipose Tissue Redox Status and Adipocyte Differentiation
Fat mass expansion occurs via two processes concomitantly occurring during WAT expansion: hypertrophia (by increasing size of fat cells) and hyperplasia (by increasing numbers of fat cells linked to differentiation of adipocyte precursors). Many studies have demonstrated a close relationship between ROS and adipogenesis. Adipocyte differentiation of 3T3-L1 or rat bone-marrow derived-mesenchymal stem cells (MSC) is accompanied by a marked increase in ROS production dependent on NAPDH oxidases. In agreement, silencing of NOX4 isoform of NAPDH oxidases inhibits insulin-dependent adipocyte differentiation [80] whereas lipid storage is favored following NOX4 overexpression or exogenous application of H2O2 [81]. Conversely, scavenging of ROS production inhibits adipogenesis process [14, 82]. However, controversial data have been reported regarding ROS effects on preadipocyte recruitment and differentiation. Proadipogenic effects of ROS have been reported following physiological elevation of mitochondrial generated ROS in human MSC [83]. The latter could be related to the promotion of early mitotic clonal expansion phase of adipogenesis essential for later adipocyte conversion by ROS [36]. Conversely, antiadipogenic ROS effects have been observed when drugs increasing mitochondrial ROS production are applied to 3T3-FF42A preadipocytes, through reduction of C/EBP (CAAT/enhanced binding protein) transcription factor DNA-binding activity [84]. One hypothesis, which might reconcile such conflicting results, is based on differential effects depending on ROS intracellular levels. On one hand, “physiological” tightly regulated redox changes drive beneficial adaptative responses. On the other hand increased and uncontrolled production of free radicals might result in “noxious effects” in WAT (Figure 2). In this view, an initial burst in ROS production will switch committed preadipocytes from proliferation to differentiation. This switch can be seen as an adaptative response to nutrient overload by accelerating differentiation of adjacent preadipocytes. In contrast, excessive or inappropriate redox balance will detrimentally affect adipocyte precursors conversion therefore limiting “adipose tissue expandability” [85] and favoring adipocyte hypertrophy. Hypertrophic obesity is commonly accepted as more deleterious and associated with insulin-resistant state [86, 87]. Hypertrophied adipocytes will indeed impair adipose tissue function by inducing local inflammation, mechanical stress, and altered metabolism due to dysregulated adipokine secretion. Thus, impairment of adipocyte conversion instigated by high levels of ROS might consequently favor adipose tissue inflammation and other related metabolic dysfunctions [66].
3.3. Intimate Cross Talk between Oxidative Stress and Inflammation in WAT
Direct evidences from in vitro studies have established that adipocytes exposed to ROS upregulate expression of proinflammatory cytokines (PAI-1, IL-6) and macrophage chemoattractive molecule (MCP-1) [14]. Also, exposure of adipocytes to high levels of ROS decreases secretion of adiponectin [72]. Indeed, treatment of obese mice with antioxidants corrects adipokine dysregulation and improves diabetes, hyperlipemia, and hepatic steatosis [14]. These findings suggest that increased fat ROS in obesity is an upstream factor for adipokines dysregulation often observed during metabolic syndrome.
Additionally, many proinflammatory cytokines produced by adipocytes are able to stimulate ROS and nitrogen production by macrophages, monocytes, and endothelial cells [88]. Mitochondrial ROS production can be induced by TNF receptors signaling, following binding of TNFα, through TRAF2 to promote NF-κB signaling [89]. Furthermore, glutathionylated lipid aldehydes, by-products of oxidative stress found to be elevated in obese WAT, are potent activators of macrophage inflammation [90]. This inflammatory state is moreover accentuated by the inhibitory effect of inflammatory cytokines on many antioxidant enzymes expression [91] or transporter selenoprotein P (SeP) known to play a role in the regulation of antioxidant enzyme activity [92]. Alternatively, immune cells infiltration might contribute to enhancing WAT-oxidative stress since ROS generated by professional phagocytes are essential components of the innate immune response against intracellular bacteria [93]. Moreover, phenotypic switch toward M1 macrophages in detriment of M2 macrophages occurring in obese WAT [94] might contribute to enhancing ROS production. Indeed, M1 macrophages secrete greater amount of proinflammatory cytokines and ROS production leading to enhance microbicidal capacity [95].
Altogether, ROS-derived hypertrophied adipocytes might initiate an inflammatory state in WAT establishing a systematic feedback-loop between inflammation and oxidative stress in obese adipose tissue concurring to WAT dysfunction.
3.4. WAT-Derived Oxidative Stress Could Promote Endothelial Dysfunction
Perivascular adipose tissue (PVAT) has been recently highlighted for its role in the regulation of vascular tone. PVAT can attenuate vasoconstriction based on the release of adipose tissue-derived relaxing factors [96]. In contrast, PVAT may enhance vascular contractile responses by preventing endothelium-dependent vasodilatation through inhibition of endothelial NOS (eNOS) [97]. Recent report demonstrates that PVAT promotes endothelial dysfunction in diet-induced obese C57Bl/6 mice via mechanisms linked to increased NADPH oxidase-derived oxidative stress and increased production of proinflammatory cytokines [98]. Therefore, defects in vascular tone associated with obesity might be directly related to ROS-derived PVAT on endothelium.
A major deleterious effect of ROS on endothelial cells is the decreased bioavailability of NO•, resulting from eNOS uncoupling [25]. NO•, as a potent vasodilator, is a critical component of hemodynamic regulation. Moreover, eNOS-derived NO• also plays a crucial role in angiogenesis by upregulating vascular endothelial growth factors and increasing mobilization of progenitor cells from the bone marrow. Therefore, ROS-induced endothelial dysfunction will not only impair blood flow regulation but also limit capillary network formation. Such alterations will result ultimately in attenuation of microcirculatory network in metabolic active tissues and subsequent decrease in glucose utilization, particularly in WAT, muscle, and liver, key organs in the establishment of insulin-resistant state. Alteration of NO•-related angiogenic processes might therefore participate in inadequate vasculature observed upon fat expansion in obesity giving rise to hypoxia [99]. Another effect of oxidative stress-mediating endothelial dysfunction might rely on the deleterious effects of ROS on insulin signaling by activating stress-sensitive pathways including JNK, p38 MAPK, IKKβ kinases, and nuclear factor-kappaB in endothelial cells (for review, [100]). Activated kinases can act on a number of potential targets in the insulin-signaling pathways, including the insulin receptor and the family of IRS proteins. For IRS-1 and IRS-2, an increase in serine phosphorylation decreases the extent of the activating tyrosine phosphorylation and is consistent with the attenuation of insulin action [101]. However, despite established roles of these different kinases in the insulin-resistant state [102–104] contribution of oxidative stress in the exacerbation of insulin resistance via IRS-1 serine phosphorylation still needs to be established. Oxidative stress, especially derived from perivascular adipose tissue, can therefore mediate endothelial dysfunction and initiate a feedback loop participating in WAT dysfunction.
4. Obesity-Associated Dysregulated Metabolic Parameters: Contributors and Amplifiers of Oxidative Stress
Overproduction of proinflammatory adipokines is a trait of adipose tissue dysfunction in obesity leading to systemic inflammation in obese patients [7]. Since inflammation and oxidative stress are closely interconnected, systemic oxidative stress also appears as hallmark of the metabolic syndrome [8]. Clinical evidences for obesity-associated oxidative stress have been provided by measurement of either biomarkers or end-products of free radical-mediated oxidative processes. For instance, lipid peroxidation markers such as malondialdehyde (MDA), lipid hydroperoxides, conjugated dienes, 4-HNE, and F2-isoprostanes (8-epiPGF2α) are found to be increased in plasma from obese subjects in many clinical studies [9]. Of note, urinary 8-epi-PGF2α is strongly associated with visceral fat accumulation and BMI suggesting an important role of oxidative stress in the deleterious impact of obesity on metabolic diseases [77, 105]. Accordingly, oxidative stress has been shown to play critical roles in the development of pathologies such as diabetes, atherosclerosis, and hypertension [106, 107]. Furthermore, dysregulated metabolic parameters such as hyperglycemia, hyperlipidemia, and hyperleptinemia are important contributors and amplifiers of systemic oxidative stress (Figure 3).
Figure 3.
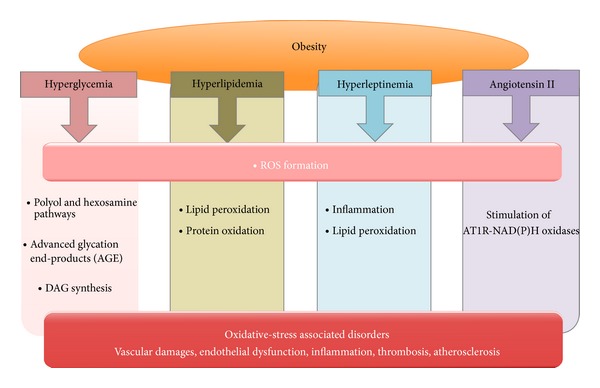
Systemic metabolic alterations associated with obesity contribute to the increase in oxidative stress. Hyperglycemia as a hallmark of type II diabetes, a metabolic complication of obesity, induces oxidative stress through activation of the polyol and hexosamine pathways, production of AGE, and increase of DAG synthesis. Excess of circulating lipids induces ROS formation pathways, which contribute to the increase in lipid oxidation and protein carbonylation. Leptin and angiotensin II, secreted at high levels by adipocytes, are inducers of ROS generation and might therefore promote inflammation and lipid peroxidation. Altogether, dysregulation of metabolic parameters occurring with fat mass expansion will contribute to inducing oxidative-stress damages notably at the vascular level.
4.1. Hyperglycemia
Diabetes mellitus is a group of metabolic diseases characterized by hyperglycemia, impaired insulin sensitivity, and developments of diabetes-induced pathologies. Hyperglycemia has been identified as the initiating cause of diabetic tissue damages seen in clinical situations including nephropathy, retinopathy, neuropathy, or vascular damages [108]. Importantly, these tissue-damaging effects of hyperglycemia are particularly noticeable in endothelial cells, since they retain expression of non-insulin-dependent GLUTs allowing intracellular glucose to rise concomitantly with extracellular glucose concentrations.
In the vascular endothelium, hyperglycemia induces overproduction of O2 •− by the mitochondrial ETC and reduces the activity of the key glycolytic enzyme glyceraldehyde-3 phosphate dehydrogenase (GAPDH) leading to the activation of three major pathways of hyperglycemic damage [109]. (1) Decreasing GAPDH activity leads to an enhancement of glucose catabolism by glycolysis and increased fluxes in the polyol and hexosamine pathways. Thereby, excess of sorbitol generated has been shown to cause cellular damage and to activate inflammatory pathways including p38 MAPK and JNK [110]. In addition, increase of N-acetyl glucosamine modifies the activity of key transcription factors leading to increased expression plasminogen activator inhibitor-1 (PAI-1) [111]. (2) Increased levels of the glycolic intermediate glyceraldehyde-3-phosphate (G3P), following GAPDH inhibition, activate intracellular production of advanced glycation end-products (AGE) precursors. Intracellular production of AGE will modify intracellular proteins or diffuse out the cells and modify matrix proteins as well as circulating proteins such albumin [16]. By binding to specific cell surface receptors (RAGE), AGE induce the production of inflammatory cytokines and growth factors, which in turn may cause vascular pathology [112]. (3) G3P elevation also leads to increased diacylglycerols (DAG) synthesis, critical activating cofactors of protein kinase-C (PKC) [113]. Increased PKC activity has been involved in vascular permeability, blood flow, and neovascularization, processes altered in diabetic patients [114]. The importance of PKC pathway in the development of diabetic complications is supported by studies showing that inhibition of PKCβ ameliorates abnormal retina and renal hemodynamics in diabetic rats [115]. Importantly, normalization of glucose-induced mitochondrial ROS by oxidative phosphorylation uncouplers or overexpression of manganese superoxide dismutase (MnSOD) in endothelial cells prevents the activation of these three pathways of hyperglycaemic damage [116].
Alternatively, hyperglycemia also amplifies ROS production by adipocytes since high-glucose levels induce mitochondrial ROS production in 3T3-L1 adipocytes as noted previously [37, 38]. Indeed, primary adipocytes exposed to hyperglycemic conditions result in increased ROS production either in vitro [117] or in vivo [35]. Adipocytes exposed to hyperglycemia display reduced insulin sensitivity [117], partly explained by reduced insulin-stimulated glucose uptake [118] and increased inflammatory response including PAI-1 and IL-1β [35, 119]. Moreover, hyperglycemic clamp triggers the induction of a proinflammatory response in WAT from rats that can effectively be reduced by coinfusion of the antioxidant N-acetylcysteine (NAC) [35].
4.2. Hyperlipidemia
Plasma FFA concentrations are commonly elevated in obese individuals and associate with fat mass expansion [120, 121]. In healthy subjects, infusion of FFAs causes increased oxidative stress and insulin resistance, which can be reversed by infusion with antioxidants such as glutathione [122]. Elevated FFAs occurring during obesity enhance oxidative stress via increased β-oxidation of cytosolic long-chain acyl-CoA esters, the metabolically active form of FFAs [123]. Moreover, FFAs can also stimulate ROS production through PKC-dependent activation of NAD(P)H oxidase in aortic smooth muscle cells and endothelial cells [22]. Since ROS elicit lipid peroxidation, systemic oxidative stress will likely increase susceptibility of increased lipid pools to lipid oxidation. Accordingly, higher concentrations of 4-HNE have been found in skeletal muscles of obese persons compared to lean patients [67]. Moreover, in vitro oxidation of LDL and VLDL lipoproteins is significantly increased in obese, nondiabetic subjects and related to increased body weight and might be a direct consequence of lipid peroxidation-associated obesity favoring the development of atherosclerosis [124].
4.3. Hyperleptinemia
Leptin, WAT-derived hormone, has been shown to increase the generation of ROS in endothelial cells [125, 126]. NF-κB is also activated by leptin in an oxidant-dependent manner. This effect is associated with an enhanced expression of monocyte chemoattractant protein-1 (MCP-1), which promotes atherosclerosis by favoring migration of inflammatory cells [125]. Stimulation of ROS by leptin occurs through PKC-dependent activation of NAD(P)H oxidase in vascular smooth muscle cells [127]. In addition, leptin, at concentrations similar to those found in the plasma of diabetic patients, stimulates the release of active macrophage LPL through an oxidative stress-dependent pathway suggesting a proatherogenic effect of leptin on macrophages in diabetes [128].
Another mechanism reported for leptin to induce oxidative stress is the decrease of paraoxonase 1 (PON-1) activity following its exogenous administration in Wistar rats [129]. PON-1, transported in plasma HDL, normally protects plasma lipoproteins from oxidative modification by ROS. In addition, plasma concentrations of isoprostanes as well as lipid peroxidation products are markedly increased following leptin infusion [129]. Therefore, prooxidant and PON1-reducing effects of leptin may contribute to accelerated atherogenesis in hyperleptinemic obese subjects.
4.4. Activation of the Renin-Angiotensin System
The renin-angiotensin system (RAS), including angiotensinogen, renin, angiotensin-converting enzyme (ACE), angiotensin II (Ang II), and its receptors, is involved in the maintenance of systemic blood pressure. In pathological state, Ang II also functions as a local biologically active mediator in the progression of cardiovascular remodeling and inflammation through Ang II type-1 receptors (AT1R). Adipocytes are a suggested source of all components of the RAS, in which production is related to obesity-associated hypertension [130]. Plasma levels of renin activity, angiotensinogen, Ang II, and aldosterone values are elevated during obesity [131]. Ang II can stimulate ROS production through AT1R-mediated NAD(P)H oxidases in different types of vascular cells [132, 133]. Moreover, AT1R blockers treatment of obese mice significantly reduces ROS originating from fat pads, attenuates the expression of NAPDH subunits in WAT, and ameliorates cytokines dysregulation [134]. Ang II-induced production of ROS is therefore an important initiator and contributor of oxidative stress-associated obesity.
5. Therapeutical Strategies to Reduce Oxidative Stress-Associated Obesity
Oxidative stress appears as a major contributor in the development of many metabolic complications associated obesity. Therefore, therapeutics designed to lower ROS production may have beneficial effects on health. Practically, many therapeutical strategies used currently to treat obesity-associated metabolic disorders have the potential to decrease oxidative stress, which might, at least partially, participate in their beneficial effects.
5.1. Caloric Restriction and Exercise
Several studies indicate that weight loss in obese patients considerably decreases oxidative stress and inflammation as evidenced by a major decrease in oxidative stress markers and decline of inflammatory cytokines [135, 136]. At the molecular level, caloric restriction activates sirtuins, NAD+-dependent deacetylases that will drive antioxidant and anti-inflammatory responses [137]. In addition, scarcity of nutrients illustrated by a low insulin/IGF-1 signaling activity enhances FOXO (forkhead transcription factor) activity by downregulating insulin-mediated FOXO phosphorylation [138] and upregulating SIRT1-mediated deacetylation [139]. Activated FOXO modulates transcription of genes involved in energy homeostasis, cell survival, inflammatory, and oxidative stress responses [140]. Finally, caloric restriction has also been shown to induce PGC-1α increase in mitochondria capable of efficient and balanced bioenergetics to reduce oxidative stress and ROS-associated damages [141].
Despite the paradox that exhaustive exercise might induce ROS formation, mild oxidative stress produced by regular exercise appears indeed to be able to reduce oxidative damage. Physical exercise exerts ameliorating effects on insulin resistance by increasing mitochondrial formation of ROS in skeletal muscle to induce expression of PGC1 α, PGC1 β, and PPAR γ as inducers of insulin sensitivity as well as SODs 1 and 2 and glutathione peroxidase 1, key enzymes of ROS defense [50, 142]. Noteworthy, regular exercise considerably improves glucose homeostasis [143] likely by restoring mitochondrial content and functional capacity of the skeletal muscle [144]. Finally, physical activity contributes per se to adipocyte lipid mobilization resulting in fat mass loss and repartitioning of intracellular fat, thereby improving its utilization and insulin sensitivity [145].
5.2. Antioxidants Diet Supplementation
Another strategy to reduce obesity-associated oxidative stress might be to supplement diet with molecules, which have proven their antioxidant capacities.
5.2.1. ω-3-Polyunsaturated Fatty Acids
Fish oil, a major source of ω-3-polyunsaturated fatty acids (PUFAs), is recommended for the management of hypertriglyceridemia and to prevent from secondary cardiovascular disorders [146]. Anti-inflammatory actions of ω-3 PUFAs are likely mediated by the formation of their active metabolites (eicosanoids and other lipid mediators) as well as their regulation of the production of inflammatory mediators [147]. Anti-inflammatory effects of ω-3 PUFAs are likely linked to an inhibition of the production of eicosanoids from arachidonic acid [148].
ω-3 PUFAs might also exert antioxidative effects [149]. For instance, ω-3 PUFAS can modulate ROS production in adipocytes by impacting NADPH oxidase [19]. Moreover, supplementation of individuals with ω-3 PUFAs increases the expression of antioxidant enzymes and reduces the expression of prooxidant and tissue enzymes, such as cytochrome P450 enzymes and matrix metalloproteinases [150]. In adipocytes, treatment with EPA (eicosapentaenoic acid) and DHA (docosahexaenoic acid) induces heme-oxygenase 1 through the activation of Nrf-2 and prevents oxidative stress [151]. Finally, a recent study reports that the reduced macrophage infiltration is associated with increased capillaries in insulin-resistant patients following ω-3-acid ethyl esters supplementation. This effect contributes to damper WAT inflammation [152].
Altogether, ω-3 PUFA antioxidant effects associated with their anti-inflammatory properties may be of particular therapeutic value to lower obesity-associated oxidative stress.
5.2.2. Vitamins
Increased oxidative stress in obesity may be exacerbated by decreased availability of antioxidants [153, 154]. This has led to the hypothesis that vitamin supplementation might benefit obese people. In agreement, daily consumption of mixed fruits and vegetable supplements, as generally preconized by dietary recommendations, significantly increases serum levels of antioxidant provitamins and vitamins (β-carotene, vitamins C and E) but also reduces markers of oxidative stress [155]. However, results from clinical trials regarding the beneficial effects of vitamins E, C or carotenoids in reducing the risk of cardiovascular disease are controversial since many trials failed to demonstrate any positive effects (reviewed by [156]). Conversely, adverse effects of antioxidant supplementation have even been reported such as increased risk to develop mortality [157] and cancers [158, 159]. Therefore, further studies will be needed to clearly establish the beneficial effect of vitamins supplementation on obesity-associated metabolic complications.
5.2.3. Polyphenols
Polyphenols constitute the most abundant phytochemicals provided by food due to their enrichment in fruits, vegetables, and seeds-derived products. Many studies have pointed out their antioxidant and free radical scavenging properties leading to consider them as powerful antioxidants [160]. The antioxidant activity of polyphenols notably relies on their ability to inhibit ROS-generating enzymes in addition to upregulation of multiple antioxidant enzymes.
As we previously reviewed [161], polyphenols including resveratrol, quercetin, delphinidin, and red wine polyphenol extracts are beneficial in obesity-associated alterations in several animal models. We have reported that red wine polyphenol (RWP) supplementation prevents metabolic and cardiovascular alterations associated with obesity in Zucker fatty rats [162]. Particularly, RWP supplementation decreases hyperglycemia and hyperlipidemia and improves vasodilation by enhancing eNOS activity and reducing O2 •− release via decreased expression of the NADPH oxidase membrane subunit Nox-1 [162]. Resveratrol supplementation extends the lifespan of high-fat diet mice by reducing fat accumulation and improving glucose tolerance and insulin sensitivity [163]. Additionally, long-term resveratrol administration also improved metabolic disorders in obese Zucker rats and moreover produced anti-inflammatory effects in visceral adipose tissue through AMPK activation [164]. Quercetin has also been shown to improve dyslipidemia, hypertension, and hyperinsulinemia in obese Zucker rats, but only the high dose evokes the anti-inflammatory effects in visceral adipose tissue [165]. Comparing the same doses (21 mg/kg) of polyphenolic molecules (catechin, resveratrol, delphinidin, and gallic acid) in a rat model of the metabolic syndrome reveals that all molecules prevent cardiac ROS overproduction and NADPH overexpression, whereas only delphinidin and gallic acid correct insulin resistance revealing specific properties for each polyphenolic compound.
Besides their beneficial effects on lowering ROS levels, polyphenols might also influence adipose tissue mass by acting on multiple metabolic pathways (for review, [166]). Evaluation of antioxidant properties of major dietary polyphenols on 3T3-L1 adipocytes reveals that most of them inhibit proliferation of preadipocytes [167]. Curcumin and resveratrol have moreover been shown to inhibit adipocyte differentiation [168, 169]. Furthermore, polyphenol-rich grape skin extract supplementation significantly suppresses the activities of lipogenic enzymes in both adipose and liver tissues, which is concomitant with β-oxidation activation [170]. Polyphenols compounds also exert an anti-inflammatory effect on preadipocytes particularly by reducing inflammatory cytokines such as IL6 [167, 169]. Finally, green tea polyphenols reduce fat deposits in high fat-fed rats via erk1/2-PPARγ-adiponectin pathway [171] whereas dietary black tea polyphenols prevent diet-induced obesity by inhibiting intestinal lipid absorption [172]. Mechanistically, these dietary polyphenols exert their additive and/or synergistic effects through one or more signaling and transcriptional pathways including those mediated by NF-κB, AMPK, PPARγ, and PGC-1α [166].
Despite persuasive data showing reduced body weight following treatment of obese rodents with polyphenols, only a limited number of clinical trials with supplementation of dietary polyphenols have been conducted (for review, [161]). Indeed, emerging evidence has been provided on the beneficial role of polyphenols in reducing body weight/body fat through its effects on fat and carbohydrate metabolism as well as on satiety, yet results from clinical trials are still conflicting although some results are promising. Further human long-term intervention studies are needed before recommending polyphenols in obesity management programs.
Such inconsistencies might be related to differences in chemical structure, bioavailability, and metabolism of each polyphenol tested, which may all account for different physiological effects. Moreover, despite well-established antioxidant in vitro activity, the biological relevance of antioxidant effects in humans is questionable in regard to their low degree of absorption and rapid metabolism within the organism. While most in vitro studies used polyphenol concentration range between 10 and 200 μM, physiological polyphenols concentrations in humans do not exceed 10 μM following a polyphenol-rich diet or after polyphenols supplementation [163]. Gut microbiota, which appeared recently to play a critical role in the development of obesity [173, 174], might moreover play a key role in bioactivity of polyphenols [175]. Taking into account qualitative changes and lack of diversities of gut microbiota associated with the obese phenotype, disturbances in polyphenol-metabolizing microbes might participate in the interindividual variability observed upon polyphenol uptake. Conversely, polyphenols might affect microbiota subpopulations by modifying redox state thereby influencing energy harvest, storage, and expenditure.
5.3. Thiazolidinediones (TZD)
TZD are effective oral medications for type 2 diabetes due to their insulin-sensitizing potential. The mechanism of actions of these synthetic ligands is centered on the activation of the transcription factor PPARγ, a master regulator of adipocyte differentiation. Due to pleiotropic effects of PPARγ activation, it is likely that the insulin-sensitizing effect would result from synergized effects of transcriptional activation of adipogenesis, glucose homeostasis, and lipid metabolism by PPARγ-RXR heterodimers. PPARγ also controls the expression of many adipose-secreted factors such as adiponectin, resistin, leptin, and TNF-α, which influence insulin sensitivity (for review, [176]). In addition, in vitro and in vivo studies have revealed that TZD enhance mitochondrial biogenesis, notably by upregulating PGC1-α [177]. Importantly, rosiglitazone-treated mice display marked increased oxygen consumption and palmitate oxidation [178]. Together with increased adiponectin levels, the antioxidant properties of TZD likely participate in part in their beneficial effects.
However, safety concerns due to side effects of TZD based on increased risk of cardiovascular disorders have led to a tightly restricted access of these drugs in the United States and a recommendation for market withdrawal in Europe. Consequently, development of a new class of highly targeted and effective drugs that would preserve the strong antidiabetic efficacy of TZD but yet eliminate many of the unwanted side effects such as weight gain, fluid retention, bone loss, and heart problems is awaited. Importantly, TZD have recently been shown to induce a white-to-brown conversion through stabilization of PRDM16 protein (PRD-BF-1-RIZ1 homologous domain containing protein 16) [179]. These findings raise the possibility to transform substantial amounts of WAT to BAT, which will participate in energy dissipation rather than energy storage through UCP-1 mediated uncoupling. In this context, brown remodeling of WAT induced by SIRT1-dependent deacetylation of PPARγ is promising [180]. Of particular note, activation of SIRT1 allows the cell to adapt to situations of energy stress. For instance, deacetylation of FOXO transcription factors by SIRT1 drives their actions towards the induction of oxidative stress resistance genes [139].
5.4. Apelin
Apelin has been recently identified as a novel peptide hormone abundantly secreted by adipocytes [181]. By interacting with G-coupled apelin receptors (APJ), apelin is implicated in various physiological functions including regulation of cardiovascular functions, fluid homeostasis, vessel formation, and cell proliferation. Of particular importance, apelin has been shown to mediate antiobesity and antidiabetics properties particularly by promoting glucose utilization and β-oxidation in skeletal muscles [182] and by suppressing lipolysis and adipogenesis [183, 184]. Interestingly, apelin is shown to prevent cardiomyocytes, vascular smooth muscle cells, and neurons from oxidative stress [185–187]. Antioxidant properties of apelin have been also recently reported in adipocytes [184] and excess of ROS in fat cells enhances apelin release. Apelin promotes the expression of antioxidant enzymes and suppresses the expression of prooxidant ones especially via AMPK pathway [184]. Moreover, apelin stimulates the release of adiponectin and enhances mitochondrial biogenesis. These effects contribute to counteracting oxidative-stress induced dysregulation in adipocytes.
Given the antiobesity and antioxidant properties of apelin, modulating APJ signaling appears as a promising target in the treatment of metabolic complications associated with obesity. Of particular note, ACE inhibitors or AT1R antagonists enhance apelin expression and secretion in 3T3-L1 adipocytes [188]. Therefore, the antioxidant effect reported in adipocytes following blockade of AT1R might be related to apelin action [134]. However, ACE2 enzyme has also been reported to degrade apelin-13 and apelin-36 [189]. EPA has also been described as a potent stimulator of apelin secretion in adipocytes [190]. Apelin receptors agonists are currently under development and have already proven their efficiency as vasodilator in mice [191]. Nonetheless, the use of apelin as a novel therapeutic target would need further investigations in order to better characterize properties of apelin isoforms (apelin-13, apelin-17, or apelin-36) and their bioavailability.
6. Concluding Remarks
Obesity is nowadays considered as a top risk factor in the development of cardiometabolic diseases and is causative of morbidity of patients suffering from metabolic syndrome. Oxidative stress has appeared in recent years as a hallmark of the obese state, intrinsically linked to chronic low-grade inflammation. High levels of ROS generated by hypertrophied adipocytes impact many metabolic signaling pathways as well as neighboring environment, such as perivascular endothelium or immune-residing WAT cells. WAT-oxidative stress therefore triggers and/or contributes to installing WAT dysfunction. Such impairment is further amplified by altered systemic metabolic parameters (hyperglycemia, hyperlipemia, hyperleptinemia, etc.) that also enhance ROS generation. Overall, systemic oxidative stress-associated obesity directly impacts insulin sensitivity of metabolic organs, promotes inflammation, and alters lipid metabolism or endothelial dysfunction. Conversely, vascular damages and inflammation participate actively in ROS generation therefore entertaining a vicious circle, which contributes to maintaining high levels of oxidative stress. Altogether, oxidative stress appears as an important contributor of metabolic diseases associated obesity such as type II diabetes or atherosclerosis.
Lowering oxidative stress to prevent such metabolic disorders therefore constitutes an interesting target. Lifestyle interventions, including dietary restriction or physical activity, have been proven to be essential in the treatment and prevention of obesity but also beneficial for ROS reduction. However, such recommendations are often difficult to implement in obese patients. An interesting aspect to explore is the potential of already available pharmacological tools (including TZD or ACE inhibitors) to reduce oxidative stress, concomitantly to their efficiency in preventing cardiometabolic risk. Whether reduction of oxidative stress might participate in their beneficial effects is however not clearly established. Future design of molecules that will further promote ROS decrease combined with their initial therapeutical properties might reveal particular interest. Another perspective is the use of antioxidants supplementation. Such a strategy can be easily undertaken since they can be extracted from natural products. Polyphenols appear as exciting molecules but further exploration would be needed to better understand their bioavailability and their molecular targets, in particular in term of genes modulated via nutragenomic approaches. Despite promising results from clinical trials, long-term studies to clearly evaluate their beneficial potential and to avoid adverse effects such as those reported for vitamins supplementation are needed [157]. Finally, one has to keep in mind that physiological levels of ROS also act as essential second messengers to maintain normal cellular functioning and intracellular signaling. Additionally, the spatiotemporal distribution of ROS and cell-antioxidant defense capacities both balance ROS intracellular levels adding to the complexity to target them. One possibility would be to target specifically ROS sources in a cell-type dependent manner. Development of mitochondria-probes molecules based on selective lipophilic cation carrier further conjugated to α-tocopherol (Mito VitE) or to ubiquinone (MitoQ) has already proven their efficiency to intercept mitochondrial ROS [192]. One challenge still resides in targeting specifically fat cells.
Overall, further studies are needed to clearly understand and control degree of ROS generation, typology, and distribution in metabolic tissues as well as at the whole organism level. This can lead to develop new strategies to specifically decrease ROS levels in adipocytes.
Acknowledgment
Soazig Le Lay is funded by a grant from “Region Pays de la Loire.”
Abbreviations
- ACE:
Angiotensin-converting enzyme
- AGE:
Advanced glycation end-products
- AT1R:
Ang II type-1 receptors
- BH4:
Tetrahydrobiopterin
- BMI:
Body mass index
- DAG:
Diacylglycerol
- DHA:
Docosahexaenoic acid
- EPA:
Eicosapentaenoic acid
- eNOS:
Endothelial nitric oxide synthase
- ETC:
Electron transport chain
- FFA:
Free fatty acids
- FOXO:
Forkhead transcription factor
- GAPDH:
Glyceraldehyde-3 phosphate dehydrogenase
- Gpx1:
Glutathione peroxidase 1
- H2O2:
Hydrogen peroxide
- 4-HNE:
4-Hydroxynonenal
- iNOS:
Inducible nitric oxide synthase
- LDL:
Low density lipoprotein
- MDA:
Malondialdehyde
- MSC:
Mesenchymal stem cells
- NAC:
N-Acetylcysteine
- NOX:
NAD(P)H oxidase
- NO•:
Nitric oxide
- PKC:
Protein kinase C
- PUFA:
Polyunsaturated fatty acids
- PVAT:
Perivascular adipose tissue
- RAS:
Renin-angiotensin system
- RNS:
Reactive nitrogen species
- ROS:
Reactive oxygen species
- SOD:
Superoxide dismutase
- O2•−:
Superoxide anions
- OH•:
Hydroxyl radical
- ONOO−:
Peroxynitrite
- TGs:
Triacylglycerols
- VLDL:
Very low density lipoprotein
- WAT:
White adipose tissue.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 2.Cao H. Adipocytokines in obesity and metabolic disease. Journal of Endocrinology. 2014;220(2):T47–T59. doi: 10.1530/JOE-13-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spalding KL, Arner E, Westermark PO, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 4.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr. Obesity is associated with macrophage accumulation in adipose tissue. Journal of Clinical Investigation. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kintscher U, Hartge M, Hess K, et al. T-lymphocyte infiltration in visceral adipose tissue: A primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arteriosclerosis, Thrombosis, and Vascular Biology. 2008;28(7):1304–1310. doi: 10.1161/ATVBAHA.108.165100. [DOI] [PubMed] [Google Scholar]
- 6.Qatanani M, Lazar MA. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes & Development. 2007;21(12):1443–1455. doi: 10.1101/gad.1550907. [DOI] [PubMed] [Google Scholar]
- 7.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annual Review of Immunology. 2011;29:415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 8.Roberts CK, Sindhu KK. Oxidative stress and metabolic syndrome. Life Sciences. 2009;84(21-22):705–712. doi: 10.1016/j.lfs.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 9.Vincent HK, Taylor AG. Biomarkers and potential mechanisms of obesity-induced oxidant stress in humans. International Journal of Obesity. 2006;30(3):400–418. doi: 10.1038/sj.ijo.0803177. [DOI] [PubMed] [Google Scholar]
- 10.Sankhla M, Sharma TK, Mathur K, et al. Relationship of oxidative stress with obesity and its role in obesity induced metabolic syndrome. Clinical Laboratory. 2012;58(5-6):385–392. [PubMed] [Google Scholar]
- 11.Chrysohoou C, Panagiotakos DB, Pitsavos C, et al. The implication of obesity on total antioxidant capacity in apparently healthy men and women: the ATTICA study. Nutrition, Metabolism & Cardiovascular Diseases. 2007;17(8):590–597. doi: 10.1016/j.numecd.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. 2006;440(7086):944–948. doi: 10.1038/nature04634. [DOI] [PubMed] [Google Scholar]
- 13.Konrad T, Vicini P, Kusterer K, et al. α-lipoic acid treatment decreases serum lactate and pyruvate concentrations and improves glucose effectiveness in lean and obese patients with type 2 diabetes. Diabetes Care. 1999;22(2):280–287. doi: 10.2337/diacare.22.2.280. [DOI] [PubMed] [Google Scholar]
- 14.Furukawa S, Fujita T, Shimabukuro M, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. Journal of Clinical Investigation. 2004;114(12):1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 17.Frayn KN, Langin D, Karpe F. Fatty acid-induced mitochondrial uncoupling in adipocytes is not a promising target for treatment of insulin resistance unless adipocyte oxidative capacity is increased. Diabetologia. 2008;51(3):394–397. doi: 10.1007/s00125-007-0901-z. [DOI] [PubMed] [Google Scholar]
- 18.Katz J, Rognstad R. The metabolism of tritiated glucose by rat adipose tissue. Journal of Biological Chemistry. 1966;241(15):3600–3610. [PubMed] [Google Scholar]
- 19.Han CY, Umemoto T, Omer M, et al. NADPH oxidase-derived reactive oxygen species increases expression of monocyte chemotactic factor genes in cultured adipocytes. The Journal of Biological Chemistry. 2012;287(13):10379–10393. doi: 10.1074/jbc.M111.304998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bedard K, Krause K-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 21.Mahadev K, Motoshima H, Wu X, et al. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Molecular and Cellular Biology. 2004;24(5):1844–1854. doi: 10.1128/MCB.24.5.1844-1854.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 23.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radical Biology and Medicine. 2011;51(7):1289–1301. doi: 10.1016/j.freeradbiomed.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dikalova AE, Bikineyeva AT, Budzyn K, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation Research. 2010;107(1):106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vásquez-Vivar J, Kalyanaraman B, Martásek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. The Journal of Biological Chemistry. 2003;278(25):22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 27.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113(13):1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 28.Heitzer T, Brockhoff C, Mayer B, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circulation Research. 2000;86(2):E36–E41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 29.Shinozaki K, Kashiwagi A, Nishio Y, et al. Abnormal biopterin metabolism is a major cause of impaired endothelium-dependent relaxation through nitric oxide/O2- imbalance in insulin- resistant rat aorta. Diabetes. 1999;48(12):2437–2445. doi: 10.2337/diabetes.48.12.2437. [DOI] [PubMed] [Google Scholar]
- 30.Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nature Medicine. 2001;7(10):1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- 31.Elizalde M, Rydén M, Van Harmelen V, et al. Expression of nitric oxide synthases in subcutaneous adipose tissue of nonobese and obese humans. Journal of Lipid Research. 2000;41(8):1244–1251. [PubMed] [Google Scholar]
- 32.Engeli S, Janke J, Gorzelniak K, et al. Regulation of the nitric oxide system in human adipose tissue. Journal of Lipid Research. 2004;45(9):1640–1648. doi: 10.1194/jlr.M300322-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Curtis JM, Grimsrud PA, Wright WS, et al. Downregulation of adipose glutathione S-tansferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes. 2010;59(5):1132–1142. doi: 10.2337/db09-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Talior I, Tennenbaum T, Kuroki T, Eldar-Finkelman H. PKC-δ-dependent activation of oxidative stress in adipocytes of obese and insulin-resistant mice: Role for NADPH oxidase. American Journal of Physiology: Endocrinology and Metabolism. 2005;288(2):E405–E411. doi: 10.1152/ajpendo.00378.2004. [DOI] [PubMed] [Google Scholar]
- 35.Lin Y, Berg AH, Iyengar P, et al. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. Journal of Biological Chemistry. 2005;280(6):4617–4626. doi: 10.1074/jbc.M411863200. [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Lee YJ, Choi H, Ko EH, Kim J. Reactive oxygen species facilitate adipocyte differentiation by accelerating mitotic clonal expansion. The Journal of Biological Chemistry. 2009;284(16):10601–10609. doi: 10.1074/jbc.M808742200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao C-L, Zhu C, Zhao Y-P, et al. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Molecular and Cellular Endocrinology. 2010;320(1-2):25–33. doi: 10.1016/j.mce.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 38.Han CY, Kargi AY, Omer M, et al. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: dissociation of adipocyte hypertrophy from inflammation. Diabetes. 2010;59(2):386–396. doi: 10.2337/db09-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Pauw A, Tejerina S, Raes M, Keijer J, Arnould T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. American Journal of Pathology. 2009;175(3):927–939. doi: 10.2353/ajpath.2009.081155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Visentin V, Prévot D, De Saint Front VD, et al. Alteration of amine oxidase activity in the adipose tissue of obese subjects. Obesity Research. 2004;12(3):547–555. doi: 10.1038/oby.2004.62. [DOI] [PubMed] [Google Scholar]
- 41.Galinier A, Carrière A, Fernandez Y, et al. Adipose tissue proadipogenic redox changes in obesity. The Journal of Biological Chemistry. 2006;281(18):12682–12687. doi: 10.1074/jbc.M506949200. [DOI] [PubMed] [Google Scholar]
- 42.Galinier A, Carriere A, Fernandez Y, et al. Site specific changes of redox metabolism in adipose tissue of obese Zucker rats. FEBS Letters. 2006;580(27):6391–6398. doi: 10.1016/j.febslet.2006.10.052. [DOI] [PubMed] [Google Scholar]
- 43.Landrier J-F, Marcotorchino J, Tourniaire F. Lipophilic micronutrients and adipose tissue biology. Nutrients. 2012;4(11):1622–1649. doi: 10.3390/nu4111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuno T, Hozumi M, Morinobu T, Murata T, Mingci Z, Tamai H. Antioxidant vitamin levels in plasma and low density lipoprotein of obese girls. Free Radical Research. 1998;28(1):81–86. doi: 10.3109/10715769809097878. [DOI] [PubMed] [Google Scholar]
- 45.Neuhouser ML, Rock CL, Eldridge AL, et al. Serum concentrations of retinol, α-tocopherol and the carotenoids are influenced by diet, race and obesity in a sample of healthy adolescents. Journal of Nutrition. 2001;131(8):2184–2191. doi: 10.1093/jn/131.8.2184. [DOI] [PubMed] [Google Scholar]
- 46.Blum M, Dolnikowski G, Seyoum E, et al. Vitamin D 3 in fat tissue. Endocrine. 2008;33(1):90–94. doi: 10.1007/s12020-008-9051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker RS. Carotenoids in human blood and tissues. Journal of Nutrition. 1989;119(1):101–104. doi: 10.1093/jn/119.1.101. [DOI] [PubMed] [Google Scholar]
- 48.Traber MG, Kayden HJ. Tocopherol distribution and intracellular localization in human adipose tissue. American Journal of Clinical Nutrition. 1987;46(3):488–495. doi: 10.1093/ajcn/46.3.488. [DOI] [PubMed] [Google Scholar]
- 49.D'Autréaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature Reviews Molecular Cell Biology. 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 50.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 51.Mao L, Franke J. Hormesis in aging and neurodegeneration-a prodigy awaiting dissection. International Journal of Molecular Sciences. 2013;14(7):13109–13128. doi: 10.3390/ijms140713109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Desaint S, Luriau S, Aude J, Rousselet G, Toledano MB. Mammalian antioxidant defenses are not inducible by H2O2 . The Journal of Biological Chemistry. 2004;279(30):31157–31163. doi: 10.1074/jbc.M401888200. [DOI] [PubMed] [Google Scholar]
- 53.Abuin E, Lissi E, Ahumada M. Diffusion of hydrogen peroxide across DPPC large unilamellar liposomes. Chemistry and Physics of Lipids. 2012;165(6):656–661. doi: 10.1016/j.chemphyslip.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 54.Cordeiro RM. Reactive oxygen species at phospholipid bilayers: distribution, mobility and permeation. Biochimica et Biophysica Acta. 2014;1838(1, part B):438–444. doi: 10.1016/j.bbamem.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 55.May JM, De Haen C. Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells. Journal of Biological Chemistry. 1979;254(7):2214–2220. [PubMed] [Google Scholar]
- 56.Krieger-Brauer HI, Medda PK, Kather H. Insulin-induced activation of NADPH-dependent H2O2 generation in human adipocyte plasma membranes is mediated by Gα(i2) Journal of Biological Chemistry. 1997;272(15):10135–10143. doi: 10.1074/jbc.272.15.10135. [DOI] [PubMed] [Google Scholar]
- 57.Czech MP, Lawrence JC, Jr., Lynn WS. Evidence for the involvement of sulfhydryl oxidation in the regulation of fat cell hexose transport by insulin. Proceedings of the National Academy of Sciences of the United States of America. 1974;71(10):4173–4177. doi: 10.1073/pnas.71.10.4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hayes GR, Lockwood DH. Role of insulin receptor phosphorylation in the insulinomimetic effects of hydrogen peroxide. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(22):8115–8119. doi: 10.1073/pnas.84.22.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kono T, Robinson FW, Blevins TL, Ezaki O. Evidence that translocation of the glucose transport activity is the major mechanism of insulin action on glucose transport in fat cells. Journal of Biological Chemistry. 1982;257(18):10942–10947. [PubMed] [Google Scholar]
- 60.Little SA, de Haen C. Effects of hydrogen peroxide on basal and hormone-stimulated lipolysis in perifused rat fat cells in relation to the mechanism of action of insulin. Journal of Biological Chemistry. 1980;255(22):10888–10895. [PubMed] [Google Scholar]
- 61.Mahadev K, Wu X, Zilbering A, Zhu L, Lawrence JTR, Goldstein BJ. Hydrogen peroxide generated during cellular insulin stimulation is integral to activation of the distal insulin signaling cascade in 3T3-L1 adipocytes. The Journal of Biological Chemistry. 2001;276(52):48662–48669. doi: 10.1074/jbc.M105061200. [DOI] [PubMed] [Google Scholar]
- 62.Loh K, Deng H, Fukushima A, et al. Reactive oxygen species enhance insulin sensitivity. Cell Metabolism. 2009;10(4):260–272. doi: 10.1016/j.cmet.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rudich A, Kozlovsky N, Potashnik R, Bashan N. Oxidant stress reduces insulin responsiveness in 3T3-L1 adipocytes. American Journal of Physiology: Endocrinology and Metabolism. 1997;272(5):E935–E940. doi: 10.1152/ajpendo.1997.272.5.E935. [DOI] [PubMed] [Google Scholar]
- 64.Wang CH, Huang H, Wei Y. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS Journal. 2013;280(4):1039–1050. doi: 10.1111/febs.12096. [DOI] [PubMed] [Google Scholar]
- 65.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. Journal of Clinical Investigation. 2009;119(3):573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murdolo G, Piroddi M, Luchetti F, et al. Oxidative stress and lipid peroxidation by-products at the crossroad between adipose organ dysregulation and obesity-linked insulin resistance. Biochimie. 2013;95(3):585–594. doi: 10.1016/j.biochi.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 67.Russell AP, Gastaldi G, Bobbioni-Harsch E, et al. Lipid peroxidation in skeletal muscle of obese as compared to endurance-trained humans: a case of good vs. bad lipids? FEBS Letters. 2003;551(1–3):104–106. doi: 10.1016/s0014-5793(03)00875-5. [DOI] [PubMed] [Google Scholar]
- 68.Ingram KH, Hill H, Moellering DR, et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. The Journal of Clinical Endocrinology and Metabolism. 2012;97(7):E1182–E1186. doi: 10.1210/jc.2011-2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pillon NJ, Croze ML, Vella RE, Soulère L, Lagarde M, Soulage CO. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology. 2012;153(5):2099–2111. doi: 10.1210/en.2011-1957. [DOI] [PubMed] [Google Scholar]
- 70.Miwa I, Ichimura N, Sugiura M, Hamada Y, Taniguchi S. Inhibition of glucose-induced insulin secretion by 4-hydroxy-2-nonenal and other lipid peroxidation products. Endocrinology. 2000;141(8):2767–2772. doi: 10.1210/endo.141.8.7614. [DOI] [PubMed] [Google Scholar]
- 71.Demozay D, Mas J, Rocchi S, Van Obberghen E. FALDH reverses the deleterious action of oxidative stress induced by lipid peroxidation product 4-hydroxynonenal on insulin signaling in 3T3-L1 adipocytes. Diabetes. 2008;57(5):1216–1226. doi: 10.2337/db07-0389. [DOI] [PubMed] [Google Scholar]
- 72.Soares AF, Guichardant M, Cozzone D, et al. Effects of oxidative stress on adiponectin secretion and lactate production in 3T3-L1 adipocytes. Free Radical Biology and Medicine. 2005;38(7):882–889. doi: 10.1016/j.freeradbiomed.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 73.Grimsrud PA, Picklo MJ, Sr., Griffin TJ, Bernlohr DA. Carbonylation of adipose proteins in obesity and insulin resistance: Identification of adipocyte fatty acid-binding protein as a cellular target of 4-hydroxynonenal. Molecular and Cellular Proteomics. 2007;6(4):624–637. doi: 10.1074/mcp.M600120-MCP200. [DOI] [PubMed] [Google Scholar]
- 74.Zhang X, Wang Z, Li J, et al. Increased 4-hydroxynonenal formation contributes to obesity-related lipolytic activation in adipocytes. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0070663.e70663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zarrouki B, Soares AF, Guichardant M, Lagarde M, Géloën A. The lipid peroxidation end-product 4-HNE induces COX-2 expression through p38MAPK activation in 3T3-L1 adipose cell. FEBS Letters. 2007;581(13):2394–2400. doi: 10.1016/j.febslet.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 76.Ye R, Scherer PE. Adiponectin, driver or passenger on the road to insulin sensitivity? Molecular Metabolism. 2013;2(3):133–141. doi: 10.1016/j.molmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fujita K, Nishizawa H, Funahashi T, Shimomura I, Shimabukuro M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circulation Journal. 2006;70(11):1437–1442. doi: 10.1253/circj.70.1437. [DOI] [PubMed] [Google Scholar]
- 78.Tao L, Gao E, Jiao X, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115(11):1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 79.Li R, Wang W-Q, Zhang H, et al. Adiponectin improves endothelial function in hyperlipidemic rats by reducing oxidative/nitrative stress and differential regulation of eNOS/iNOS activity. American Journal of Physiology: Endocrinology and Metabolism. 2007;293(6):E1703–E1708. doi: 10.1152/ajpendo.00462.2007. [DOI] [PubMed] [Google Scholar]
- 80.Schröder K, Wandzioch K, Helmcke I, Brandes RP. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(2):239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 81.Higuchi M, Dusting GJ, Peshavariya H, et al. Differentiation of human adipose-derived stem cells into fat involves reactive oxygen species and forkhead box o1 mediated upregulation of antioxidant enzymes. Stem Cells and Development. 2013;22(6):878–888. doi: 10.1089/scd.2012.0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kanda Y, Hinata T, Kang SW, Watanabe Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sciences. 2011;89(7-8):250–258. doi: 10.1016/j.lfs.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 83.Tormos KV, Anso E, Hamanaka RB, et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metabolism. 2011;14(4):537–544. doi: 10.1016/j.cmet.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carrière A, Carmona M, Fernandez Y, et al. Mitochondrial reactive oxygen species control the transcription factor CHOP-10/GADD153 and adipocyte differentiation: a mechanism for hypoxia-dependent effect. Journal of Biological Chemistry. 2004;279(39):40462–40469. doi: 10.1074/jbc.M407258200. [DOI] [PubMed] [Google Scholar]
- 85.Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the metabolic syndrome. Endocrinologia y Nutricion. 2013;60(1):39–43. doi: 10.1016/s1575-0922(13)70026-3. [DOI] [PubMed] [Google Scholar]
- 86.Stern JS, Batchelor BR, Hollander N, Cohn CK, Hirsch J. Adipose-cell size and immunoreactive insulin levels in obese and normal-weight adults. The Lancet. 1972;2(7784):948–951. doi: 10.1016/s0140-6736(72)92474-9. [DOI] [PubMed] [Google Scholar]
- 87.Lundgren M, Svensson M, Lindmark S, Renström F, Ruge T, Eriksson JW. Fat cell enlargement is an independent marker of insulin resistance and “hyperleptinaemia”. Diabetologia. 2007;50(3):625–633. doi: 10.1007/s00125-006-0572-1. [DOI] [PubMed] [Google Scholar]
- 88.Volk T, Hensel M, Schuster H, Kox WJ. Secretion of MCP-1 and IL-6 by cytokine stimulated production of reactive oxygen species in endothelial cells. Molecular and Cellular Biochemistry. 2000;206(1-2):105–112. doi: 10.1023/a:1007059616914. [DOI] [PubMed] [Google Scholar]
- 89.Chandel NS, Schumacker PT, Arch RH. Reactive oxygen species are downstream products of TRAF-mediated signal transduction. Journal of Biological Chemistry. 2001;276(46):42728–42736. doi: 10.1074/jbc.M103074200. [DOI] [PubMed] [Google Scholar]
- 90.Frohnert BI, Long EK, Hahn WS, Bernlohr DA. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes. 2013;63(1):89–100. doi: 10.2337/db13-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Long EK, Olson DM, Bernlohr DA. High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radical Biology and Medicine. 2013;63:390–398. doi: 10.1016/j.freeradbiomed.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhang Y, Chen X. Reducing selenoprotein P expression suppresses adipocyte differentiation as a result of increased preadipocyte inflammation. The American Journal of Physiology—Endocrinology and Metabolism. 2011;300(1):E77–E85. doi: 10.1152/ajpendo.00380.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jay Forman H, Torres M. Redox signaling in macrophages. Molecular Aspects of Medicine. 2001;22(4-5):189–216. doi: 10.1016/s0098-2997(01)00010-3. [DOI] [PubMed] [Google Scholar]
- 94.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. Journal of Clinical Investigation. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dale DC, Boxer L, Conrad Liles W. The phagocytes: neutrophils and monocytes. Blood. 2008;112(4):935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 96.Löhn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM. Periadventitial fat releases a vascular relaxing factor. FASEB Journal. 2002;16(9):1057–1063. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- 97.Payne GA, Bohlen HG, Dincer ÜD, Borbouse L, Tune JD. Periadventitial adipose tissue impairs coronary endothelial function via PKC-β-dependent phosphorylation of nitric oxide synthase. The American Journal of Physiology—Heart and Circulatory Physiology. 2009;297(1):H460–H465. doi: 10.1152/ajpheart.00116.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ketonen J, Shi J, Martonen E, Mervaala E. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57BL/6 mice. Circulation Journal. 2010;74(7):1479–1487. doi: 10.1253/circj.cj-09-0661. [DOI] [PubMed] [Google Scholar]
- 99.Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–911. doi: 10.2337/db06-0911. [DOI] [PubMed] [Google Scholar]
- 100.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress—activated signaling pathways mediators of insulin resistance and β-cell dysfunction? Diabetes. 2003;52(1):1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 101.Archuleta TL, Lemieux AM, Saengsirisuwan V, et al. Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of p38 MAPK. Free Radical Biology and Medicine. 2009;47(10):1486–1493. doi: 10.1016/j.freeradbiomed.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka T. Involvement of oxidative stress and the JNK pathway in glucose toxicity. The Review of Diabetic Studies. 2004;1(4):165–174. doi: 10.1900/RDS.2004.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Arkan MC, Hevener AL, Greten FR, et al. IKK-β links inflammation to obesity-induced insulin resistance. Nature Medicine. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 104.Hirosumi J, Tuncman G, Chang L, et al. A central, role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 105.Keaney JF, Jr., Larson MG, Vasan RS, et al. Obesity and systemic oxidative stress: clinical correlates of oxidative stress in the Framingham study. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(3):434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 106.Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nature Reviews Endocrinology. 2012;8(2):92–103. doi: 10.1038/nrendo.2011.138. [DOI] [PubMed] [Google Scholar]
- 107.Wang C, Wei Y. Mitochondrial dysfunction in insulin insensitivity: implication of mitochondrial role in type 2 diabetes. Annals of the New York Academy of Sciences. 2010;1201:157–165. doi: 10.1111/j.1749-6632.2010.05625.x. [DOI] [PubMed] [Google Scholar]
- 108.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The New England Journal of Medicine. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 109.Du X, Matsumura T, Edelstein D, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. The Journal of Clinical Investigation. 2003;112(7):1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gabbay KH, Merola LO, Field RA. Sorbitol pathway: presence in nerve and cord with substrate accumulation in diabetes. Science. 1966;151(3707):209–210. doi: 10.1126/science.151.3707.209. [DOI] [PubMed] [Google Scholar]
- 111.Du XL, Edelstein D, Dimmeler S, Ju Q, Sui C, Brownlee M. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. Journal of Clinical Investigation. 2001;108(9):1341–1348. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bierhaus A, Hofmann MA, Ziegler R, Nawroth PP. AGEs and their interaction with AGE-receptors in vascular disease and diabetes mellitus. I. The AGE concept. Cardiovascular Research. 1998;37(3):586–600. doi: 10.1016/s0008-6363(97)00233-2. [DOI] [PubMed] [Google Scholar]
- 113.Lee TS, Saltsman KA, Ohashi H, King GL. Activation of protein kinase C by elevation of glucose concentration: proposal for a mechanism in the development of diabetic vascular complications. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(13):5141–5145. doi: 10.1073/pnas.86.13.5141. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 114.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circulation Research. 2010;106(8):1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC β inhibitor. Science. 1996;272(5262):728–731. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 116.Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 117.Lu B, Ennis D, Lai R, et al. Enhanced sensitivity of insulin-resistant adipocytes to vanadate is associated with oxidative stress and decreased reduction of vanadate (+5) to vanadyl (+4) Journal of Biological Chemistry. 2001;276(38):35589–35598. doi: 10.1074/jbc.M106783200. [DOI] [PubMed] [Google Scholar]
- 118.Tang S, Le-Tien H, Goldstein BJ, Shin P, Lai R, Fantus IG. Decreased in situ insulin receptor dephosphorylation in hyperglycemia-induced insulin resistance in rat adipocytes. Diabetes. 2001;50(1):83–90. doi: 10.2337/diabetes.50.1.83. [DOI] [PubMed] [Google Scholar]
- 119.Koenen TB, Stienstra R, van Tits LJ, et al. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1β transcription in human adipose tissue. Diabetes. 2011;60(2):517–524. doi: 10.2337/db10-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Björntorp P, Bergman H, Varnauskas E. Plasma free fatty acid turnover rate in obesity. Acta Medica Scandinavica. 1969;185(4):351–356. doi: 10.1111/j.0954-6820.1969.tb07347.x. [DOI] [PubMed] [Google Scholar]
- 121.Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles JM. Influence of body fat distribution on free fatty acid metabolism in obesity. Journal of Clinical Investigation. 1989;83(4):1168–1173. doi: 10.1172/JCI113997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paolisso G, Di Maro G, Pizza G, et al. Plasma GSH/GSSG affects glucose homeostasis in healthy subjects and non- insulin-dependent diabetics. American Journal of Physiology: Endocrinology and Metabolism. 1992;263(3, part 2):E435–E440. doi: 10.1152/ajpendo.1992.263.3.E435. [DOI] [PubMed] [Google Scholar]
- 123.Wojtczak L, Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochimica et Biophysica Acta—Bioenergetics. 1993;1183(1):41–57. doi: 10.1016/0005-2728(93)90004-y. [DOI] [PubMed] [Google Scholar]
- 124.van Gaal LF, Vertommen J, de Leeuw IH. The in vitro oxidizability of lipoprotein particles in obese and non- obese subjects. Atherosclerosis. 1998;137(supplement):S39–S44. doi: 10.1016/s0021-9150(97)00316-x. [DOI] [PubMed] [Google Scholar]
- 125.Bouloumié A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB Journal. 1999;13(10):1231–1238. [PubMed] [Google Scholar]
- 126.Yamagishi S, Edelstein D, Du X, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. The Journal of Biological Chemistry. 2001;276(27):25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- 127.Li L, Mamputu J, Wiernsperger N, Renier G. Signaling pathways involved in human vascular smooth muscle cell proliferation and matrix metalloproteinase-2 expression induced by leptin: inhibitory effect of metformin. Diabetes. 2005;54(7):2227–2234. doi: 10.2337/diabetes.54.7.2227. [DOI] [PubMed] [Google Scholar]
- 128.Maingrette F, Renier G. Leptin increases lipoprotein lipase secretion by macrophages: involvement of oxidative stress and protein kinase C. Diabetes. 2003;52(8):2121–2128. doi: 10.2337/diabetes.52.8.2121. [DOI] [PubMed] [Google Scholar]
- 129.Bełtowski J, Wójcicka G, Jamroz A. Leptin decreases plasma paraoxonase 1 (PON1) activity and induces oxidative stress: The possible novel mechanism for proatherogenic effect of chronic hyperleptinemia. Atherosclerosis. 2003;170(1):21–29. doi: 10.1016/s0021-9150(03)00236-3. [DOI] [PubMed] [Google Scholar]
- 130.Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Current Hypertension Reports. 2008;10(2):93–98. doi: 10.1007/s11906-008-0019-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Engeli S, Schling P, Gorzelniak K, et al. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? International Journal of Biochemistry and Cell Biology. 2003;35(6):807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 132.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation Research. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 133.Brandes RP, Kreuzer J. Vascular NADPH oxidases: molecular mechanisms of activation. Cardiovascular Research. 2005;65(1):16–27. doi: 10.1016/j.cardiores.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 134.Kurata A, Nishizawa H, Kihara S, et al. Blockade of angiotensin II type-1 receptor reduces oxidative stress in adipose tissue and ameliorates adipocytokine dysregulation. Kidney International. 2006;70(10):1717–1724. doi: 10.1038/sj.ki.5001810. [DOI] [PubMed] [Google Scholar]
- 135.Dandona P, Mohanty P, Ghanim H, et al. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. The Journal of Clinical Endocrinology & Metabolism. 2001;86(1):355–362. doi: 10.1210/jcem.86.1.7150. [DOI] [PubMed] [Google Scholar]
- 136.Crujeiras AB, Parra D, Milagro FI, et al. Differential expression of oxidative stress and inflammation related genes in peripheral blood mononuclear cells in response to a low-calorie diet: a nutrigenomics study. OMICS. 2008;12(4):251–261. doi: 10.1089/omi.2008.0001. [DOI] [PubMed] [Google Scholar]
- 137.Cantó C, Auwerx J. Targeting sirtuin 1 to improve metabolism: All you need is NAD+? Pharmacological Reviews. 2012;64(1):166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Perrot V, Rechler MM. Characterization of insulin inhibition of transactivation by a C-terminal fragment of the forkhead transcription factor Foxo1 in rat hepatoma cells. The Journal of Biological Chemistry. 2003;278(28):26111–26119. doi: 10.1074/jbc.M212750200. [DOI] [PubMed] [Google Scholar]
- 139.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 140.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 141.López-Lluch G, Hunt N, Jones B, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(6):1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gomez-Cabrera M-C, Domenech E, Viña J. Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radical Biology and Medicine. 2008;44(2):126–131. doi: 10.1016/j.freeradbiomed.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 143.Boulé NG, Weisnagel SJ, Lakka TA, et al. Effects of exercise training on glucose homeostasis: the HERITAGE family study. Diabetes Care. 2005;28(1):108–114. doi: 10.2337/diacare.28.1.108. [DOI] [PubMed] [Google Scholar]
- 144.Toledo FGS, Menshikova EV, Ritov VB, et al. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes. 2007;56(8):2142–2147. doi: 10.2337/db07-0141. [DOI] [PubMed] [Google Scholar]
- 145.Balkau B, Mhamdi L, Oppert J, et al. Physical activity and insulin sensitivity the RISC study. Diabetes. 2008;57(10):2613–2618. doi: 10.2337/db07-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kris-Etherton PM, Harris WS, Appel LJ. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(2):151–152. doi: 10.1161/01.atv.0000057393.97337.ae. [DOI] [PubMed] [Google Scholar]
- 147.Calder PC. n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. The American Journal of Clinical Nutrition. 2006;83(6) doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- 148.González-Périz A, Horrillo R, Ferré N, et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by ω-3 fatty acids: A role for resolvins and protectins. The FASEB Journal. 2009;23(6):1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fan C, Zirpoli H, Qi K. N-3 fatty acids modulate adipose tissue inflammation and oxidative stress. Current Opinion in Clinical Nutrition and Metabolic Care. 2013;16(2):124–132. doi: 10.1097/MCO.0b013e32835c02c8. [DOI] [PubMed] [Google Scholar]
- 150.Schmidt S, Stahl F, Mutz K, Scheper T, Hahn A, Schuchardt JP. Transcriptome-based identification of antioxidative gene expression after fish oil supplementation in normo- and dyslipidemic men. Nutrition and Metabolism. 2012;9(1, article 45) doi: 10.1186/1743-7075-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kusunoki C, Yang L, Yoshizaki T, et al. Omega-3 polyunsaturated fatty acid has an anti-oxidant effect via the Nrf-2/HO-1 pathway in 3T3-L1 adipocytes. Biochemical and Biophysical Research Communications. 2013;430(1):225–230. doi: 10.1016/j.bbrc.2012.10.115. [DOI] [PubMed] [Google Scholar]
- 152.Spencer M, Finlin BS, Unal R, et al. Omega-3 fatty acids reduce adipose tissue macrophages in human subjects with insulin resistance. Diabetes. 2013;62(5):1709–1717. doi: 10.2337/db12-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Moor De Burgos A, Wartanowicz M, Ziemlariski S. Blood vitamin and lipid levels in overweight and obese women. European Journal of Clinical Nutrition. 1992;46(11):803–808. [PubMed] [Google Scholar]
- 154.Strauss RS. Comparison of serum concentrations of α-tocopherol and β-carotene in a cross-sectional sample of obese and nonobese children (NHANES III) Journal of Pediatrics. 1999;134(2):160–165. doi: 10.1016/s0022-3476(99)70409-9. [DOI] [PubMed] [Google Scholar]
- 155.Esfahani A, Wong JMW, Truan J, et al. Health effects of mixed fruit and vegetable concentrates: a systematic review of the clinical interventions. Journal of the American College of Nutrition. 2011;30(5):285–294. doi: 10.1080/07315724.2011.10719971. [DOI] [PubMed] [Google Scholar]
- 156.Savini I, Catani MV, Evangelista D, Gasperi V, Avigliano L. Obesity-associated oxidative stress: strategies finalized to improve redox state. International Journal of Molecular Sciences. 2013;14(5):10497–10538. doi: 10.3390/ijms140510497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. The Journal of the American Medical Association. 2007;297(8):842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 158.Bjelakovic G, Nagorni A, Nikolova D, Simonetti RG, Bjelakovic M, Gluud C. Meta-analysis: Antioxidant supplements for primary and secondary prevention of colorectal adenoma. Alimentary Pharmacology and Therapeutics. 2006;24(2):281–291. doi: 10.1111/j.1365-2036.2006.02970.x. [DOI] [PubMed] [Google Scholar]
- 159.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10(3):175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 160.Andriantsitohaina R, Duluc L, García-Rodríguez JC, et al. Systems biology of antioxidants. Clinical Science. 2012;123(3):173–192. doi: 10.1042/CS20110643. [DOI] [PubMed] [Google Scholar]
- 161.Andriantsitohaina R, Auger C, Chataigneau T, et al. Molecular mechanisms of the cardiovascular protective effects of polyphenols. The British Journal of Nutrition. 2012;108(9):1532–1549. doi: 10.1017/S0007114512003406. [DOI] [PubMed] [Google Scholar]
- 162.Agouni A, Lagrue-Lak-Hal A, Mostefai HA, et al. Red wine polyphenols prevent metabolic and cardiovascular alterations associated with obesity in Zucker fatty rats (Fa/Fa) PLoS ONE. 2009;4(5) doi: 10.1371/journal.pone.0005557.e5557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Rivera L, Morón R, Zarzuelo A, Galisteo M. Long-term resveratrol administration reduces metabolic disturbances and lowers blood pressure in obese Zucker rats. Biochemical Pharmacology. 2009;77(6):1053–1063. doi: 10.1016/j.bcp.2008.11.027. [DOI] [PubMed] [Google Scholar]
- 165.Rivera L, Morón R, Sánchez M, Zarzuelo A, Galisteo M. Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity. 2008;16(9):2081–2087. doi: 10.1038/oby.2008.315. [DOI] [PubMed] [Google Scholar]
- 166.Wang S, Moustaid-Moussa N, Chen L, et al. Novel insights of dietary polyphenols and obesity. The Journal of Nutritional Biochemistry. 2014;25(1):1–18. doi: 10.1016/j.jnutbio.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hatia S, Septembre-Malaterre A, Le Sage F, et al. Evaluation of antioxidant properties of major dietary polyphenols and their protective effect on 3T3-L1 preadipocytes and red blood cells exposed to oxidative stress. Free Radical Research. 2014;48(4):387–401. doi: 10.3109/10715762.2013.879985. [DOI] [PubMed] [Google Scholar]
- 168.Ejaz A, Wu D, Kwan P, Meydani M. Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. Journal of Nutrition. 2009;139(5):919–925. doi: 10.3945/jn.108.100966. [DOI] [PubMed] [Google Scholar]
- 169.Fischer-Posovszky P, Kukulus V, Tews D, et al. Resveratrol regulates human adipocyte number and function in a Sirt1-dependent manner. The American Journal of Clinical Nutrition. 2010;92(1):5–15. doi: 10.3945/ajcn.2009.28435. [DOI] [PubMed] [Google Scholar]
- 170.Park H-J, Jung UJ, Lee M-K, et al. Modulation of lipid metabolism by polyphenol-rich grape skin extract improves liver steatosis and adiposity in high fat fed mice. Molecular Nutrition & Food Research. 2013;57(2):360–364. doi: 10.1002/mnfr.201200447. [DOI] [PubMed] [Google Scholar]
- 171.Tian C, Ye X, Zhang R, et al. Green Tea Polyphenols Reduced Fat Deposits in High Fat-Fed Rats via erk1/2-PPARγ-Adiponectin Pathway. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0053796.e53796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Uchiyama S, Taniguchi Y, Saka A, Yoshida A, Yajima H. Prevention of diet-induced obesity by dietary black tea polyphenols extract in vitro and in vivo. Nutrition. 2011;27(3):287–292. doi: 10.1016/j.nut.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 173.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 174.Ridaura VK, Faith JJ, Rey FE, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150) doi: 10.1126/science.1241214.1241214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bolca S, van de Wiele T, Possemiers S. Gut metabotypes govern health effects of dietary polyphenols. Current Opinion in Biotechnology. 2013;24(2):220–225. doi: 10.1016/j.copbio.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 176.Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nature Medicine. 2013;19(5):557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wilson-Fritch L, Burkart A, Bell G, et al. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Molecular and Cellular Biology. 2003;23(3):1085–1094. doi: 10.1128/MCB.23.3.1085-1094.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wilson-Fritch L, Nicoloro S, Chouinard M, et al. Mitochondrial remodeling in adipose tissue associated with obesity and treatment with rosiglitazone. The Journal of Clinical Investigation. 2004;114(9):1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ohno H, Shinoda K, Spiegelman BM, Kajimura S. PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metabolism. 2012;15(3):395–404. doi: 10.1016/j.cmet.2012.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Qiang L, Wang L, Kon N, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ . Cell. 2012;150(3):620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Boucher J, Masri B, Daviaud D, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146(4):1764–1771. doi: 10.1210/en.2004-1427. [DOI] [PubMed] [Google Scholar]
- 182.Dray C, Knauf C, Daviaud D, et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metabolism. 2008;8(5):437–445. doi: 10.1016/j.cmet.2008.10.003. [DOI] [PubMed] [Google Scholar]
- 183.Yue P, Jin H, Xu S, et al. Apelin decreases lipolysis via Gq, Gi, and AMPK-dependent mechanisms. Endocrinology. 2011;152(1):59–68. doi: 10.1210/en.2010-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Than A, Cheng Y, Foh L, et al. Apelin inhibits adipogenesis and lipolysis through distinct molecular pathways. Molecular and Cellular Endocrinology. 2012;362(1-2):227–241. doi: 10.1016/j.mce.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 185.Foussal C, Lairez O, Calise D, et al. Activation of catalase by apelin prevents oxidative stress-linked cardiac hypertrophy. FEBS Letters. 2010;584(11):2363–2370. doi: 10.1016/j.febslet.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 186.Chun HJ, Ali ZA, Kojima Y, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. The Journal of Clinical Investigation. 2008;118(10):3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zeng XJ, Yu SP, Zhang L, Wei L. Neuroprotective effect of the endogenous neural peptide apelin in cultured mouse cortical neurons. Experimental Cell Research. 2010;316(11):1773–1783. doi: 10.1016/j.yexcr.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hung W, Hsieh T, Lin T, et al. Blockade of the renin-angiotensin system ameliorates apelin production in 3T3-L1 adipocytes. Cardiovascular Drugs and Therapy. 2011;25(1):3–12. doi: 10.1007/s10557-010-6274-4. [DOI] [PubMed] [Google Scholar]
- 189.Vickers C, Hales P, Kaushik V, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. Journal of Biological Chemistry. 2002;277(17):14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 190.Lorente-Cebrián S, Bustos M, Marti A, Martinez JA, Moreno-Aliaga MJ. Eicosapentaenoic acid up-regulates apelin secretion and gene expression in 3T3-L1 adipocytes. Molecular Nutrition and Food Research. 2010;54(1):S104–S111. doi: 10.1002/mnfr.200900522. [DOI] [PubMed] [Google Scholar]
- 191.Iturrioz X, Alvear-Perez R, de Mota N, et al. Identification and pharmacological properties of E339-3D6, the first nonpeptidic apelin receptor agonist. The FASEB Journal. 2010;24(5):1506–1517. doi: 10.1096/fj.09-140715. [DOI] [PubMed] [Google Scholar]
- 192.Addabbo F, Montagnani M, Goligorsky MS. Mitochondria and reactive oxygen species. Hypertension. 2009;53(6):885–892. doi: 10.1161/HYPERTENSIONAHA.109.130054. [DOI] [PMC free article] [PubMed] [Google Scholar]