

Abstract
Stress kinases can be activated by hyperthermia and modify the expression level and properties of membranous and intercellular channels. We examined the role of c-Jun NH2-terminal kinase (JNK) in hyperthermia-induced changes of connexin43 (Cx43) expression and permeability of Cx43 gap junctions (GJs) in the rabbit skeletal myoblasts (SkMs) and Cx43-EGFP transfected HeLa cells. Hyperthermia (42°C for 6 h) enhanced the activity of JNK and its target, the transcription factor c-Jun, in both SkMs and HeLa cells. In SkMs, hyperthermia caused a 3.2-fold increase in the total Cx43 protein level and enhanced the efficacy of GJ intercellular communication (GJIC). In striking contrast, hyperthermia reduced the total amount of Cx43 protein, the number of Cx43 channels in GJ plaques, the density of hemichannels in the cell membranes, and the efficiency of GJIC in HeLa cells. Both in SkMs and HeLa cells, these changes could be prevented by XG-102, a JNK inhibitor. In HeLa cells, the changes in Cx43 expression and GJIC under hyperthermic conditions were accompanied by JNK-dependent disorganization of actin cytoskeleton stress fibers while in SkMs, the actin cytoskeleton remained intact. These findings provide an attractive model to identify the regulatory players within signalosomes, which determine the cell-dependent outcomes of hyperthermia.
1. Introduction
Skeletal myoblasts (SkMs) have been investigated during the last decade for their potential in several fields of regenerative medicine. SkMs have been applied for the treatment of myocardial infarction (MI), Duchenne's muscular dystrophy, Chagas' disease, muscle trauma, and so forth [1–5]. Thus, SkMs are considered to be appropriate candidates for stem cell therapy due to their high proliferative potential, resistance to ischemia, simple isolation from muscular biopsy, and absence of tumorigenicity as well as of immunological and ethical concerns. Animal studies have shown positive effects of autologous SkM transplantation on the cardiac function [1, 6–8], but controversial data were obtained from phase I clinical trials, which failed to demonstrate the functionally effective postinfarctional heart regeneration with SkMs [9]. A number of issues need to be resolved concerning stem cell transplantation. Implanted cells show a low rate of incorporation and viability in the ischemic environment. For instance, Suzuki et al. have shown that only 7.4% of SkMs survived in mice hearts 72 h after injection [10]. MI is accompanied by adverse side effects such as inflammation, hypoxia, and impaired metabolism [11–13]. The main disadvantage of SkM application is the increased risk of ventricular tachyarrhythmias [14]. For the proper excitation of the heart, engrafted cells need to establish functional intercellular communication with host cardiomyocytes [15]. Gap junction (GJ) channels are composed of two opposing hemichannels in contiguous cells and provide a direct pathway for electrical and metabolic intercellular communication [16]. Six connexin (Cx) subunits oligomerize into connexon, which is called a hemichannel after insertion into plasma membrane. The family of connexin genes consists of 21 genes in the human genome. Cx43 is the most abundant connexin protein in the ventricular myocardium, responsible for gap junction intercellular communication (GJIC) between working myocytes [17, 18]. Nondifferentiated SkMs also express endogenous Cx43 that is important not only in coupling with cardiac myocytes but also in the differentiation of SkMs and the regeneration of skeletal muscle [19]. Unfortunately, Cx43 is downregulated during SkM differentiation [20, 21]. Induced expression of Cx43 in SkMs may serve as an appropriate strategy to improve their therapeutic benefit. At least, genetically modified myoblasts expressing Cx43 have been shown to decrease the arrhythmogenicity [22–24]. Many other factors, such as antiapoptotic or angiogenesis-initiating genes [25, 26], preconditioning with growth factors [15, 27–29], or heat treatment [30–33], have been shown to contribute to the improvement of the efficiency of stem cell therapy. Inflammation and cell survival during cardiac ischemia/reperfusion (I/R) injury is essentially regulated by mitogen activated protein kinase (MAPK) signaling pathways. Three MAPK subfamilies are known to play a major role in the I/R heart: extracellular signal-regulated kinases 1/2 (ERK1/2), c-Jun NH2-terminal kinase (JNK), and p38 MAPK [34]. The exposure of rat cardiomyocytes to ischemia activates ERK, p38, and JNK [35]. The activation of ERK protects cardiomyocytes from apoptosis and reduces infarct size [36, 37], but the data on the impact of p38 and JNK activation on the cardiac function during I/R are conflicting. On the one hand, the activation of p38 and JNK induces apoptosis of cardiomyocytes and exacerbates heart injury after I/R [35, 38–40], but on the other hand, there is evidence of their protective mechanisms [41–43]. Moreover, the activation of ERK or the inhibition of JNK and p38 pathways has been reported to improve the heart function after MI (or I/R) [34, 44–46].
JNK can be activated by inflammatory cytokines and numerous stressors such as heat shock, oxidative stress, or DNA damage, which follow I/R [47–50]. I/R and surgical interventions evoke inflammatory responses that activate JNK and/or other kinases. Consequently, the expression of Cxs and the properties of membranous and intercellular channels can be modified by stress kinases. Indeed, the activation of JNK up- or downregulates the expression of Cx43 depending on the cell type [51, 52].
Here we found that hyperthermia induced JNK-dependent changes in Cx43 expression and GJIC in HeLa cells expressing exogenous Cx43-EGFP and in SkMs expressing endogenous Cx43.
In parallel, hyperthermia caused JNK-dependent disorganization of the F-actin network in HeLa Cx43-EGFP cells, while in SkMs it remained unaltered.
2. Methods
The investigation conforms to the European Community guidelines for the care and use of animals (86/609/CEE, CE Off J no. L358, December 18, 1986). The license for the use of laboratory animals (no. 0171, October 31, 2007) was received from the Lithuanian Food and Veterinary Service.
2.1. Isolation and Culturing of Skeletal Myoblasts
New Zealand white rabbits of both genders, aged 6 to 12 months and weighing 3.0 to 3.5 kg, were used in this study. A piece of femoral skeletal muscle (0.5 cm3) was taken under general anesthesia and placed into a transportation medium (Iscove's modified Dulbecco's medium (IMDM) with antibiotics, penicillin 300 U/mL, and streptomycin 300 μg/mL). The tissue was mechanically cut into small pieces and treated with an enzyme mixture: 1 mg/mL collagenase (type V), 0.3 mg/mL hyaluronidase (type IV-S), 0.125% trypsin, and 0.1% EDTA in PBS. After washing with IMDM, cells were seeded into flasks with a growing medium (IMDM with 10% FBS, penicillin 100 U/mL, and streptomycin 100 μg/mL). All chemicals were purchased from Sigma Aldrich Corp. (Steinheim, Germany).
2.2. HeLa Cx43-EGFP Growing Conditions
Cx43-EGFP-expressing HeLa cells were kindly donated by Professor Feliksas Bukauskas (Yeshiva University, New York, NY, USA). HeLa Cx43-EGFP cells were grown in a Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% FBS and antibiotics (penicillin 100 U/mL and streptomycin 100 μg/mL).
2.3. Immunocytochemistry
Cells were grown on glass coverslips 1 cm in diameter and fixed with 4% paraformaldehyde for 15 min. Then cells were washed with PBS and incubated in PBS with 0.2% Triton X-100 for 3 min to permeabilize membranes. Cells were incubated with primary antibodies against desmin, myogenin (Abcam, Cambridge, UK), Cx43 (Transduction Laboratories, Lexington, KY, USA), or Cx45 (Invitrogen, CA, USA) for 1 h or with Alexa Fluor 594 phalloidin (Invitrogen, CA, USA) for 30 min at 37°C. Then cells were rinsed in PBS with 1% BSA and incubated with secondary goat anti-mouse IgG H&L antibodies conjugated with Cy5 or with donkey anti-rabbit IgG H&L Alexa Fluor 488 (Abcam, Cambridge, UK). Coverslips were attached with a Vectashield Mounting Medium with DAPI (Vector Laboratories, CA, USA). The analysis was performed with an inverted fluorescence microscope Olympus IX81 (Olympus Europa holding Gmbh, Hamburg, Germany) equipped with an Orca-R2 cooled digital camera (Hamamatsu Photonics K.K., Japan), the fluorescence excitation system MT10 (Olympus Life Science Europa Gmbh, Hamburg, Germany), and the fluorescence imaging system XCELLENCE (Olympus Soft Imaging Solutions Gmbh, München, Germany).
2.4. Immunoblot Assay
Cells were lysed in ice-cold lysis buffer (50 mM Tris pH 7.5, 50 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 1% Triton X-100, 1 mM β-glycerophosphate, 1 mM Na3VO4, and protease inhibitor cocktail 1 : 50). Equal amounts of protein were separated by SDS-PAGE on 10% acrylamide gels. Protein concentrations were estimated by the Bradford assay (Bio-Rad, CA, USA). Proteins separated in gel were transferred to the PVDF membrane (GE Healthcare, CA, USA) and then were blocked with blocking buffer containing 5% low fat milk or 5% BSA in TBST (50 mM Tris pH 7.6, 150 mM NaCl, 0.05% Tween-20) for 1 h at room temperature. Membranes were probed with primary antibodies against Cx43 (Transduction Laboratories, Lexington, KY, USA), GFP (Invitrogen, CA, USA), p-JNK, p-c-Jun, and AKT β (Cell Signalling Technology, MA, USA) in 5% low fat milk or 5% BSA in TBST for 24 h at 4°C and after washing with TBST solution were incubated with secondary antibodies, goat anti-rabbit Fab fragment of IgG, and goat anti-mouse conjugated with alkaline phosphatase (Invitrogen, CA, USA), for 1 h at room temperature. The proteins were visualized by the alkaline phosphatase method (Carl Roth GmbH & Co., KG Karlsruhe, Germany).
2.5. Hyperthermic Conditions and Inhibition of JNK Activity
Two hours before the beginning of experiments, cells were washed with freshly equilibrated growth media (37°C, 5% CO2). Subconfluent cultured cells (SkMs and HeLa cells) were subjected to hyperthermia (42°C for 2, 6 or 24 h) in the 5% CO2 incubator. XG-102, a selective JNK inhibitor [53, 54], was applied 30 min before the induction of hyperthermia treatment (4 μM, 37°C, 5% CO2). For the respective control measurements, cells were kept at 37°C for the same periods as those under hyperthermic conditions.
2.6. Measurement of GJIC
For fluorescence recording, cells grown onto glass coverslips were transferred to an experimental chamber with constant flow-through perfusion mounted on the stage of an inverted microscope Olympus IX81. Appropriate excitation and emission filters (Chroma technology, Brattleboro, VT) were used to image Lucifer Yellow CH dilithium salt dye (LY; MW = 457.25; net charge −2, λ ex = 436 nm, λ em = 480 nm). For dye transfer studies, LY was introduced into the first cell of a contiguous cell row through a patch pipette in the whole-cell voltage-clamp mode. Typically, this resulted in rapid loading of the first cell, followed by dye transfer via GJs to the neighboring cells. Fluorescence kinetics was evaluated using the XCELLENCE software (background subtracted). Microelectrode resistance was 2-3 MΩ. Internal solution: NaCl 130 mM, KCl 30 mM, NaAsp 10 mM, CaCl2 0.26 mM, Hepes 5 mM, EGTA 2 mM, TEA 5 mM, MgCl2 1 mM, MgATP 3 mM (pH 7.3), and 2 mM LY; external solution: NaCl 140 mM, KCl 4 mM, CaCl2 2 mM, MgCl2 1 mM, Hepes 5 mM, glucose 5 mM, and pyruvate 2 mM (pH 7.4). LY diffusion was registered for 30 min. To minimize dye bleaching, images were taken every 2 min at low excitation intensity and 200-ms exposure.
2.7. Scrape-Loaded Dye Transfer Measurements
GJIC was studied by measuring scrape-loaded LY transfer. Grown to confluence on the glass coverslips, cells were washed with Ca2+-free PBS. LY solution (100 μL, 2 mM) was applied on each coverslip, and several cuts were made on the monolayer with a surgical blade to introduce the dye into the damaged cells. After incubation for 3 min at room temperature, cells were rinsed three times with PBS containing 2 mM Ca2+, before fixing with 4% paraformaldehyde in PBS for 15 min [55]. LY transfer between cells was evaluated by measuring the exponential decay of LY fluorescence (FD50) perpendicularly from the scrape line. Fluorescence imaging and measurements were performed using the same hardware and software as described in Section 2.3.
2.8. Statistical Analysis
Data are expressed as mean of at least 3 independent experiments ± standard error. The unpaired Student's t-test was used for western blot (WB) quantitative evaluation. For comparison of GJIC data, the density of hemichannels in cell membranes, and the number of channels in junctional plaques (JPs), we used ANOVA analysis with the post hoc Dunnett's or Bonferroni's test. Differences were considered statistically significant at P < 0.05.
3. Results
3.1. Characterization of Skeletal Myoblasts
Desmin, a muscle-specific marker, demonstrated the purity of the experimental cultures of SkMs obtained from the rabbit femoral muscle [56]. Almost all cells (99%) were desmin positive (Figure 1(a)). Myogenin, a marker of myogenic differentiation, was present in around 80% of the cell nuclei (Figure 1(b)). The majority of the SkMs expressed endogenous Cx43 (Figures 1(a) and 1(b)). Araya et al. have suggested that, in addition to Cx43, the skeletal muscle and primary cultures of myoblasts of the mice may express Cx45 [19]; however, in our SkMs, Cx45 expression was not detected by immunofluorescence experiments (data not shown).
Figure 1.
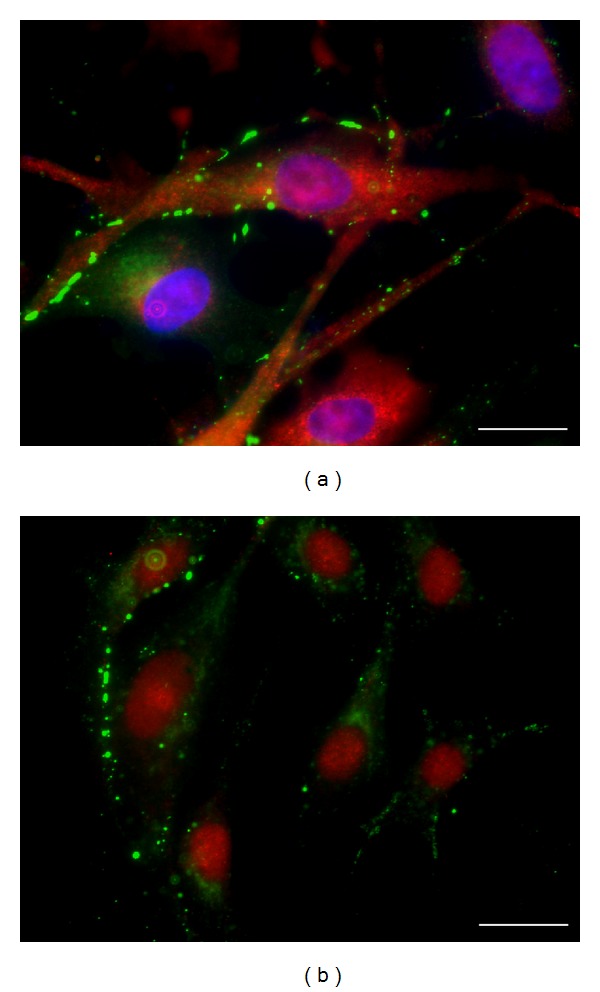
Characterization of rabbit SkMs. (a) Immunostaining of desmin (red) and endogenous Cx43 (green). Nuclei are labeled with DAPI (blue). (b) Immunostaining of myogenin (red) and endogenous Cx43 (green). Scale bar 20 μm.
3.2. Hyperthermia and SkM Intercellular Permeability
Since undifferentiated SkMs express the endogenous Cx43 protein, we used a scrape-loaded dye transfer technique to evaluate GJIC. FD50 of LY was measured perpendicularly from the scrape line (Figures 2(a) and 2(b)). The distance of LY diffusion was 2 times longer in hyperthermically incubated cells compared with control cells. FD50 was 15.7 ± 2.4 μm and 30.3 ± 5.4 μm in control and after 6 h at 42°C, respectively (P < 0.05). Treatment with XG-102 (4 μM), a JNK-inhibitor, had no effect on the FD50 under control conditions but prevented the hyperthermia-induced increase in FD50 (Figure 2(c)).
Figure 2.
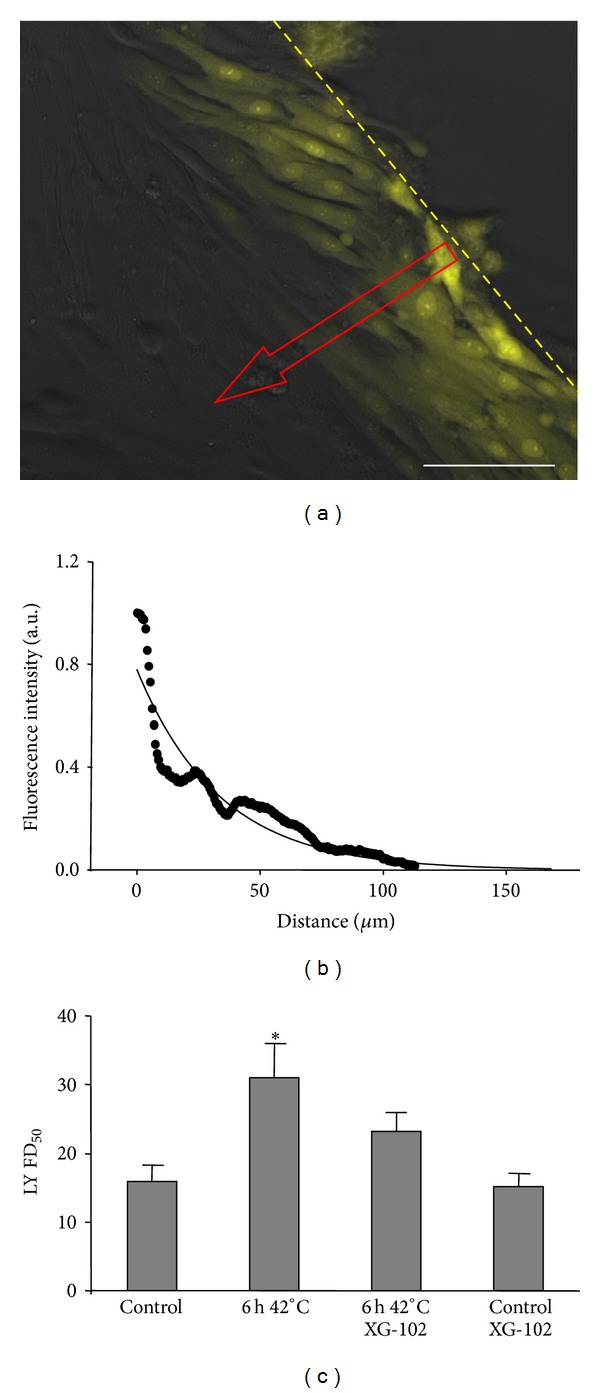
Effect of hyperthermia on Cx43 GJIC in SkMs. (a) LY diffusion between SkMs was measured by the scrape-loaded dye transfer technique (dashed line indicates the scrape margin; arrow shows the measurement direction of the LY diffusion). (b) Kinetics of scrape-loaded LY diffusion under control conditions, measured 3 min after LY application. Raw data were fitted with an exponential decay function. (c) Comparison of scrape-loaded LY diffusion under control and hyperthermia conditions in the presence and absence of XG-102 (4 μM). FD50 was 15.7 ± 2.4, 30.3 ± 5.4, 15.1 ± 1.6, and 22.5 ± 2.8 μm, respectively. n = 10 in each set of experiments; * = P < 0.05, ANOVA followed by post hoc Dunnet's test. Scale bar 100 μm.
3.3. Hyperthermia and Total Cx43 Protein Expression in SkMs
Thus, hyperthermia treatment caused the JNK-dependent amelioration of GJIC in the SkM culture. To determine whether these changes were caused by an increase in total Cx43 protein expression, the total amounts of activated JNK (p-JNK), activated c-Jun (p-c-Jun), and Cx43 protein were measured by WB under all experimental conditions described above. Hyperthermia activated JNK signaling in SkMs resulting in significantly increased levels of phosphorylated JNK, phosphorylated c-Jun, and Cx43 protein by 1.3-, 1.6-, and 2.8-fold, respectively (Figure 3). XG-102 did not affect JNK phosphorylation since the phosphorylation domain differs from the catalytic domain. In contrast, XG-102 significantly inhibited the basal and hyperthermia-stimulated activity of the JNK substrate c-Jun (Figure 3(b)). In addition, XG-102 prevented the upregulation of Cx43 by hyperthermia (Figure 3(c)).
Figure 3.
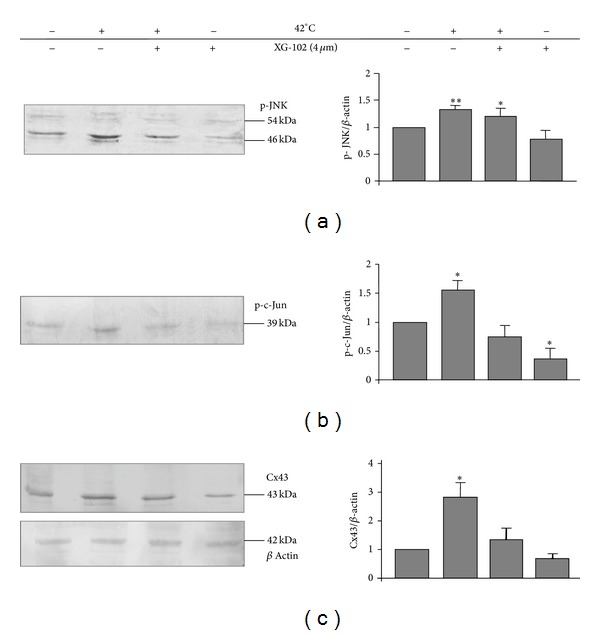
Effect of hyperthermia on the total expression of p-JNK (a), p-c-Jun (b), and endogenous Cx43 protein (c) in SkMs. n = 3 in each set of experiments; * = P < 0.05; ** = P < 0.01, Student's t-test.
3.4. The Effect of Hyperthermia and JNK on Cx43-EGFP GJ Plaque Size and Membranous Hemichannel Density in HeLa Cells
HeLa cells do not express the Cx43 protein. Thus, the transfection of HeLa cells with Cx43 conjugated with enhanced green fluorescent protein (Cx43-EGFP) allows the analysis of GJ channel and hemichannel formation. By conventional fluorescence microscopy and TIRF microscopy, we determined the hyperthermia-induced changes in GJ plaque size and membranous hemichannel density, respectively. We also measured changes in LY transfer between several contiguous cells in a row, as described previously [57, 58], and changes in total Cx43-EGFP protein expression by WB.
JPs varied in size from small puncta with a diameter of less than 1 μm to very large JPs, up to ~10 μm in length, occupying almost the entire region of the cell-cell contact (Figure 4(a)). To determine the number of GJ channels in HeLaCx43-EGFP cells, initially, we searched for overlapping cell pairs that showed JPs in contact regions and the JP could be seen en face, as it is illustrated in Figure 4(b). Imaging at different focus planes showed that some of the selected JPs were large, oriented parallel to the focal plane, and exhibited nearly homogeneous fluorescence intensity. To obtain fluorescence per unit area, emitted light was integrated over a given area of uniform fluorescence (region of interest, ROI). Fluorescence per unit area in en face JPs (F JP) was evaluated in arbitrary fluorescence units, a.u. (background fluorescence measured outside the JP was subtracted). In all experiments, we used the same objective, light intensity, and exposure time for fluorescence imaging. We estimated fluorescence produced by a single GJ channel (F γ) using F JP and the density of channel packing. It was assumed that each Cx43-EGFP channel occupied 100 nm2 (corresponding to 10 nm center-to-center in a square array) as it was shown in electron microscopy studies of junctions in fixed tissues [59, 60] or isolated gap junctions imaged in aqueous media by atomic force microscopy [61]. In the present study, F JP was equal to 503 ± 14 a.u. per 1 μm2 on average (n = 14). From the ratio, 503 a.u./10 000 channels per 1 μm2, F γ = 0.05 a.u. was calculated. Furthermore, the total fluorescence intensity of JPs (F T) was measured independently on their spatial orientation. F T was estimated by measuring the total fluorescence in the ROI enclosing JP as it is shown in Figure 4(a) (background subtracted). The number of physical channels (N T) present in each JP was determined from the following ratio: N T = F T/F γ. We found that under control conditions (37°C), JPs of various size, ranging from 1 to 12 μm, contained 120 935 ± 21 288 (n = 73) channels.
Figure 4.
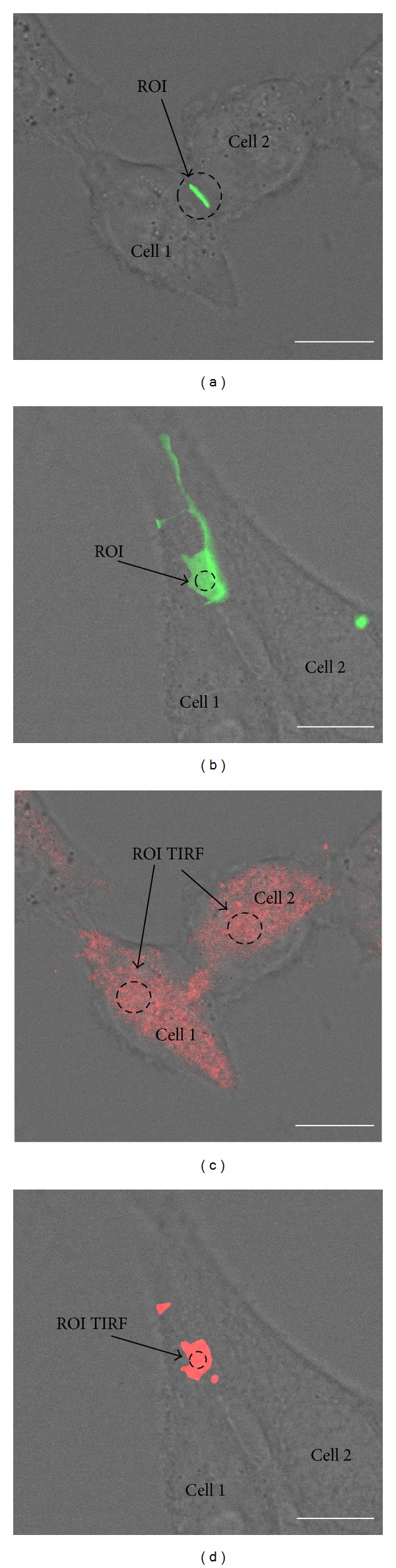
Measurement of Cx43-EGFP GJ channel number in JPs and hemichannel density in the plasma membrane of HeLa cells. (a) To estimate the total fluorescence of Cx43-EGFP JP (green), emitted light was integrated over the ROI encircling JP. (b) To estimate fluorescence intensity of the single Cx43-EGFP GJ channel, emitted light was integrated over the ROI situated in the horizontal JP of overlapping cells. (c) TIRF microscopy was used to estimate the density of Cx43-EGFP hemichannels (red) on the cell surface. Emitted light was integrated over the ROI situated in the site with most homogenous fluorescence. (d) To estimate the fluorescence intensity of the single Cx43-EGFP GJ hemichannel, emitted light was integrated by a TIRF microscope over the ROI situated in the horizontal JP of overlapping cells. Details are presented in the main text. Scale bar 10 μm.
In order to determine the density of hemichannels in HeLaCx43-EGFP cell membranes by TIRF (Figure 4(c)), we analyzed JPs that could be seen en face at the maximal proximity to coverslip surface (Figure 4(d)). To obtain fluorescence per unit area, we used plaques where fluorescence in the region of interest (ROI TIRF) was homogeneous. We estimated fluorescence produced by single hemichannel (Fhγ = F JP/2) using the same density of channel packing as above. We found that F JP was equal to 239 ± 6 a.u. per 1 μm2 on average (n = 21). From the ratio, 239 a.u./20 000 hemichannels per 1 μm2, we found that Fhγ = 0.012 a.u. Further, we measured the total fluorescence intensity of ROI TIRF (FhT, background subtracted, Figure 4(c)). The density of hemichannels (DhT) on HeLa cell surface was determined from the following ratio: DhT = FhT/Fhγ. Under control conditions, the Cx43-EGFP hemichannel density on HeLa cell surface was 1514 ± 69 (n = 236) per 1 μm2.
In the next set of experiments, HeLa cells were preincubated at 42°C for 2, 6, and 24 hours. Surprisingly, hyperthermia caused the opposite effects in HeLa cells compared with SkMs. The maximal effect of hyperthermia on Cx43-EGFP expression was achieved after 6 h; therefore, here we do not show respective results obtained after 2 and 24 hours of incubation under hyperthermic conditions. After 6 h of hyperthermic incubation, the number of Cx43-EGFP channels in JPs decreased by 34% (P < 0.05), and the density of hemichannels decreased by 52% (P < 0.005) (Figure 5).
Figure 5.
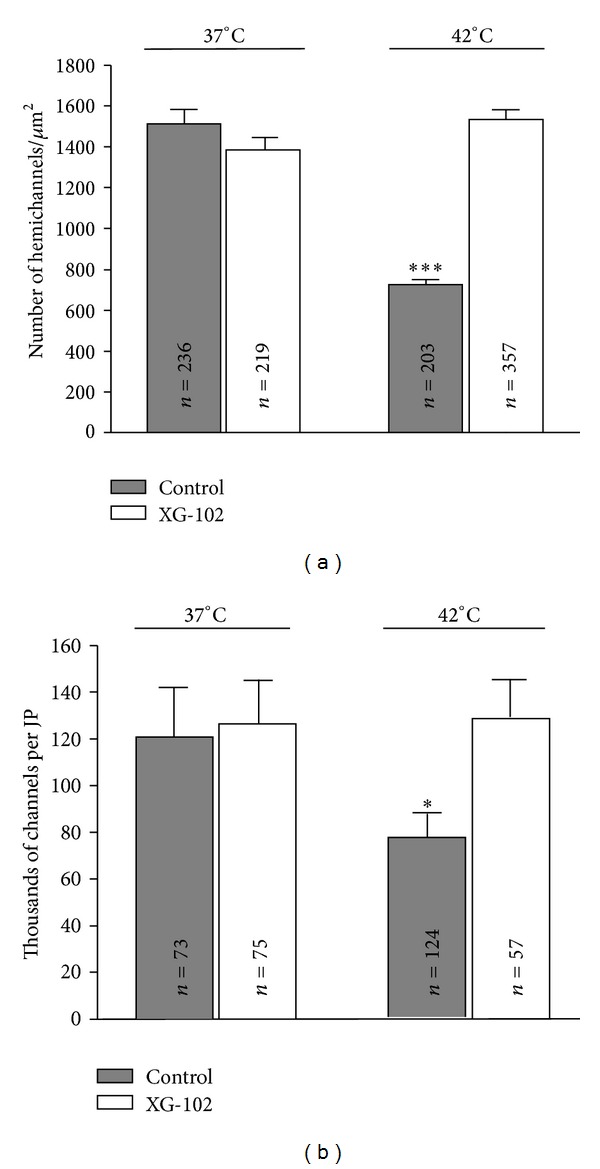
The effect of hyperthermia on the hemichannel density in Cx43-EGFP HeLa cell membranes (a) and number of channels in the JPs (b) in the absence and presence of JNK inhibitor XG-102 (4 μM). n is indicated on the respective bar; * = P < 0.05; *** = P < 0.005, ANOVA followed by post hoc Bonferroni's test.
Preincubation of HeLa cells at 37°C with XG-102 (4 μM) had no effect on the number of Cx43-EGFP channels in JPs and on the density of hemichannels (Figure 5). However, the inhibition of JNK completely prevented the effect of hyperthermia; that is, the density of Cx43-EGFP hemichannels and the number of GJ channels were almost identical as in untreated controls at 37°C or 42°C.
Again, the inhibition of basal JNK activity at 37°C did not affect these parameters suggesting that hyperthermia activates different signal cascades as it does at the normal physiological temperature.
3.5. The Effect of Hyperthermia and JNK on Cx43 GJ Permeability in HeLa Cells
To evaluate the effect of hyperthermia on GJIC, LY was introduced through the patch-pipette into the first cell, and diffusion of LY was measured in at least 5 cells in a row with Cx43-EGFP GJ plaques between them (Figure 6(a)). The experiments were carried out under control (37°C) and hyperthermic conditions (6 h, 42°C) with and without XG-102 (4 μM). Hyperthermia slowed down the LY diffusion between cells through GJs (Figures 6(b) and 6(c)). Under hyperthermic conditions, LY fluorescence in the fifth cell after 3 min from its introduction into the first cell was 2.9-fold lower compared with control (P < 0.05). XG-102, a JNK inhibitor, did not affect GJIC under control conditions but completely abolished the effect of hyperthermia on GJIC.
Figure 6.
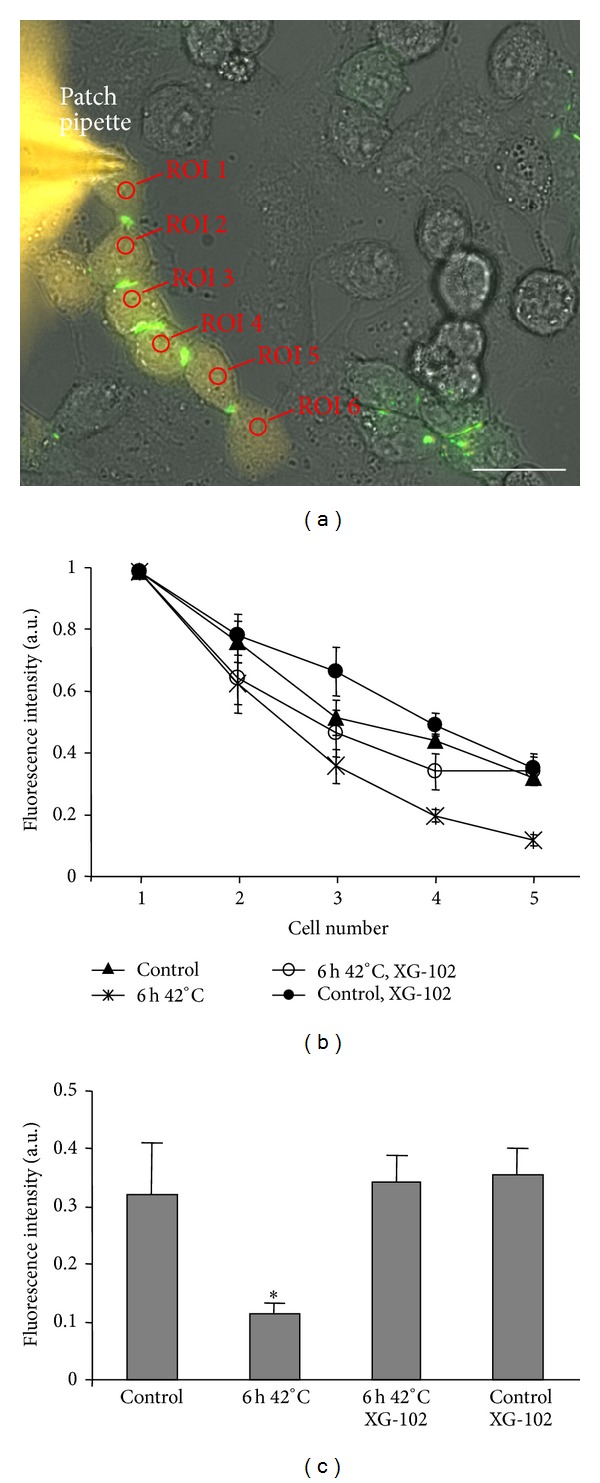
The effect of hyperthermia on Cx43-EGFP GJIC in HeLa cells. (a) LY was introduced through the patch pipette into the first cell of six cells in a row. (b) Kinetics of LY diffusion under different conditions. (c) LY fluorescence intensity in the fifth cell 3 min after its introduction into the first cell under control and hyperthermic conditions in the absence and presence of XG-102 (4 μM). n = 8 in each set of experiments. Scale bar 25 μm. * = P < 0.05, ANOVA followed by post hoc Dunnet's test.
3.6. The Effect of Hyperthermia and JNK on Cx43 Protein Expression in HeLa Cells
In HeLa cells, hyperthermia enhanced the activity of JNK and c-Jun by 1.4- and 1.8-fold, respectively (P < 0.05) (Figures 7(a) and 7(b)), similarly as seen in SkMs (Figures 3(a) and 3(b)). In contrast to SkMs, hyperthermia reduced the expression of Cx43-EGFP by ~35% (P < 0.005). XG-102 did not change the effect of hyperthermia on JNK phosphorylation but completely abolished the increase in phosphorylated c-Jun and a decline in Cx43-EGFP expression (Figures 7(b) and 7(c)). Under control conditions, XG-102 decreased the basal activity of c-Jun to ~20% of control (P < 0.01) but did not change the basal expression of Cx43.
Figure 7.
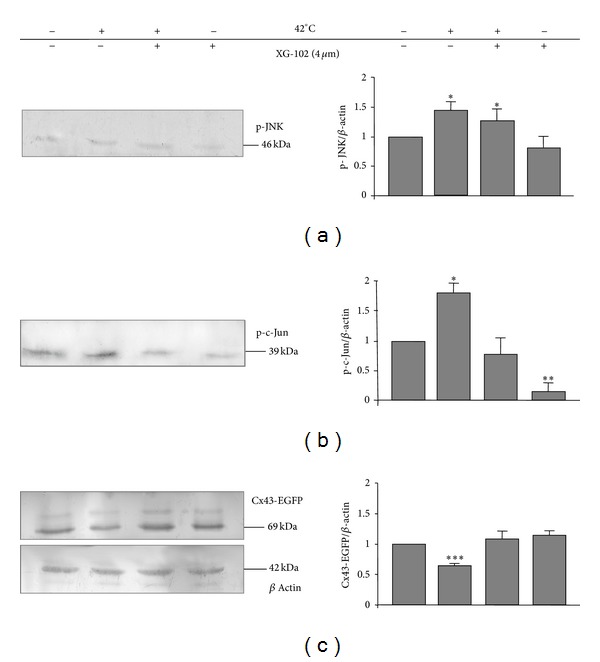
Effect of hyperthermia on the total expression of p-JNK (a), p-c-Jun (b), and Cx43-EGFP proteins (c) in HeLa cells. n = 3 in each set of experiments; * = P < 0.05; ** = P < 0.01; *** = P < 0.005, Student's t test.
Thus, in SkMs (Figure 3) and HeLa cells (Figure 7), hyperthermia induced JNK activation that exerted the opposite effects on Cx43 expression.
3.7. Impact of Hyperthermia on Actin Cytoskeleton in HeLa Cells and SkMs
Hemichannels from the Golgi apparatus are delivered to the plasma membrane through the tubulin and actin network, and this delivery can be affected by inflammation and heat shock [62, 63]. JNK could also be involved into remodeling and polymerization of F-actin underlying changes in cell motility [64]. Moreover, the changes in Cx expression and GJIC could affect the structure of the cytoskeleton [65]. Therefore, we wanted to know to which extent hyperthermia and JNK signaling are involved in actin cytoskeleton organization.
HeLa Cells. The exposure of HeLa cells expressing Cx43-EGFP to 42°C for 6 h drastically changed the actin filament network. Under control conditions, 4–6 thick integral actin filaments can be detected along HeLa cells (Figure 8(a)). After hyperthermia treatment, no actin filament in the center of the cells was observed, and only a few cells contained 1-2 actin filaments near the cell borders (Figure 8(b)). Since under hyperthermic conditions, the total fluorescence of actin did not change, we assume that the ratio of polymerized/nonpolymerized forms of actin was modified. The actin remodeling under hyperthermic conditions was substantially prevented by XG-102 (Figures 8(c) and 8(d)).
Figure 8.
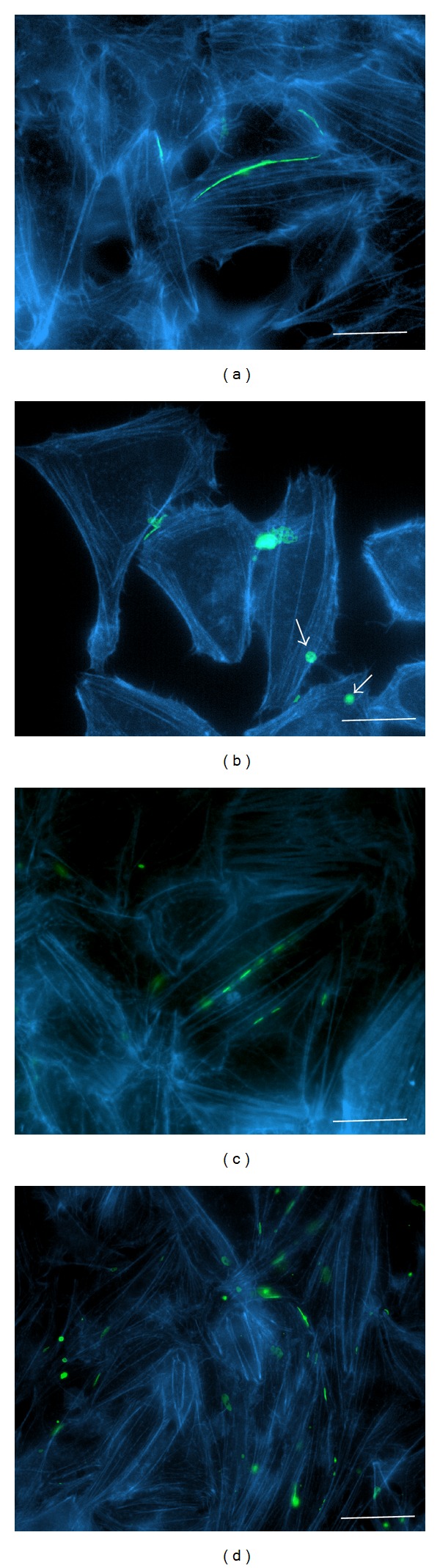
Impact of hyperthermia on actin filament organization in HeLa cells expressing Cx43-EGFP under control conditions (a); after hyperthermia (6 h at 42°C) (b); under control conditions with XG-102 (4 μM) (c); after hyperthermia (6 h at 42°C) with XG-102 (4 μM) (d). Blue: F-actin labeled with Alexa Fluor 594 phalloidin; green: Cx43-EGFP; white arrows indicate annular junctions containing internalized GJ plaques. Scale bar 25 μm.
SkM Cells. In contrast, actin filaments remained intact and formed an integral actin network in SkMs after 6 h incubation at 42°C (Figure 9). XG-102 had no impact on the SkM actin network under control or hyperthermic conditions.
Figure 9.
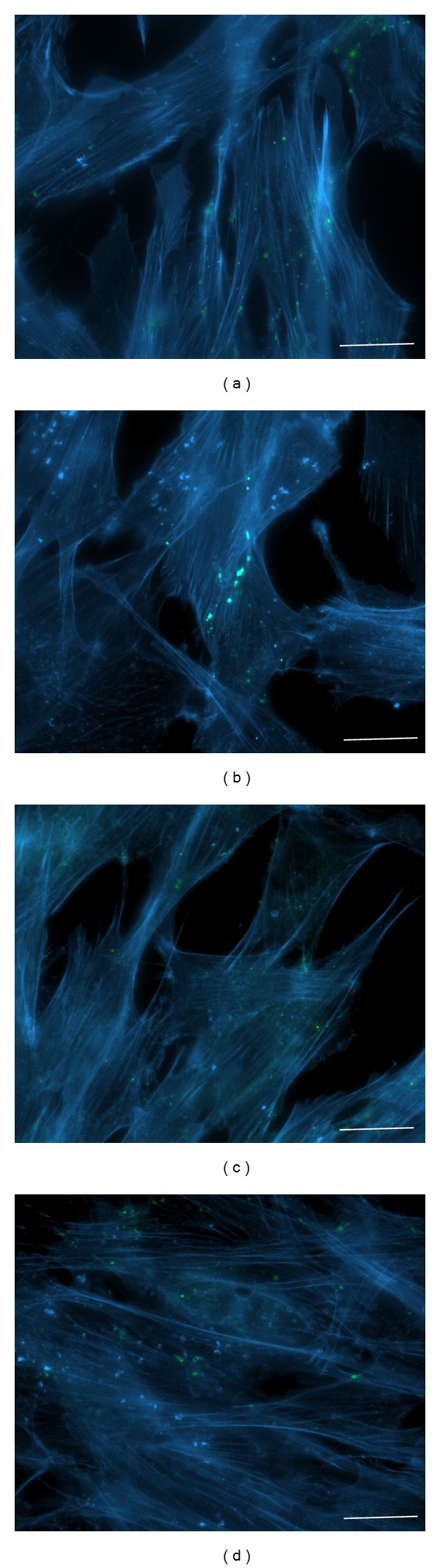
Impact of hyperthermia on actin filament organization in SkMs under control conditions (a); after hyperthermia (6 h at 42°C) (b); under control conditions with XG-102 (4 μM) (c); after hyperthermia (6 h at 42°C) with XG-102 (4 μM) (d). Blue: F-actin labeled with Alexa Fluor 594 phalloidin; green: Cx43-EGFP. Scale bar 25 μm.
4. Discussion
The present study demonstrates that (i) in SkMs, hyperthermia induced the expression of Cx43 and enhanced the efficiency of GJIC but did not affect the actin cytoskeleton; (ii) in Cx43-EGFP-transfected HeLa cells, hyperthermia reduced the Cx43-EGFP expression and the efficiency of GJIC and disrupted the actin cytoskeleton. These opposite effects could be both antagonized by the inhibition of JNK/c-Jun stress pathway, which was activated by hyperthermia. These data support the idea that MAPK signaling may exert divergent, even opposite, effects depending on the cellular context.
4.1. JNK Mediate the “Positive” Effects of Hyperthermia in SkMs
In SkMs, hyperthermia increased the expression of Cx43 protein and improved GJIC, whereas the actin cytoskeleton was insensitive to hyperthermia. It is known that the stress-activated protein kinase JNK pathway can be activated upon the exposure of mammalian cells to hyperthermic stress [66]. For example, heat shock improves resistance to hypoxia-reoxygenation injury in vitro and enhances the survival of SkMs engrafted into the heart [33]. Impaired cell-to-cell coupling correlates with reduced Cx43 expression [67] and vice versa Cx43 upregulation improves GJIC [68]. However, the total Cx43 protein amount and even the total channel number in JPs are not sufficient indicators of the efficiency of cell-to-cell communication because only very small fraction of channels in JPs is functional for yet unknown reasons [69, 70]. Many other factors, such as transjunctional voltage, intracellular Ca2+, pH, and phosphorylation, also contribute to the gating and permeability properties of GJ channels [71–75]. The resistance of actin cytoskeleton to hyperthermia in SkMs could contribute to therapeutic effects of hyperthermia since actin filaments have been shown to be involved in the trafficking of Cx43 hemichannels to the plasma membrane [76]. In addition, the actin cytoskeleton may affect GJIC indirectly by targeting kinases, phosphatases, and other regulatory molecules to GJs [77].
4.2. JNK Mediate the “Negative” Effects of Hyperthermia in HeLa Cx43-EGFP Cells
In HeLa cells, hyperthermia diminished Cx43 protein levels, size of Cx43 GJ plaques, density of membranous Cx43 hemichannels, and efficiency of GJIC. These changes were blocked by the inhibition of JNK. JNK plays a major role in the cellular response to stress induced by inflammation and this is also true in myocardial I/R [11, 78, 79]. Controversial results were reported in different cell types concerning JNK activation, Cx43 expression, and Cx43 GJIC depending on whether pro- or antiapoptotic signaling pathways were activated [80]. While JNK mediated apoptosis in adult rat myocytes [81], it demonstrated antiapoptotic properties associated with the Akt signaling pathway in neonatal rat cardiomyocytes after I/R [41]. Interestingly, infarction size and lesions were smaller in the rat heart, when the JNK activation pathway was blocked by XG-102 [44]. The divergent effects of JNK actions can be mediated by individual isoforms and specific signalosomes activated in different cellular compartments. The improvement of Cx43-mediated intercellular communication possibly could have protective functions in the ischemic heart due to improved homeostasis. On the other hand, GJs may transmit cytotoxic materials as well [82], and consequently, the administration of GJ blockers might reduce the infarction size [83].
The role of JNK in the regulation of Cx43 expression in cardiac myocytes is also ambiguous. For instance, the inactivation of the JNK/c-Jun pathway decreased Cx43 expression in the cardiomyocytes of transgenic animals lacking MKK4 (JNK kinase) after phenylephrine stimulation [84], or JNK activation after amphetamine application increased the expression of Cx43 protein in cardiomyocytes [51]. In contrast, JNK activation mediated the downregulation of Cx43 expression in cardiomyocytes [52]. There are no data available on the JNK-dependent regulation of Cx43 protein expression in SkMs; however, the proliferation and differentiation processes in these cells may depend on the variations of Cx43 expression [85, 86].
4.3. JNK Inhibition Effectively Blocks the Outcomes of Hyperthermia
The inhibition of JNK by XG-102, a small-molecule inhibitor, was specific in two aspects. First, it did not affect the phosphorylation of JNK, since XG-102 blocks the binding domain but not the phosphorylated activation domain. Second, XG-102 inhibited the JNK-dependent activation, that is, the phosphorylation of c-Jun. XG-102 blocked hyperthermia-induced “positive” outcomes in SkMs and “negative” outcomes in Cx43-transfected HeLa cells. Thus, the signal-transcription coupling apparently differs between SkMs and Cx43-transfected HeLa cells. Also, the exogenous Cx43-EGFP might be differently more coupled to the JNK pathway than endogenous Cx43 proteins. However, in Cx43-EGFP-transfected SkMs, hyperthermia had a similar effect on GJIC as in wild type SkMs (our unpublished observation). These findings are in line with the concept of individual biological JNK signaling. Not only JNK isoforms (JNK1, 2, and 3) differ in their actions, but also one isoform can exert opposite actions depending on its intracellular localization and position within a given signalosome. In this context, the upstream MKK signaling with its spliced isoforms is an essential conductor for the orchestration of context-specific cellular responses [50, 87].
JNK has a pronounced basal activity, which contributes to normal cell functionality including proliferation [50, 88]. However, for the modulation of Cx43 expression and GJIC, much higher activity of JNK was required since the inhibition of basal activity of JNK by XG-102 had only a minor effect on these parameters whereas the hyperthermia-induced changes were blocked by XG-102. Also, in HeLa cells and SkMs, different JNK isoforms may realize JNK actions. We observed only one 46-kDa band in the lysates of HeLa cells, but typical 46- and 56-kDa (indicating p-JNK1 and p-JNK2) bands in the SkM samples.
4.4. Cytoskeleton and Hyperthermia
A close interaction between Cx43 and F-actin was demonstrated in various studies. For instance, on the one hand, cytochalasin B, a F-actin disrupter, was shown to inhibit Cx transport and GJ assembly [89] and vice versa GJ inhibitors (octanol, 18β-glycyrrhetinic acid) caused the discordance of actin stress fibers in embryonic rat astrocytes [90]. The functional blockade of GJ impaired F-actin fiber alignment and consequently reduced the migration of breast cancer cells [91]. Since hyperthermia may trigger the rearrangement of cytoskeleton proteins [92, 93], it could modify the Cx43 trafficking to the plasma membrane and GJ plaques. We demonstrate that the same hyperthermic conditions differently affected the F-actin network in HeLa cells and SkMs: after incubation of cells for 6 h at 42°C, the actin cytoskeletal network was disrupted in HeLa cells but remained intact in SkMs. These findings confirm previous observations that, depending on the cellular context and the type of stress stimuli [92, 93], the individual effects are modulated by different JNK isoforms [54, 94, 95].
Contradictory results can be found in the literature concerning the effect of inflammation and/or heat stress on Cx43 expression levels. For instance, after the exposure of neonatal rat cardiomyocytes to 43.5°C for 30 min, a drastic degradation of Cx43 was observed [96], while in human HE49 fibroblasts, the Cx43 expression level remained unchanged even after 8 h preincubation at 43°C [97]. Repeated heat shock does not alter Cx43 expression, possibly due to protective activity of Hsp70. Another heat shock protein Hsp72 was associated with increased resistance to hypoxia in rat L6 myoblasts [33]. The incubation of L6 cells at 42°C for 1 hour before transplantation into the heart increased the viability of preconditioned cells. An increase in Cx43 expression and intercellular communication under hyperthermic conditions might facilitate SkM integration into the damaged myocardium, presumably due to improved electrical communication between SkMs and cardiac myocytes. In turn, the reduction of Cx43 expression and impairment of GJ communication in cancer cells is closely related with the disruption of the actin filament network. A number of carcinogenesis studies have shown that levels of Cx43 or other connexin GJs are downregulated in cancer cells; decreased GJIC is considered as an important factor in oncogenesis [98–100] as well as the increased cytoplasmic pool of Cxs when Cx protein synthesis remains unchanged but connexon trafficking to the plasma membrane is inhibited due to disruption of the actin network [76]. In our experiments with HeLa cells, the decreased Cx43 GJ plaque size and hemichannel density after hyperthermic treatment clearly correlated with the breakdown of the actin network and possibly with a decrease in protein synthesis.
For many years hyperthermia has been known as a therapeutic tool for the treatment of certain cancer types [101], because cancer cells are particularly sensitive to hyperthermia [102]. Our finding that the effects of moderate hyperthermia on the cytoskeletal structure differ in normal SkMs and cancer (HeLa) cells indicate that molecular mechanisms underlying the sensitivity of cellular response to hyperthermia are complex and involve cell type-dependent changes of membrane properties, signaling pathways, ion homeostasis, protein expression, ROS generation, redox disbalance, and possible energy crisis due to mitochondrial dysfunction [103–105].
5. Conclusions
In SkMs, hyperthermia had no effect on the actin filament network and increased the total amount of Cx43 and the efficiency of GJIC. Thus, hyperthermia pretreatment of SkMs may improve their efficiency in the strategies of heart regeneration.
In Cx43-EGFP-expressing HeLa cells, hyperthermia decreased the total amount of the Cx43-EGFP protein, number of channels in GJ plaques, density of hemichannels in the cell membranes, and efficiency of GJIC. These effects correlated with the downregulation of the actin filament network.
JNK is a central mediator of hyperthermia-induced cellular responses in both SkMs and HeLa cells.
The comparison of SkMs and HeLa cells provides an attractive model to identify the regulatory players within signalosomes, which determine the cell-specific outcomes of hyperthermia.
Acknowledgments
This work was supported by Lithuanian State Science and Studies Foundation Grant no. B-26/2007. The authors thank Professor Feliksas Bukauskas for providing HeLa cells expressing Cx43-EGFP.
Abbreviations
- Cx43:
Connexin 43
- DhT:
Density of physical hemichannels
- EGFP:
Enhanced green fluorescent protein
- ERK1/2:
Extracellular-signal-regulated kinases 1/2
- FD50:
Exponential decay constant estimated as a time after which dye fluorescence was equal to half of maximal
- FJP:
Fluorescence per unit area in en face JPs
- Fγ:
Fluorescence produced by single GJ channel
- Fhγ:
Fluorescence produced by single hemichannel
- FhT:
Total fluorescence intensity of ROI TIRF
- GJ:
Gap junction
- GJIC:
Gap junction intercellular communication
- I/R:
Ischemia/reperfusion
- JNK:
c-Jun NH2-terminal kinase
- JP:
Junctional plaque
- LY:
Lucifer yellow CH dilithium salt
- MAPK:
Mitogen activated protein kinase
- MI:
Myocardial infarction
- NT:
Number of physical channels
- ROI:
Region of interest
- p-c-Jun:
Phosphorylated c-jun
- p-JNK:
Phosphorylated JNK
- SkM:
Skeletal myoblast
- TIRF:
Total internal reflection fluorescence
- VEGF:
Vascular endothelial growth factor
- WB:
Western blot.
Conflict of Interests
The authors declare no conflict of interests.
References
- 1.Menasche P. Skeletal myoblasts as a therapeutic agent. Progress in Cardiovascular Diseases. 2007;50(1):7–17. doi: 10.1016/j.pcad.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Camirand G, Rousseau J, Ducharme ME, Rothstein DM, Tremblay JP. Novel Duchenne muscular dystrophy treatment through myoblast transplantation tolerance with anti-CD45RB, anti-CD154 and mixed chimerism. American Journal of Transplantation. 2004;4(8):1255–1265. doi: 10.1111/j.1600-6143.2004.00501.x. [DOI] [PubMed] [Google Scholar]
- 3.Mouly V, Aamiri A, Perie S, et al. Myoblast transfer therapy: is there any light at the end of the tunnel? Acta Myologica. 2005;24(2):128–133. [PubMed] [Google Scholar]
- 4.Tremblay JP, Malouin F, Roy R, et al. Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplantation. 1993;2(2):99–112. doi: 10.1177/096368979300200203. [DOI] [PubMed] [Google Scholar]
- 5.Smythe GM, Hodgetts SI, Grounds MD. Problems and solutions in myoblast transfer therapy. Journal of Cellular and Molecular Medicine. 2001;5(1):33–47. doi: 10.1111/j.1582-4934.2001.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McConnell PI, del Rio CL, Jacoby DB, et al. Correlation of autologous skeletal myoblast survival with changes in left ventricular remodeling in dilated ischemic heart failure. The Journal of Thoracic and Cardiovascular Surgery. 2005;130(4):1001.e1001–1001.e1012. doi: 10.1016/j.jtcvs.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 7.Hata H, Matsumiya G, Miyagawa S, et al. Grafted skeletal myoblast sheets attenuate myocardial remodeling in pacing-induced canine heart failure model. The Journal of Thoracic and Cardiovascular Surgery. 2006;132(4):918–924. doi: 10.1016/j.jtcvs.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 8.Gavira JJ, Nasarre E, Abizanda G, et al. Repeated implantation of skeletal myoblast in a swine model of chronic myocardial infarction. European Heart Journal. 2010;31(8):1013–1021. doi: 10.1093/eurheartj/ehp342. [DOI] [PubMed] [Google Scholar]
- 9.Menasche P, Alfieri O, Janssens S, et al. The Myoblast Autologous Grafting in Ischemic Cardiomyopathy (MAGIC) trial: first randomized placebo-controlled study of myoblast transplantation. Circulation. 2008;117(9):1189–1200. doi: 10.1161/CIRCULATIONAHA.107.734103. [DOI] [PubMed] [Google Scholar]
- 10.Suzuki K, Murtuza B, Beauchamp JR, et al. Dynamics and mediators of acute graft attrition after myoblast transplantation to the heart. The FASEB Journal. 2004;18(10):1153–1155. doi: 10.1096/fj.03-1308fje. [DOI] [PubMed] [Google Scholar]
- 11.Ren G, Dewald O, Frangogiannis NG. Inflammatory mechanisms in myocardial infarction. Current Drug Targets: Inflammation & Allergy. 2003;2(3):242–256. doi: 10.2174/1568010033484098. [DOI] [PubMed] [Google Scholar]
- 12.Anderson JD, Honigman B. The effect of altitude-induced hypoxia on heart disease: do acute, intermittent, and chronic exposures provide cardioprotection? High Altitude Medicine and Biology. 2011;12(1):45–55. doi: 10.1089/ham.2010.1021. [DOI] [PubMed] [Google Scholar]
- 13.van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovascular Research. 2004;61(2):218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 14.Fernandes S, Amirault J-C, Lande G, et al. Autologous myoblast transplantation after myocardial infarction increases the inducibility of ventricular arrhythmias. Cardiovascular Research. 2006;69(2):348–358. doi: 10.1016/j.cardiores.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Haider H, Ashraf M. Preconditioning and stem cell survival. Journal of Cardiovascular Translational Research. 2010;3(2):89–102. doi: 10.1007/s12265-009-9161-2. [DOI] [PubMed] [Google Scholar]
- 16.Rackauskas M, Neverauskas V, Skeberdis VA. Diversity and properties of connexin gap junction channels. Medicina. 2010;46(1):1–12. [PubMed] [Google Scholar]
- 17.Davis LM, Kanter HL, Beyer EC, Saffitz JE. Distinct gap junction protein phenotypes in cardiac tissues with disparate conduction properties. Journal of the American College of Cardiology. 1994;24(4):1124–1132. doi: 10.1016/0735-1097(94)90879-6. [DOI] [PubMed] [Google Scholar]
- 18.Kanter HL, Laing JG, Beyer EC, Green KG, Saffitz JE. Multiple connexins colocalize in canine ventricular myocyte gap junctions. Circulation Research. 1993;73(2):344–350. doi: 10.1161/01.res.73.2.344. [DOI] [PubMed] [Google Scholar]
- 19.Araya R, Eckardt D, Maxeiner S, et al. Expression of connexins during differentiation and regeneration of skeletal muscle: functional relevance of connexin43. Journal of Cell Science. 2005;118, part 1:27–37. doi: 10.1242/jcs.01553. [DOI] [PubMed] [Google Scholar]
- 20.Reinecke H, MacDonald GH, Hauschka SD, Murry CE. Electromechanical coupling between skeletal and cardiac muscle: implications for infarct repair. The Journal of Cell Biology. 2000;149(3):731–740. doi: 10.1083/jcb.149.3.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menasche P. Stem cell therapy for heart failure: are arrhythmias a real safety concern? Circulation. 2009;119(20):2735–2740. doi: 10.1161/CIRCULATIONAHA.108.812693. [DOI] [PubMed] [Google Scholar]
- 22.Abraham MR, Henrikson CA, Tung L, et al. Antiarrhythmic engineering of skeletal myoblasts for cardiac transplantation. Circulation Research. 2005;97(2):159–167. doi: 10.1161/01.RES.0000174794.22491.a0. [DOI] [PubMed] [Google Scholar]
- 23.Roell W, Lewalter T, Sasse P, et al. Engraftment of connexin 43-expressing cells prevents post-infarct arrhythmia. Nature. 2007;450(7171):819–824. doi: 10.1038/nature06321. [DOI] [PubMed] [Google Scholar]
- 24.Fernandes S, van Rijen HV, Forest V, et al. Cardiac cell therapy: overexpression of connexin43 in skeletal myoblasts and prevention of ventricular arrhythmias. Journal of Cellular and Molecular Medicine. 2009;13(9):3703–3712. doi: 10.1111/j.1582-4934.2009.00740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangi AA, Noiseux N, Kong D, et al. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nature Medicine. 2003;9(9):1195–1201. doi: 10.1038/nm912. [DOI] [PubMed] [Google Scholar]
- 26.Haider H, Ye L, Jiang S, et al. Angiomyogenesis for cardiac repair using human myoblasts as carriers of human vascular endothelial growth factor. Journal of Molecular Medicine. 2004;82(8):539–549. doi: 10.1007/s00109-004-0546-z. [DOI] [PubMed] [Google Scholar]
- 27.Kanemitsu N, Tambara K, Premaratne GU, et al. Insulin-like growth factor-1 enhances the efficacy of myoblast transplantation with its multiple functions in the chronic myocardial infarction rat model. The Journal of Heart and Lung Transplantation. 2006;25(10):1253–1262. doi: 10.1016/j.healun.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Kofidis T, de Bruin JL, Yamane T, et al. Insulin-like growth factor promotes engraftment, differentiation, and functional improvement after transfer of embryonic stem cells for myocardial restoration. Stem Cells. 2004;22(7):1239–1245. doi: 10.1634/stemcells.2004-0127. [DOI] [PubMed] [Google Scholar]
- 29.Haider HK, Jiang S, Idris NM, Ashraf M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1α/CXCR4 signaling to promote myocardial repair. Circulation Research. 2008;103(11):1300–1308. doi: 10.1161/CIRCRESAHA.108.186742. [DOI] [PubMed] [Google Scholar]
- 30.Kojima A, Goto K, Morioka S, et al. Heat stress facilitates the regeneration of injured skeletal muscle in rats. Journal of Orthopaedic Science. 2007;12(1):74–82. doi: 10.1007/s00776-006-1083-0. [DOI] [PubMed] [Google Scholar]
- 31.Yang S, Laumonier T, Menetrey J. Heat shock pretreatment enhances porcine myoblasts survival after autotransplantation in intact skeletal muscle. Science in China C: Life Sciences. 2007;50(4):438–446. doi: 10.1007/s11427-007-0065-6. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura Y, Yasuda T, Weisel RD, Li R-K. Enhanced cell transplantation: preventing apoptosis increases cell survival and ventricular function. American Journal of Physiology: Heart and Circulatory Physiology. 2006;291(2):H939–H947. doi: 10.1152/ajpheart.00155.2006. [DOI] [PubMed] [Google Scholar]
- 33.Suzuki K, Smolenski RT, Jayakumar J, Murtuza B, Brand NJ, Yacoub MH. Heat shock treatment enhances graft cell survival in skeletal myoblast transplantation to the heart. Circulation. 2000;102(19, supplement 3):III216–III221. doi: 10.1161/01.cir.102.suppl_3.iii-216. [DOI] [PubMed] [Google Scholar]
- 34.Vassalli G, Milano G, Moccetti T. Role of mitogen-activated protein kinases in myocardial ischemia-reperfusion injury during heart transplantation. Journal of Transplantation. 2012;2012:18 pages. doi: 10.1155/2012/928954.928954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yue T-L, Wang C, Gu J-L, et al. Inhibition of extracellular signal-regulated kinase enhances ischemia/reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circulation Research. 2000;86(6):692–699. doi: 10.1161/01.res.86.6.692. [DOI] [PubMed] [Google Scholar]
- 36.Lips DJ, Bueno OF, Wilkins BJ, et al. MEK1-ERK2 signaling pathway protects myocardium from ischemic injury in vivo. Circulation. 2004;109(16):1938–1941. doi: 10.1161/01.CIR.0000127126.73759.23. [DOI] [PubMed] [Google Scholar]
- 37.Das A, Salloum FN, Xi L, Rao YJ, Kukreja RC. ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. American Journal of Physiology: Heart and Circulatory Physiology. 2009;296(5):H1236–H1243. doi: 10.1152/ajpheart.00100.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meldrum DR, Dinarello CA, Cleveland JC, Jr., et al. Hydrogen peroxide induces tumor necrosis factor α-mediated cardiac injury by a P38 mitogen-activated protein kinase-dependent mechanism. Surgery. 1998;124(2):291–297. doi: 10.1067/msy.1998.90570. [DOI] [PubMed] [Google Scholar]
- 39.Remondino A, Kwon SH, Communal C, et al. β-adrenergic receptor-stimulated apoptosis in cardiac myocytes is mediated by reactive oxygen species/c-Jun NH2-terminal kinase-dependent activation of the mitochondrial pathway. Circulation Research. 2003;92(2):136–138. doi: 10.1161/01.res.0000054624.03539.b4. [DOI] [PubMed] [Google Scholar]
- 40.Kim SJ, Jeong CW, Bae HB, et al. Protective effect of sauchinone against regional myocardial ischemia/reperfusion injury: inhibition of p38 MAPK and JNK death signaling pathways. Journal of Korean Medical Science. 2012;27(5):572–575. doi: 10.3346/jkms.2012.27.5.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shao Z, Bhattacharya K, Hsich E, et al. c-Jun N-terminal kinases mediate reactivation of Akt and cardiomyocyte survival after hypoxic injury in vitro and in vivo. Circulation Research. 2006;98(1):111–118. doi: 10.1161/01.RES.0000197781.20524.b9. [DOI] [PubMed] [Google Scholar]
- 42.Dougherty CJ, Kubasiak LA, Prentice H, Andreka P, Bishopric NH, Webster KA. Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochemical Journal. 2002;362, part 3:561–571. doi: 10.1042/0264-6021:3620561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zechner D, Craig R, Hanford DS, McDonough PM, Sabbadini RA, Glembotski CC. MKK6 activates myocardial cell NF-κB and inhibits apoptosis in a p38 mitogen-activated protein kinase-dependent manner. The Journal of Biological Chemistry. 1998;273(14):8232–8239. doi: 10.1074/jbc.273.14.8232. [DOI] [PubMed] [Google Scholar]
- 44.Milano G, Morel S, Bonny C, et al. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. American Journal of Physiology: Heart and Circulatory Physiology. 2007;292(4):H1828–H1835. doi: 10.1152/ajpheart.01117.2006. [DOI] [PubMed] [Google Scholar]
- 45.Gao F, Yue T-L, Shi D-W, et al. p38 MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation. Cardiovascular Research. 2002;53(2):414–422. doi: 10.1016/s0008-6363(01)00488-6. [DOI] [PubMed] [Google Scholar]
- 46.Ikeda Y, Miura T, Sakamoto J, et al. Activation of ERK and suppression of calcineurin are interacting mechanisms of cardioprotection afforded by σ-opioid receptor activation. Basic Research in Cardiology. 2006;101(5):418–426. doi: 10.1007/s00395-006-0595-2. [DOI] [PubMed] [Google Scholar]
- 47.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiological Reviews. 2010;90(4):1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- 49.Himmetoglu S, Dincer Y, Bozcali E, Vural VA, Akcay T. Oxidative DNA damage and antioxidant defense after reperfusion in acute myocardial infarction. Journal of Investigative Medicine. 2009;57(4):595–599. doi: 10.2310/JIM.0b013e31819d87c1. [DOI] [PubMed] [Google Scholar]
- 50.Haeusgen W, Herdegen T, Waetzig V. The bottleneck of JNK signaling: molecular and functional characteristics of MKK4 and MKK7. European Journal of Cell Biology. 2011;90(6-7):536–544. doi: 10.1016/j.ejcb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 51.Shyu K-G, Wang B-W, Yang Y-H, Tsai S-C, Lin S, Lee C-C. Amphetamine activates connexin43 gene expression in cultured neonatal rat cardiomyocytes through JNK and AP-1 pathway. Cardiovascular Research. 2004;63(1):98–108. doi: 10.1016/j.cardiores.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 52.Petrich BG, Gong X, Lerner DL, et al. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circulation Research. 2002;91(7):640–647. doi: 10.1161/01.res.0000035854.11082.01. [DOI] [PubMed] [Google Scholar]
- 53.Hirt L, Badaut J, Thevenet J, et al. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke. 2004;35(7):1738–1743. doi: 10.1161/01.STR.0000131480.03994.b1. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y, Spigolon G, Bonny C, Culman J, Vercelli A, Herdegen T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Molecular and Cellular Neuroscience. 2012;49(3):300–310. doi: 10.1016/j.mcn.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 55.Statuto M, Bianchi C, Perego R, del Monte U. Drop of connexin 43 in replicative senescence of human fibroblasts HEL-299 as a possible biomarker of senescence. Experimental Gerontology. 2002;37(8-9):1113–1120. doi: 10.1016/s0531-5565(02)00089-x. [DOI] [PubMed] [Google Scholar]
- 56.Zhu W, Wang Y, Qiu G, Chen B. Characterization of the purification and primary culture of adult canine myoblasts in vitro. Molecular Medicine Reports. 2010;3(3):463–468. doi: 10.3892/mmr_00000281. [DOI] [PubMed] [Google Scholar]
- 57.Rackauskas M, Verselis VK, Bukauskas FF. Permeability of homotypic and heterotypic gap junction channels formed of cardiac connexins mCx30.2, Cx40, Cx43, and Cx45. American Journal of Physiology: Heart and Circulatory Physiology. 2007;293(3):H1729–H1736. doi: 10.1152/ajpheart.00234.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanaporis G, Brink PR, Valiunas V. Gap junction permeability: selectivity for anionic and cationic probes. American Journal of Physiology: Cell Physiology. 2011;300(3):C600–C609. doi: 10.1152/ajpcell.00316.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caspar DL, Goodenough DA, Makowski L, Phillips WC. Gap junction structures. I. Correlated electron microscopy and X-ray diffraction. The Journal of Cell Biology. 1977;74(2):605–628. doi: 10.1083/jcb.74.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peracchia C. Gap junctions. Structural changes after uncoupling procedures. The Journal of Cell Biology. 1977;72(3):628–641. doi: 10.1083/jcb.72.3.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lal R, John SA, Laird DW, Arnsdorf MF. Heart gap junction preparations reveal hemiplaques by atomic force microscopy. American Journal of Physiology: Cell Physiology. 1995;268(4):C968–C977. doi: 10.1152/ajpcell.1995.268.4.C968. [DOI] [PubMed] [Google Scholar]
- 62.Coss RA, Linnemans WA. The effects of hyperthermia on the cytoskeleton: a review. International Journal of Hyperthermia. 1996;12(2):173–196. doi: 10.3109/02656739609022507. [DOI] [PubMed] [Google Scholar]
- 63.Grzanka D, Stepien A, Grzanka A, Gackowska L, Helmin-Basa A, Szczepanski MA. Hyperthermia-induced reorganization of microtubules and microfilaments and cell killing in CHO AA8 cell line. Neoplasma. 2008;55(5):409–415. [PubMed] [Google Scholar]
- 64.Altan ZM, Fenteany G. c-Jun N-terminal kinase regulates lamellipodial protrusion and cell sheet migration during epithelial wound closure by a gene expression-independent mechanism. Biochemical and Biophysical Research Communications. 2004;322(1):56–67. doi: 10.1016/j.bbrc.2004.07.079. [DOI] [PubMed] [Google Scholar]
- 65.Stout C, Goodenough DA, Paul DL. Connexins: functions without junctions. Current Opinion in Cell Biology. 2004;16(5):507–512. doi: 10.1016/j.ceb.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 66.Han SI, Ha K-S, Kang KI, Kim HD, Kang HS. Heat shock-induced actin polymerization, SAPK/JNK activation, and heat-shock protein expression are mediated by genistein-sensitive tyrosine kinase(s) in K562 cells. Cell Biology International. 2000;24(7):447–457. doi: 10.1006/cbir.2000.0512. [DOI] [PubMed] [Google Scholar]
- 67.Sato T, Haimovici R, Kao R, Li A-F, Roy S. Downregulation of connexin 43 expression by high glucose reduces gap junction activity in microvascular endothelial cells. Diabetes. 2002;51(5):1565–1571. doi: 10.2337/diabetes.51.5.1565. [DOI] [PubMed] [Google Scholar]
- 68.Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. The Journal of Biological Chemistry. 2000;275(33):25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- 69.Bukauskas FF, Bukauskiene A, Verselis VK. Conductance and permeability of the residual state of connexin43 gap junction channels. The Journal of General Physiology. 2002;119(2):171–186. doi: 10.1085/jgp.119.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Palacios-Prado N, Bukauskas FF. Modulation of metabolic communication through gap junction channels by transjunctional voltage; synergistic and antagonistic effects of gating and ionophoresis. Biochimica et Biophysica Acta. 2012;1818(8):1884–1894. doi: 10.1016/j.bbamem.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annual Review of Biochemistry. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 72.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. The International Journal of Biochemistry & Cell Biology. 2004;36(7):1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saez JC, Retamal MA, Basilio D, Bukauskas FF, Bennett MVL. Connexin-based gap junction hemichannels: gating mechanisms. Biochimica et Biophysica Acta. 2005;1711(2):215–224. doi: 10.1016/j.bbamem.2005.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Palacios-Prado N, Briggs SW, Skeberdis VA, Pranevicius M, Bennett MVL, Bukauskas FF. pH-dependent modulation of voltage gating in connexin45 homotypic and connexin45/connexin43 heterotypic gap junctions. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(21):9897–9902. doi: 10.1073/pnas.1004552107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Skeberdis VA, Rimkute L, Skeberdyte A, Paulauskas N, Bukauskas FF. pH-dependent modulation of connexin-based gap junctional uncouplers. The Journal of Physiology. 2011;589, part 14:3495–3506. doi: 10.1113/jphysiol.2011.209072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smyth JW, Vogan JM, Buch PJ, et al. Actin cytoskeleton rest stops regulate anterograde traffic of connexin 43 vesicles to the plasma membrane. Circulation Research. 2012;110(7):978–989. doi: 10.1161/CIRCRESAHA.111.257964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dbouk HA, Mroue RM, El-Sabban ME, Talhouk RS. Connexins: a myriad of functions extending beyond assembly of gap junction channels. Cell Communication and Signaling. 2009;7, article 4 doi: 10.1186/1478-811X-7-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Engelbrecht A-M, Niesler C, Page C, Lochner A. p38 and JNK have distinct regulatory functions on the development of apoptosis during simulated ischaemia and reperfusion in neonatal cardiomyocytes. Basic Research in Cardiology. 2004;99(5):338–350. doi: 10.1007/s00395-004-0478-3. [DOI] [PubMed] [Google Scholar]
- 79.Ruparelia N, Digby JE, Jefferson A, et al. Myocardial infarction causes inflammation and leukocyte recruitment at remote sites in the myocardium and in the renal glomerulus. Inflammation Research. 2013;62(5):515–525. doi: 10.1007/s00011-013-0605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Decrock E, Vinken M, de Vuyst E, et al. Connexin-related signaling in cell death: to live or let die? Cell Death and Differentiation. 2009;16(4):524–536. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- 81.Aoki H, Kang PM, Hampe J, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. The Journal of Biological Chemistry. 2002;277(12):10244–10250. doi: 10.1074/jbc.M112355200. [DOI] [PubMed] [Google Scholar]
- 82.Garcia-Dorado D, Rodriguez-Sinovas A, Ruiz-Meana M. Gap junction-mediated spread of cell injury and death during myocardial ischemia-reperfusion. Cardiovascular Research. 2004;61(3):386–401. doi: 10.1016/j.cardiores.2003.11.039. [DOI] [PubMed] [Google Scholar]
- 83.Shintani-Ishida K, Unuma K, Yoshida K. Ischemia enhances translocation of connexin43 and gap junction intercellular communication, thereby propagating contraction band necrosis after reperfusion. Circulation Journal. 2009;73(9):1661–1668. doi: 10.1253/circj.cj-09-0079. [DOI] [PubMed] [Google Scholar]
- 84.Zi M, Kimura TE, Liu W, et al. Mitogen-activated protein kinase kinase 4 deficiency in cardiomyocytes causes connexin 43 reduction and couples hypertrophic signals to ventricular arrhythmogenesis. The Journal of Biological Chemistry. 2011;286(20):17821–17830. doi: 10.1074/jbc.M111.228791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trovato-Salinaro A, Belluardo N, Frinchi M, et al. Regulation of connexin gene expression during skeletal muscle regeneration in the adult rat. American Journal of Physiology: Cell Physiology. 2009;296(3):C593–C606. doi: 10.1152/ajpcell.00458.2008. [DOI] [PubMed] [Google Scholar]
- 86.von Maltzahn J, Wulf V, Willecke K. Spatiotemporal expression of connexin39 and -43 during myoblast differentiation in cultured cells and in the mouse embryo. Cell Communication and Adhesion. 2006;13(1-2):55–60. doi: 10.1080/15419060600631508. [DOI] [PubMed] [Google Scholar]
- 87.Haeusgen W, Tueffers L, Herdegen T, Waetzig V. Map2k4σ—identification and functional characterization of a novel Map2k4 splice variant. Biochimica et Biophysica Acta. 2014;1843(5):875–884. doi: 10.1016/j.bbamcr.2014.01.028. [DOI] [PubMed] [Google Scholar]
- 88.Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochimica et Biophysica Acta. 1997;1333(2):F85–F104. doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- 89.Thomas T, Jordan K, Laird DW. Role of cytoskeletal elements in the recruitment of Cx43-GFP and Cx26-YFP into gap junctions. Cell Communication and Adhesion. 2001;8(4-6):231–236. doi: 10.3109/15419060109080729. [DOI] [PubMed] [Google Scholar]
- 90.Yamane Y, Shiga H, Asou H, Ito E. GAP junctional channel inhibition alters actin organization and calcium propagation in rat cultured astrocytes. Neuroscience. 2002;112(3):593–603. doi: 10.1016/s0306-4522(02)00095-7. [DOI] [PubMed] [Google Scholar]
- 91.Zhao K, Wang W, Guan C, Cai J, Wang P. Inhibition of gap junction channel attenuates the migration of breast cancer cells. Molecular Biology Reports. 2012;39(3):2607–2613. doi: 10.1007/s11033-011-1013-x. [DOI] [PubMed] [Google Scholar]
- 92.Bensaude O, Bellier S, Dubois MF, Giannoni F, Nguyen VT. Heat-shock induced protein modifications and modulation of enzyme activities. EXS. 1996;77:199–219. doi: 10.1007/978-3-0348-9088-5_13. [DOI] [PubMed] [Google Scholar]
- 93.Gabai VL, Sherman MY. Invited review: interplay between molecular chaperones and signaling pathways in survival of heat shock. Journal of Applied Physiology. 2002;92(4):1743–1748. doi: 10.1152/japplphysiol.01101.2001. [DOI] [PubMed] [Google Scholar]
- 94.Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): differences revealed by gene targeting. BioEssays. 2006;28(9):923–934. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- 95.Reinecke K, Eminel S, Dierck F, et al. The JNK inhibitor XG-102 protects against TNBS-induced colitis. PLoS ONE. 2012;7(3, article e30985) doi: 10.1371/journal.pone.0030985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Laing JG, Tadros PN, Green K, Saffitz JE, Beyer EC. Proteolysis of connexin43-containing gap junctions in normal and heat- stressed cardiac myocytes. Cardiovascular Research. 1998;38(3):711–718. doi: 10.1016/s0008-6363(98)00060-1. [DOI] [PubMed] [Google Scholar]
- 97.Hamada N, Kodama S, Suzuki K, Watanabe M. Gap junctional intercellular communication and cellular response to heat stress. Carcinogenesis. 2003;24(11):1723–1728. doi: 10.1093/carcin/bgg135. [DOI] [PubMed] [Google Scholar]
- 98.Leithe E, Sirnes S, Omori Y, Rivedal E. Downregulation of gap junctions in cancer cells. Critical Reviews in Oncogenesis. 2006;12(3-4):225–256. doi: 10.1615/critrevoncog.v12.i3-4.30. [DOI] [PubMed] [Google Scholar]
- 99.Czyz J, Szpak K, Madeja Z. The role of connexins in prostate cancer promotion and progression. Nature Reviews Urology. 2012;9(5):274–282. doi: 10.1038/nrurol.2012.14. [DOI] [PubMed] [Google Scholar]
- 100.Yamasaki H, Krutovskikh V, Mesnil M, Tanaka T, Zaidan-Dagli ML, Omori Y. Role of connexin (gap junction) genes in cell growth control and carcinogenesis. Comptes Rendus de l’Academie des Sciences Serie III. 1999;322(2-3):151–159. doi: 10.1016/s0764-4469(99)80038-9. [DOI] [PubMed] [Google Scholar]
- 101.Vertrees RA, Leeth A, Girouard M, Roach JD, Zwischenberger JB. Whole-body hyperthermia: a review of theory, design and application. Perfusion. 2002;17(4):279–290. doi: 10.1191/0267659102pf588oa. [DOI] [PubMed] [Google Scholar]
- 102.Calderwood S, Dickson JA. Thermosensitivity of neoplastic tissues in vivo. In: Storm FK, editor. Hyperthermia in Cancer Therapy. Boston, Mass, USA: GK Hall Medical; 1983. pp. 63–140. [Google Scholar]
- 103.Despa F, Orgill DP, Neuwalder J, Lee RC. The relative thermal stability of tissue macromolecules and cellular structure in burn injury. Burns. 2005;31(5):568–577. doi: 10.1016/j.burns.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 104.Park HG, Han SI, Oh SY, Kang HS. Cellular responses to mild heat stress. Cellular and Molecular Life Sciences. 2005;62(1):10–23. doi: 10.1007/s00018-004-4208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Roti JL. Cellular responses to hyperthermia (40–46°C): cell killing and molecular events. International Journal of Hyperthermia. 2008;24(1):3–15. doi: 10.1080/02656730701769841. [DOI] [PubMed] [Google Scholar]