

Abstract
Ca2+-Calmodulin kinase II (CaMKII) activation is deleterious in cardiac ischemia/reperfusion (I/R). Moreover, inhibition of CaMKII-dependent phosphorylations at the sarcoplasmic reticulum (SR) prevents CaMKII-induced I/R damage. However, the downstream targets of CaMKII at the SR level, responsible for this detrimental effect, remain unclear.
In the present study we aimed to dissect the role of the two main substrates of CaMKII at the SR level, phospholamban (PLN) and ryanodine receptors (RyR2), in CaMKII-dependent I/R injury.
In mouse hearts subjected to global I/R (45/120 min), phosphorylation of the primary CaMKII sites, S2814 on cardiac RyR2 and of T17 on PLN, significantly increased at the onset of reperfusion whereas PKA-dependent phosphorylation of RyR2 and PLN did not change. Similar results were obtained in vivo, in mice subjected to regional myocardial I/R (1/24 hrs). Knock-in mice with an inactivated serine 2814 phosphorylation site on RyR2 (S2814A), significantly improved post-ischemic mechanical recovery, reduced infarct size and decreased apoptosis. Conversely, knock-in mice, in which CaMKII site of RyR2 is constitutively activated (S2814D), significantly increased infarct size and exacerbated apoptosis. In S2814A and S2814D mice subjected to regional myocardial ischemia, infarct size was also decreased and increased respectively. Transgenic mice with double-mutant non-phosphorylatable PLN (S16A/T17A) in the PLN knockout background (PLNDM) also showed significantly increased post-ischemic cardiac damage. This effect cannot be attributed to PKA-dependent PLN phosphorylation and was not due to the enhanced L-type Ca2+ current, present in these mice.
Our results reveal a major role for the phosphorylation of S2814 site on RyR2 in CaMKII-dependent I/R cardiac damage. In contrast, they showed that CaMKII-dependent increase in PLN phosphorylation during reperfusion opposes rather than contributes to I/R damage.
Keywords: RyR2, PLN, CaMKII, ischemia/reperfusion injury, apoptosis, necrosis, myocardium
1. Introduction
Myocardial infarction (MI) is the leading cause of morbidity and mortality in the Western world with >700,000 heart attacks diagnosed each year only in the United States [1]. Ischemic injury results from severe impairment of coronary blood supply and oxygen to the myocardium and prompt restoration of coronary flow can limit the infarct size and reduce mortality. Currently, primary reperfusion therapies, including percutaneous coronary intervention and thrombolysis, are the standard treatments for patients suffering from MI. However, the strategy of rapid reperfusion of ischemic myocardium is usually associated with additional cell damage, cardiac dysfunction and ventricular arrhythmias as a result of reperfusion injury [2-4] The causes of these deleterious effects are multifactorial, but altered Ca2+ handling has emerged as a major contributor to post-ischemic dysfunction and injury [3, 5-7].
In the heart, Ca2+ handling during the excitation-contraction coupling process is mediated by a mechanism known as Ca2+-induced Ca2+release [8], by which a small influx of external Ca2+ through the L-type Ca2+channel binds to and opens the cardiac Ca2+ release channel/ryanodine receptor (RyR2), producing a large release of Ca2+ from the sarcoplasmic reticulum (SR). Cytosolic Ca2+ is then re-sequestered into the SR by the SR Ca2+-ATPase (SERCA2a). The function of this Ca2+pump is regulated by the phosphorylation level of a regulatory protein, also associated with the SR, named phospholamban (PLN). PLN in turn, is subject of additional regulation by interacting partners like HAX-1 (an anti-apoptotic protein) and Gm or the anchoring subunit of protein phosphatase 1, which affect the overall SERCA-mediated Ca2+-transport [9, 10].
Recent studies strongly suggested that the phosphorylation of SR proteins by the Ca2+/calmodulin-dependent protein kinase II (CaMKII), is involved in reperfusion damage (necrosis, apoptosis, functional impairment) following prolonged ischemia. Conversely, inhibition of CaMKII activity at the SR level protects from the injury caused by ischemia/reperfusion (I/R) [11]. However, the downstream targets of CaMKII responsible for the detrimental effects of I/R, remain unclear. For instance, it has been shown that CaMKII-dependent phosphorylation of the threonine 17 site of PLN (T17), occurs at the onset of reperfusion [11, 12], yet the functional consequences of this phosphorylation events are uncertain. Indeed, the effects of enhanced SR Ca2+ uptake in the face of an ischemic insult are controversial. On one hand, experimental evidence indicated that gene transfer of SERCA2a alleviated post-ischemic cardiac injury in rat and porcine animal models[13, 14], suggesting a beneficial effect of increasing SR Ca2+ uptake in I/R. On the other hand, several studies indicated that inhibiting SR Ca2+ uptake is cardioprotective whereas increasing it, is harmful [15-18]. In line with these previous findings, more recent studies have shown that deletion of PLN eliminated the protective effects of a CaMKII-inhibitory peptide in mice (AC3-I) subjected to myocardial infarction. PLN deletion also exacerbated the deleterious cardiac effects of CaMKII transgenic mice [19]. Interestingly, the recently described protective effect of HAX-1/Hsp90 complex on I/R damage [10] can also be related to the inhibitory effect of the combined action of these compounds on SR Ca2+ uptake [10, 20].
Referent to RyR2, it is known that the increased SR Ca2+ leak resulting from CaMKII-dependent phosphorylation of RyR2 is a main contributor to heart failure and reperfusion arrhythmias [4, 21-23].
The main goal of the present study was to dissect the role of the two primary targets of CaMKII at the SR level, PLN and RyR2, in CaMKII-dependent I/R injury. We demonstrate here that RyR2 channels are phosphorylated by CaMKII during I/R and play a crucial role in CaMKII-dependent cardiac damage. Knock-in mice with a genetically inactivated CaMKII-phosphorylation site on RyR2 (S2814A) [22] are protected from necrosis, apoptosis and impaired cardiac function, following irreversible I/R injury. Conversely, knock-in mice with a phosphomimetic mutation on RyR2 (S2814D) [21] are more susceptible to cardiac damage following I/R. In contrast, PLN phosphorylation during reperfusion opposes rather than contributes to I/R damage.
Thus, CaMKII-dependent phosphorylation of RyR2 during reperfusion is crucial in determining cardiac damage and the main factor at the SR level, responsible for the deleterious effect of CaMKII in I/R injury.
2. Materials and Methods
An expanded Methods section is available in the online Supplementary material.
2.1 Animals
Experiments were performed in knock-in male mice (4 months old) in which the S2814 site of RyR2 was either replaced by alanine (S2814A mice), to genetically inhibit RyR2 phosphorylation by CaMKII [22] or aspartic acid (S2814D mice), to mimic constitutive phosphorylation of RyR2 by CaMKII [21]. An additional experimental group was performed in transgenic male mice, expressing a mutant PLN in which both phosphorytables residues (S16 and T17) were replaced by Ala (PLNDM) [24], (Obtained from MMRRC, University of Missouri/Harlan, Mouse Regional Resource Center, NCRR, NIH) [24]. The corresponding wild type (WT) littermates were utilized as controls for the S2814A/D mice [21]. C57BL/6 mice were used as controls for the PLNDM mice. Finally, a small group of global CaMKIIδ-knockout mice [25] was also used in the in vivo experiments (See Results). Animals were inbreeded and maintained in our animal facilities in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 1996). The protocols were approved by the Ethics Committee of the Cardiovascular Research Center, National Research Council (CONICET).
2.2 Preparations
a. Ex-vivo experiments: Langendorff perfusion and experimental protocol
Isolated hearts were perfused according to the Langendorff technique [26]. After a 10 min stabilization period, hearts were subjected to 45 min of global ischemia followed by 120 min of reperfusion [11].
b. Isolated Myocytes
Enzymatic isolation of ventricular myocytes and fluorescence measurements were performed as previously described [27, 28]. The standard whole-cell configuration of the patch-clamp technique was used for voltage-clamp recordings with a patch-clamp amplifier [29].
c. In-vivo experiments
Mice were anesthetized by sodium pentobarbital (45 mg/kg, i.p.) and then subjected to myocardial ischemia induced by left anterior descending (LAD) coronary artery occlusion for 1 hr followed by reperfusion for 24 hrs. Sham operated mice were subjected to the same surgical procedures without LAD ligation [25].
2.3 Infarct size
Infarct size was assessed by the triphenyltetrazolium chloride (TTC) technique [12, 25].
2.4 LDH determinations
Cardiac injury was evaluated by LDH released in the perfusion effluent. LDH samples were taken every minute during the first 10 min of reperfusion [11, 12].
2.5 Terminal deoxynucleotidyltransferase-mediated dUTP nick-end (TUNEL) labeling
TUNEL assay was performed on myocardial slices fixed in buffered formalin and processed for histological examination[11, 12]
2.6 Electrophoresis and Western blot analysis
Cardiac homogenates were prepared as previously described [11, 30]. Proteins were probed with antibodies raised against S16 and T17-phosphorylated PLN, total PLN, S2814 and S2808-phosphorylated RyR2, total RyR2, Bcl-2 and Bax [3, 11]. GAPDH was used as control loading.
2.7 Statistical analysis
Data are expressed as mean± SEM. Unpaired, paired Student t-test or ANOVA followed by Tukey post hoc test were used for statistical comparisons when appropriate. Differences were considered significant at P<0.05.
3. Results
3.1 Increased CaMKII, but not PKA-dependent phosphorylation of RyR2 and PLN during I/R
To define the cardioprotective mechanisms associated with CaMKII-inhibition at the SR level, we performed ex-vivo experiments in Langendorff perfused mouse hearts subjected to a protocol of 45 min ischemia followed by reperfusion. At different times during reperfusion (3 and 15 min), hearts were freeze-clamped to measure the phosphorylation status of RyR2, at S2814 and S2808, and PLN, at T17 and S16, using phospho-epitope specific antibodies. Selection of these reperfusion times for phosphorylation assessment was based on previous results that showed that maximal CaMKII-dependent phosphorylation of PLN occurred at the onset of reperfusion and then declined [11, 26, 31], consistent with the abrupt increase in diastolic Ca2+ observed at the onset of reperfusion [32]. Western blotting of ventricular lysates revealed a significant increase in S2814 phosphorylation (CaMKII site) in perfused hearts subjected to myocardial infarction following by 3 min of reperfusion, compared to pre-ischemia (Figure 1A). After 15 min of reperfusion, phosphorylation of S2814 site was still higher than pre-ischemic values. In contrast, there were no significant changes in S2808 phosphorylation (PKA site) of RyR2 in either group. Moreover, we also observed a decrease in RyR2 abundance at the onset of reperfusion, similar to previous findings [11, 33]. Phosphorylation of PLN at T17 (CaMKII site), also significantly increased in hearts subjected to 3 and 15 min of reperfusion, whereas phosphorylation of PLN at S16 (PKA site) remained unchanged in reperfused hearts versus pre-ischemic controls (Figure 1B), consistent with previous reports [11, 12, 25, 26, 31]. Control experiments in which hearts were continuously perfused for a period of time identical to that of hearts submitted to I/R protocol, did not show any significant change in either the expression or phosphorylation of RyR2 or PLN (results not shown).
Figure 1. Reperfusion significantly increases CaMKII dependent phosphorylation of RyR2 and PLN.

A. Western blots and summary results of CaMKII and PKA-dependent phosphorylation of RyR2 at S2814 and S2808 respectively, and of RyR2 expression, in Langendorff perfused WT mouse hearts subjected to 45 min ischemia followed by various reperfusion times. Comparing with pre-ischemic values (Pre-Isch) there was a significant increase in S2814 phosphorylation at 3 min of reperfusion, without significant changes in the phosphorylation of S2808. A decrease in the expression of RyR2 was present at 3 and 15 min of reperfusion. B. Results of PLN phosphorylation sites during reperfusion. A significant increase in the phosphorylation of the CaMKII-dependent site, T17, was evident at 3 and 15 min of reperfusion with no significant changes in the PKA dependent site, S16. PLN expression did not change relative to pre-ischemic values. GAPDH was used as a loading control. Data represent the average ± SEM of values from 4-9 hearts per group.* P<0.05 vs. the corresponding pre-ischemic values.
To further confirm the role of RyR2 phosphorylation on I/R in vivo we performed an additional series of experiments in WT mice submitted to regional myocardial ischemia followed by reperfusion (1/24 hrs). Supplementary Figure 1 shows immunoblots and averaged results of these experiments. No changes in total expression/degradation of RyR2 were apparent after 24 hrs of reperfusion in the in vivo experiments, which suggests that RyR2 alterations during global I/R might be rescued during reperfusion. Moreover, and similar to the results in the intact heart, no significant changes in S2808 phosphorylation were observed in WT mice after the 1/24 hrs I/R protocol. However, WT mice showed significantly increased RyR2 phosphorylation at S2814 site, whereas there were no significant changes in the phosphorylation of this site in parallel I/R experiments performed in CaMKIIδ-KO mice [25].
3.2 Inhibition of CaMKII-dependent Ser2814 phosphorylation on RyR2 protects against necrosis and apoptosis following I/R injury
To determine whether genetic inhibition of CaMKII phosphorylation of RyR2 prevented cardiac damage following I/R, we studied RyR2 knock-in mice [22] in which the S2814 phosphorylation site was mutated to alanine (S2814A). CaMKII-phosphorylation patterns of PLN in these mice were similar to those in WT mice (Supplementary Figure 2). At baseline, twitch shortening and Ca2+ transient amplitude during electric field stimulation (0.5 Hz) and SR Ca2+ load (assessed by rapid application of 25mmol/L caffeine) in isolated myocytes, were similar to those of WT mice (Supplementary Figure 3). Basal contractility was also similar in WT vs. S2814A mice in the intact heart (Figure 2). At the onset of global ischemia, isovolumic left ventricular developed pressure (LVDP) dropped essentially to 0 in both groups. However, on reperfusion, S2814A mice exhibited better post-ischemic mechanical recovery, i.e. LVDP was higher in S2814A mice compared to WT (Figure 2A). The vast contractile depression observed in both groups of mice after 45 min ischemia may not only be attributed to the infarct areas (See below) but also to the presence of myocardial stunning areas peripheral to the infarct size zone. Postischemic dysfunction (stunned myocardium) has been shown to revert after approximately 48/72 hs of reperfusion [34]. This would greatly influence contractility depression observed in acute experiments after prolonged ischemic periods, as used herein. By comparing Figure 2 A and B, it is clear than in this model of I/R with a prolonged ischemic period, the alteration of contractile (systolic) activity is trivial contrasted with the marked elevation of diastolic pressure. However, the increase in LVEDP was not significantly different between the WT and S2814A animals. In spite of that, it was possible to detect a significant difference in LVDP. To better study mechanical recovery, an additional series of experiments was performed in which ischemia was reduced to 20 min. This ischemic period also produced infarct damage in mouse hearts (results not shown). Supplementary Figure 4 indicates that the mechanical recovery was significantly more important in S2814A than in WT mice, similar to the results obtained with prolonged ischemia.
Figure 2. Lack of CaMKII-phosphorylation of RyR2 protects against I/R injury.
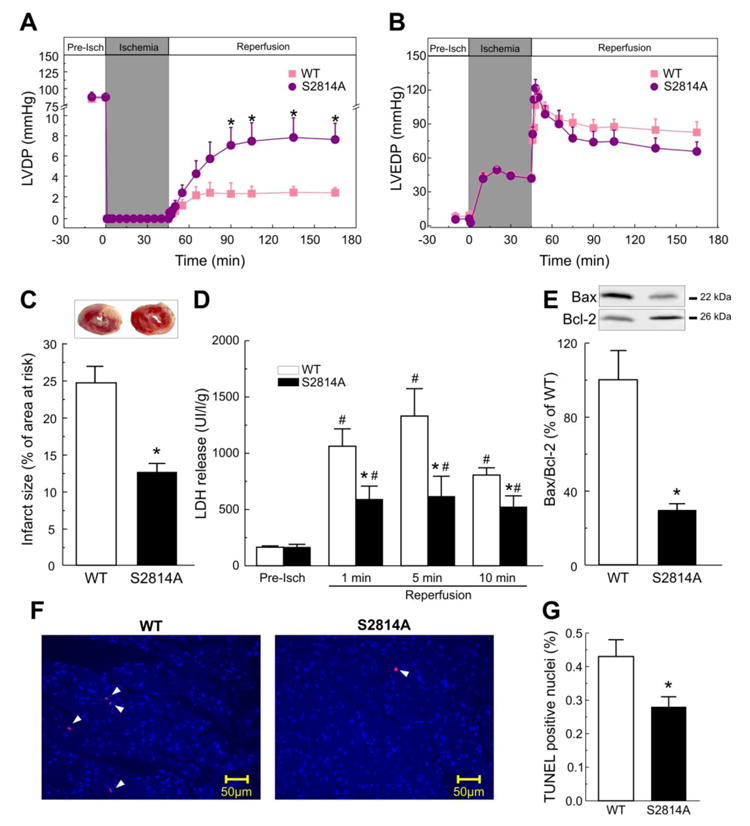
Perfused hearts from S2814A mice subjected to I/R (45/120 min), showed a significant improvement of contractile parameters during reperfusion when compared to WT mice, i.e. left ventricular developed pressure (LVDP), was increased (A). Left ventricular end diastolic pressure (LVEDP), showed a trend to be lower in S2814A vs. WT mice, although without reaching significant levels (B). Myocardial viability was enhanced in S2814A vs. WT mice, as reflected by a decrease in the infarct size at the end of reperfusion (C) and in LDH released during the first 10 min of reperfusion (D). Apoptosis was also diminished in S2814A vs. WT mice: Typical immunoblots and overall results after 120 min of reperfusion, depicted in panel (E), showed a decrease in the ratio between the proapoptotic protein Bax and the antiapoptotic protein Bcl2, (Bax/Bcl2). TUNEL assay confirmed apoptotic death decrease in S2814A mice hearts relative to WT. Examples of TUNEL stained cardiac tissue are shown in (F). Total nuclei were stained with DAPI (light blue). Apoptotic nuclei, in red, are marked with arrow heads. Original magnification is 40x. Overall results of this assay are shown in (G). Data represent the average ± SEM of n = 5 - 12 hearts per group.*P<0.05 vs. WT mice. # P<0.05 vs. the corresponding pre-ischemic values.
To further delineate the effects of inhibition of RyR2 phosphorylation in post-ischemic damage, the extent of I/R-induced myocardial infarction was assessed. The infarct size post I/R (45/120 min) was 24.6±2.2% in WT hearts, whereas it was significantly attenuated in S2814A mice (12.6±1.2%; P< 0.05) (Figure 2C). Moreover the extent of necrotic cell death examined by lactate dehydrogenase (LDH) efflux during the first 10 min of reperfusion was decreased by approximately 40% in S2814A mice relative to WT (Figure 2D). Finally, the extent of apoptotic cell death examined by the ratio between the proapoptotic protein Bax and the antiapoptotic protein Bcl2 (Bax/Bcl2) and TUNEL assay, were also significantly lower in S2814A than in WT mice after reperfusion (Figure 2E-G).
Taken together these results indicate that prevention of S2814 phosphorylation diminished I/R injury, i.e. necrotic and apoptotic death, and improved functional recovery in the intact heart.
3.3 Constitutive phosphorylation of S2814 does not affect contractile recovery but enhances I/R damage
To further assess the role of RyR2 in the deleterious effect of CaMKII in I/R, we performed experiments in mice, with a S2814D mutation in RyR2 to mimic constitutive phosphorylation [21]. CaMKII-phosphorylation patterns of PLN in these mice were similar to those in WT mice (Supplementary Figure 2). Supplementary Figure 3A and B shows that twitch shortening and Ca2+ transient amplitude were similar in S2814D compared with WT and S2814A mice, although SR Ca2+ load was lower than in the other two strains. The decrease in SR Ca2+ load in S2814D is presumably the consequence of the higher diastolic SR Ca2+ leak observed in these mice [21]. Thus, S2814D mice exhibited enhanced fractional SR Ca2+ release (ratio of twitch/caffeine-induced Ca2+ transient), compared with WT or S2814A mice, supporting the notion that RyR2 phosphorylation at S2814 activates both diastolic and systolic RyR2 Ca2+ release [21, 35]. Basal contractility in the intact heart was also similar in S2814D and WT mice and post-ischemic mechanical recovery (LVDP) was not significantly altered. (Figure 3A and B). Similar results were obtained when the ischemic period was reduced to 20 min (Supplementary Figure 4). In contrast to the contractile behavior, infarct size measured at the end of 45/120 min I/R, was significantly greater in S2814D than in WT mice, as shown in the typical examples and overall results of Figure 3C. LDH release was slightly higher in S2814D compared with WT mice at baseline and significantly increased in the first minutes of reperfusion in S2814D mice compared to WT mice. This difference disappeared however in samples obtained at the end of 10min of reperfusion (Figure 3D). These results suggest that necrosis was primarily enhanced at the onset of reperfusion in S2814D hearts. Apoptotic cell death was also increased in S2814D mice. Figures 3E to G shows a significantly higher Bax/Bcl2 ratio and more TUNEL positive nuclei after reperfusion in S2814D mice than in WT animals.
Figure 3. Constitutive activation of CaMKII-dependent S2814 site of RyR2 does not change post-ischemic mechanical recovery but enhances I/R cardiac damage.
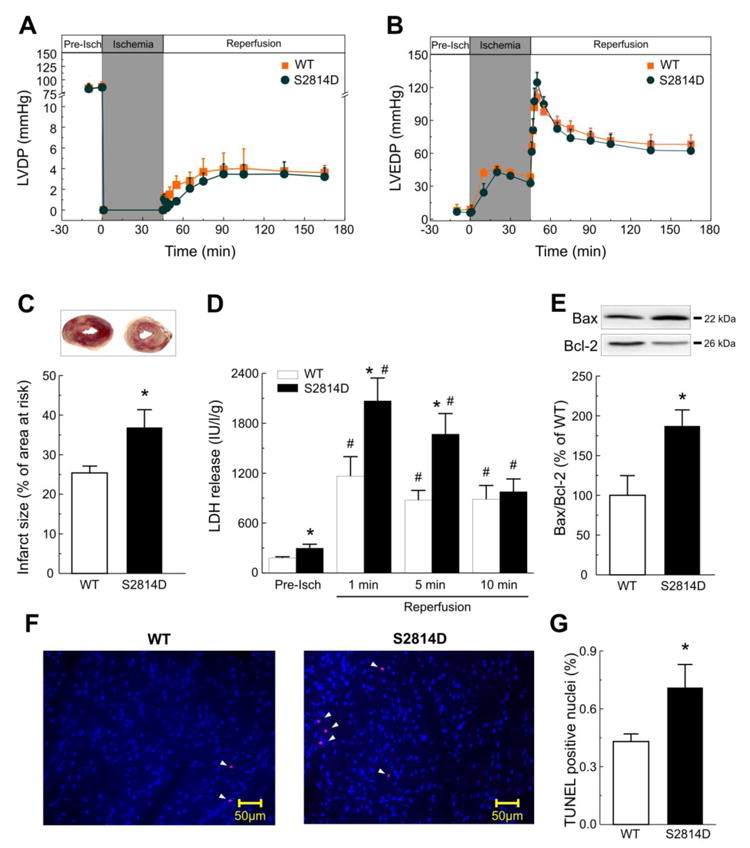
Perfused hearts from mice with constitutive phosphorylation of S2814 on RyR2 (S2814D mice), showed a similar post-ischemic mechanical recovery (LVDP and LVEDP) compared with WT mice (A, B). In contrast, constitutive activation of S2814 increased infarct size (C) and LDH release at 1 and 5 min of reperfusion (D). The apoptotic death was also enhanced at the end of reperfusion in S2814D mice relative to WT, when evaluated by the increase in either Bax/Bcl2 ratio (E) or TUNEL staining. Typical examples and overall results of the TUNEL assay are shown in panels (F) and (G) respectively. Apoptotic nuclei, in red, are marked with arrowheads. Data represent the average ± SEM of n = 7 - 12 hearts per group.*P<0.05 vs. WT mice. # P<0.05 vs. the corresponding pre-ischemic values.
Interestingly, Ser2814D mice showed a dissociation between infarct size and mechanical recovery, i.e. whereas the mechanical recovery was not significantly different relative to WT, the infarct size was significantly increased. A possible explanation for these somewhat discordant results may lie on the increased fractional release observed in these mice (See above and Supplementary Figure 3), which would be able to counteract the deleterious mechanical effect of I/R.
To further test the impact of CaMKII-dependent phosphorylation of RyR2, we performed an additional series of in vivo experiments in S2814A, S2814D and WT mice subjected to regional myocardial ischemia induced by left anterior descending (LAD) coronary artery occlusion for 1 h followed by 24hs of reperfusion. Supplementary Figure 5 shows that the infarct size relative to the area at risk was significantly decreased in S2814A mice and increased in S2814D compared to WT mice. Taken together, the data in S2814A and S2814D mice demonstrate that CaMKII-dependent phosphorylation of RyR2 is an essential signaling event that promotes cardiac damage and contributes to irreversible I/R injury.
3.4 CaMKII-dependent PLN phosphorylation does not participate in the deleterious effect of CaMKII in I/R
To determine whether CaMKII-dependent phosphorylation of PLN also contributes to I/R damage, we performed experiments in transgenic mice in which the PKA and CaMKII-dependent phosphorylation sites of PLN (T17 and S16) were both mutated to Ala, and thereby rendered non-phosphorylatable. Supplemental Figure 6A shows that the ability of isoproterenol to increase PLN phosphorylation at T17 was blocked in these PLN double mutant (PLNDM) mice. The relaxant effect of isoproterenol was likewise blunted although the positive inotropic effect of the β-agonist was similar in both groups (Supplementary Figure 6B and C). The similar inotropic action of isoproterenol in PLNDM and WT mice was attributed to enhanced L-type Ca2+ current in these animals [24].
Figure 4A shows that LVDP was significantly impaired during reperfusion after ischemia in PLNDM mice after 120 and of ischemia, whereas LVEDP did not significantly changed (B), consistent with previous findings in which perfused hearts were submitted to a shorter ischemic period [6]. Infarct size and LDH release were not reduced but, on the contrary they were significantly enhanced in PLNDM compared to WT mice (Figure 4C and D). The finding that preventing PLN phosphorylation exacerbates the functional and structural heart damage after MI, suggests that the CaMKII-dependent phosphorylation of PLN observed during reperfusion (Figure 1B) favors post-ischemic recovery and protects from I/R cardiac damage.
Figure 4. Lack of PLN phosphorylation increases I/R injury.
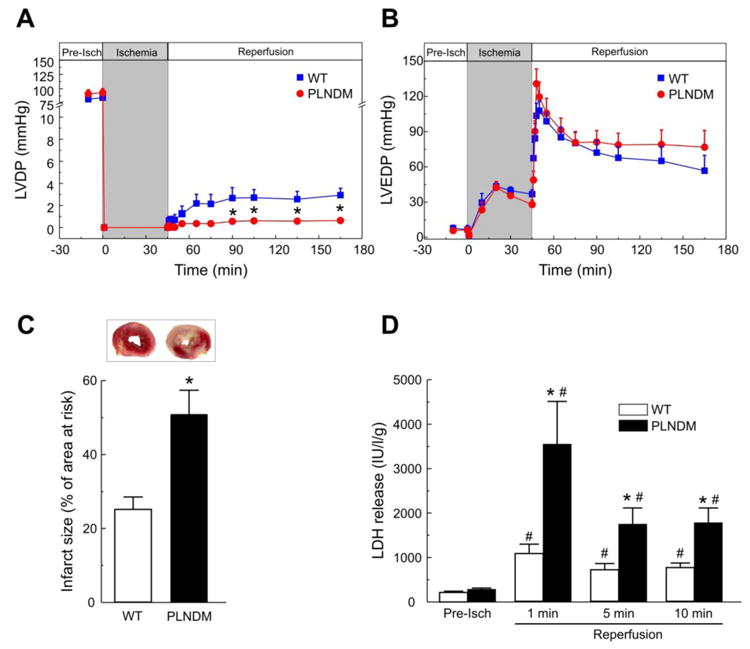
Double PLN mutant mice (PLNDM) exhibited a significant decrease in contractile recovery vs. WT mice (A), without significant changes in LVEDP (B). Infarct size at the end of reperfusion (C) and LDH released during the first 10 min of reperfusion (D) were also significantly increased when compared with WT mice. Data represent the average ± SEM of n = 7 - 14 per group. *P<0.05 vs. WT mice. # P<0.05 vs. the corresponding pre-ischemic values.
An important limitation of PLNDM mice is that they develop an increase in L-type Ca2+ current [24] which was interpreted to be a compensatory mechanism since basal contractility was not affected (Figure 4 and Supplementary Figure 6). To further explore whether this increase in L-type Ca2+ current contributes to the increased infarct size observed in PLNDM mice, additional experiments were performed to determine the effect of returning this current to basal levels. In patch-clamp experiments performed in isolated myocytes from PLNDM and WT mice, we observed an increase in L-type Ca2+ current of approximately 20%, in agreement with previous findings [24]. This increase was inhibited in the presence of 10 nmol/L of the Ca2+ channel blocker nifedipine. Figure 5A and B show typical records and overall results of these experiments. Figure 5C to F show that contractile recovery, infarct size and LDH release were not significantly changed in the presence of a nifedipine concentration sufficient to block the increase in L-type Ca2+ current in PLNDM myocytes. Of note, this nifedipine concentration slightly decreased LVDP under basal conditions although without reaching significant levels. These findings indicate that the increase in L-type Ca2+ current does not significantly contribute to the detrimental effect of I/R in PLNDM mice. Taken together the above results reveal that phosphorylation of T17 of PLN at the beginning of reperfusion is not detrimental. On the contrary, this response serves to oppose to the deleterious action of RyR2 phosphorylation.
Figure 5. Enhanced I/R injury in PLNDM mice is not due to the increased L-type Ca2+ current.
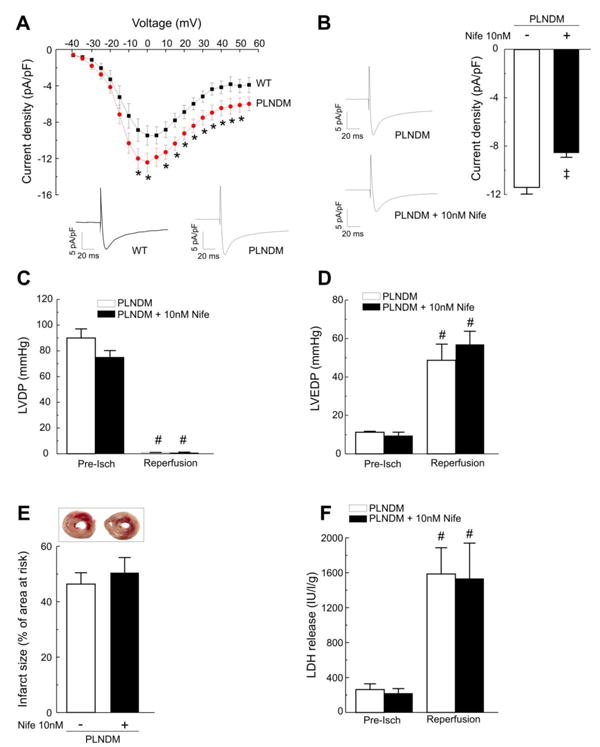
A. Current density-voltage relations for average data of peak current density collected from WT mice (n=8) and from PLNDM mice (n=11). Representative traces of ICa, compensated for cell capacitance evoked by the voltage-clamp depolarizing step to 0 mV, recorded in WT and PLNDM myocytes, are shown below. B. Representative traces of ICa, compensated for cell capacitance, recorded in a PLNDM myocyte before and after application of nifedipine (Nife) to the bath solution (left) and average data of peak ICa density of 6 cells recorded at 0 mV before and after nifedipine (right), are displayed. The currents were evoked by a voltage-clamp depolarizing step to 0 mV (250 ms) from a prepulse potential of −40 mV. C. There were not significant differences in left ventricular developed pressure, (LVDP, C), left ventricular end-diastolic pressure, (LVEDP, D), infarct size at the end of reperfusion (E) and LDH released during the first 10 min of reperfusion (F) between PLNDM with or without nifedipine. * P< 0.05 vs. WT; ‡ P< 0.05 PLNDM vs. PLNDM + Nife; # P< 0.05 vs. the corresponding pre-ischemic values.
4. Discussion
Previous works from our laboratory has shown that CaMKII-dependent phosphorylation of Ca2+ handling proteins at the SR level is responsible for the detrimental effects of CaMKII during irreversible I/R injury [11, 12]. The salutary effects of CaMKII-inhibition at the SR level resulted from normalization of SR Ca2+ homeostasis that contributes to post-ischemic cell death and mechanical dysfunction. We have further shown that I/R-induced activation of CaMKII and phosphorylation of SR Ca2+ handling proteins contribute to mitochondrial dysfunction and subsequent necrotic and apoptotic cell death [11, 12]. However the specific targets phosphorylated by CaMKII at the SR level and responsible for this effect, were not dissected in those previous papers. In the present work, we explored two main candidates for this action: RyR2 and PLN, based on the facts that they are the two main proteins involved in SR Ca2+ handling and that both are targets of CaMKII.
Here we provide evidence that CaMKII-dependent phosphorylation of RyR2 during reperfusion is a critical determinant of the severity of detrimental effects of CaMKII (necrosis, apoptosis and contractile depression) following I/R injury. We used knock-in mice which express either the CaMKII-phosphomimetic S2814D or the non-phosphorylatable S2814A mutants on RyR2, which enabled us to evaluate the importance of CaMKII phosphorylation of RyR2 [21, 22]. Our results provide direct evidence showing that CaMKII-dependent phosphorylation of RyR2 is the main target responsible for I/R damage at the SR level. The in vivo experiments, in which I/R was produced by left coronary artery ligation, -the gold standard model of I/R-, also revealed the critical role of CaMKII-dependent RyR2 phosphorylation in I/R injury, giving further support to the above conclusion. Furthermore, CaMKII-dependent PLN phosphorylation at the onset of reperfusion did not contribute to the deleterious effect of I/R. To the best of our knowledge, this is the first report that shows that RyR2 phosphorylation is a major mediator of CaMKII-induced injury in I/R.
4.1 The RyR2 is a major downstream target of CaMKII, mediating its detrimental effects during I/R
A major conclusion from the present work is that when RyR2 cannot be phosphorylated by CaMKII, the irreversible damage associated with I/R (necrosis, apoptosis) as well as the associated mechanical dysfunction, are largely prevented. A second major conclusion is that constitutive CaMKII-phosphorylation of RyR2 increases the susceptibility to post-ischemic myocardial damage, in both ex-vivo and in-vivo models. An important corollary of these two findings is that the increase in CaMKII-dependent phosphorylation of RyR2 that occurs during reperfusion (Figure 1A and Supplementary Figure 1) is a primary determinant of CaMKII-induced necrosis and apoptosis following I/R injury. Although we did not assess either diastolic Ca2+ or SR Ca2+ leak upon reperfusion in these mice, it is possible to speculate that the increase in SR Ca2+ leak produced by RyR2 phosphorylation may favor mitochondria Ca2+ overload and cardiac damage. Thus, preventing this phosphorylation event could ameliorate cardiac reperfusion injury. In previous experiments performed in the rat heart [11], we demonstrated that CaMKII-dependent cardiac damage during irreversible I/R was reliant on a CaMKII-phosphorylation at the SR level. In these experiments there was a trend to increase in the ratio of phosphorylated S2814/RyR2. However, this increase failed to reach statistical significance. The reason for the discrepancy between these previous findings and the present results is not apparent to us. Although we cannot discard that species differences may underlie this inconsistency, it is also possible that the simultaneous changes in RyR2 expression and Ser2814 phosphorylation that occurs at the onset of reperfusion, enhances data spreading, making the detection of a significant increase in the Ser2814/RyR2 ratio, more difficult. The present experiments in mouse hearts, showing a significant increase in the ratio Ser2814/RyR2 and in knock-in mice revealing a significant decrease and increase of I/R injury in Ser2814A and Ser2814D mice, respectively, together with the fact that T17 phosphorylation seems not to contribute to cardiac damage, clearly show however that the damaging effect of CaMKII in I/R is chiefly dependent on the phosphorylation of S2814 on RyR2. The previously described diminished expression levels of RyR2 at the onset of reperfusion [33], was also found in the present experiments (Figure 1A). Although this alteration seems to be reversible, since it was not apparent in the in vivo experiments (Supplementary Figure 2), a possible contribution of RyR2 degradation to I/R damage cannot not be discarded from the present results. Current experiments in our laboratory are exploring this possibility.
4.2 Beneficial effects of CaMKII-dependent PLN phosphorylation in I/R injury
The present experiments also indicate that preventing CaMKII-dependent phosphorylation of PLN at T17 site at the onset of reperfusion, as assessed in mice in which PLN phosphorylation sites S16 and T17 are ablated, increased cardiac injury. This finding cannot be attributed to inhibition of S16 phosphorylation, since phosphorylation of this site did not increase during reperfusion (Figure 1B). Neither can it be due to the 20% increase in L-type Ca2+ current, the typical compensation observed in these transgenic mice [24], since similar results were obtained in the presence of a nifedipine concentration that blocked the Ca2+ current increase observed in isolated myocytes from PLNDM mice (Figure 5). These experiments therefore imply that there is a beneficial effect of CaMKII-dependent PLN phosphorylation in I/R injury.
The role of PLN phosphorylation in I/R has been difficult to ascertain and indeed previous results support either a beneficial or a detrimental effect of PLN phosphorylation [11, 36, 37]. These contradictory observations may reflect the unique effect of PLN phosphorylation on accelerating SR Ca2+ uptake, which could result in two opposite consequences. On the one hand, it would counteract the diastolic Ca2+ elevation resulting from SR Ca2+ leak, thus avoiding an increase in mitochondrial Ca2+ and favoring contractile recovery. Indeed, previous studies in PLNDM mice submitted to I/R revealed an increase in diastolic Ca2+ during reperfusion relative to WT mice [6]. On the other hand, by increasing SR Ca2+ load, it may favor SR Ca2+ leak due to the regulatory effect of intra-SR Ca2+on RyR2 function. This would eventually produce a futile circle in which the continuous SR Ca2+ leak would evoke mitochondrial Ca2+ overload and cardiac injury. This latter sequence may underlie the detrimental effect of maximally increasing SR Ca2+ uptake in I/R, when interbreeding AC3-I mice (which were protected against I/R) with PLNKO mice [38] and may also explain the cardioprotective effect of inhibiting SR Ca2+ uptake, like the one produced by HAX-1/hsp90 complex [10]. The role of PLN phosphorylation on I/R would therefore depend on several factors among which are the status of the different PLN protein partners, the degree and time course of PLN phosphorylation, the Ca2+ threshold for SR Ca2+ leak and the status (integrity, phosphorylation, redox alterations) of RyR2. This latter factor appears to be crucial, since it is clear that in hearts of PLNKO mice, which mimic a maximum degree of PLN phosphorylation but conserve intact RyR2 under resting conditions, there is no evident necrosis or apoptosis [38].
Previous experiments from our laboratory using perfused mouse hearts that express an inhibitory CaMKII peptide at the SR level (SR-AIP mice), reported that infarct size was reduced by approximately 60% [11], not different from the reduction observed in S2814A mice (present results). Notably in SR-AIP mice, CaMKII-dependent phosphorylation of PLN and of RyR2 are both inhibited while in the S2814A mice only the CaMKII-dependent phosphorylation of RyR2 was prevented. This finding indicates that when CaMKII-dependent RyR2 phosphorylation is precluded, prevention of PLN phosphorylation failed to increase cardiac injury as should be expected from the results in PLNDM mice. Thus, CaMKII-dependent inhibition of RyR2 phosphorylation seems to be necessary and sufficient to prevent the CaMKII-dependent cardiac damage that originates at the SR level in I/R.
In summary, we demonstrate for the first time that phosphorylation of S2814 of RyR2 plays a major role in the detrimental effect of CaMKII in I/R. Our studies using knock-in mice which express either the CaMKII-phosphomimetic S2814D or the non-phosphorylatable S2814A mutants of RyR2 provide direct evidence showing that RyR2 are the main CaMKII targets at the SR responsible for I/R damage. Our previous results indicated that CaMKII promotes mitochondrial dysfunction and subsequent necrotic and apoptotic cell death during I/R through a phosphorylation occurring at the SR level. The present findings revealed that CaMKII-dependent phosphorylation of RyR2 by enhancing SR Ca2+ leak is the main responsible of this post-ischemic cardiac damage. In contrast, CaMKII–dependent PLN phosphorylation does not contribute to this injury but rather opposes to it. Our experiments further indicate that inhibition of RyR2 phosphorylation may be necessary and sufficient to prevent the CaMKII-dependent cardiac damage that originates at the SR level in I/R. Given the critical role of RyR2 in I/R damage, the strategy of impairing CaMKII phosphorylation at these Ca2+ release sites would represents a promising therapeutic strategy for the prevention/treatment of I/R damage.
Supplementary Material
Highlights.
Ischemia/reperfusion (I/R) enhances CaMKII phosphorylation of RyR2 and PLN.
Inactivation of CaMKII phosphorylation of RyR2 reduced I/R-induced cardiac damage.
Constitutive activation of CaMKII phosphorylation of RyR2 increased I/R-induced damage.
Inactivation of CaMKII phosphorylation of PLN increased I/R-induced cardiac damage.
The results reveal the prominent role CaMKII phosphorylation of RyR2 in I/R damage.
Acknowledgments
M.S., C.A.V., V.DG., J.P., A.E.A. G.R and A.M. are Established Investigators of National Research Council, (CONICET), Argentina. M.A.S. is an Established Investigator of the University of La Plata, Argentina. X.H.T.W. is an Established Investigator of the American Heart Association (AHA). M.N. D C. is a fellow of the National Agency for the Promotion of Science and Technology; L.S. is a fellow from Buenos Aires Province Research Council.
The authors are grateful to Mónica Rando, Omar Castillo, Juan Manuel Lofeudo and Inés Vera for their skilled technical assistance.
Source of Funding: This work was supported by PICT 1795 to M.A.S., PICT 1903 (FONCyT) and PIP 02139 and 0890 (CONICET) to A.M., and NIH grants HL089598 and HL091947 to X.H.T.W, and NIH grant T32-HL07676 to J.R. H. Ling was supported by an American Heart Association (AHA) Postdoctoral Fellowship. This work was also supported by the Muscular Dystrophy Association and in part by Fondation Leducq (‘Alliance for CaMKII Signaling in Heart’) to X.H.T.W.
Abbreviations
- AC3-I
Ca2+-calmodulin kinase II-inhibitory peptide
- Ca2+
Calcium
- CaMKII
Ca2+-Calmodulin kinase II
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- I/R
Ischemia/reperfusion
- LAD
Left anterior descending
- LDH
Lactodehydrogenase
- MI
Myocardial infarction
- PLN
Phospholamban
- PKA
Protein kinase A
- PLNDM
double-mutant non-phosphorylatable PLN
- SR
Sarcoplasmic reticulum
- SR-AIP mice
Inhibitory CaMKII peptide at SR level
- RyR2
Isoform 2 of ryanodine receptors
- S16A/T17A mice
Serine16 mutated to Ala/Threonine 17 mutated to Ala
- S2814A mice
Serine 2814 mutated to Ala (RyR2 constitutively inactivated)
- S2814D mice
Serina 2814 mutated to Asp (RyR2 constitutively activated)
- SERCA2a
Isoform 2a of sarco/endoplasmic reticulum Ca2+-ATPase
- S
Serine
- T
Threonine
- TTC
Triphenyltetrazolium chloride
- TUNEL
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- WT
Wild-type
Footnotes
Disclosure Statement: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, et al. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713–9. doi: 10.1172/JCI112160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res. 2012;94:168–80. doi: 10.1093/cvr/cvs116. [DOI] [PubMed] [Google Scholar]
- 4.Said M, Becerra R, Valverde CA, Kaetzel MA, Dedman JR, Mundina-Weilenmann C, et al. Calcium-calmodulin dependent protein kinase II (CaMKII): a main signal responsible for early reperfusion arrhythmias. J Mol Cell Cardiol. 2011;51:936–44. doi: 10.1016/j.yjmcc.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piper HM, Kasseckert S, Abdallah Y. The sarcoplasmic reticulum as the primary target of reperfusion protection. Cardiovasc Res. 2006;70:170–3. doi: 10.1016/j.cardiores.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 6.Valverde CA, Mundina-Weilenmann C, Reyes M, Kranias EG, Escobar AL, Mattiazzi A. Phospholamban phosphorylation sites enhance the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart. Cardiovasc Res. 2006;70:335–45. doi: 10.1016/j.cardiores.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 7.Steenbergen C, Murphy E, Watts JA, London RE. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat heart. Circ Res. 1990;66:135–46. doi: 10.1161/01.res.66.1.135. [DOI] [PubMed] [Google Scholar]
- 8.Fabiato A, Fabiato F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, and frog hearts and from fetal and new-born rat ventricles. Ann N Y Acad Sci. 1978;307:491–522. doi: 10.1111/j.1749-6632.1978.tb41979.x. [DOI] [PubMed] [Google Scholar]
- 9.Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ Res. 2012;110:1646–60. doi: 10.1161/CIRCRESAHA.111.259754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam CK, Zhao W, Cai W, Vafiadaki E, Florea SM, Ren X, et al. Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ Res. 2013;112:79–89. doi: 10.1161/CIRCRESAHA.112.279935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salas MA, Valverde CA, Sanchez G, Said M, Rodriguez JS, Portiansky EL, et al. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48:1298–306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, et al. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res. 2007;73:689–98. doi: 10.1016/j.cardiores.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 13.del Monte F, Lebeche D, Guerrero JL, Tsuji T, Doye AA, Gwathmey JK, et al. Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc Natl Acad Sci USA. 2004;101:5622–7. doi: 10.1073/pnas.0305778101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prunier F, Kawase Y, Gianni D, Scapin C, Danik SB, Ellinor PT, et al. Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation. 2008;118:614–24. doi: 10.1161/CIRCULATIONAHA.108.770883. [DOI] [PubMed] [Google Scholar]
- 15.Kumada Y, Yamamoto F, Yamamoto H, Ishikawa T, Kagisaki K, Hirose H. Decreasing sarcoplasmic reticular calcium gives rise to myocardial protection--the effect of thapsigargin for myocardial protection under conditions of normothermia. Jpn J Thorac Cardiovasc Surg. 1998;46:368–74. doi: 10.1007/BF03217757. [DOI] [PubMed] [Google Scholar]
- 16.Zucchi R, Ronca F, Ronca-Testoni S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther. 2001;89:47–65. doi: 10.1016/s0163-7258(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 17.Avellanal M, Rodriguez P, Barrigon S. Protective effects of cyclopiazonic acid on ischemia-reperfusion injury in rabbit hearts. J Cardiovasc Pharmacol. 1998;32:845–51. doi: 10.1097/00005344-199811000-00022. [DOI] [PubMed] [Google Scholar]
- 18.Cross HR, Kranias EG, Murphy E, Steenbergen C. Ablation of PLB exacerbates ischemic injury to a lesser extent in female than male mice: protective role of NO. Am J Physiol. 2003;284:H683–90. doi: 10.1152/ajpheart.00567.2002. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Guo T, Mishra S, Dalton ND, Kranias EG, Peterson KL, et al. Phospholamban ablation rescues sarcoplasmic reticulum Ca2+ handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ Res. 2010;106:354–62. doi: 10.1161/CIRCRESAHA.109.207423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao W, Waggoner JR, Zhang ZG, Lam CK, Han P, Qian J, et al. The anti-apoptotic protein HAX-1 is a regulator of cardiac function. Proc Natl Acad Sci USA. 2009;106:20776–81. doi: 10.1073/pnas.0906998106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Oort RJ, McCauley MD, Dixit SS, Pereira L, Yang Y, Respress JL, et al. Ryanodine receptor phosphorylation by calcium/calmodulin-dependent protein kinase II promotes life-threatening ventricular arrhythmias in mice with heart failure. Circulation. 2010;122:2669–79. doi: 10.1161/CIRCULATIONAHA.110.982298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J Clin Invest. 2009;119:1940–51. doi: 10.1172/JCI37059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload-induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brittsan AG, Kranias EG. Phospholamban and cardiac contractile function. J Mol Cell Cardiol. 2000;32:2131–9. doi: 10.1006/jmcc.2000.1270. [DOI] [PubMed] [Google Scholar]
- 25.Ling H, Gray CB, Zambon AC, Grimm M, Gu Y, Dalton N, et al. Ca2+/Calmodulin-dependent protein kinase II delta mediates myocardial ischemia/reperfusion injury through nuclear factor-kappaB. Circ Res. 2013;112:935–44. doi: 10.1161/CIRCRESAHA.112.276915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Said M, Vittone L, Mundina-Weilenmann C, Ferrero P, Kranias EG, Mattiazzi A. Role of dual-site phospholamban phosphorylation in the stunned heart: insights from phospholamban site-specific mutants. Am J Physiol. 2003;285:H1198–205. doi: 10.1152/ajpheart.00209.2003. [DOI] [PubMed] [Google Scholar]
- 27.Palomeque J, Petroff MV, Sapia L, Gende OA, Mundina-Weilenmann C, Mattiazzi A. Multiple alterations in Ca2+ handling determine the negative staircase in a cellular heart failure model. J Card Fail. 2007;13:143–54. doi: 10.1016/j.cardfail.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, et al. Angiotensin II-induced oxidative stress resets the Ca2+ dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105:1204–12. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 29.Aiello EA, Cingolani HE. Angiotensin II stimulates cardiac L-type Ca2+ current by a Ca2+- and protein kinase C-dependent mechanism. Am J Physiol. 2001;280:H1528–36. doi: 10.1152/ajpheart.2001.280.4.H1528. [DOI] [PubMed] [Google Scholar]
- 30.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, et al. The cardiac-specific nuclear delta(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 31.Vittone L, Mundina-Weilenmann C, Said M, Ferrero P, Mattiazzi A. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J Mol Cell Cardiol. 2002;34:39–50. doi: 10.1006/jmcc.2001.1488. [DOI] [PubMed] [Google Scholar]
- 32.Valverde CA, Kornyeyev D, Ferreiro M, Petrosky AD, Mattiazzi A, Escobar AL. Transient Ca2+ depletion of the sarcoplasmic reticulum at the onset of reperfusion. Cardiovasc Res. 2010;85:671–80. doi: 10.1093/cvr/cvp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pedrozo Z, Sanchez G, Torrealba N, Valenzuela R, Fernandez C, Hidalgo C, et al. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Bioch Biophys Acta. 2010;1802:356–62. doi: 10.1016/j.bbadis.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Cohen MV, Yang XM, Downey JM. Smaller infarct after preconditioning does not predict extent of early functional improvement of reperfused heart. Am J Physiol. 1999;277:H1754–61. doi: 10.1152/ajpheart.1999.277.5.H1754. [DOI] [PubMed] [Google Scholar]
- 35.Ferrero P, Said M, Sanchez G, Vittone L, Valverde C, Donoso P, et al. Ca2+/calmodulin kinase II increases ryanodine binding and Ca2+-induced sarcoplasmic reticulum Ca2+ release kinetics during beta-adrenergic stimulation. J Mol Cell Cardiol. 2007;43:281–91. doi: 10.1016/j.yjmcc.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nicolaou P, Rodriguez P, Ren X, Zhou X, Qian J, Sadayappan S, et al. Inducible expression of active protein phosphatase-1 inhibitor-1 enhances basal cardiac function and protects against ischemia/reperfusion injury. Circ Res. 2009;104:1012–20. doi: 10.1161/CIRCRESAHA.108.189811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Y, Zhu WZ, Joiner ML, Zhang R, Oddis CV, Hou Y, et al. Calmodulin kinase II inhibition protects against myocardial cell apoptosis in vivo. Am J Physiol. 2006;291:H3065–75. doi: 10.1152/ajpheart.00353.2006. [DOI] [PubMed] [Google Scholar]
- 38.Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, et al. Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta-agonist stimulation. Circ Res. 1994;75:401–9. doi: 10.1161/01.res.75.3.401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.