

Abstract
The mutation γ375Arg → Trp (fibrinogen Aguadilla) is one of four mutations (Brescia, Aguadilla, Angers, and AI duPont) capable of causing hepatic storage of fibrinogen. It has been observed in four children from the Caribbean, Europe, and Japan, suffering from cryptogenic liver disease. We report the first case of hepatic fibrinogen storage disease in Arabs due to a mutation in the fibrinogen γ-chain gene in a 3-year-old Syrian girl presenting with elevated liver enzymes. The finding of an impressive accumulation of fibrinogen in liver cells raised the suspicion of endoplasmic reticulum storage disease. Sequencing of the fibrinogen genes revealed a γ375Arg → Trp mutation (fibrinogen Aguadilla) in the child and in her father. In conclusion, when confronted with chronic hepatitis of unknown origin, one should check the plasma fibrinogen level and look carefully for the presence of hepatocellular intracytoplasmic globular inclusions to exclude hepatic fibrinogen storage disease.
Keywords: Endoplasmic reticulum storage disease, fibrinogen, fibrinogen gamma chain, hepatocyte intracytoplasmic inclusions, hypofibrinogenemia
Fibrinogen is a high-molecular-weight plasma protein synthesized exclusively in the liver. The circulating molecule is a dimer, with each half comprising three polypeptide chains (Aα, Bβ, and γ) encoded by three different genes.[1] Numerous missense mutations in the three fibrinogen genes have been reported, leading to hypofibrinogenemia, afibrinogenemia, or dysfibrinogenemia. The clinical consequences reported for these genetic defects include bleeding, thrombosis, and renal amyloidosis, but no liver disease.[2] In the last decade, a novel subgroup of fibrinogen disorders has emerged in which mutations in the fibrinogen γ-chain gene (FGG) result in inherited hypofibrinogenemia with storage of fibrinogen in the rough endoplasmic reticulum (RER) of hepatocytes, leading to liver disease of variable severity in both children and adults.[3,4,5,6] Mutations in FGG have been confirmed in families from Europe, the United States, and Japan,[3,4,5,6,7,8,9] but no such genetic abnormalities have been identified in Arabs.
Here, we report the case of an Arabic family in which a young child and her father were found to be heterozygous for the FGG Arg375Trp (fibrinogen Aguadilla) mutation, causing hypofibrinogenemia and hepatic endoplasmic reticulum storage disease (ESRD). This report emphasizes the appropriate diagnostic approach for recognition of this entity and discusses the correlation between the type of mutation in the fibrinogen γ-chain gene and the phenotypic presentation.
CASE REPORT
A 3-year-old Syrian girl, born to healthy parents with no consanguinity, was diagnosed at age 2.5 years with nephrotic syndrome. At the time of diagnosis of nephrotic syndrome, her liver enzymes were observed to be elevated: Alanine aminotransferase level was 250 IU/L and aspartate aminotransferase level was 185 IU/L. She received steroids to induce remission and was maintained on levamisole. Six months later, because of persistently elevated liver enzymes, the child was transferred to the pediatric gastroenterology department. There was no history of herbal ingestion and no family history of liver disease or coagulopathy. The liver was palpable at 2 cm below the right costal margin, with a span of 10 cm, soft consistency, and a smooth edge; the spleen was not palpable. Abdominal ultrasonography showed mild hepatomegaly. Liver function tests revealed elevated levels of aspartate aminotransferase (190 IU/L; normal range, 15-35 IU/L), alanine aminotransferase (200 IU/L; normal range, 30-60 IU/L), and alkaline phosphatase (358 IU/L; normal range, 150-350 IU/L); total serum bilirubin level of 8 μmol/L (normal range, 3-17 μmol/L); γ-glutamyltransferase level of 25 U/L (normal range, 7-32 U/L); albumin level of 32 g/L (normal range, 35-50 g/L); partial thromboplastin time of 40 seconds; and an international normalized ratio (INR) of 1. Extensive laboratory evaluations excluded Wilson disease, viral hepatitis, and metabolic and endocrine diseases (including normal serum ceruloplasmin, urinary copper, negative hepatitis B virus surface antigen and hepatitis C virus antibody, normal plasma amino acids and urinary organic acids, and normal levels of thyroid hormones and cortisol hormone). Antinuclear, smooth muscle, anti-liver/kidney microsome, anti-mitochondrial, and perinuclear antineutrophilic cytoplasmic antibodies were negative. The serum α-1-antitrypsin level was normal (125 mg/dL; normal range, 94-150 mg/dL) with a PiMM phenotype. The serum fibrinogen level was 0.89 (normal range, 1.8-3.5 g/L). A diagnostic percutaneous needle liver biopsy was performed.
Histopathologic studies
The liver specimen was processed for light microscopy and electron microscopy. Light microscopy studies included routine stains [i.e., hematoxylin and eosin, periodic acid-Schiff (PAS) with and without diastase, and van Gieson], as well as immunohistochemistry, using commercially available polyclonal antibodies against fibrinogen (1:1000; Dako, Glostrup, Denmark), α-1-antitrypsin (1:500; Novocastra, Newcastle upon Tyne, UK), and α-1-antichymotrypsin (1:500; Dako). For electron microscopy, biopsy tissues were fixed in 2% or 3% buffered glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated in graded concentrations of ethyl alcohol followed by propylene oxide, and embedded in Epon 1 semi-thin sections. Next, the sections were stained with methylene blue and azure II and counterstained with sodium borate and basic fuchsine for light microscopy orientation. Ultrathin sections were stained with uranyl acetate and Sato's lead citrate, mounted on copper grids, and examined using a Zeiss 109 electron microscope. Histologic examination of the liver biopsy specimen showed mild portal fibrosis associated with a mild proliferation of bile ductules and mild inflammatory infiltration. The lobular architecture was intact and comprised regular cords of hepatocytes containing multiple, irregularly rounded, deeply eosinophilic globules surrounded by a clear halo [Figure 1]. These globules failed to stain with PAS following pretreatment with diastase. By the immunoperoxidase method, the globules were found to be negative for α-1-antitrypsin and α-1-antichymotrypsin. Intracytoplasmic material in the hepatocytes reacted strongly with anti-fibrinogen antisera [Figure 2]. Electron microscopy of the liver tissue showed that most hepatocytes contained dilated granular endoplasmic reticulum with the lumen engorged with densely packed tubular structures, arranged in curved bundles and resulting in a fingerprint pattern [Figure 3].
Figure 1.
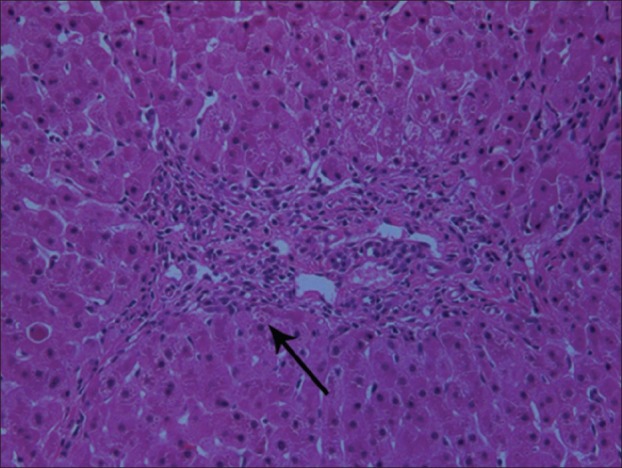
Liver tissue section shows hepatocytes containing multiple, irregularly rounded, deeply eosinophilic globules surrounded by a clear halo (arrow) (hematoxylin and eosin, ×40)
Figure 2.
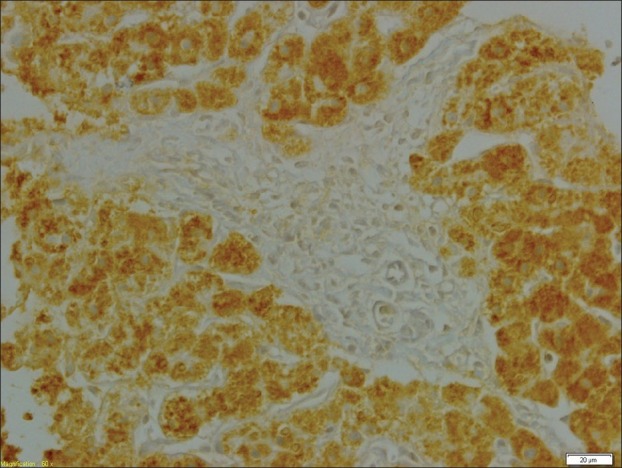
Intracytoplasmic hepatocytic inclusions react strongly with anti-fibrinogen antisera (immunoperoxidase staining with fibrinogen antibody), ×60
Figure 3.
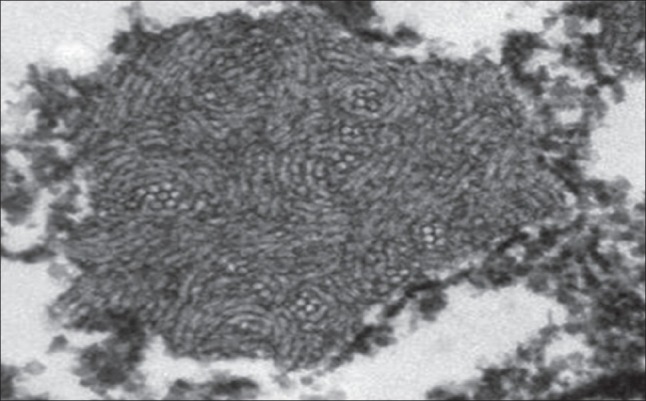
Electron microscopy of liver tissue shows densely packed tubular structures in the rough endoplasmic reticulum, arranged in curved bundles, resulting in a fingerprint pattern (×7000)
Molecular genetic studies
Genomic DNA was isolated from total blood obtained from the patient and parents using extraction protocols from the QIAMP DNA mini kit (QIAGEN, Hilden, Germany). The coding exons and exon-intron boundaries of FGG (MIM FNx01134850) were amplified using reported oligonucleotide primers[6] and 30 polymerase chain reaction (PCR) cycles with cycling parameters of denaturation at 94°C for 30 seconds, annealing at 55°C for 30 seconds, and extension at 72°C for 30 seconds, followed by a final extension of 7 minutes at 72°C. PCR products were gel-purified using MinElute Spin Columns (QIAGEN) and sequenced on both strands using BigDye 1.1 chemistry on an ABI 3130xl system (Applied Biosystems, Foster City, CA, USA). FGG analysis demonstrated a heterozygous c. 1201C>T mutation in the propositus [Figure 4]. The mutation in exon 9 replaced tryptophan 375 with arginine (p.Arg375Trp, fibrinogen Aguadilla mutation). Familial analyses for the fibrinogen γ-chain gene revealed that the patient's father also had the Aguadilla mutation and had normal liver function tests and a low serum fibrinogen level (1.3 g/L). The mother and two other siblings did not have the Aguadilla mutation. The nephrotic syndrome in the index case had run a frequent relapsing course that necessitated a renal biopsy. Histopathologic examination of the renal specimen revealed changes consistent with minimal-change nephrotic syndrome, the absence of intracytoplasmic inclusions, and a negative reaction with anti-fibrinogen antisera. Until the writing of the present manuscript, at age 11 years, the patient is clinically asymptomatic and has shown normal growth and development. Laboratory investigations disclosed the persistence of hypofibrinogenemia and mild elevation of liver enzymes.
Figure 4.
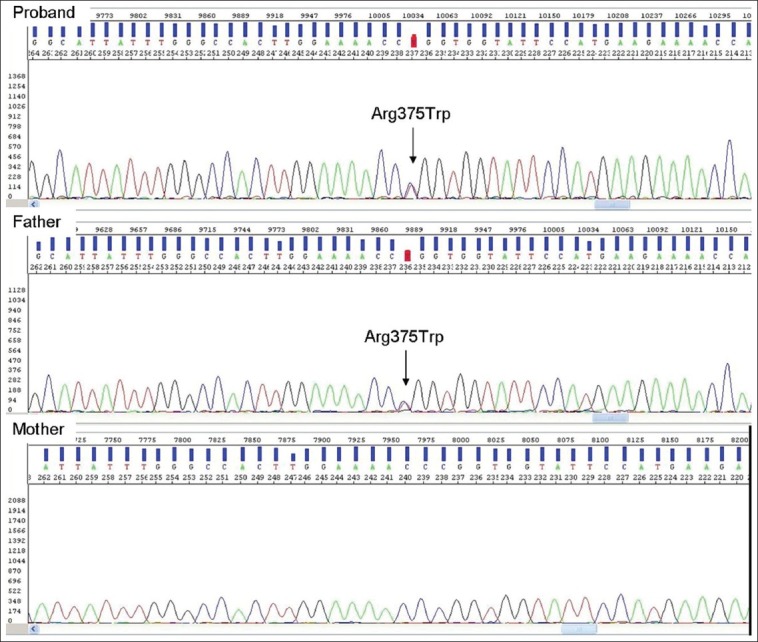
DNA sequence from exon 9 of the fibrinogen γ-chain gene. DNA analysis shows heterozygous CGG>TGG mutation (arrow) at codon 375 of the γ-chain gene in this patient and her father. Lower panel shows normal sequence in mother
DISCUSSION
We have reported a child with hereditary hypofibrinogenemia and type I fibrinogen hepatic storage associated with the γ375 Arg → Trp mutation (fibrinogen Aguadilla). This represents the first report of an ERSD and, consequently, a genetically determined fibrinogen storage disease in an Arab.
ERSDs have the property of exclusively and selectively retaining mutant secretory proteins due to their aggregation within the RER.[10]α-1-Antitrypsin deficiency is the prototype of ERSD. α-1-Antichymotrypsin, fibrinogen, α-2-antiplasmin, and C3-C4 complement Lebanese low-density lipoprotein are the other proteins susceptible to storage.[10] In recent years, the evidence that misfolding of proteins can occur in cells other than hepatocytes, such as Alzheimer protein in neurons, or in the extracellular space, such as in amyloidosis, has generated the more comprehensive term “conformational diseases” for these conditions.[11]
The storage of fibrinogen in liver cells at light microscopy and electron microscopy levels was first observed in German families.[12] However, definite evidence for a genetic basis of the storage phenomenon came from an Italian family identified in 1987 in which a mutation in the γ-fibrinogen chain (284Gly → Arg, fibrinogen Brescia) was found to be the cause of the conformational abnormality of the molecule, leading to aggregation of the mutant protein within the RER.[13] The fibrinogen Brescia mutation had remained solitary until the discovery of other mutations, designated Aguadilla,[4] Angers,[5] and AI duPont,[6] which were observed in Caribbean, French, and US families, respectively. The Aguadilla mutation has been subsequently detected in Italian,[7] Swiss,[8] and Japanese[9] families, as well as, from the present report, an Arab family.
The FGG mutations, responsible for inclusion of fibrinogen in the liver and for hypofibrinogenemia, are summarized in Table 1. The first mutation, fibrinogen Brescia (γ284GlyArg), occurred in an Italian woman whose cirrhosis was initially noted during abdominal surgery.[13] The second mutation, fibrinogen Aguadilla (γ375ArgTrp), gained attention because of chronically elevated liver enzymes in two asymptomatic young girls from Puerto Rico.[4] This latter mutation has subsequently been found in additional cases reported from Switzerland, Japan, and Italy.[7,8,9] Fibrinogen Anger, the third causative mutation (g346-350 D GVYYQ), was reported in a French family.[5] It affected a woman with cirrhosis; three other heterozygotes had mild, chronically elevated liver transaminases. The fourth recently reported causative mutation, γ314ThrPro mutation (fibrinogen AI duPont), occurred in a young Caucasian boy from the United States.[6]
Table 1.
Literature review for confirmed cases of fibrinogen gamma chain mutations
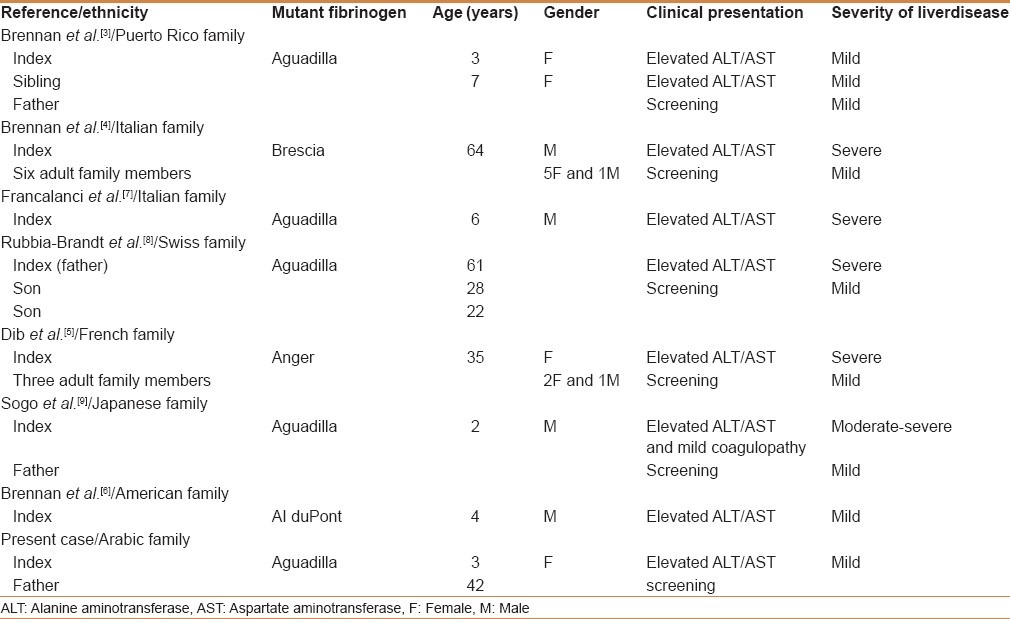
Our review of the medical literature [Table 1] has identified 21 individuals (from seven families) with confirmed mutations in FGG (16 adults and 5 children; 11 males and 10 females). Fibrinogen Aguadilla affects both adults and children, whereas fibrinogen Brescia and fibrinogen Anger affect adults only. It appears that the liver is the primary organ affected by this particular fibrinogen abnormality, whereas coagulation is only mildly disordered. Hepatic fibrinogen storage disease is often reflected by abnormal liver aminotransferases in an asymptomatic patient. Hepatomegaly may or may not be present. Most reported patients had mild liver disease; however, hepatic fibrinogen storage has been shown to result in significant liver injury, leading to cirrhosis in adults and children.[5,7,8,9] Adults with hepatic fibrinogen storage disease and clinical signs of decompensated cirrhosis have been described.[13] There are few reports of FGG mutations to allow comments on the natural history of the liver disease. However, reports of cirrhosis in adults with FGG mutations and mild liver disease in younger asymptomatic heterozygote carriers in the same family[4,5,7,8,9] suggest that hepatic fibrinogen storage disease might insidiously progress to cirrhosis over time. The progression to severe liver disease could be perpetuated by an additional injury such as alcohol abuse, obesity, or viral hepatitis.[8] It is possible that the steroid had perpetuated the liver injury in our case, and this might explain the significant improvement in transaminases observed off steroids. Fibrinogen Aguadilla is the only FGG mutation that has been reported to cause severe liver disease in children.[7,9] An interesting feature of α1-antitrypsin deficiency with Z mutation, the most well-recognized and characterized ERSD, is the variation in the number and density of liver inclusions and the severity of liver disease that varies considerably among individuals.[14] Only a subset of patients homozygous for α1PiZ or heterozygous for the FGG mutation develops liver disease due to the accumulation of aggregates of aberrant protein in the endoplasmic reticulum of hepatocytes. Similarly, the FGG mutations do not interfere with protein synthesis but are predicted to cause an altered conformation of the fibrinogen molecule that cannot be exported from the endoplasmic reticulum, accounting for reduced fibrinogen levels and possible evolution toward chronic cryptogenic liver disease. Based on mammalian and yeast model systems, it was speculated that the subsets of individuals who develop liver disease are predisposed to less efficient endoplasmic reticulum quality control mechanisms, perhaps through a second mutation in a gene important for the prevention or clearance of protein aggregates within the endoplasmic reticulum.[15,16]
Four main points for discussion are raised by our case and from the literature review:
Hereditary hypofibrinogenemia with hepatic storage appears to be invariably associated with mutations in the gamma chain of the fibrinogen molecule
The storage process predisposes to the development of severe chronic liver disease that shows an early onset with the Aguadilla mutation
The four mutations (Brescia, Aguadilla, Angers, and AI duPont) share light, immunohistochemical, and ultrastructural features that characterize type I fibrinogen inclusions;[10] and
The four gamma chain mutations exclusively affect the liver, in contrast to all previously described mutations,[2] which lead to hypofibrinogenemia, afibrinogenemia, or dysfibrinogenemia and coagulation problems in the absence of liver involvement.
Although reports of detailed morphologic and genetic analysis of this particular metabolic storage disease are scarce, fibrinogen-positive globules have been detected in 8 of 700 liver biopsies with chronic hepatitis, indicating that the FGG mutation is an underdiagnosed entity.[17] Therefore, when confronted with chronic hepatitis or cirrhosis of unknown origin, one should either check the plasma fibrinogen level and look carefully for the presence of weakly stained hepatocellular intracytoplasmic globular inclusions or eventually perform immunohistochemistry for fibrinogen to exclude hepatic fibrinogen storage disease. In addition to type I inclusions, fibrinogen hepatic storage in liver cells can present in the form of type II (ground-glass fibrinogen)[17] or type III (round eosinophilic) inclusions.[10] Type II and type III inclusions display a different light or ultrastructural appearance compared with type I inclusions.[10,18] Moreover, patients with type II or III inclusions do not show hypofibrinogenemia or mutations.[10] The combination of low plasma fibrinogen levels and type I inclusions in liver cells can be considered virtually diagnostic of hereditary hypofibrinogenemia with hepatic storage. The diagnosis is corroborated by the demonstration of fingerprint-like or fiberglass-like inclusions within the RER and is definitely established by molecular genetics analysis. Thus, the phenotype-genotype correlation is restricted to type I fibrinogen inclusions that fulfill all criteria for ERSD[10] and conformational diseases.[11]
To summarize, the present report of hereditary fibrinogen storage disease shows the occurrence of this genetic disease in another ethnic population (i.e., an Arab family), in addition to families of Europe, America, and East Asia. The present report also suggests an independent origin (possibly ancient) of the mutation in various ethnic groups. The disease manifests in the heterozygous condition, suggesting an autosomal dominant modality of transmission and indirectly indicating the incompatibility with life in the homozygous condition. That would explain why the disease is so exceedingly rare.
At the time of the original detection, our patient was a 3-year-old child who presented with nephrotic syndrome. The possibility of a novel kidney-liver disease association was suggested because of the recent findings of renal pathology (amyloidosis) in patients with α-fibrinogen gene mutations.[19] However, in our case, the kidney biopsy failed to show any amyloid or fibrinogen deposition; the long follow-up (8 years now) has proven that the two pathologies were unrelated, thus allowing the interpretation of a fortuitous association.
In conclusion, our report emphasizes the occurrence of the Aguadilla mutation in individuals of Arab ethnicity that can cause chronic, otherwise mild liver disease with an early onset in children. Our findings reinforce that low fibrinogen levels with no (or an irrelevant) coagulation alteration profile should invariably require a diagnostic work-up, as outlined herein, for hereditary hypofibrinogenemia.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Huang S, Mulvhill ER, Farrell DH, Chung DW, Davie EW. Biosynthesis of human fibrinogen. Subunit interactions and potential intermediates in assembly. J Biol Chem. 1993;268:8919–26. [PubMed] [Google Scholar]
- 2.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 3.Brennan SO, Wyatt J, Medicina D, Callea F, George PM. Hepatic endoplasmic reticulum storage and hypofibrinogenemia because of a γ284 Gly → Arg mutation. Am J Pathol. 2000;157:189–96. doi: 10.1016/s0002-9440(10)64530-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan SO, Maghzal G, Shneider BL, Gordon R, Magid MS, George PM. Novel fibrinogen γ375 Arg → Trp mutation (Fibrinogen Aguadilla) causes hepatic endoplasmic reticulum storage and hypofibrinogenemia. Hepatology. 2002;36:652–8. doi: 10.1053/jhep.2002.35063. [DOI] [PubMed] [Google Scholar]
- 5.Dib N, Quelin F, Ternisien C, Hanss M, Michalak S, De Mazancourt P, et al. Fibrinogen angers with a new deletion (c GVYYQ 346-350) causes hypofibrinogenemia with hepatic storage. J Thromb Haemost. 2007;5:1999–2005. doi: 10.1111/j.1538-7836.2007.02713.x. [DOI] [PubMed] [Google Scholar]
- 6.Brennan SO, Davis RL, Conard K, Savo A, Furuya KN. Novel fibrinogen mutation γ314Thr → Pro (fibrinogen AI duPont) associated with hepatic fibrinogen storage disease and hypofibrinogenaemia. Liver Int. 2010;30:1541–7. doi: 10.1111/j.1478-3231.2010.02312.x. [DOI] [PubMed] [Google Scholar]
- 7.Francalanci P, Santorelli FM, Talini I, Boldrini R, Devito R, Camassei FD, et al. Severe liver disease in early childhood due to fibrinogen storage disease and de novo Gamma 375 Arg → Trp Mutation. J Pediatr. 2006;148:396–8. doi: 10.1016/j.jpeds.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 8.Rubbia-Brandt L, Neerman-Arbez M, Rougemont AL, Male× PJ, Spahr L. Fibrinogen Gamma375 Arg → Trp Mutation (Fibrinogen Aguadilla) Causes Hereditary Hypofibrinogenemia, Hepatic Endoplasmic Reticulum Storage Disease and Cirrhosis. Am J Surg Pathol. 2006;30:906–11. doi: 10.1097/01.pas.0000209848.59670.2c. [DOI] [PubMed] [Google Scholar]
- 9.Sogo T, Nagasaka H, Komatsu H, Inui A, Miida T, Callea F, et al. Fibrinogen storage disease caused by aguadilla mutation presenting with hypobeta-lipoproteinemia and considerable liver disease. J Pediatr Gastroenterol Nutr. 2009;49:133–6. doi: 10.1097/MPG.0b013e31817ed7ea. [DOI] [PubMed] [Google Scholar]
- 10.Callea F, Brisigotti M, Fabbretti G, Bonino F, Desmet VJ. Hepatic endoplasmic reticulum storage diseases. Liver. 1992;12:357–62. doi: 10.1111/j.1600-0676.1992.tb00589.x. [DOI] [PubMed] [Google Scholar]
- 11.Carrell RW, Lomas DA. Alpha1-antitrypsin deficiency--a model for conformational diseases. N Engl J Med. 2002;346:45–53. doi: 10.1056/NEJMra010772. [DOI] [PubMed] [Google Scholar]
- 12.Pfeifer U, Ormanns W, Klinge O. Hepatocellular fibrinogen storage in familial hypofibrinogenemia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1981;36:247–55. doi: 10.1007/BF02912070. [DOI] [PubMed] [Google Scholar]
- 13.Callea F, De Vos R, Pinackat J. Hereditary hypofibrinogenemia with hepatic storage of fibrinogen: A new endoplasmic reticulum storage disease, in Fibrinogen 2. In: Lowe GDO, Douglas JT, Forbes CD, Henschen A, editors. Biochemistry, Physiology, and Clinical Relevance. Amsterdam: Elsevier; 1987. pp. 75–8. [Google Scholar]
- 14.Hinds R, Hadchouel A, Shanmugham NP, Al-Hussaini A, Chambers S, Cheeseman P, et al. Variable degree of liver involvement in siblings with PiZZ Alpha-1-Antitrypsin Deficiency-related liver disease. J Pediatr Gastroenterol Nutr. 2006;43:136–8. doi: 10.1097/01.mpg.0000226370.09085.39. [DOI] [PubMed] [Google Scholar]
- 15.Kruse KB, Dear A, Kaltenbrun ER, Crum BE, George PM, Brennan SO, et al. Mutant fibrinogen cleared from the endoplasmic reticulum via endoplasmic reticulum-associated protein degradation and autophagy: An explanation for liver disease. Am J Pathol. 2006;168:1299–308. doi: 10.2353/ajpath.2006.051097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, Perlmutter DH. The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem. 2001;276:44865–72. doi: 10.1074/jbc.M103703200. [DOI] [PubMed] [Google Scholar]
- 17.Callea F, deVos R, Togni R, Tardanico R, Vanstapel MJ, Desmet VJ. Fibrinogen inclusions in liver cells: A new type of ground-glass hepatocyte. Immune light and electron microscopic characterization. Histopathology. 1986;10:65–73. doi: 10.1111/j.1365-2559.1986.tb02461.x. [DOI] [PubMed] [Google Scholar]
- 18.Callea F, Tortora O, Kojima T. Hypofibrinogenemia and fibrinogen storage disease, in Fibrinogen 3. In: Mosesson MW, Amrani DL, Siebenlist KR, DiOrio JP, editors. Biochemistry, Biological Functions, Gene Regulation and Expression. Amsterdam: Elsevier; 1988. pp. 247–250. [Google Scholar]
- 19.Benson MD, Liepricks J, Uemichi R, Wheeler G, Correa R. Hereditary renal amyloidosis associated with a mutant fibrinogen a chain. Nat Genet. 1993;3:252–63. doi: 10.1038/ng0393-252. [DOI] [PubMed] [Google Scholar]