

Abstract
Inappropriate or chronic detection of self nucleic acids by the innate immune system underlies a number of human autoimmune diseases. We discuss here an unexpected source of endogenous immunostimulatory nucleic acids: the reverse transcribed cDNAs of endogenous retroelements. The interplay between innate immune sensing and clearance of retroelement cDNAs has important implications for understanding immune responses to infectious retroviruses like human immunodeficiency virus (HIV). Further, cDNA detection by the innate immune system reveals an evolutionary tradeoff: selection for a vigorous, sensitive response to infectious retroviruses may predispose to inappropriate detection of endogenous retroelements. We propose that this tradeoff has placed unique constraints on the sensitivity of the DNA-activated antiviral response, with implications for the interactions of DNA viruses and retroviruses with their hosts. Finally, we discuss how a better understanding of the intersection of retroelement biology and innate immunity can guide the way to novel therapies for specific autoimmune diseases.
INTRODUCTION
Innate immune sensing of nucleic acids has emerged as the primary means by which host cells detect the presence of viral infection1. Since 2004, two key intracellular sensing pathways have been discovered and characterized that mediate innate immune detection of either RNA or DNA. The RNA sensors are the RIG-I and MDA5 helicases2; they detect structural features of viral RNA that are distinct from host RNAs, and they activate a potent antiviral response that includes the inducible production of the type I interferon (IFN) family of cytokines1. Activation of these RIG-I-like receptors (RLRs) results in their binding to MAVS, a transmembrane protein on the surface of mitochondria that serves as an adapter between the sensors and the TBK1 kinase and IRF3 transcription factor that activates type I IFN production1. In contrast, innate immune receptors for intracellular DNA (which are discussed in detail below) activate STING, a transmembrane protein on the endoplasmic reticulum that fulfills a similar adapter function to link DNA detection to the TBK1/IRF3-dependent antiviral response (Fig. 1;1,3). In addition to its role as an adapter protein in DNA detection, STING is also directly activated by cyclic dinucleotides produced as second messengers by many species of bacteria4. Finally, detection of intracellular DNA by the AIM2 receptor activates the ASC inflammasome5–8, a signaling platform for production of the cytokines IL-1β and IL-18 and a proinflammatory form of cell death called pyroptosis1. A number of recent reviews outline the details of intracellular RNA and DNA sensing that are only summarized here9,10.
Figure 1. Types of Endogenous Retroelements in the Mammalian Genome.

Autonomous retroelements encode all the proteins necessary for replication. These include LTR type endogenous retroviruses and LINE elements. Nonautonomous retroelements include SINEs, and require LINEs for their mobility. (LTR, Long terminal repeat; GAG, group-specific antigen; PRT, protease; Pol, polymerase; ENV, envelope)
These intracellular nucleic acid sensing pathways are crucial for signaling the presence of viruses within infected cells, and as such are essential for the activation of a productive antiviral response. However, the same nucleic acid sensors that protect us from viral infection are also responsible for a number of human autoimmune diseases. Perhaps the most well studied example of this is the involvement of the nucleic acid-sensing Toll-like receptors (TLRs) in the pathological autoimmune response to self nucleic acid-protein complexes11–13. Another, recently described source of endogenous immunosimulatory nucleic acids is derived from a class of viruses that is present within our own genomes: the endogenous retroelements. Indeed, innate immune sensing of the reverse transcribed DNA (cDNA) intermediates of these viruses does occur, and exciting recent studies have shown that this mechanism of detection is also highly relevant for immune responses to human immunodeficiency virus (HIV).
In this review, we will first describe the dynamics of endogenous retroelements within cells. We highlight the discovery of enzymes that metabolize endogenous retroelement cDNAs, and how key insights into the genetics of a rare human autoimmune disease illuminated a new area of biology. We summarize fascinating recent advances in our understanding of innate immune responses to infectious retroviruses. Next, we discuss the implications of cDNA sensing for the evolution of DNA-activated antiviral responses. Finally, we propose that interventions that prevent the formation of retroelement cDNAs may hold promise as treatments for a number of human autoimmune disorders.
The life and times of endogenous retroelements
Retrotransposons replicate through a copy and paste mechanism, inserting new copies of themselves into unique genomic locations. They have undergone several episodes of dramatic expansion in mammals and comprise over 40% of the base content of the human genome14–16. Two families of retrotansposons are autonomous in that they encode all of the proteins required to reverse transcribe an RNA intermediate into a dsDNA product that can be inserted into a new place in the genome. The retrovirus-like long terminal repeat (LTR) retrotransposons are relics of ancient retroviruses that once integrated into the germline but then lost their ability to infect other cells. Their genomic organization and replication cycle resembles that of extant infectious retroviruses like HIV, with reverse transcription of the viral mRNA primed by a specific cellular tRNA (Figs. 1, 3). LTR retrotransposons make up about 8% of the human genome and are a stable population that is transcriptionally active, but not expanding in number through new integration. However, these human endogenous retroviruses (HERVs) continue to influence genome organization, in large part through the high rate of recombination between their identical LTRs17. The LINE1 (L1) retrotransposon is the only actively replicating retroelement in the human genome. L1s are about 6kb in length and encode two proteins, ORF1 and ORF2, that coordinate replication through target primed reverse transcription (TPRT). TPRT involves generation of a nick in the genome and cDNA synthesis through reverse transcription mediated by ORF2 directly on genomic DNA (Figs 1, 3). A third family of retrotransposons called short interspersed nuclear elements (SINEs) are nonautonomous, and do not encode for any proteins (Fig. 1). Most of the human SINEs belong to a single type known as Alu, which at 300 bp in length are much smaller than L1s15,16. However, Alu elements are very successful at replicating themselves, and they hijack the L1 reverse transcriptase to mediate their retrotransposition in trans. There are over one million copies of Alu in the human genome, meaning there is one Alu for every ~3,000 bp of genomic sequence14–16. In addition to reverse transcribing Alu RNAs, the L1 reverse transcriptase can act on any polyadenylated RNA to generate a processed pseudogene, which is an insertion of an intronless, promoterless cDNA into a unique genomic location. According to the latest estimates18, there are 145 full-length, functional L1 elements in the human genome, together with 103 additional L1s with an intact ORF2 but a mutated ORF1. These “ORF2 only” L1s may be functionally relevant: whereas both ORF1 and ORF2 are required for retrotransposition of the L1 RNA, ORF2 can mediate the retrotransposition of Alu elements independent of ORF119. A number of excellent recent reviews summarize the biology and fascinating properties of endogenous retroelements15,16; we will focus here on their potential to trigger the innate immune system and cause specific autoimmune diseases.
Figure 3. AGS Genes Restrict Replication of Endogenous Retroelements and Exogenous Retroviruses.

Schematic of LINE and Retrovirus Replication. AGS genes act throughout the steps of replication to restrict products of reverse transcriptase. SAMHD1 inhibits reverse transcription through degradation of cellular dNTPs. TREX1 metabolizes DNA products of reverse transcription, and can block retrotransposition of endogenous retroelements. RNASEH2 can degrade RNA of RNA/DNA hybrids and can also block retrotransposition of endogenous retroelements, and we hypothesize that it acts by degrading viral RNA in the context of reverse transcription.
The intersection of retroelement biology and innate immunity
We fortuitously discovered innate immune sensing of retroelement cDNAs when we were looking for candidate sensors of the antiviral response to intracellular DNA that we named the interferon stimulatory DNA (ISD) pathway20,21. We recovered proteins that bound to transfected ISD with the hope of finding “the ISD sensor”, and the first protein we identified by mass spectrometry was 3′ repair exonuclease 1 (Trex1), the activity of which was first described over 40 years ago by Lindahl and colleagues22. Our work on Trex1 and its relation to the ISD pathway was crucially influenced by the discovery of human mutations in the TREX1 gene that cause a rare and severe autoimmune disease called Aicardi-Goutieres Syndrome (AGS23). AGS is an early-onset, type I IFN-associated disorder characterized by neurological dysfunction, psychomotor retardation, and skin inflammation24,25. The finding of TREX1 mutations in AGS, and the subsequent identification of all other known AGS genes by Yanick Crow and his colleagues26–28, established the framework within which we continue to interpret the functions of these enzymes and their roles in innate immunity. In addition to its association with AGS, TREX1 mutations were also identified in a monogenic form of cutaneous lupus called familial chilblain lupus29,30. Finally, heterozygous mutations in TREX1 were identified in a small percentage of patients with systemic lupus erythematosus (SLE31,32), but very rarely in healthy controls. In fact, the odds ratio of the association of Trex1 mutations with SLE remains the strongest single gene association with this autoimmune disorder identified to date33.
Using Trex1 deficient mice as a model of AGS disease mechanisms, we found that the lethal autoimmune disease in these mice requires STING, IRF3, type I IFNs, and lymphocytes34,35. We then purified the intracellular DNAs that accumulated within Trex1-deficient cells and devised a way to identify them by sequencing. We were surprised to find a strong over-representation of DNA fragments that mapped to endogenous retroelements in Trex1-deficient cells compared to control cells. We then showed that Trex1 potently blocks retrotransposition of model endogenous retroelements by metabolizing reverse-transcribed cDNAs34. Based on these findings, we proposed that innate immune detection of retroelement cDNAs caused AGS and that the AGS enzymes functioned as anti-retroviral proteins. It is important to note that at the time we identified the connections between the DNA sensing pathway, retroelement cDNA metabolism, and autoimmunity, very little was known about how retrovirus infection is sensed by the innate immune system, or whether a mechanism for detecting retroviruses within infected cells even existed36. As discussed below, several studies in the last few years have dramatically confirmed and extended these findings in the context of HIV infection, establishing a new and exciting area of interactions between HIV and its human host with clear implications for the future design of better HIV vaccines.
The first description of a role for cDNA sensing during HIV infection came from Lieberman and colleagues, who found that, in addition to its role in metabolizing the reverse-transcribed cDNA of endogenous retroelements, Trex1 can also degrade cDNAs during HIV infection37. Interestingly, Trex1-depleted cells triggered a STING-dependent type I IFN response to HIV infection, whereas control cells did not. At almost precisely the same time, Littman and colleagues reported that HIV-1 infection of human dendritic cells activated a potent, IRF3-dependent type I IFN response38. As HIV-1 does not normally infect myeloid cells, Manel et al used a “trick” to enable productive HIV-1 infection of these refractory cells: they included virus-like particles from simian immunodeficiency virus of macaques (SIVmac), which overcome the restriction of HIV because of a SIV-encoded accessory factor called Vpx39. Finally, Greene and colleagues reported that HIV-1 infection of human lymphoid aggregate cultures (HLACs) resulted in a massive depletion of abortively infected human CD4 T cells40. They provided compelling evidence that this cell death response was caused by the accumulation of incomplete reverse transcription intermediates in these non-permissive cells. Together, these three important papers established that HIV is indeed detected by the innate immune system and raised a number of important questions. How is the Vpx accessory protein enabling the infection of otherwise refractory cells by HIV-1, and how is this tied to activation of the IFN response? What are the sensors of HIV-1 cDNAs and how are they connected to IFNs and cell death?
A key insight came several months later, when two groups identified SAMHD1, first described as an AGS gene by Crow and colleagues in 200927, as the key myeloid HIV-1 restriction factor that is targeted for degradation by Vpx41,42. SAMHD1 was then found to be a dNTP phosphohydrolase that functions to “starve” the HIV reverse transcriptase of the nucleotides that are required for cDNA synthesis43,44. This function of SAMHD1 in preventing HIV reverse transcription extends beyond myeloid cells to resting CD4 T cells45, which have long been known to be refractory to HIV-1 infection. Importantly, the identification of a second AGS gene as a potent anti-retroviral enzyme provided crucial independent support for our model that defective retroelement cDNA metabolism causes AGS.
As it became clear that innate immune detection of HIV converged on its reverse transcription intermediates, attention turned to identifying the relevant DNA sensors involved in this detection. Many candidate receptors for intracellular DNA have been proposed, and considerable controversy remains regarding the relative contributions of these diverse proteins to the antiviral response46. The field enjoyed a major breakthrough when James Chen and colleagues purified an enzyme called cyclic GMP-AMP synthase (cGAS;47). cGAS binds to immunostimulatory DNA and catalyzes the formation of cyclic GMP-AMP (cGAMP), which then binds to STING and triggers the IFN response48. The importance of this discovery cannot be overstated, and it has already spawned an entirely new area of investigation of the nature of the cyclic dinucleotide created by cGAS49–51, as well as structural insight into DNA binding and catalysis by this enzyme49,52–54. Importantly, Chen and colleagues found that cGAS is essential for the IFN response to DNA viruses, HIV, and other retroviruses, providing important evidence for the essential role of the cGAS-cGAMP-STING pathway in the DNA-activated antiviral response55,56.
Interestingly, Greene and colleagues recently reported that the death of abortively infected CD4 T cells during HIV infection is mediated by pyroptosis57, dependent on the IFI16 receptor58, which was identified by Bowie and colleagues as a key DNA sensor in 201059. Moreover, Paludan and colleagues found that IFI16 is essential for the IFN response to HIV-1 infection in human monocytes60. Thus, cGAS and IFI16 have emerged as important sensors that mediate the type I IFN response to HIV infection. Further work will be required to clarify the relative contributions of cGAS, IFI16, and other potential DNA sensors to immune defense against HIV.
All of these remarkable studies raise the important question of how this response to HIV reverse transcription intermediates was only recently discovered, given that scientists have been scrutinizing the immune response to HIV infection for decades. One key reason is that HIV has evolved to avoid and manipulate innate immune detection61. Indeed, the HIV accessory factor Vpu targets the IRF3 transcription factor for degradation62,63, severely blunting the DNA-activated antiviral response. Moreover, two recent studies revealed that the HIV-1 capsid acts to specifically shield its reverse transcription intermediates from innate immune sensing. The Manel and Towers groups found that certain capsid mutations reveal a potent, IFN-mediated response to HIV infection, presumably by destabilizing its structure and allowing access of innate immune sensors to the cDNA intermediates64,65. These intriguing studies suggest that engineered capsid mutations or pharmacologic inhibition of the interactions between the capsid and its cellular partners may enable the design of an HIV-based vaccine that efficiently stimulates a potent innate immune response.
An evolutionary tradeoff between responses to infectious and endogenous retroviruses
The exciting recent studies described above prove definitively that innate immune detection of HIV indeed occurs within infected cells, that this detection is based primarily on sensing of cDNA intermediates, and that HIV has evolved to avoid and/or manipulate this antiviral response. These findings reveal a long-standing evolutionary arms race that has unfolded between infectious retroviruses and their mammalian hosts. It is clear that this arms race has impacted the evolutionary trajectory of the APOBEC3 family of cytidine deaminases, the SAMHD1 dNTP phosphohydrolase, the TRIM5α capsid-binding protein, and other host restriction factors that target retroviruses66,67. We can therefore infer that similar pressures have shaped the intracellular DNA sensing pathways, with important implications for a number of autoimmune diseases. Specifically, we propose an evolutionary tradeoff with two opposing forces (Fig. 2). The first is the benefit of a more sensitive and robust innate immune response to cDNAs of infectious retroviruses. This could in principle be enabled by higher expression of DNA sensors, by an increased affinity of these sensors for DNA, or by a reduction in the function of key negative regulators that impact the DNA-activated antiviral response. More broadly, this selective pressure extends beyond retroviruses to include all viruses that are sensed by the same mechanism, including herpesviruses59,68, adenoviruses69, and presumably all other classes of DNA viruses. This evolutionary drive towards a more robust antiviral response is balanced by the need to avoid excessive responses to the cDNAs of endogenous retroelements. We speculate that this requirement for minimal autoreactivity to retroelements places a limit on the sensitivity of the DNA-activated antiviral response (Fig. 2). Moreover, unlike the RLRs, which detect key structural features of viral RNAs that are scarce within host RNAs10, all available structural evidence suggests that the key DNA sensors simply detect double stranded DNA, independent of its sequence52,70,71. Thus, the limits on the sensitivity of the DNA-activated antiviral response are imposed not only by endogenous retroelements, but also by the identical structures of foreign and self-DNA. Taken one step further, we speculate that these unique evolutionary constraints may have enabled the development of DNA viruses that establish latency and lifelong infections of their hosts. In other words, the need to “ignore” low levels of retroelement cDNAs may provide an opportunity, for example, for a herpesvirus to maintain a copy of its ~100–250 kilobase dsDNA genome within cells without triggering a potent immune response (Fig. 2). These latent DNA viruses probably actively antagonize innate immune detection, which could further raise the threshold of DNA sensing within latently infected cells (Fig. 2). In contrast, just as there are no known endogenous RNA viruses, there are also no infectious RNA viruses that establish latency: RNA viruses are either cleared or establish chronic, symptomatic infections. We propose that this is because the RLRs are not constrained by the presence of abundant endogenous ligands, so they have evolved to be more sensitive than the innate immune receptors of DNA. Consequently, RNA viruses cannot persist below the threshold of innate immune detection (Fig. 2). The details of this evolutionary tradeoff hypothesis in DNA sensing are now testable because we know many of the key sensors and how they signal activation of the antiviral response.
Figure 2. Model: Coevolution with Endogenous Retroelements Shapes the DNA Sensing Pathway.
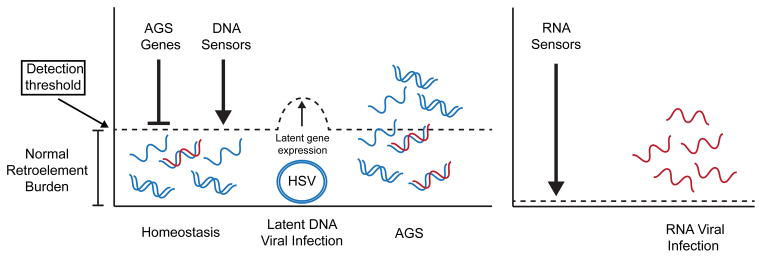
Low levels of nucleic acids arising from reverse transcription of endogenous retroelements are kept in check by AGS genes. We predict that the sensitivity of DNA sensors must be above this level to prevent inappropriate activation of the DNA sensing pathway and autoimmune disease. This level of detection would aid DNA viruses to maintain their genome in the nucleus during latent infection. We predict that some DNA viruses during latency would act to inhibit the DNA sensing pathway, thereby raising the threshold needed to initiate an immune response. During AGS the burden of nucleic acids from endogenous retroelements reaches a level now detectable by the DNA sensors and initiates a chronic immune response leading to autoimmune disease. The threshold of sensitivity for RNA sensors can be much lower because of a lack of endogenous RNA ligands.
Interestingly, at least two of the AGS genes (Trex1 and SAMHD1) are positioned at the pivot point of this evolutionary tradeoff (Figs 2, 3). Loss of function mutations in either of these genes enables a more robust immune response to infectious retroviruses37,38 but causes autoimmunity because of inappropriate detection of retroelement cDNA34. It would be very interesting to explore whether individuals who are heterozygous for mutations in these enzymes are protected from infection by DNA viruses and/or retroviruses. Such a benefit would provide a rationale for the existence and maintenance of these relatively rare alleles.
The retroelement model and tissue-specific autoimmune diseases
Our hypothesis that defective retroelement cDNA metabolism underlies AGS is in keeping with the clearly defined anti-retroviral function of two of the AGS enzymes34,37,41,42. Moreover, we have similarly found that RNase H2, another AGS enzyme26, potently restricts retrotransposition of endogenous retroelements (unpublished data). Thus, five of the AGS genes can be placed within a common pathway of metabolism of retroelement cDNA intermediates (Fig. 3). Within the framework of this model, there are two key questions that have not yet been adequately addressed. First, if the AGS enzymes can each function as anti-retroviral enzymes, why are they not redundant with each other? Second, how can retroelements, which are ubiquitously present in the genomes of all of our cells, cause tissue-specific autoimmune disease if their cDNAs are not metabolized?
Our model for AGS enzyme interactions with retroelements is summarized in Figure 3. As described above, SAMHD1 starves the reverse transcriptase of the dNTPs that are required to generate cDNA, preventing the formation of potentially immunostimulatory nucleic acids. We speculate that RNase H2 attacks the RNA strand of the RNA/DNA hybrid that is an essential intermediate of reverse transcription. Finally, Trex1 can metabolize the cDNAs of retroelements and retroviruses. Why, then, do mutations in each individual enzyme cause AGS? For example, mutations in SAMHD1 would result in the production of more cDNAs, but RNase H2 and Trex1 should still (in principle) be able to metabolize them. Moreover, SAMHD1 and Trex1 are IFN inducible genes27,34, so their levels should increase in response to ISD pathway activation to enable more efficient metabolism of accumulated cDNAs. We envision two possible reasons for the lack of redundancy among AGS enzymes. First, each enzyme may contribute uniquely to disposal of nascent cDNAs. Based on the sensitivity of cDNA detection dictated by the evolutionary tradeoff model described above, mutation of each enzyme may increase the levels of endogenous retroelement cDNAs above the threshold of detection, resulting in a chronic antiviral response (Fig. 3). Second, each AGS enzyme may be part of a linear pathway for cDNA metabolism such that the product of one enzyme is the substrate for the next. For example, SAMHD1 may need to be recruited to sites of early reverse transcription, and this in turn may be required for subsequent RNase H2 activity on the RNA/DNA hybrid. Trex1 may then require access to DNA that has been exposed as a result of RNase H2 activity. In this way, each enzyme may depend on the activity of the others for its function, and mutation of one enzyme prevents the anti-retroviral function of the others.
It is important to note that an alternative model exists in which mutations in AGS genes result in a chronic DNA damage response72. The most compelling evidence for this model was recently found by Jackson and colleagues, who demonstrated an essential role for RNase H2 in the removal of ribonucleotides that are misincorporated into genomic DNA during replication73. Mice deficient in Rnaseh2b suffer early embryonic lethality that is caused by massive genome instability73,74, and this is partially relieved by simultaneous deletion of the p53 tumor suppressor that controls many aspects of the DNA damage response73. Lee-Kirsch and colleagues recently reported that SAMHD1 depletion in fibroblasts results in increased genomic DNA damage75, and Schuh and colleagues reported an AGS patient with SAMHD1 mutations who developed chronic lymphocytic leukemia (CLL), as well as recurrent somatic SAMHD1 mutations in CLL in non-AGS patients76. While these findings clearly show a link between AGS genes and the DNA damage response, the mechanistic connections to the autoimmune disorder remain undefined. Indeed, the Rnaseh2b-null mice have no evidence for an aberrant type I IFN response that is present in Trex1 deficient mice34,73, and, to a lesser extent, in SAMHD1-deficient mice77,78. Interestingly, RNase H2 in AGS patients is still enzymatically active79, suggesting an interesting dichotomy between null alleles of AGS genes and AGS mutations. Moreover, it is unclear how chronic DNA damage could lead to IFN-dependent autoimmune disease. For these reasons, we suggest that retroelement cDNA detection represents a more plausible scenario for autoimmune disease in this setting, although further work will be required to reconcile these two models.
The ubiquitous presence of retroelements in our genomes raises the question of how defective metabolism of these elements could result in an autoimmune disease that has clear tissue specificity in mice and in humans34,35,80,81. We speculate that the tissue specificity of the autoimmune disease reflects tissue-specific expression of functional retroelements. Most retroelement sequences in the genome are silenced by epigenetic mechanisms that prevent their transcription; these include specific recruitment of chromatin remodeling factors that form heterochromatin on these sequences15,16. Because of this, retroelement activity is maintained at a very low level in most cells, most of the time. As mentioned above, there are 248 functional copies of the L1 reverse transcriptase in the human genome, among thousands of non-functional RT genes. However, tissue-specific expression of a functional retroelement could occur if that retroelement is contained within an intron of an abundantly expressed, essential, tissue-specific gene. In this context, it will be very interesting to compare the genomic locations of all of the functional retroelements with an atlas of mRNA transcript levels and genome-wide chromatin states in specific tissues. All of these datasets are currently available for many tissues. We predict that a limited number of functional retroelements will be found within tissue-specific genes that coincide with affected tissues in AGS and related diseases. Such an analysis is simpler for humans than for mice because mice have over ten times as many functional copies of the L1 RT18, along with a number of functional endogenous LTR retroviruses that do not exist in humans.
If tissue-specific retroelement expression exists, we can extend this model to include environmental stimuli that cause the transient derepression of retroelements. Intriguingly, DNA damage resulting from exposure to ultraviolet light induces the massive transcription of retroelements and a dramatic increase in cellular reverse transcriptase activity82,83. If the level of retroelement cDNAs in these damaged cells exceeds the threshold for detection by DNA sensors, an inflammatory response would be initiated, particularly in those cells with defective metabolism of these retroelement cDNAs. Trex1 mutations cause a monogenic form of cutaneous lupus called familial chilblain lupus and are strongly associated with SLE29–31, and photosensitivity to ultraviolet light is a common feature of SLE. It is tempting to speculate that retroelement cDNA metabolism is linked to these episodic cutaneous features, and more broadly, that environmental stimuli that impact retroelement expression may similarly drive inflammation through the same mechanism.
Implications for novel therapies
The identification of a role for retroelement cDNA metabolism in AGS, together with the delineation of the key innate immune pathways that mediate autoimmunity, provides a number of new opportunities to develop novel therapeutic interventions for AGS, which is currently untreatable and incurable. It was discovered over 25 years ago that AGS is associated with elevated levels of type I IFNs25, and mouse models of AGS confirmed the central pathogenic role for these cytokines in disease progression34,35. Biologics that antagonize IFN signaling are in development and hold great promise to ameliorate the IFN-dependent aspects of disease. Similarly, pharmacological inhibition of the TBK1 kinase that is essential for DNA-activated IFN production84–86 would blunt the chronic antiviral response. The recent identification of the cGAS/cGAMP pathway of DNA sensing offers a very appealing target for specific inhibition47,48. However, these approaches are not ideal because cGAS- and TBK1-dependent IFN production is essential for antiviral immunity against many infectious viruses, so long-term treatment of patients with these drugs would likely increase susceptibility to infections. In the context of AGS, the severity of the disease may outweigh these potential complications87, so they should be considered as viable therapeutic options.
A novel approach to the treatment of AGS and related diseases is to prevent the immunostimulatory nucleic acids from being formed in the first place. In the case of cDNAs from endogenous retroelements, there are twelve FDA-approved reverse transcriptase inhibitors (RTIs) that are effective against HIV. Wabl and colleagues tested a cocktail of three of these RTIs in Trex1-deficient mice and found a remarkable rescue from mortality in drug-treated mice88. This important experiment opens the door to a potential therapeutic approach that has four important advantages. First, RTIs would block the disease at its most proximal source by preventing the accumulation of immunostimulatory nucleic acids. Second, this treatment would be highly specific for AGS, without the potential immunosuppressive effects of global IFN blockade. Third, these drugs are already FDA approved, and many of them have years of clinical data detailing safety, tolerability, and side effects. Finally, unlike HIV, which can easily mutate away from sensitivity to a specific RTI, endogenous retroelements cannot rapidly evolve drug resistance because they are not infectious.
Going forward, a number of key questions must be answered in order to inform the potential utility of using RTIs to treat AGS and related diseases. Most importantly, which reverse transcriptase is relevant for disease? The L1 RT is the most likely candidate because hundreds of functional copies of this enzyme exist in the human genome and it can reverse transcribe a variety of polyadenylated RNAs, including the L1 RNAs, SINE RNAs, and certain cellular RNAs15,16. In contrast, there are no known endogenous LTR retroviruses that are fully functional in humans. This point is particularly important because the need to inhibit the L1 RT would eliminate all non-nucleoside RTIs (NNRTIs) from consideration, as NNRTIs specifically target HIV RT and related retroviral enzymes but are not effective against L1 (unpublished data). The specific RTIs must not only target the appropriate RT enzyme(s), but they must also be able to access the relevant tissues and cells to inhibit cDNA formation in situ. In AGS, the brain is a prominent site of autoimmune attack. Thus, these drugs must be capable of crossing the blood-brain barrier. Finally, if we extend the possible utility of RTIs to SLE, a more common autoimmune disorder, it will be important to know which SLE patients could potentially benefit from this therapeutic approach. This would require a means to distinguish SLE patients with chronic ISD pathway activation from those with disease that is driven by distinct mechanisms. If such stratification of SLE patients is possible, this would go a long way towards “personalizing” therapies based on the underlying basis of the autoimmune response.
In summary, we have outlined recent advances that have established innate immune detection of retroelement cDNAs as a major contributor to specific autoimmune diseases. We propose an evolutionary tradeoff between immune detection of exogenous retroviruses and endogenous retroelements that places unique constraints on the sensing of foreign DNA. We place the AGS enzymes in the context of this model, and we suggest that reverse transcriptase inhibitors hold considerable promise for the treatment of AGS and related diseases. Further understanding of the mechanisms that underlie these autoimmune disorders will undoubtedly reveal new and exciting avenues for the development of useful therapies to treat these diseases.
Figure 4. Therapeutic Strategies to Mitigate Cell Intrinsic Activation of the ISD Pathway.

Failure to metabolize reverse transcribed products causes inappropriate activation of the DNA sensing pathway, chronic type I IFN production, and autoimmune disease. Disease intervention strategies include blockade of type I IFNs, preventing signaling through the ISD pathway through TBK1 specific inhibitors, and elimination of reverse transcribed products through the use of reverse transcriptase inhibitors (RTIs).
Acknowledgments
We are grateful to members of the Stetson lab for helpful discussions, and to Yanick Crow for continued collaborations. We apologize to those colleagues whose work we were unable to cite because of space considerations. H.E.V. was supported by a Jane Coffin Childs postdoctoral fellowship. D.B.S. is a scholar of the Rita Allen Foundation. Our work is supported by the National Institute of Allergy and Infectious Disease (AI084914), the European Union (FP7/2007-2013) grant agreement number 241779 (NIMBL: http://www.NIMBL.eu), and the Lupus Research Institute.
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Barbalat R, Ewald SE, Mouchess ML, Barton GM. Nucleic acid recognition by the innate immune system. Annu Rev Immunol. 2011;29:185–214. doi: 10.1146/annurev-immunol-031210-101340. [DOI] [PubMed] [Google Scholar]
- 2.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 3.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. nature07317 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burdette DL, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. nature10429 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muruve DA, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. nature06664 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Hornung V, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. nature07725 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. nature07710 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burckstummer T, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. ni.1702 [pii] [DOI] [PubMed] [Google Scholar]
- 9.Xiao TS, Fitzgerald KA. The cGAS-STING Pathway for DNA Sensing. Mol Cell. 2013;51:135–139. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goubau D, Deddouche S, Reis ESC. Cytosolic sensing of viruses. Immunity. 2013;38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leadbetter EA, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 12.Barrat FJ, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–1139. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christensen SR, et al. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 14.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 15.Goodier JL, Kazazian HH., Jr Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135:23–35. doi: 10.1016/j.cell.2008.09.022. [DOI] [PubMed] [Google Scholar]
- 16.Levin HL, Moran JV. Dynamic interactions between transposable elements and their hosts. Nat Rev Genet. 2011;12:615–627. doi: 10.1038/nrg3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hughes JF, Coffin JM. Human endogenous retrovirus K solo-LTR formation and insertional polymorphisms: implications for human and viral evolution. Proc Natl Acad Sci U S A. 2004;101:1668–1672. doi: 10.1073/pnas.0307885100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Penzkofer T, Dandekar T, Zemojtel T. L1Base: from functional annotation to prediction of active LINE-1 elements. Nucleic Acids Res. 2005;33:D498–500. doi: 10.1093/nar/gki044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nat Genet. 2003;35:41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 20.Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Ishii KJ, et al. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl T, Gally JA, Edelman GM. Properties of deoxyribonuclease 3 from mammalian tissues. J Biol Chem. 1969;244:5014–5019. [PubMed] [Google Scholar]
- 23.Crow YJ, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 24.Aicardi J, Goutieres F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- 25.Lebon P, et al. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- 26.Crow YJ, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 27.Rice GI, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829–832. doi: 10.1038/ng.373. ng.373 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice GI, et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet. 2012;44:1243–1248. doi: 10.1038/ng.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rice G, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811–815. doi: 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee-Kirsch MA, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med. 2007;85:531–537. doi: 10.1007/s00109-007-0199-9. [DOI] [PubMed] [Google Scholar]
- 31.Lee-Kirsch MA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065–1067. doi: 10.1038/ng2091. [DOI] [PubMed] [Google Scholar]
- 32.Namjou B, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun. 2011;12:270–279. doi: 10.1038/gene.2010.73. gene201073 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harley IT, Kaufman KM, Langefeld CD, Harley JB, Kelly JA. Genetic susceptibility to SLE: new insights from fine mapping and genome-wide association studies. Nat Rev Genet. 2009;10:285–290. doi: 10.1038/nrg2571. nrg2571 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008;134:587–598. doi: 10.1016/j.cell.2008.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gall A, et al. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-Dependent Autoimmune Disease. Immunity. 2012;36:120–131. doi: 10.1016/j.immuni.2011.11.018. S1074-7613(12)00008-8 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Medzhitov R, Littman D. HIV immunology needs a new direction. Nature. 2008;455:591. doi: 10.1038/455591a. [DOI] [PubMed] [Google Scholar]
- 37.Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol. 2010;11:1005–1013. doi: 10.1038/ni.1941. ni.1941 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manel N, et al. A cryptic sensor for HIV-1 activates antiviral innate immunity in dendritic cells. Nature. 2010;467:214–217. doi: 10.1038/nature09337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goujon C, et al. With a little help from a friend: increasing HIV transduction of monocyte-derived dendritic cells with virion-like particles of SIV(MAC) Gene therapy. 2006;13:991–994. doi: 10.1038/sj.gt.3302753. [DOI] [PubMed] [Google Scholar]
- 40.Doitsh G, et al. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell. 2010;143:789–801. doi: 10.1016/j.cell.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laguette N, et al. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hrecka K, et al. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature. 2011;474:658–661. doi: 10.1038/nature10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldstone DC, et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 44.Powell RD, Holland PJ, Hollis T, Perrino FW. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J Biol Chem. 2011;286:43596–43600. doi: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Descours B, et al. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology. 2012;9:87. doi: 10.1186/1742-4690-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Unterholzner L. The interferon response to intracellular DNA: why so many receptors? Immunobiology. 2013;218:1312–1321. doi: 10.1016/j.imbio.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 47.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu J, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi: 10.1126/science.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao P, et al. Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ablasser A, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diner EJ, et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Civril F, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi: 10.1038/nature12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell reports. 2013;3:1362–1368. doi: 10.1016/j.celrep.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell reports. 2014;6:421–430. doi: 10.1016/j.celrep.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li XD, et al. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science. 2013 doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gao D, et al. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science. 2013;341:903–906. doi: 10.1126/science.1240933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doitsh G, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Monroe KM, et al. IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV. Science. 2014;343:428–432. doi: 10.1126/science.1243640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Unterholzner L, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. ni.1932 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jakobsen MR, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc Natl Acad Sci U S A. 2013;110:E4571–4580. doi: 10.1073/pnas.1311669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manel N, Littman DR. Hiding in plain sight: how HIV evades innate immune responses. Cell. 2011;147:271–274. doi: 10.1016/j.cell.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doehle BP, et al. Vpu-Deficient HIV Strains Stimulate Innate Immune Signaling Responses in Target Cells. J Virol. 2012;86:8499–8506. doi: 10.1128/JVI.00424-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Doehle BP, et al. Vpu Mediates Depletion of Interferon Regulatory Factor 3 during HIV Infection by a Lysosome-Dependent Mechanism. J Virol. 2012;86:8367–8374. doi: 10.1128/JVI.00423-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rasaiyaah J, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lahaye X, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39:1132–1142. doi: 10.1016/j.immuni.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Daugherty MD, Malik HS. Rules of engagement: molecular insights from host-virus arms races. Annu Rev Genet. 2012;46:677–700. doi: 10.1146/annurev-genet-110711-155522. [DOI] [PubMed] [Google Scholar]
- 67.Lim ES, et al. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe. 2012;11:194–204. doi: 10.1016/j.chom.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li T, Chen J, Cristea IM. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe. 2013;14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lam E, Stein S, Falck-Pedersen E. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol. 2014;88:974–981. doi: 10.1128/JVI.02702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin T, et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao JC, et al. Interferon-inducible protein 16: insight into the interaction with tumor suppressor p53. Structure. 2011;19:418–429. doi: 10.1016/j.str.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell. 2007;131:873–886. doi: 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 73.Reijns MA, et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012;149:1008–1022. doi: 10.1016/j.cell.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hiller B, et al. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med. 2012;209:1419–1426. doi: 10.1084/jem.20120876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kretschmer S, et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann Rheum Dis. 2014 doi: 10.1136/annrheumdis-2013-204845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clifford R, et al. SAMHD1 is mutated recurrently in chronic lymphocytic leukemia and is involved in response to DNA damage. Blood. 2014;123:1021–1031. doi: 10.1182/blood-2013-04-490847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rehwinkel J, et al. SAMHD1-dependent retroviral control and escape in mice. Embo J. 2013;32:2454–2462. doi: 10.1038/emboj.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Behrendt R, et al. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell reports. 2013;4:689–696. doi: 10.1016/j.celrep.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perrino FW, Harvey S, Shaban NM, Hollis T. RNaseH2 mutants that cause Aicardi-Goutieres syndrome are active nucleases. J Mol Med. 2009;87:25–30. doi: 10.1007/s00109-008-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morita M, et al. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–6727. doi: 10.1128/MCB.24.15.6719-6727.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rice G, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rudin CM, Thompson CB. Transcriptional activation of short interspersed elements by DNA-damaging agents. Genes Chromosomes Cancer. 2001;30:64–71. [PubMed] [Google Scholar]
- 83.Hagan CR, Sheffield RF, Rudin CM. Human Alu element retrotransposition induced by genotoxic stress. Nat Genet. 2003;35:219–220. doi: 10.1038/ng1259. [DOI] [PubMed] [Google Scholar]
- 84.Fitzgerald KA, et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 85.Sharma S, et al. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 86.Ishii KJ, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. nature06537 [pii] [DOI] [PubMed] [Google Scholar]
- 87.Crow YJ, Vanderver A, Orcesi S, Kuijpers TW, Rice GI. Therapies in Aicardi-Goutieres syndrome. Clinical and experimental immunology. 2014;175:1–8. doi: 10.1111/cei.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Beck-Engeser GB, Eilat D, Wabl M. An autoimmune disease prevented by anti-retroviral drugs. Retrovirology. 2011;8:91. doi: 10.1186/1742-4690-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]