

Abstract
Fluorinated organic compounds have a long history in medicinal chemistry, and synthetic methods to access target fluorinated compounds are undergoing a revolution. One powerful strategy for the installation of fluorine-containing functional groups includes decarboxylative reactions. Benefits of decarboxylative approaches potentially include: 1) readily available substrates or reagents 2) mild reaction conditions; 3) simplified purification. This focus review highlights the applications of decarboxylation strategies for fluorination reactions to access compounds with biomedical potential. The manuscript highlights on two general strategies, fluorination by decarboxylative reagents and by decarboxylation of substrates. Where relevant, examples of medicinally useful compounds that can be accessed using these strategies are highlighted.
Keywords: decarboxylation, fluorination, difluoromethylation, trifluoromethylation, copper
1) Introduction
Synthetic methods for the efficient incorporation of fluorinated functional groups enable the rapid construction of biologically interesting molecules and therefore, are important for medicinal chemistry [1–2]. Although many creative and elegant fluorination strategies have recently been developed [3–6], a need remains for new transformations that grant access to unique products and that rely on principles of green chemistry [7]. One emerging approach that has gained appreciation in non-fluorine chemistry involves decarboxylative coupling [8–9]. This strategy typically exhibits several appealing features, including the: 1) use of inexpensive and readily accessible starting materials; 2) ability to selectively generate reactive species under mild reaction conditions; 3) release of CO2 as a benign and easily removed by-product [9]. Given these benefits, decarboxylative coupling represents an attractive approach for the installation of fluorine and fluorinated functional groups into organic compounds. The following manuscript discusses historical strategies for decarboxylative fluorination to provide a framework for presenting recent developments in the field. The discussion is divided into two general sections: 1) decarboxylation of reagents to generate reactive fluorinated species; 2) decarboxylation of substrates to generate reactive species that can be converted to fluorinated functional groups. Within these two topics, only reactions that utilize decarboxylation to form new C–F or C–C(F)n bonds are considered.
2) Fluorination by Decarboxylative Reagents
2.1 Introduction to Reagents
The development of reactions that utilize inexpensive, atom-economical reagents represents a goal of green chemistry [7]. Halodifluoroacetic acids represent an attractive class of reagents that undergo decarboxylation to release reactive fluorinated species. While trifluoroacetic acid is the least expensive and most desirable reagent in this class, it decarboxylates slowly, and has an estimated half-life of 40,000 years at 15 °C in aqueous solution [10]. Trifluoroacetates have found synthetic utility as fluorinating reagents and will be discussed later in this section; however, decarboxylation typically requires stoichiometric Cu and reaction temperatures >150 °C [11–13]. Other halodifluoroacetic acids decarboxylate more rapidly (Fig. 1); however, metal catalysts, other reagents, and/or high temperatures are required to facilitate synthetically useful transformations.
Fig. (1).
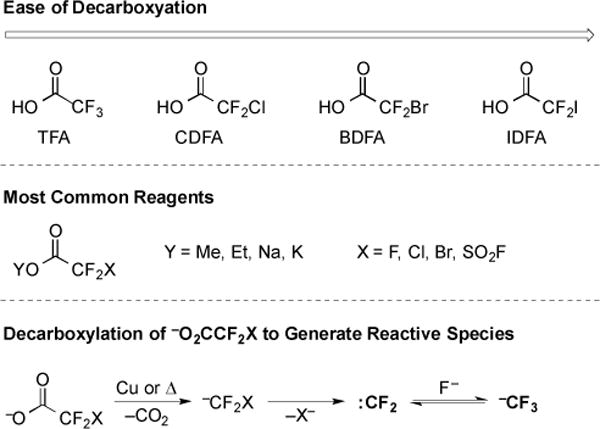
Reactivity of halodifluoroacetates.
In contrast, halodifluoroacetates decarboxylate to generate both :CF2, and in the presence of F−, −CF3, which can be utilized in a variety of synthetic transformations. The reactivity trend of halodifluoroacetic acids is consistent for both catalyzed and non-catalyzed conditions. Bromo-and chlorodifluoroacetates decarboxylate at lower temperatures (50–130 °C), and therefore, are more commonly employed than trifluoroacetates. These milder conditions allow for increased functional group compatibility, which in turn enables the installation of fluorinated groups into a broader array of molecules.
The present section focuses on reactions involving the use of halodifluoroacetates as sources of :CF2 and −CF3, and concludes with discussion of other decarboxylative fluorination reagents closely related to halodifluoroacetates.
2.2 Decarboxylative Trifluoromethylation of Halodifluoroacetate Esters
Activated alcohols have been converted to trifluoromethanes via a two-step procedure involving formation of a halodifluoroacetic ester followed by reaction with stoichiometric CuI in the presence of fluoride (Fig. 2) [14]. Substrates were limited to simple primary allylic, benzylic, and propargylic alcohols. Allylic and benzylic halodifluoroacetates underwent formal SN2 substitution, while propargylic chlorodifluoroacetates underwent formal SN2′ substitution to yield trifluoromethylallenes [14]. Bromodifluoroacetic esters displayed greater reactivity than chlorodifluoroacetic esters, and produced higher yields (ca. 10%) at 20–30 °C lower reaction temperatures [14]. Most commonly, a two-step procedure was employed in which an activated alcohol was converted to a halodifluoroacetate, purified, and then subjected to stoichiometric CuI and KF. A two-step, one-pot procedure was also developed that utilized ethyl halodifluoroacetates and afforded the product in moderate to low yield [14].
Fig. (2).
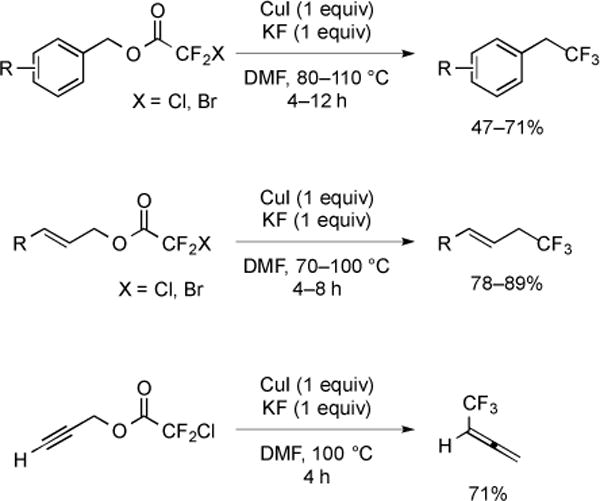
Decarboxylative trifluoromethylation of activated halodifluoroacetates.
The mechanism of Cu-mediated decarboxylative trifluoromethylation was proposed to proceed by a multi-step process that involves both CuI and the I− counterion (Fig. 3) [14]. Initially, the halodifluoroacetic ester reacted with CuI to yield an organic iodide and CuO2CCF2X (Fig. 3A). Next, CuO2CCF2X decarboxylated to generate CuX, CO2, and :CF2 (Fig. 3B). In the presence of KF, an equilibrium was established that formed −CF3 (Fig. 3C). CuI subsequently reacted with −CF3 to form Cu–CF3 (Fig. 3D). Finally, Cu–CF3 reacted with the organic iodide to yield the trifluoromethyl product (Fig. 3E). Product was not formed in the absence of either CuI or KF [14].
Fig. (3).
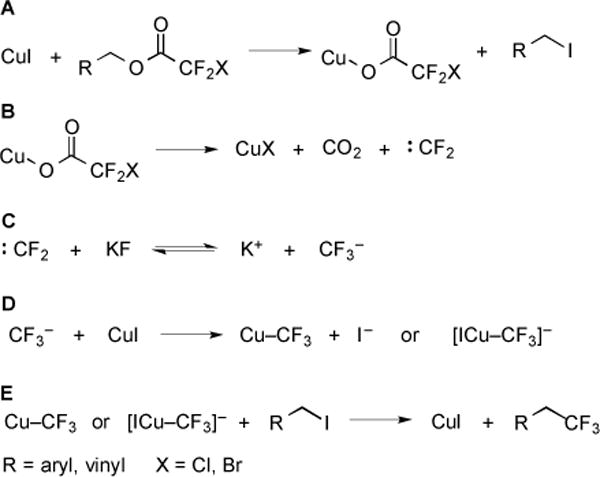
Proposed mechanism for CuI-mediated conversion of halodifluoroacetic esters to trifluoromethanes.
This procedure has been employed to access an allylic trifluoromethyl building block, as a precursor to biologically relevant molecules. In one example, a one-pot, two-step conversion of an allylic alcohol to an allylic trifluoromethane was employed for the synthesis of the COX-2 inhibitor, L-784,512 (Fig. 4) [15]. Initial treatment of alcohol 1 with chlorodifluoroacetic anhydride in the presence of N,N-diisopropylethylamine (DIPEA) yielded intermediate ester 2. Addition of KF and stoichiometric CuI and heating at 90 °C for 1 h, provided the product trifluoromethane 3 in high yield. Interestingly, DIPEA was critical for the reaction, as the use of NEt3, pyridine, or K2CO3 resulted in formation of the corresponding allylic chloride, and 30–60% lower yield of desired product. The one-pot procedure was more efficient than a two-step procedure in which alcohol 1 was first converted to an allylic iodide, and subsequently treated with CuI and MeO2CCF2Cl. Subjection of trifluoromethane 3 to Sharpless asymmetric dihydroxylation, alcohol oxidation, and coupling/cyclization reactions afforded L-784,512 [15].
Fig. (4).
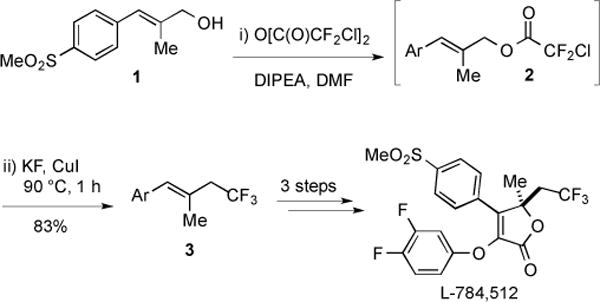
Decarboxylative trifluoromethylation employed in the synthesis of L-784,512.
Recent research has focused on the development of more green reaction sequences that utilize catalytic quantities of Cu. Towards this end, the decarboxylative trifluoromethylation of primary allylic bromodifluoroacetates was conducted in the presence of catalytic CuI (Fig. 5) [16]. A variety of functional groups were tolerated, including aryl halides, N- and O-containing functional groups, and a variety of substitution patterns on the alkene. The system benefited from the use of N,N’-dimethylethylenediamine (DMEDA) as a ligand, and a catalyst activation procedure in which a solution of CuI, DMEDA, and NaO2CCF2Br were heated before the addition of substrate. The procedure presumably generated a DMEDA–Cu–CF3 species that facilitated entry into the catalytic cycle (Fig. 5). The Cu–CF3 species generated during activation presumably underwent substitution with allylic bromodifluoroacetate, resulting in the formation of product and DMEDA–Cu–O2CCF2Br. Decarboxylation and formal halogen exchange allowed catalyst turnover and regeneration of a DMEDA–Cu–CF3 species. Unlike the proposed mechanism for the Cu-mediated allylic trifluoromethylation, the mechanism of the Cu-catalyzed reaction did not invoke an allylic iodide intermediate; control reactions provided comparable yields in the presence and absence of iodide. For substrates containing Z-alkenes, isomerization to more stable E-alkene products was observed (Fig. 5). Therefore, this allylic substitution might proceed via a π–allyl intermediate, which has been invoked to explain substitution reactions of allylic halides and trifluoroacetates from well defined (PPh3)3Cu–CF3 complexes [17] and Cu–CF3 complexes generated in situ [18].
Fig. (5).
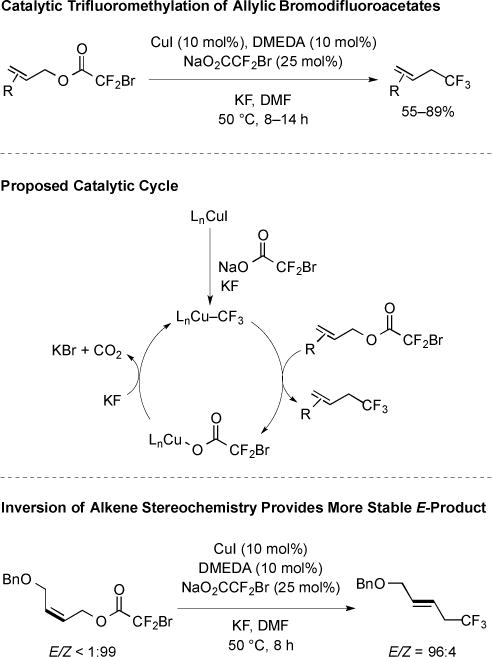
Cu-catalyzed decarboxylative trifluoromethylation.
2.3 Aromatic Trifluoromethylation
A series of trifluoromethylating reagents, including halodifluoroacetate salts and esters, and fluorosulfonyldifluoroacetate salts and esters were developed [19–27], and have been applied to the trifluoromethylation of aryl and heteroaryl halides (Fig. 6) [19–30]. Generally, these reagents undergo Cu-mediated decarboxylation of the reagent in situ to release :CF2, and then react with F− to generate −CF3. Most of these reagents are commercially available, and those that are not, such as KO2CCF2SO2F, are easily accessible [26]. Historically, aryl trifluoromethylation reactions with these reagents required high temperatures, stoichiometric CuI, and polar solvents such as N,N-dimethylformamide (DMF) or N,N-dimethylacetamide (DMAc). For these reactions, reactivity trends of aryl and heteroaryl halides mimic trends of other Cu-based coupling reactions (Ar–I > Ar–Br ≫ Ar–Cl) [31]. The introduction of electron-withdrawing groups, such as NO2, to the aromatic ring can improve the reactivity of aryl halides, especially aryl chlorides.
Fig. (6).
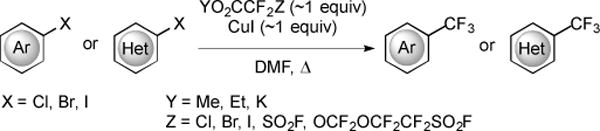
Trifluoromethylation of aromatic halides.
Traditionally, decarboxylative aromatic trifluoromethylation required stoichiometric CuI; however recently, reactions utilizing catalytic CuI were reported [29–30]. In one example, the Weinreb amide derived from a 5-bromo-2-iodobenzoic acid underwent trifluoromethylation in the presence of MeO2CCF2SO2F and catalytic CuBr (Fig. 7) [29]. In this unique example, the amide may have served as a directing group to help facilitate the reaction. For substrates lacking directing groups, Cu-catalyzed reactions have been achieved through the use of 1,10-phenanthroline as a ligand (Fig. 7) [30]. Unfortunately, when this Cu-catalyzed reaction was conducted on a multi-kilogram scale, the formation and separation of Ar(CF2)nCF3 side products proved problematic [30]. These byproducts resulted from insertion of :CF2 into Cu–CF3, and were suppressed by reverting to the use of stoichiometric CuI.
Fig. (7).
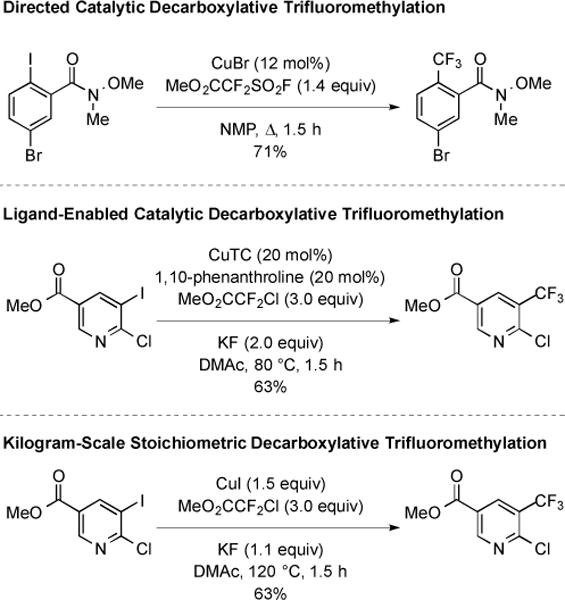
Recent developments in decarboxylative aromatic trifluoromethylation.
Fluorine-18-labelled organic molecules are used for positron emission tomography (PET) [32]. Recently, a method of decarboxylative trifluoromethylation enabled the preparation of Ar–CF3[18F] based PET imaging agents (Fig. 8) [32]. This procedure generated radiolabeled Cu–[18F]CF3 in situ from MeO2CCF2Cl, CuI, N,N,N’,N’-tetramethylethylenediamine (TMEDA), and [18F]KF/kryptofix. Under the reaction conditions, a diverse set of aryl and heteroaryl iodides were rapidly trifluoromethylated. While some medicinally important functional groups, such as acids and amines, were not compatible with the reaction conditions, an example of a successful protection strategy was demonstrated by synthesis of [18F]fluoxetine (selective serotonin reuptake inhibitor) (Fig. 8). Due to the large scope of substrates and operational simplicity, this transformation should facilitate the drug discovery process. For more details on PET imaging agents, the reader is referred to an alternate article [33].
Fig. (8).
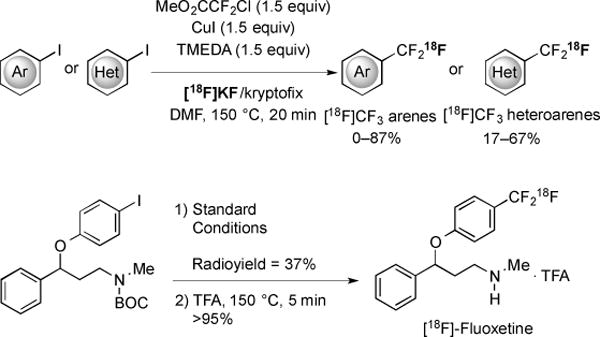
The synthesis of 18F-labelled trifluoromethylated arenes.
Compared to halodifluoroacetates, trifluoroacetic acid derivatives represent more ideal reagents for trifluoromethylation, because they are less expensive and do not require the addition of fluoride to generate −CF3. To this end MeO2CCF3 has served as a trifluoromethylating reagent in the presence of stoichiometric CuI and promoters, such as CsF or CsCl [34]. The trifluoromethylation of aryl and heteroaryl halides (the reactivity: ArI > ArBr ≫ ArCl) using MeO2CCF3 provided the corresponding benzotrifluorides in 42–79% yield, using DMF at 180 °C or sulfolane at 140–180 °C. However, the use of stoichiometric Cu limits the economic value of the transformation, and the development of a method that employs only catalytic quantities of a Cu salt represents a desirable target. The CuI-catalyzed decarboxylative trifluoromethylation of aryl and heteroaryl halides with MeO2CCF3 using CsF as initiator provided products in 47–95% yield (Fig. 9). To achieve the catalytic variant, MeO2CCF3 was added at a rate so that the slow decarboxylation step (k1) matched the consumption of CuCF3 in the aromatic trifluoromethylation step (k2). For some less active aryl bromides, the use of 1,10-phenanthroline as ligand proved helpful for the trifluoromethylation reactions [35].
Fig. (9).
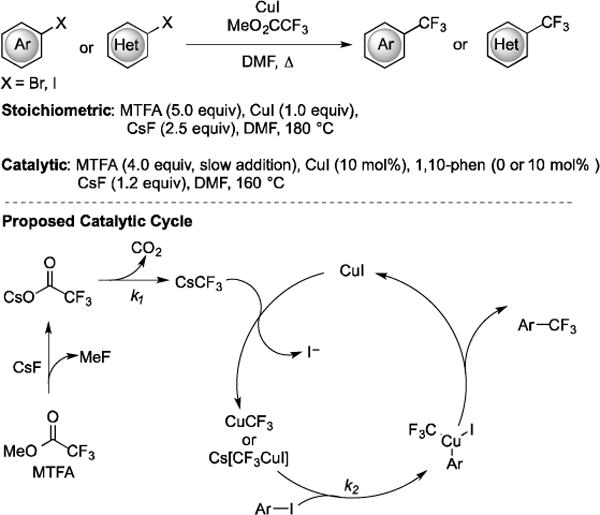
The stoichiometric and catalytic decarboxylative trifluoromethylation of aryl halides using MeO2CCF3.
Alkali trifluoroacetate salts are also inexpensive and readily available, and represent attractive sources of CF3 for large-scale processes. As early as 1981, the decarboxylative trifluoromethylation of aryl iodides and bromides occurred using NaO2CCF3 in the presence of CuI [36]. Further investigations confirmed the reactivity of a cuprate, [F3CCuI]−, with aryl halides to deliver the trifluoromethylated products (Fig. 10A) [11]. Although the use of the trifluoroacetate salts makes this a desirable protocol for trifluoromethylation, the use of stoichiometric CuI and high reaction temperature to promote decarboxylation (160–180 °C) limited the method to reactions of stable substrates. To address this problem, the use of Ag2O as an additive (30–40 mol%) allowed the analogous CuI-catalyzed reaction of aryl iodides to occur using NaO2CCF3 at reduced temperature (130 °C) [37]. For this reaction, the Ag2O was essential, and might lower the activation energy of decarboxylation of sodium trifluoroacetate to generate AgCF3 in situ (Fig. 10B).
Fig. (10).
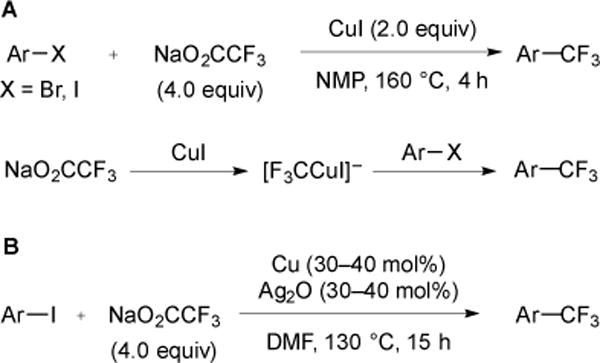
The stoichiometric (A) and catalytic (B) decarboxylative trifluoromethylation of aryl halides using NaO2CCF3.
Another commercially available trifluoroacetate salt, KO2CCF3, can also be used in the trifluoromethylation of aryl and heteroaryl halides. The CuI-mediated trifluoromethylation of a bromotetrahydronaphthalene with KO2CCF3 afforded the product in 35% yield [38], and the CuI-mediated trifluoromethylation of N-protected 5-bromoisoindoline with KO2CCF3, using toluene as co-solvent, provided the desired product in 90% yield [39]. However, the reaction time of the latter was far less than the former (1.5 h vs. 2 d), which may be attributed to the electron-withdrawing group at the meta-position of the bromide or the coordinating ability of the –OMe group (Fig. 11).
Fig. (11).
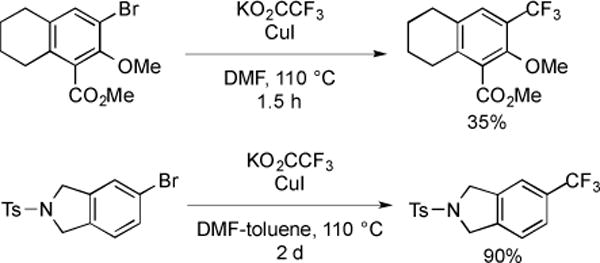
The decarboxylative trifluoromethylation of aryl bromides using KO2CCF3.
Chemical reactions conducted in flow microreactors have several advantages over traditional reaction vessels including: 1) precise control of reaction variables, such as temperature and rates of mixing; 2) increased safety profiles; 3) the ability to perform multiple transformations without isolating potentially reactive species [40]. Flow chemistry has facilitated various fluorination reactions, and recently, enabled the rapid and efficient trifluoromethylation of aryl and heteroaryl iodides using KO2CCF3 [41]. In the flow reactor, a solution of KO2CCF3, CuI and pyridine in NMP was mixed with a solution of aryl or heteroaryl iodides, and the mixture was introduced into a stainless steel tube reactor submerged in a preheated 200 °C bath using an optimized 16 minutes residence time (Fig. 12). At this point, the mixture was diluted with a stream of ethyl acetate (controlled with a back regulator), and the resulting mixture was collected and purified to afford trifluoromethylated arenes in good to excellent yields [41].
Fig. (12).
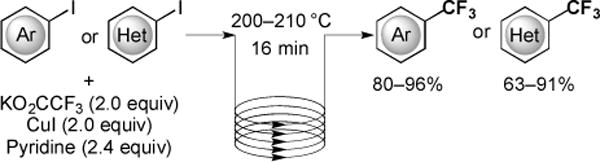
The decarboxylative trifluoromethylation of aromatic halides using KO2CCF3 in a flow reactor.
In addition to the systems described above, trifluoromethylation of aryl halides has been accomplished using well-defined NHC–Cu–O2CCF3 and –O2CCF2Cl complexes. Upon heating, the complexes released CO2 and generated NHC–Cu–CF3, which reacted with aryl electrophiles [42]. When aryl halide was used as the solvent, the ligated copper complex provided trifluoromethylated products in higher yields than “ligandless” CuI. However, when the aryl halide was used as only a co-solvent with DMAc, the copper complexes did not perform superior to reaction of CuI with MO2CCF3 and MO2CCF2Cl (Fig. 13).
Fig. (13).
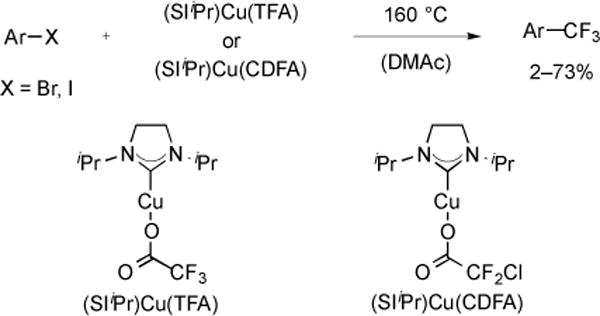
Decarboxylative trifluoromethylation of aryl halides using well-defined copper complexes.
2.4 Difluoroolefination
Halodifluoroacetate salts can also serve as regents for difluoroolefination reactions (Fig. 14). An early one-step synthesis of l,l-difluoroolefins involved heating a solution of an aldehyde, PPh3 and NaO2CCF2Cl [43].
Fig. (14).
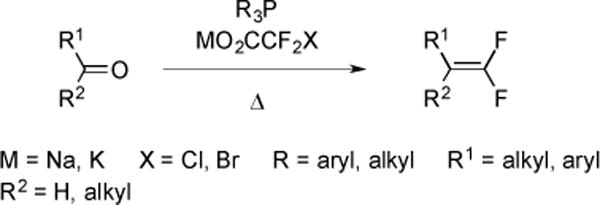
General strategy of difluoroolefination.
Three mechanisms were proposed for the conversion of carbonyl compounds to difluoroolefins (Fig. 15) [44]. Mechanism A would involve the decomposition of NaO2CCF2Cl to generate :CF2, which would then react with PPh3 to form a phosphonium ylide. In the presence of a carbonyl compound, the phosphonium ylide could then react via a Wittig mechanism to form product [43]. Mechanism B involved initial reaction of NaO2CCF2Cl and PPh3 to generate Ph3P+CF2CO2−. This species would then decarboxylate to form the common phosphonium ylide species. Finally, mechanism C would involve a reaction between NaO2CCF2Cl and PPh3 to form an enolate and a phosphonium species, which would decompose to form a phosphonium ylide.
Fig. (15).
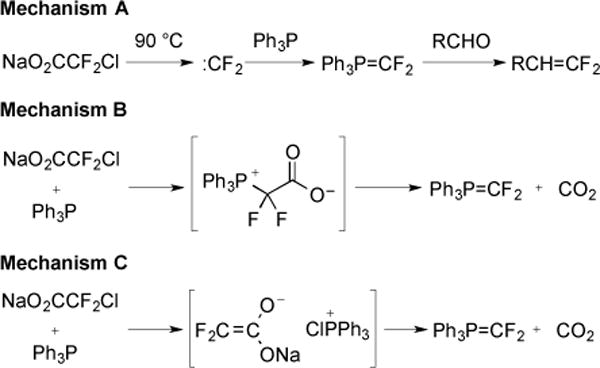
Mechanisms for the formation of l, l-difluoroolefins.
In order to distinguish between these potential reaction pathways, a series of experiments were conducted. First, NaO2CCF2Cl and PPh3 were heated in the presence of two :CF2-trapping reagents, tetramethylethylene and isopropyl alcohol. Formation of difluorocyclopropane was not observed, which suggested that free :CF2 was not present in solution [45]. A second experiment monitored the evolution of CO2, when NaO2CCF2Cl was heated in the presence and absence of PPh3 [45]. The decomposition of the salt was accelerated in the presence of PPh3, and required only 4–6 h to evolve 70% of the theoretical amount of CO2. In contrast, the decomposition of the salt in the absence of PPh3 was slow, and required 17 h to achieve a comparable result. These data were able to discount mechanism A; however, could not be used to differentiate mechanisms B and C.
The difluoroolefination reaction was later applied to various classes of ketones. For activated ketones (Fig. 16A), good to excellent yields of difluoroalkenes were obtained using the standard reaction conditions (PPh3, NaO2CCF2Cl, diglyme, 105 °C) [46]; however, non-activated substrates did not provide substantial quantities of products. Replacement of PPh3 and diglyme with P(nBu)3 and N-methylpyrrolidone (NMP) (Fig. 16B) allowed for the extension of the difluoromethylation reaction to some non-activated ketones [47]; however in some cases, transfer of a butylidine group to the ketone formed a butylidine side product [48]. Formation of this butylidene product occurred during the reaction with various electrophilic substrates, including cyclohexanone, heptanal, and trifluoroacetophenone. In the latter case, only the butylidene product was isolated from the reaction mixture [48].
Fig. (16).
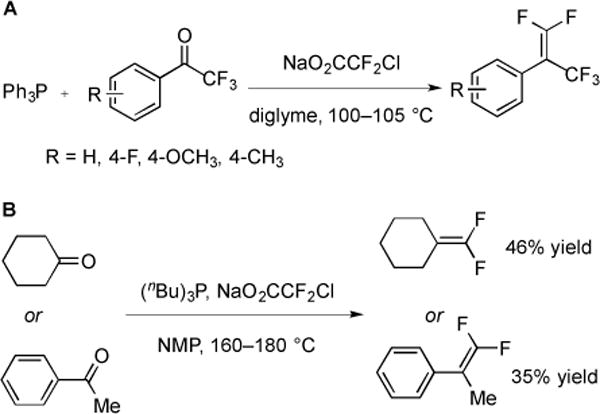
Synthesis of 1,l-difluoroolefins from ketones.
A rearrangement mechanism was proposed to explain the formation of butylidene product (Fig. 17) [48]. However, further studies revealed that the butylidene product was directly formed via reaction of P(nBu)3 and trifluoroacetophenone (Fig. 18) [49].
Fig. (17).
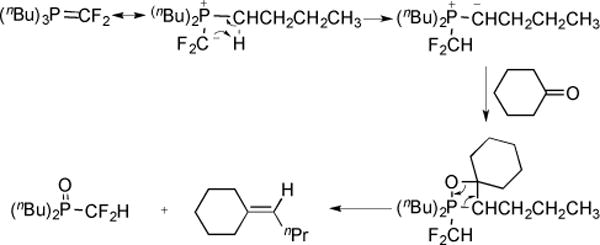
Mechanism for the formation of butylidenecyclohexane.
Fig. (18).
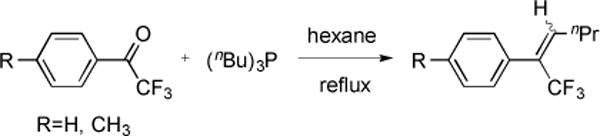
Olefination of aryl trifluoromethyl ketones with n-trialkylphosphines.
While earlier researchers favored mechanism B (Fig. 15) (formation of Ph P3P+CF2CO−2, decarboxylation to provide Ph3P=CF2, then Wittig difluoromethylenation), they were unable to isolate the key intermediate, Ph3P+CF2CO2−. Recently, the synthesis and characterization of the key difluoromethylene phosphobetaine provided direct evidence for this reaction mechanism [50]. To identify the proposed intermediate, PPh3 was reacted with KO2CCF2Br instead of NaO2CCF2Cl, which allowed for the substitution reaction to proceed under mild conditions, which allowed for the isolation of Ph3P+CF2CO2− in good yield (Fig. 19). Ph3P+CF2CO2− was stable to storage in under ambient conditions, and provided a convenient new reagent for the difluoromethylenation of aldehydes. Moreover, replacement of PPh3 with P(NMe2)3 provided an alternate reagent, (Me2N)3P+CF2CO2− that reacted well with non-activated ketones [50].
Fig. (19).
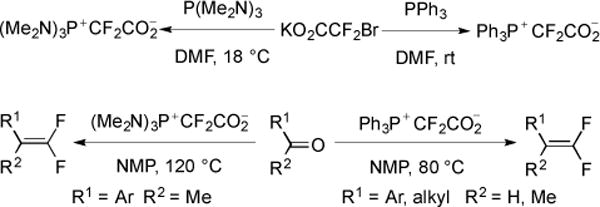
gem-Difluoroolefination of aldehydes and ketones.
2.5 Cyclopropanation
Decarboxylative strategies have also been employed for the preparation of fluorinated cyclopropanes. Early studies showed that thermolysis of NaO2CCF2Cl generated :CF2, which reacted with cyclohexene to provide 60–65% of the theoretical amount of CO2 in addition to 22% of difluoronorcarane (Fig. 20) [51].
Fig. (20).
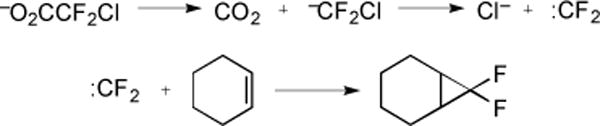
Synthesis of difluoronorcarane via :CF2 intermediate.
Later, this strategy was applied to the difluorocyclopropanation of conjugated steroidal dienes [52]. Both di- and tri-substituted alkenes reacted with :CF2 to provide a mixture of isomeric adducts (Fig. 21). In addition, the conjugated double bond of an enone system was also underwent difluorocyclopropanation [53]. However, high temperature 165–225 °C and large amount of NaO2CCF2Cl (20–50 equiv) were required. Control studies showed that cyclopropanation did not occur below 150 °C, even though decomposition of the salt occurred at 125 °C.
Fig. (21).
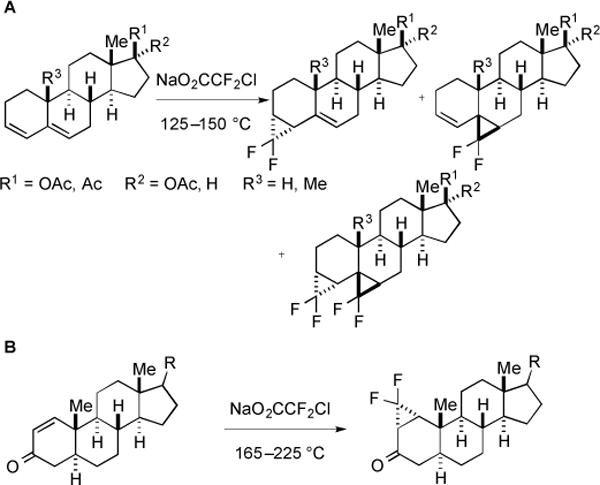
Addition of :CF2 to conjugated dienes and enones.
This strategy was also applied for difluorocyclopropanation of other nonsteroidal alkenes, including (Z)-4-(benzyloxy)-2-butenylacetate [54]. However, high temperature (190 °C) and excesses NaO2CCF2Cl (11 equiv) were required to obtain reasonable yields. Subsequent Zemplén deacetylation and Mitsunobu reactions allowed for the preparation of a large number of nucleoside analogues as potential chemotherapeutic agents (Fig. 22A). This strategy was also amenable to the stereospecific preparation of boron-substituted gem-difluorocyclopropanes [55], which could be rapidly transformed to afford functionalized gem-difluorocyclopropanes via lithium carbenoids or by subsequent oxidation (Fig. 22B). Moreover, this strategy was used to synthesize cis- or trans-4,5-difluoromethanoproline through difluorocyclopropanation of the aminoacyl derivatives of 4,5-dehydroproline [56]. The two building blocks were stable and could be easily incorporated into the proline-rich SAP peptide (Fig. 22C). Subsequently, a convenient synthesis of fluorinated cyclopropanes and cyclopropenes was developed using NaO2CCF2Br as a source of :CF2 [57]. Compared to NaO2CCF2Cl, NaO2CCF2Br was less hygroscopic at room temperature, and thus, easier to handle. Further reactions using NaO2CCF2Br were more efficient, and tolerated the presence of electron-deficient substrates (Fig. 22D).
Fig. (22).
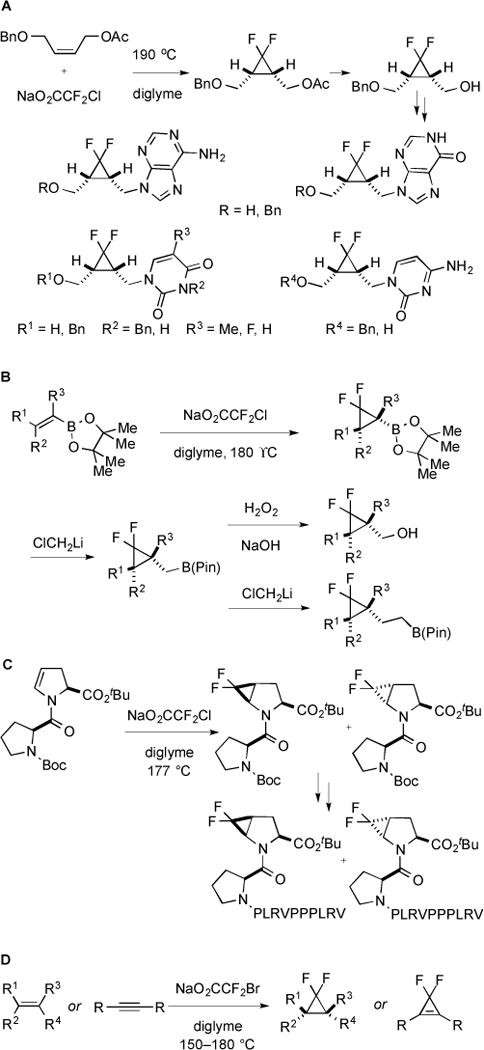
Synthesis derivatives of difluorocyclopropanes.
The use of an alternative source of :CF2, trimethylsilyl fluorosulfonyldifluoroacetate (TFDA), provided access to difluorocyclopropanes under more mild conditions [58] (Fig. 23A). In the presence of catalytic fluoride, TFDA easily decomposed to generate :CF2 (105 °C), and difluorocyclopropanes were obtained in high yields. The moisture-sensitive nature of TFDA provided a poor shelf life, and made the reagent relatively challenging to handle. Therefore, a more stable and convenient reagent was designed [59]. This new reagent, MeO2CCF2SO2F, in combination with KI and TMSCl exhibited comparable reactivity as TFDA at high concentration and high temperature [60]. Using MeO2CCF2SO2F, a broad range of alkenes was converted into the corresponding difluorocyclopropanes, even including unreactive n-butyl acrylate (Fig. 23B). To contrast the usage of MeO2CCF2SO2F and TFDA, sodium trifluoroacetate can also serve as a precursor to :CF2 [61]. NaO2CCF3 decomposed at 170 °C in the presence of AIBN to form :CF2, which then reacted with a variety of alkenes. Given the inexpensive nature of NaO2CCF3, this procedure provided an economical method for difluorocyclopropanation of robust substrates.
Fig. (23).
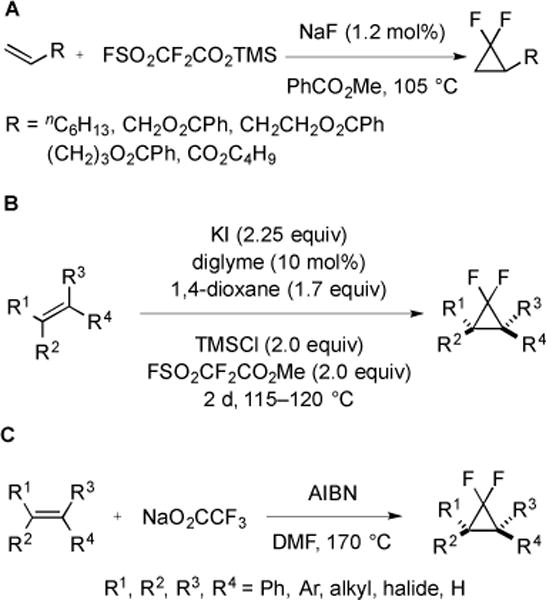
Synthesis of gem-difluorocyclopropanes using TFDA, MeO2CCF2SO2F, and NaO2CCF3 as sources of :CF2.
Historically, little overlap existed between reactions that generate :CF2 and difluoromethylene phosponium ylides. MO2CCF2X-based reagents decarboxylated to form reactive :CF2 species that were used to generate difluorocyclopropanes. In contrast, regents derived from MO2CCF2X and PPh3 generated difluoromethylene phosphonium ylides for difluoroolefination reactions. However recently, the interconversion between difluoromethylene ylide and :CF2 was successfully achieved [62]. By changing the solvent from DMF to less-polar p-xylene, Ph3P+CF2CO2− was converted from a precursor of difluoromethylene phosphonium ylide to an efficient precursor of :CF2 (Fig. 19 and 24A). In contrast, under the corresponding reaction conditions, other reagents only generated the difluoromethylene phosphonium ylide (Fig. 24B and C). Therefore, Ph3P+CF2CO2− holds unique status as a reagent that can generate reactive species for both difluoroolefination or difluorocyclopropanation reactions.
Fig. (24).
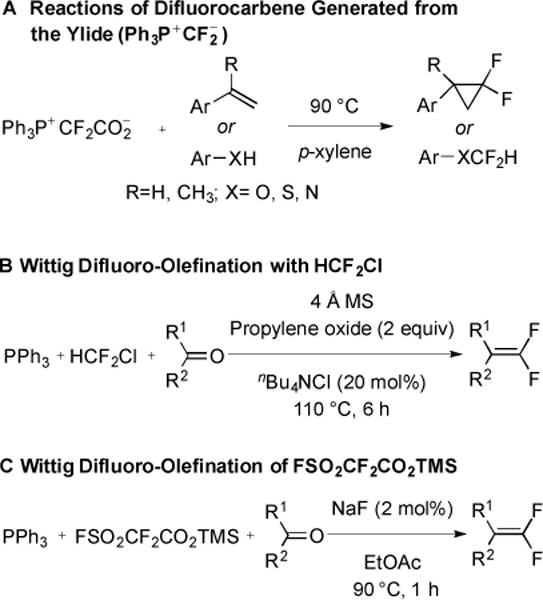
Conversion between :CF2 and difluoromethylene ylide.
The reactive :CF2 intermediate has also undergone cycloadditions with alkynes to access strained difluorocyclopropenes. Using this strategy, a number of difluorocyclopropenyl steroids were synthesized (Fig. 25A) [63]. Difluorocylopropenes were also used as precursors to biologically active compounds that showed potent insecticidal activities (Fig. 25B) [64]. Using the same strategy, a simple and efficient method for the preparation of 3,3-difluoroiodocyclopropenes provided access to interesting fluorinated building blocks [65]. In this case, the combination of TDFA and NaF (10 mol%) generated :CF2, which subsequently reacted with the alkynyl iodide. These new iodides were further functionalized by: 1) Heck type cross-coupling reactions with α,β-unsaturated compounds; 2) trifluoromethylation using CuI and TFDA; 3) hydrolysis under acidic conditions to provide ring opened β-alkyl-β-iodoacrylic acids with exclusive Z-configuration (Fig. 25C).
Fig. (25).
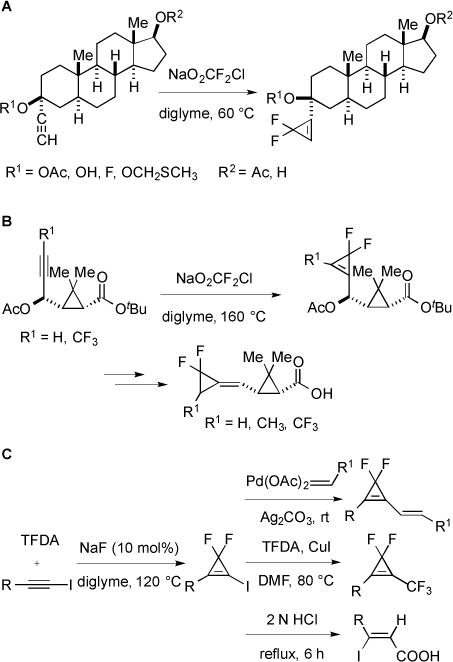
Synthesis of different difluorocyclopropene derivatives.
2.6 Heteroatom Difluoromethylation
Reactive :CF2 intermediates, generated from NaO2CCF2Cl, have also inserted into heteroatom–H bonds to provide difluoromethylated ethers and amines. At only 95 °C, NaO2CCF2Cl decarboxylated in the absence of any transition metal catalyst to afford :CF2, which was directly trapped with a variety of aromatic and heteroaromatic thiols [66]. Further, the difluoromethylation of nitrogenous heterocycles and phenylselenol were also successful (Fig. 26).
Fig. (26).
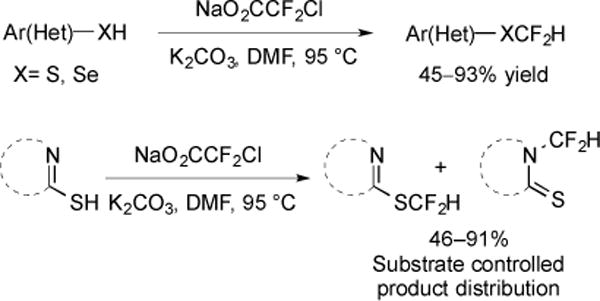
Difluoromethylation of thiophenols, nitrogenous heterocycles and phenylselenols.
3) Fluorination and Trifluoromethylation of Substrates that Undergo Decarboxylation
3.1 Decarboxylation of Acrylic Acid Derivatives
In the preceding section, trifluoromethylation and difluoromethylenation reactions were realized by decarboxylation of fluorine-based reagents to generate CF3−, :CF2 or F2C=PR3. Contrary to this strategy, fluoroalkylation and fluorination can also involve decarboxylation of the substrate in the presence of fluoroalkylating or fluorinating reagents. This developing strategy already provides medicinal chemists facile access fluoroalkyl-substituted alkenes, α-trifluoromethyl ketones, alkyl fluorides, and [18F]-labeled di- and tri-fluoromethylated arenes directly from a vast number of carboxylic acids that already exist in pharmaceutically important building blocks [67–75].
A series of Cu-mediated decarboxylative fluoroalkylation reactions were developed that converted α,β-unsaturated [67], β,γ-unsaturated [68], and propargylic [69] carboxylic acids to fluorinated species using Togni-type electrophilic fluoroalkylating reagents (Fig. 27) [76–77]. Using the combination of catalytic CuF2 and IIII–CF3 reagents, a variety of α,β-unsaturated acids were stereoselectively converted to E-vinyl trifluoromethanes [67] (Fig. 27A). Similarly, E-vinyl difluoromethylsulfones could be accessed using a DMEDA–CuII catalyst system, and an IIII–CF2SO2Ph reagent (Fig. 27B). Allylic difluoromethylsulfones could also be formed in regioselective fashion from β,γ-unsaturated carboxylic acids (Fig. 27C) [68]. Representative difluoromethylsulfone products could be subjected to reducing conditions and converted to allylic difluoromethanes, which are difficult to selectively access using other methods. Finally, propargylic alcohols were converted to trifluoroethylketones using stoichiometric Cu(OAc)2, DMEDA, and a Togni trifluoromethylating reagent (Fig. 27D) [69]. This unique result, in which an oxygen atom originating from water was incorporated into the product, was rationalized by considering inherent differences in substrate reactivity in the context of the proposed mechanisms.
Fig. (27).
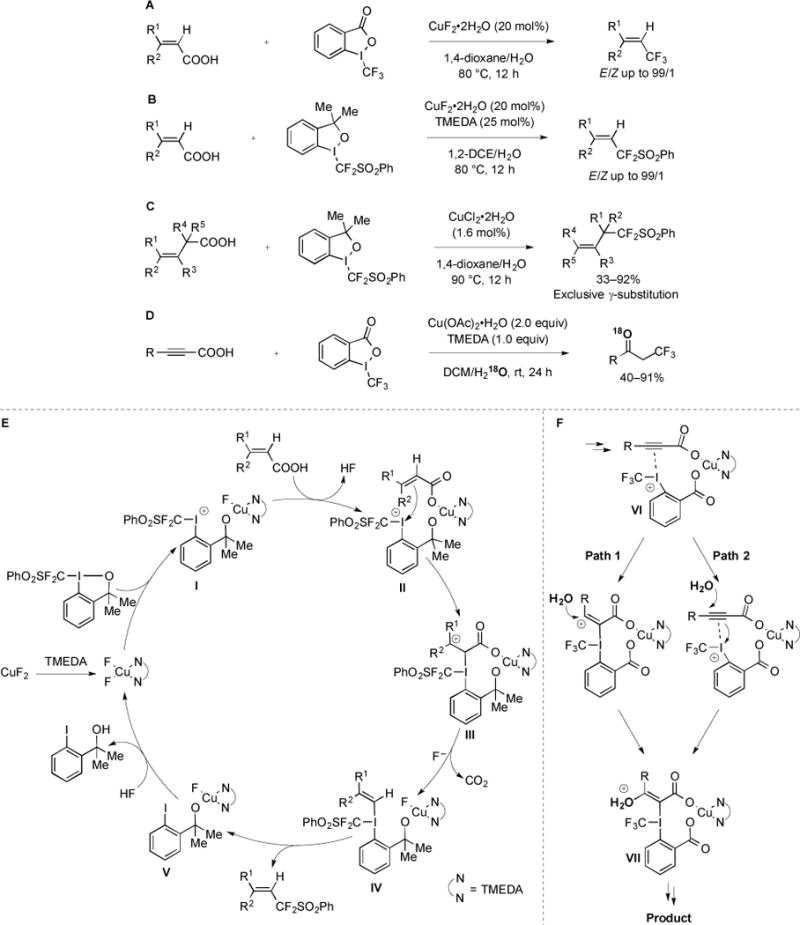
Difluoromethylation and trifluoromethylation of unsaturated carboxylic acids.
While the mechanisms of the above decarboxylative fluoroalkylation reactions might share similar intermediates, distinct properties of the substrates provided discrete products. According to the proposed mechanisms for both substrates, CuII served as a Lewis acid, and facilitated the reaction by; 1) increasing the electrophilicity of the Togni-type reagents; 2) promoting decarboxylation; 3) coordinating substrates and reagents to bring reactive centers into close proximity. The following mechanism was proposed for the decarboxylative difluoromethylsulfonylation of α,β-unsaturated acids (Fig. 27E) [67]. DMEDA–CuF2 underwent ligand exchange to form a highly electrophilic species I. Another ligand exchange would form ternary complex II, thus placing the π–system of the alkene in close proximity to the iodonium group. Nucleophilic attack by the alkene would generate carbocationic intermediate III, which could decarboxylate to form vinyl–IIII species IV. Reductive eliminate would provide product, and a series of ligand exchanges would regenerate the DMEDA–CuF2 catalyst. When propargylic carboxylic acids were used as substrates, the initial steps of the proposed mechanism would be identical, and intermediate VI would form (analogous to II) [69]. The more difficult nucleophilic attack of the alkyne in VI towards the iodonium could alter the mechanism (Fig. 27F). In one case, attack of the iodonium by π–electrons of the alkyne would provide a high-energy vinyl cation (Path 1). This species could be quenched with water to form intermediate VII, which would eventually provide product. An alternate mechanism involved the iodonium acting as a π–acid to activate the alkyne for nucleophilic attack by H2O (Path 2). Intermediate VII would then undergo subsequent tautomerization, decarboxylation, and reductive elimination to form the trifluoroethylketone product [69].
Several complementary decarboxylative methods for the construction of E-configured Cvinyl–CF3 and Cvinyl–CF2 bonds via radical addition–elimination processes were recently developed [70–71]. A variety of (heteroaryl)cinnamic acids were converted to vinyl trifluoromethanes using catalytic CuSO4·5H2O, stoichiometric TBHP, and NaSO2CF3 as a source of ·CF3 (Fig. 28A). An alternative procedure enabled the conversion of electron-rich cinnamic acids to difluoromethanes using Fe-catalysis with (CF2HSO2)2Zn (DFMS) as a source of ·CF2H. While both of these methods utilized mild reaction conditions, the substrate scope was limited to (heteroaryl)cinnamic acids. Alkyl-substituted acrylic acids failed to give the desired products, which might arise from the instability of a radical intermediate. Subsequently, a similar strategy was applied to the FeCl3-mediated decarboxylative trifluoromethylation of α,β-unsaturated carboxylic acids [71]. A variety of (heteroaryl)cinnamic acids were compatible with the reaction conditions (Fig. 28B). More importantly, the reaction could be conducted under an open atmosphere. Decarboxylative trifluoromethylation of α,β-unsaturated carboxylic acids was also accomplished by photoredox catalysis [72]. fac-Ir(ppy)3 served as a photocatalyst and generated CF3 from Togni reagent. The mild reaction conditions provided excellent functional group compatibility, and moderate to high yields of products were obtained (Fig. 28C).
Fig. (28).
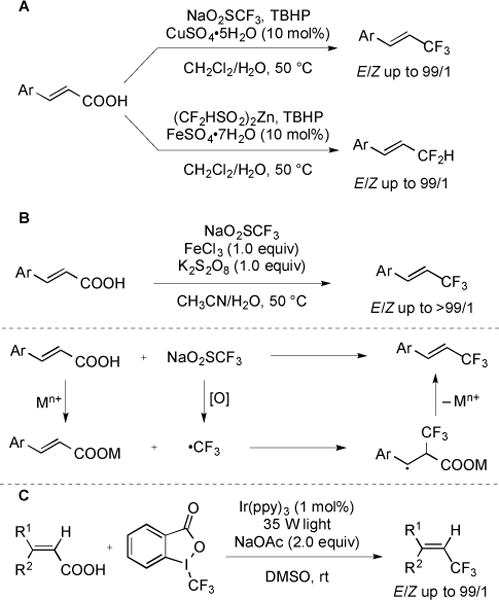
Decarboxylative trifluoromethylation of α,β-unsaturated unsaturated carboxylic acids via radical processes.
3.2 Decarboxylation of Aromatic and Aliphatic Acid Derivatives
Recently, the selective fluorination of organic compounds via a radical mechanism was successfully achieved [73]. In this reaction, decomposition of diacylperoxies or tert-butylperoxiacids generated organic radicals, which reacted with N-fluorobenzenesulfonimide (NFSI) to afford alkyl fluorides. This procedure demonstrated that alkyl radicals could undergo fluorine atom transfer to from alkyl fluorides. A broad range of alkyl radicals, including primary, secondary, tertiary, benzylic, and heteroatom-stabilized radicals underwent fluorination. In some cases, the products of the reactions would have been difficult to access using electrophilic or nucleophilic fluorination procedures.
Ag-catalyzed decarboxylative fluorinations of carboxylic acids have recently been developed [74–75]. Aliphatic carboxylic acids were converted to the corresponding alkyl fluorides using AgNO3 and Selectfluor® in aqueous acetone [74]. A catalytic cycle invoking AgI, AgII, and AgIII was proposed (Fig. 30A). The mechanism initiated with the reaction of AgI and Selectfluor® to provide AgIII–F. Single electron transfer then generated AgII–F and a carboxyl radical, which decarboxylated to form the corresponding alkyl radical. Reaction of AgII–F with the organic radical then provided the fluorinated product [74]. Under similar reaction conditions, di- and trifluoromethylated arenes were synthesized from readily available α, α-difluoro- and α-fluoroarylacetic acids (Fig. 30B) [75]. Adaptation of this system to the usage of [18F]Selectfluor bis(triflate) provided access to 18F-radiolabeled products. Compared to the usage of [18F]F2, the AgNO3/[18F]Selectfluor bis(triflate) system offered an expanded scope of substrates and in several examples, provided higher radiochemical yields.
Fig. (30).
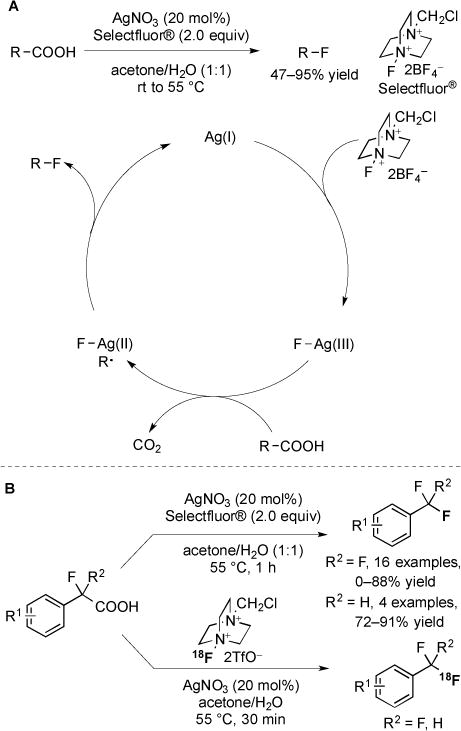
Fluorination of carboxylic acids via radical mechanisms.
CONCLUSION
Methods for the synthesis of fluorinated organic compounds are important for medicinal chemistry as they enable the facile construction of biologically interesting molecules. In recent years, new methods for the installation of fluorine-containing functional groups have been developed that utilize powerful strategies, including decarboxylative coupling. As the field of synthetic organofluorine chemistry continues to advance, decarboxylative coupling should play an important role in the development of more general, functional-group tolerant, and practical synthetic methods for the synthesis of fluorinated compounds.
Fig. (29).
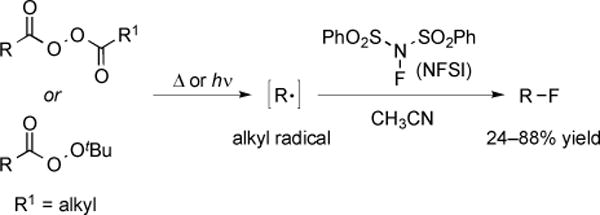
Decarboxyaltive radical fluorination of diacylperxoides and peroxides using N-fluorobenzenesulfonimide.
Acknowledgments
We thank the donors of the Herman Frasch Foundation for Chemical Research (701-HF12) and the American Chemical Society Petroleum Research Fund (52073-DNI1) for financial support, and the NIGMS Training Grant on Dynamic Aspects of Chemical Biology (T32 GM08545) for a graduate traineeship (B. R. A.). Further financial assistance from the University of Kansas Office of the Provost, Department of Medicinal Chemistry, and General Research Fund (2302264) is gratefully acknowledged.
LIST OF ABBREVIATIONS
- AIBN
Azobisisobutyronitrile
- DCE
1,2-Dichloroethane
- DCM
Dichloromethane
- DIPEA
N,N-Diisopropylethylamine
- DMAc
Dimethylacetamide
- DMEDA
N N’-Dimethylethylenediamine
- DMF
Dimethylformamide
- DMSO
Dimethyl sulfoxide
- NFSI
N-Fluorobenzenesulfonimide
- NMP
N-Methyl-2-pyrrolidone
- MDFA
Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate
- TBHP
tert-Butyl hydroperoxide
- TFA
Trifluoroacetic acid
- TFDA
Trimethylsilyl fluorosulfonyldifluoroacetate
- TMEDA
N,N,N’,N’-Tetramethylethylenediamine
- Selectfluor
1-Chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflicts of interest.
References
- 1.Bégué JP, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons; Hoboken: 2008. [Google Scholar]
- 2.Ojima I, editor. Fluorine in Medicinal Chemistry and Chemical Biology. Blackwell Publishing Ltd; West Sussex: 2009. [Google Scholar]
- 3.Liang T, Neumann CN, Ritter T. Introduction of fluorine and fluorine-containing functional groups. Angew Chem Int Ed. 2013;52:8214–8264. doi: 10.1002/anie.201206566. [DOI] [PubMed] [Google Scholar]
- 4.Liu G. Transition metal-catalyzed fluorination of multi carbon–carbon bonds: new strategies for fluorinated heterocycles. Org Biomol Chem. 2012;10:6243–6248. doi: 10.1039/c2ob25702e. [DOI] [PubMed] [Google Scholar]
- 5.Hollingworth C, Gouverneur V. Transition metal catalysis and nucleophilic fluorination. Chem Commun. 2012;48:2929–2942. doi: 10.1039/c2cc16158c. [DOI] [PubMed] [Google Scholar]
- 6.Tomashenko OA, Grushin VV. Aromatic trifluoromethylation with metal complexes. Chem Rev. 2011;111:4475–4521. doi: 10.1021/cr1004293. [DOI] [PubMed] [Google Scholar]
- 7.Bryan MC, Dillon B, Hamann LG, Hughes GJ, Kopach ME, Peterson EA, Pourashraf M, Raheem I, Richardson P, Richter D, Sneddon HF. Sustainable practices in medicinal chemistry: current state and future directions. J Med Chem. 2013;56:6007–6021. doi: 10.1021/jm400250p. [DOI] [PubMed] [Google Scholar]
- 8.Rodríguez N, Goossen LJ. Decarboxylative coupling reactions: a modern strategy for C–C-bond formation. Chem Soc Rev. 2011;40:5030–5048. doi: 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]
- 9.Weaver JD, Recio A, Grenning AJ, Tunge JA. Transition metal-catalyzed decarboxylative allylation and benzylation reactions. Chem Rev. 2011;111:1846–1913. doi: 10.1021/cr1002744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lifongo LL, Bowden DJ, Brimblecombe P. Thermal degradation of haloacetic acids in water. Int J Phys Sci. 2010;5:738–747. [Google Scholar]
- 11.Carr GE, Chambers RD, Holmes TF, Parker DG. Sodium perfluoroalkane carboxylates as sources of perfluoroalkyl groups. J Chem Soc Perkin Trans I. 1988:921–926. [Google Scholar]
- 12.Chang Y, Cai C. Trifluoromethylation of carbonyl compounds with sodium trifluoroacetate. J Fluorine Chem. 2005;126:937–940. [Google Scholar]
- 13.McReynolds KA, Lewis RS, Ackerman LKG, Dubinina GG, Brennessel WW, Vicic DA. Decarboxylative trifluoromethylation of aryl halides using well-defined copper-trifluoroacetate and –chlorodifluoroacetate precursors. J Fluorine Chem. 2010;131:1108–1112. [Google Scholar]
- 14.Duan JX, Chen QY. Novel synthesis of 2,2,2-trifluoroethyl compounds from homoallylic alcohols: a copper(I) iodide-initiated trifluoromethyl-dehydroxylation process. J Chem Soc Chem Commun. 1994:725–730. [Google Scholar]
- 15.Tan L, Chen C-y, Larsen RD, Verhoeven TR, Reider PJ. An efficient asymmetric synthesis of a potent COX-2 inhibitor L-784,512. Tetrahedron Lett. 1998;39:3961–3964. [Google Scholar]
- 16.Ambler BR, Altman RA. Copper-catalyzed decarboxylative trifluoromethylation of allylic bromodifluoroacetates. Org Lett. 2013;15:5578–5581. doi: 10.1021/ol402780k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larsson JM, Pathipati SR, Szabó KJ. Regio and stereoselective allylic trifluoromethylation and fluorination using CuCF3 and CuF reagents. J Org Chem. 2013;78:7330–7336. doi: 10.1021/jo4010074. [DOI] [PubMed] [Google Scholar]
- 18.Miyake Y, Ota S-i, Nishibayashi Y. Copper-catalyzed nucleophilic trifluoromethylation of allylic halides: a simple approach to allylic trifluoromethylation. Chem Eur J. 2012;18:13255–13258. doi: 10.1002/chem.201202853. [DOI] [PubMed] [Google Scholar]
- 19.Chen QY, Wu SW. Methyl fluorosulfonyldifluoroacetate: a new trifluoromethylating agent. J Chem Soc Chem Commun. 1989:705–706. [Google Scholar]
- 20.Chen QY, Yang GY, Wu SW. Copper electron-transfer induced trifluoromethylation with methyl fluorosulfonyldifluoroacetate. J Fluorine Chem. 1991;55:291–298. [Google Scholar]
- 21.MacNeil JG, Burton DJ. Generation of trifluoromethyl copper from chlorodifluoroacetate. J Fluorine Chem. 1991;55:225–227. [Google Scholar]
- 22.Su D-B, Duan J-X, Chen Q-Y. Methyl chlorodifluoroaceate: a convenient trifluoromethylating agent. Tetrahedron Lett. 1991;32:7689–7690. [Google Scholar]
- 23.Duan J-X, Su D-B, Wu J-P, Chen QY. Synthesis of trifluoromethyl aryl derivatives via difluorocarbene precursors and nitro-substituted aryl chlorides. J Fluorine Chem. 1994;66:167–169. [Google Scholar]
- 24.Duan J, Su D-B, Chen Q-Y. Trifluoromethylation of organic halides with methyl halodifluoroaceates—a process via difluorocarbene and trifluoromethide intermediates. J Fluorine Chem. 1993;61:279–284. [Google Scholar]
- 25.Chen QY, Duan JX. Methyl 3-oxo-ω-fluorosulfonylperfluoropentanoate: a versatile trifluoromethylating agent for organic halides. J Chem Soc Chem Commun. 1993:1389–1391. [Google Scholar]
- 26.Long Z-Y, Duan J-X, Lin Y-B, Guo C-Y, Chen Q-Y. Potassium 3-oxo-ω-fluorosulfonylperfluoropentanoate (FO2SCF2CF2OCF2CO2K), a low-temperature trifluoromethylating agent for organic halides: it’s α-carbon–oxygen bond fragmentation. J Fluorine Chem. 1996;78:177–181. [Google Scholar]
- 27.Chen QY. Trifluoromethylation of organic halides with difluorocarbene precursors. J Fluorine Chem. 1995;72:241–246. [Google Scholar]
- 28.Roy S, Gregg BT, Gribble GW, Le V-D, Roy S. Trifluoromethylation of aryl heteroaryl halides. Tetrahedron. 2011;67:2161–2195. [Google Scholar]
- 29.Nomura S, Kawanishi E, Ueta K. Preparation of glycosides as antidiabetic agents and having inhibitory activity against sodium-dependent transporter. US 2012/0258913. U.S. Patent Appl Publ. 2012;A1
- 30.Mulder JA, Frutos RP, Patel ND, Qu B, Sun X, Tampone TG, Gao J, Sarvestani M, Eriksson MC, Haddad N, Shen S, Song JJ, Senanayake CH. Development of a safe and economical synthesis of methyl 6-chloro-5-(trifluoromethyl)nicotinate: trifluoromethylation on kilogram scale. Org Process Res Dev. 2013;17:940–945. [Google Scholar]
- 31.Beletskaya IP, Cheprakov AV. Copper in cross-coupling reactions: the post-Ullmann chemistry. Coord Chem Rev. 2004;248:2337–2364. [Google Scholar]
- 32.Huiban M, Tredwell M, Mitzuta S, Wan Z, Zhang X, Collier TL, Gouverneur V, Passchier J. A broadly applicable [18F] trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nature Chem. 2013;6:941–944. doi: 10.1038/nchem.1756. [DOI] [PubMed] [Google Scholar]
- 33.Cole EL, Stewart MN, Kittich R, Hoareau R, Scott PJH. Radiosyntheses using Fluorine-18: the Art and Science of Late Stage Fluorination. Curr Top Med Chem. 2014 doi: 10.2174/1568026614666140202205035. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langlois BR, Roeques N. Nucleophilic trifluoromethylation of aryl halides with methyl trifluoroacetates. J Fluorine Chem. 2007;128:1318–1325. [Google Scholar]
- 35.Schareina T, Wu X-F, Zapf A, Cotté A, Gotta M, Beller M. Towards a practical and efficient copper-catalyzed trifluoromethylation of aryl halides. Top Catal. 2012;55:426–431. [Google Scholar]
- 36.Matsui K, Tobita E, Ando M, Kondo K. A convenient trifluoromethylation of aromatic halides with sodium trifluoroacetate. Chem Lett. 1981:1719–1720. [Google Scholar]
- 37.Li Y, Chen T, Wang H, Zhang R, Jin K, Wang X, Duan C. A ligand-free copper-catalyzed decarboxylative trifluoromethylation of aryl iodides with sodium trifluoroacetate using Ag2O as promoter. Syntlett. 2011:1713–1716. [Google Scholar]
- 38.Bernstein P, Dantzman C, Palmer W. Aryl glycinamide derivatives and their use as NK1 antagonists and serotonin reuptake inhibitors. WO 2005/100325. PCT Int Appl. 2005
- 39.Austin NE, Avenell KY, Boyfield I, Branch CL, Hadley MS, Jeffrey P, Johnson CN, Macdonald GJ, Nash DJ, Riley GJ, Smith AB, Stemp G, Thewlis KM, Vong AKK, Wood MD. Design and synthesis of novel 2,3-dihydro-1H-isoindoles with high affinity and selectivity for the dopamine D3 receptor. Bioorg Med Chem Lett. 2001;11:685–688. doi: 10.1016/s0960-894x(01)00037-3. [DOI] [PubMed] [Google Scholar]
- 40.Amii H, Nagaki A, Yoshida J-i. Flow microreactor synthesis in organo-fluorine chemistry. Beilstein J Org Chem. 2013;9:2793–2802. doi: 10.3762/bjoc.9.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M, Buchwald SL. Rapid and efficient trifluoromethylation of aromatic and heteroaromatic compounds using potassium trifluoroacetates enabled by a flow system. Angew Chem Int Ed. 2013;52:11628–11631. doi: 10.1002/anie.201306094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McReynolds KA, Lewis RS, Ackerman KG, Dubinina GG, Brennessel WW, Vicic DA. Decarboxylative trifluoromethylation of aryl halides using well-defined copper-trifluoroacetate and -chlorodifluoroacetate precursors. J Fluorine Chem. 2010;131:1108–1112. [Google Scholar]
- 43.Fuqua SA, Duncan WG, Silverstein RM. A one-step synthesis of 1,1-difluoroolefins from aldehydes by a modified Wittig synthesis. Tetrahedron Lett. 1964;5:1461–1463. [Google Scholar]
- 44.Fuqua SA, Duncan WG, Silverstein RM. A one-step synthesis of 1,1-difluoro olefins from aldehydes. J Org Chem. 1965;30:1027–1029. [Google Scholar]
- 45.Herkes FE, Burton DJ. Fluoro olefins. I. synthesis of β-substituted perfluoro olefins. J Org Chem. 1967;32:1311–1318. [Google Scholar]
- 46.Burton DJ, Herkes FE. A one-step synthesis of β-phenyl substituted perfluoroolefins. Tetrahedron Lett. 1965:1883–1887. [Google Scholar]
- 47.Fuqua SA, Duncan WG, Silverstein RM. A one-step synthesis of 1,1-difluoroolefins from ketones. Tetrahedron Lett. 1965:521–524. [Google Scholar]
- 48.Fuqua SA, Duncan WG, Silverstein RM. Synthesis of 1,1-difluoro olefins. II. reactions of ketones with tributylphosphine and sodium chlorodifluoroacetate. J Org Chem. 1965;30:2543–2545. [Google Scholar]
- 49.Burton DJ, Herkes FE, Klabunde KJ. The reaction of trifluoromethyl ketones and trialkylphosphines. J Am Chem Soc. 1966;88:5042–5043. [Google Scholar]
- 50.Zheng J, Cai J, Lin J-H, Guo Y, Xiao J-C. Synthesis and decarboxylative Wittig reaction of difluoromethylene phosphobetaine. Chem Commun. 2013;49:7513–7515. doi: 10.1039/c3cc44271c. [DOI] [PubMed] [Google Scholar]
- 51.Birchall JM, Cross GE, Haszeldine RN. Difluorocarbene. Proc Chem Soc. 1960;81 [Google Scholar]
- 52.Knox LH, Velarde E, Berger S, Cuadriello D, Landis PW, Cross AD. Steroids. CCXXVII. steroidal dihalocyclopropanes. J Am Chem Soc. 1963;85:1851–1858. [Google Scholar]
- 53.Beard C, Dyson NH, Fried JH. The methylenation of unsaturated ketones. Part 1. addition of difluoromethylene of enones. Tetrahedron Lett. 1966;28:3281–3286. [Google Scholar]
- 54.Csuk R, Eversmann L. Synthesis of difluorocyclopropyl carbocyclic homo-nucleosides. Tetrahedron. 1998;54:6445–6456. [Google Scholar]
- 55.Fujioka Y, Amii H. Boron-substituted difluorocyclopropanes: new building blocks of gem-difluorocyclopropanes. Org Lett. 2008;10:769–772. doi: 10.1021/ol702851t. [DOI] [PubMed] [Google Scholar]
- 56.Kubyshkin VS, Mykhailiuk PK, Afonin S, Ulrich AS, Komarov IV. Incorporation of cis- and trans-4,5-difluoromethanoprolines into polypeptides. Org Lett. 2012;14:5254–5257. doi: 10.1021/ol302412a. [DOI] [PubMed] [Google Scholar]
- 57.Oshiro K, Morimoto Y, Amii H. Sodium bromodifluoroacetate: a difluorocarbene source for the synthesis of gem-difluorocyclopropanes. Synthesis. 2010;12:2080–2084. [Google Scholar]
- 58.Tian F, Kruger V, Bautista O, Duan J-X, Li A-R, Dolbier WR, Jr, Chen Q-Y. A novel and highly efficient synthesis of gem-difluorocyclopropanes. Org Lett. 2000;2:563–564. doi: 10.1021/ol0055622. [DOI] [PubMed] [Google Scholar]
- 59.Dolbier WR, Jr, Tian F, Duan J-X, Li A-R, Ait-Mohand S, Bautista O, Buathong S, Baker JM, Crawford J, Anselme P, Cai XH, Modzelewska A, Koroniak H, Battiste MA, Chen Q-Y. Trimethylsilyl fluorosulfonyldifluoroacetate (TFDA): a new, highly efficient difluorocarbene reagent. J Fluorine Chem. 2004;125:459–469. [Google Scholar]
- 60.Eusterwiemann S, Martinez H, Dolbier WR., Jr Methyl 2,2-difluoro-2-(fluorosulfonyl)acetate, a difluorocarbene reagent with reactivity comparable to that of trimethylsilyl 2,2-difluoro-2-(fluorosulfonyl)acetate (TFDA) J Org Chem. 2012;77:5461–5464. doi: 10.1021/jo300876z. [DOI] [PubMed] [Google Scholar]
- 61.Chang Y, Cai C. Sodium trifluoroacetate: an efficient difluorocarbene precursor for alkenes. Chem Lett. 2005;34:1440–1441. [Google Scholar]
- 62.Zheng J, Lin J-H, Cai J, Xiao J-C. Conversion between difluorocarbene and difluoromethylene ylide. Chem Eur J. 2013;19:15261–15266. doi: 10.1002/chem.201303248. [DOI] [PubMed] [Google Scholar]
- 63.Crabbe P, Carpio H, Velarde E, Fried JH. Chemistry of difluorocyclopropenes. application to the synthesis of steroidal allenes. J Org Chem. 1973;38:1478–1483. [Google Scholar]
- 64.Bebia D, Pilorge F, Delbarre LM, Demoute JP. From difluorocyclopropene to difluorocyclopropylidene. Tetrahedron. 1995;51:9603–9610. [Google Scholar]
- 65.Xu W, Chen Q-Y. 3,3-Difluoro-1-iodocyclopropenes: a simple synthesis and their reactions. J Org Chem. 2002;67:9421–9427. doi: 10.1021/jo020431v. [DOI] [PubMed] [Google Scholar]
- 66.Mehta VP, Greaney MF. S-, N-, and Se-Difluoromethylation using sodium chlorodifluoroacetate. Org Lett. 2013;15:5036–5039. doi: 10.1021/ol402370f. [DOI] [PubMed] [Google Scholar]
- 67.He Z, Luo T, Hu M, Cao Y, Hu J. Copper-catalyzed di- and trifluoromethylation of α,β-unsaturated carboxylic acids: a protocol for vinylic fluoroalkylations. Angew Chem Int Ed. 2012;51:3944–3947. doi: 10.1002/anie.201200140. [DOI] [PubMed] [Google Scholar]
- 68.He Z, Hu M, Luo T, Li L, Hu J. Copper-catalyzed difluoromethylation of β,γ-unsaturated carboxylic acids: an efficient allylic difluoromethylation. Angew Chem Int Ed. 2012;51:11545–11547. doi: 10.1002/anie.201206556. [DOI] [PubMed] [Google Scholar]
- 69.He Z, Zhang R, Hu M, Li L, Ni C, Hu J. Copper-mediated trifluoromethylation of propiolic acids: facile synthesis of α-trifluoromethyl ketones. Chem Sci. 2013;4:3478–3483. [Google Scholar]
- 70.Li Z, Cui Z, Liu Z-Q. Copper- and Iron-catalyzed decarboxylative tri- and difluoromethylation of α,β-unsaturated carboxylic acids with CF3SO2Na and (CF2HSO2)2Zn via a radical process. Org Lett. 2013;15:406–409. doi: 10.1021/ol3034059. [DOI] [PubMed] [Google Scholar]
- 71.Patra T, Deb A, Manna S, Sharma U, Maiti D. Iron-mediated decarboxylative trifluoromethylation of α,β-unsaturated carboxylic acids with trifluoromethanesulfinate. Eur J Org Chem. 2013:5247–5250. [Google Scholar]
- 72.Xu P, Abdukader A, Hu K, Cheng Y, Zhu C. Room temperature decarboxylative trifluoromethylation of α,β-unsaturated carboxylic acids by photoredox catalysis. Chem Commun. 2013 doi: 10.1039/C3CC48598F. [DOI] [PubMed] [Google Scholar]
- 73.Rueda-Becerril M, Sazepin CC, Leung JCT, Okbinoglu T, Kennephohl P, Paquin J-F, Sammis GM. Fluorine transfer to alkyl radicals. J Am Chem Soc. 2012;134:4026–4029. doi: 10.1021/ja211679v. [DOI] [PubMed] [Google Scholar]
- 74.Yin F, Wang Z, Li Z, Li C. Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids in aqueous solution. J Am Chem Soc. 2012;134:10401–10404. doi: 10.1021/ja3048255. [DOI] [PubMed] [Google Scholar]
- 75.Mizuta S, Stenhagen ISR, O’Duill M, Wolstenhulme J, Kirjavainen AK, Forsback SJ, Tredwell M, Sandford G, Moore PR, Huiban M, Luthra SK, Passchier J, Solin O, Gouverneur V. Catalytic decarboxylative fluorination for the synthesis of tri- and difluoromethyl arenes. Org Lett. 2013;15:2648–2651. doi: 10.1021/ol4009377. [DOI] [PubMed] [Google Scholar]
- 76.Eisenberger P, Gischig S, Togni A. Novel 10-I-3 hypervalent iodine-based compounds for electrophilic trifluoromethylation. Chem Eur J. 2006;12:2579–2586. doi: 10.1002/chem.200501052. [DOI] [PubMed] [Google Scholar]
- 77.Kieltsch I, Eisenberger P, Togni Antonio. Mild electrophilic trifluoromethylation of carbon- and sulfur-centered nucleophiles by a hypervalent iodine(III)-CF3 reagent. Angew Chem Int Ed. 2007;46:754–757. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]