

Abstract
Autophagy is a series of catabolic process mediating the bulk degradation of intracellular proteins and organelles through formation of a double-membrane vesicle, known as an autophagosome, and fusing with lysosome. Autophagy plays an important role of death-survival decisions in neuronal cells, which may influence to several neurodegenerative disorders including Parkinson’s disease. Chebulagic acid, the major constituent of Terminalia chebula and Phyllanthus emblica, is a benzopyran tannin compound with various kinds of beneficial effects. This study was performed to investigate the autophagy enhancing effect of chebulagic acid on human neuroblastoma SH-SY5Y cell lines. We determined the effect of chebulagic acid on expression levels of autophago-some marker proteins such as, DOR/TP53INP2, Golgi-associated ATPase Enhancer of 16 kDa (GATE 16) and Light chain 3 II (LC3 II), as well as those of its upstream pathway proteins, AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR) and Beclin-1. All of those proteins were modulated by chebulagic acid treatment in a way of enhancing the autophagy. Additionally in our study, chebulagic acid also showed a protective effect against 1-methyl-4-phenylpyridinium (MPP+) - induced cytotoxicity which mimics the pathological symptom of Parkinson’s disease. This effect seems partially mediated by enhanced autophagy which increased the degradation of aggregated or misfolded proteins from cells. This study suggests that chebulagic acid is an attractive candidate as an autophagy-enhancing agent and therefore, it may provide a promising strategy to prevent or cure the diseases caused by accumulation of abnormal proteins including Parkinson’s disease.
Keywords: Parkinson’s disease, Autophagy, Chebulagic acid, 1-Methyl-4-phenylpyridinium (MPP+)
INTRODUCTION
As known, Parkinson’s disease (PD) is resulted by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) of midbrain (Bae et al., 2011). Preventing aggregated and misfolded proteins in brain blocked the progression of PD which may propose a potential therapeutic benefit (Pan et al., 2008a; Bae et al., 2011). Recently, the ubiquitin-proteasome system and the autophagy-lysosomal pathway are the two most important cellular mechanisms for protein degradation (Pan et al., 2008b). But, due to the size of the narrow barrel of the proteasome and the specificity of the process, many large proteins are unable to be degraded by the ubiquitinproteasome system (Pan et al., 2008a; Pan et al., 2009). On the other hand, macroautophagy (autophagy) is a diverse process, involving the formation of double membrane structures known as autophagosomes which attract aggregated or misfolded proteins regardless of the size (Pan et al., 2008a; Pan et al., 2008b; Pan et al., 2009). The autophagosome fuses with lysosome to form autophagolysosomes, and misfolded or aggregated proteins are then degraded by hydrolytic enzymes located in the lysosomes (Hara et al., 2006; Ravikumar et al., 2006; Dadakhujaev et al., 2010; Underwood et al., 2010). Through these processes, cells can remove malfunctioning proteins and extend its lifespan. Autophagy can be regulated through mammalian target of rapamycin (mTOR) signaling pathway, and also can be upregulated by AMP-activated protein kinase (AMPK) and beclin-1 (Larsen and Sulzer, 2002; Hay and Sonenberg, 2004; Liang et al., 2007; Shin et al., 2011).
The research on the PD has till now to use the neuronal toxin, such as 1-methyl-4-phenyl-pyridinium (MPP+) (Chen et al., 2003; Bollimuntha et al., 2005). MPP+, the active neurotoxic metabolite converted from MPTP by monoamine oxidase-B in astrocytes, is the proximal neurotoxin destroying the nigrostriatal pathway in animals. Therefore, MPP+ has been widely used in in vitro and in vivo models of PD (Dauer and Przedborski, 2003; Bollimuntha et al., 2005; Choi et al., 2005).
Chebulagic acid (CA), the major constituent of Terminalia chebula and Phyllanthus emblica, is a benzopyran tannin com pound with various kinds of medicinal potentials including anti-oxidative, anti-pyretic, anti-atherogenic, cardioprotective, hepatoprotective, nephroprotective, adaptogenic, anti-anemia and neuroprotective activities (Baliga and Dsouza, 2011; Park et al., 2011; Chang and Lin, 2012). However, effect of CA on autophagy is currently not reported. In this study, we used CA as an autophagy inducer to find out its neuroprotective mechanism via autophagy. Using this approach, our primary aim is to evaluate the therapeutic potential of CA for neurodegenerative diseases, especially in PD.
MATERIALS AND METHODS
Cell culture and chemicals
The human neuroblastoma cell line SH-SY5Y was from the American Type Culture Collection (Manassas, VA, USA). Cells were grown in Dulbecco’s modified Eagle medium Ham’s F12 (1:1 mixture) (Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Grand Island, NY, USA), 100 units/ml of penicillin, and 100 mg/ml of streptomycin (Grand Island, NY, USA) in a 5% CO2 incubator at 37°C. CA was purchased from the Chromadex (Irvine, CA, USA). CA was dissolved in culture medium at a concentration of 5 mM for a stock solution. For treatment of cells, CA was diluted in culture medium to the appropriate concentration. 3-methyladenine (3MA) and MPP+ were obtained from Sigma (St. Louis, MO, USA).
MTT assay
The cell viability was measured by a quantitative colorimetric assay with MTT. The MTT assay provided a sensitive measurement of the metabolic status of the cells, particularly, the status of the mitochondria, which can reflect early redox changes. Briefly, exponentially growing cells were seeded in a 96-well plate at a density of 5×104 cells/well. The next day, cells were treated with various concentrations of CA in triplicate. After incubation for 24 h, 10 μl of the MTT assay kit reagent was added to each well, and the cells were incubated for an additional hour. Another set of MTT assay was done with cells treated with MPP+. SH-SY5Y cells were plated at a density of 5×104 cells/well in 100 μl medium in 96-well plates and incubated for 24 h. The cells were then pre-treated with CA for 2 h. After the pre-treatment period, 600 μM MPP+ was added to the culture medium and incubated for 48 h. The control cells were not treated with CA or MPP+. The absorbance for each reaction product was measured with a microplate reader at a wavelength of 450 nm. The results are expressed as the percentage of MTT reduction, with the assumption that the absorbance of the control cells is 100%.
Immunoblot analysis
Whole-cell lysates were prepared by incubating cells in RIPA buffer (Beverly, MA, USA) supplemented with protease inhibitor cocktail (Roche, Mannheim, Germany) according to the manufacturer’s instructions. Briefly, cells were harvested and collected by centrifugation at 13,200 rpm for 5 min and washed in Dulbecco’s Phosphate-Buffered Saline (DPBS) (pH 7.2). The pellets were solubilized in the same volume of mitochondrial lysis buffer for 5 min and then centrifuged at 13,200 rpm for 20 min at 4°C. Equal amounts of lysate protein were loaded and separated on a 15% SDS-PAGE gel. Proteins were electrophoretically transferred to a PVDF membrane, and the membrane was blocked in 5% skim milk in tris-buffered saline containing 0.1% Tween-20 for 1 h. Then, the membranes were incubated in the presence of following primary antibodies (LC3, beclin-1, mTOR, AMPK, GAPDH from cell signaling (Beverly, MA, USA); GATE16, TP53INP2/DOR from Epitomics (Burlingame, CA, USA)) at 4°C overnight. The membranes were then washed three times with TBS Tween 20 and probed with the corresponding secondary antibodies conjugated with HRP at room temperature for 1 h. Detection was carried out using an enhanced ECL Advance Western Blotting Detection Reagents (GE Healthcare, Buckinghamshire, UK) followed by LAS-4000 film (Fujifilm, Tokyo, Japan).
Immunofluorescence analysis
To detect the expression of LC3, cells were seeded onto sterile coverslips that were placed in 12-well plates. The next day, the cells were treated with CA. At 24 h post-treatment, the cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 30 min, blocked with 2% Bovine serum albumin (BSA) in Dulbecco’s Phosphate buffered saline (DPBS) for 1 h, and incubated with the LC3B primary antibody at room temperature (RT) for 1 h and with the Alexa fluor 488 secondary antibody (Invitrogen) at RT for 1 h in the dark. This was followed by incubation with 1 mg/ ml of 40,60-diamidino-2-phenylindole (DAPI) at RT for 20 min in the dark. Slides were then prepared with one drop of Pro-Long Gold Antifade Reagent (Invitrogen), and the coverslips were sealed onto the slides with clear nail lacquer. The images were obtained using a Leica TCS SP5 confocal microscope (Leica, Mannheim, Germany) and the pictures were analyzed by Image-Pro Plus 6.0 (Bethesda, MD, USA).
Statistical analysis
Statistical data are expressed as mean ± SD and student’s t-test was used to determine statistical significance. Values of p<0.05 were considered to be statistically significant.
RESULTS
CA induced autophagy in SH-SY5Y cells without cytotoxicity
To evaluate the potential cytotoxicity of CA, we first examined the effect of CA on cell survival in SH-SY5Y cells. SHSY5Y cells were treated with CA at various concentrations (1.56, 3.13, 6.25, 12.5 and 25 μM) for 24 h. Cell viability was measured by using the MTT assay kit. MTT assay result indicated that CA treatment has no cytotoxicity on SH-SY5Y cell compared to vehicle-treated control group (Fig. 1A). To determine the induction of autophagy by CA in SH-SY5Y cells, the cells were treated with various concentrations of CA at 1.56 to 25 μM for 24 h. Immunoblot assay revealed that the ratio of LC3 II/I was significantly increased in a dose-dependent manner with CA treatment as compared with control. Also the expression of other autophagosome markers such as, TP53INP2/DOR and GATE16 showed tendency to increase with CA treatment (Fig. 1B, C).
Fig. 1.

Induction of autophagosomes by CA in SH-SY5Y cells. SH-SY5Y cells were treated with CA at the concentrations of 1.56, 3.13, 6.25, 12.5 and 25 μM for 24 hr. (A) The cell viability was measured by MTT assay and the results were expressed as the percentile of absorbance of treated samples compared to that of the control. (B, C) SH-SY5Y cells were determined by measuring expression levels, DOR early autophagosome marker, LC3 protein the autophagosome marker, and GATE16 autophagosome membrane protein using an immunoblotting assay. The expression levels of three kinds of proteins were evaluated by densitometric analysis and data was expressed as folds of the control. GAPDH was used as an equal loading control. Each data represents the mean ± SD (n=3). Significantly different (**p<0.01) from control.
CA regulated the expression of autophagy modulating proteins in SH-SY5Y cells
To determine the mechanism of CA on autophagy induction in SH-SY5Y cells, the cells were treated with CA at the concentration of 1.56, 3.13, 6.25, 12.5 and 25 μM. As shown in Fig. 2, the expression levels of p-AMPK and Beclin-1 were significantly upregulated, whereas mTOR was downregulated by CA treatment.
Fig. 2.
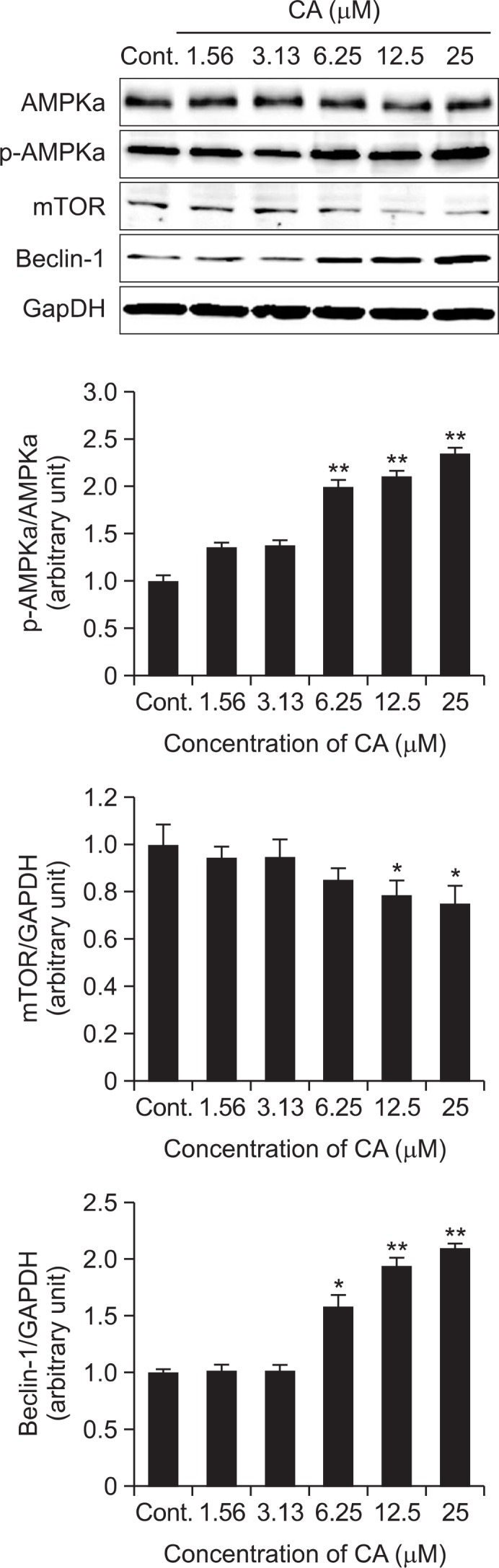
Induction of autophagosome upstream pathway by CA in SH-SY5Y cells. The induction of autophagy by CA in SH-SY5Y cells was determined by measuring the p-AMPK, mTOR and Beclin-1 protein levels using an immunoblotting assay with antibody against p-AMPK, mTOR and Beclin-1. The expression levels of three kinds of proteins were evaluated by densitometric analysis and data was expressed as folds of the control. GAPDH was used as an equal loading control. Each data represents the mean ± SD (n=3). Significantly different (*p<0.05, **p<0.01) from control.
CA-regulated autophagy expressions were inhibited by 3MA in SH-SY5Y cells
As shown in Fig. 3, the ratio of LC3 II/I and expression of Beclin-1 were increased by the treatment of rapamycin (10 μM, positive control) and CA (25 μM), while mTOR expression level showed tendency to decrease. However, increased ratio of LC3 II/I proteins by CA was inhibited with 3MA treatment (Per and Paul, 1982; Bae et al., 2011), a specific inhibitor of beclin-1 in early stage of autophagosome formation, whereas mTOR expression level showed tendency to increase.
Fig. 3.
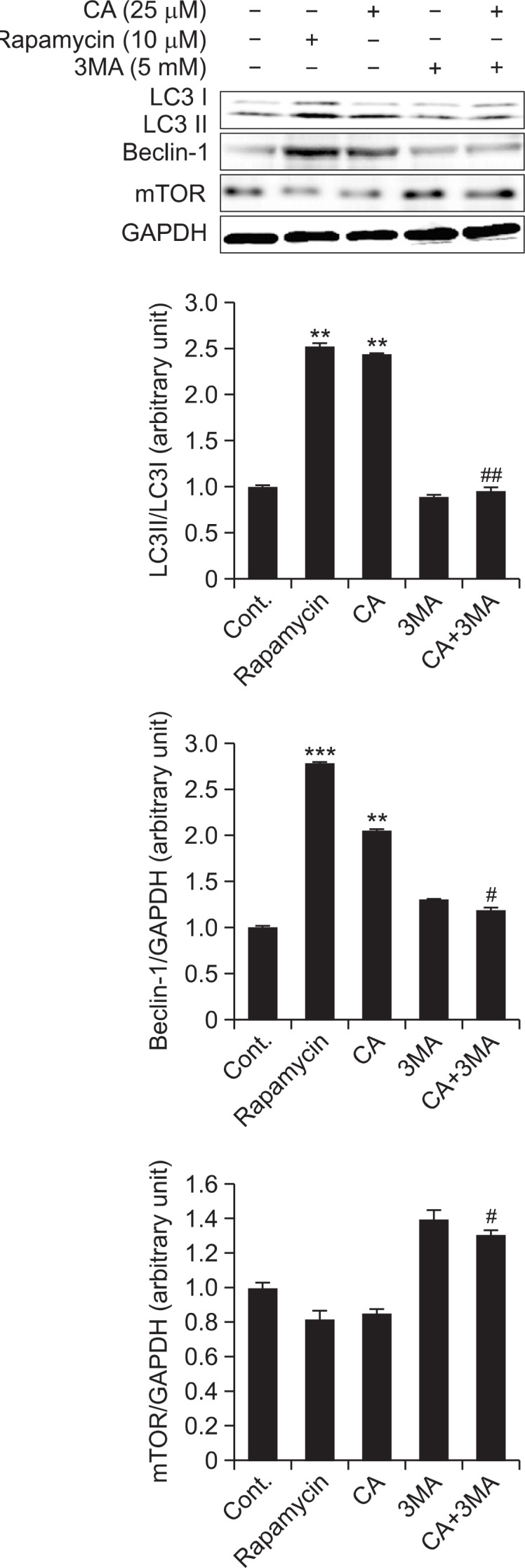
Inhibition effect of 3MA on CA-induced autophagy in SHSY5Y cells. Inhibition effect of 3MA on CA-induced autophagy was determined by measuring the LC3 protein levels using an immunoblotting assay with antibody against LC3 in SH-SY5Y cells. Rapamycin (10 μM) was used as a positive control. The expression levels of three kinds of proteins were evaluated by densitometric analysis and data were expressed as folds of the control. GAPDH used as an equal loading control. Each data represents the mean ± SD (n=3). Significantly different (**p<0.01, ***p<0.001) from control. Significantly different (#p<0.05, ##p<0.01) from CA treated group.
To visualize such effect of CA in SH-SY5Y cells with or without the treatment of 3MA, confocal imaging analysis was conducted. As shown in Fig. 4, cytosolic LC3 fluorescent puncta was significantly increased with rapamycin and CA treatments. However, 3MA treatment significantly inhibited such expression of LC3 puncta.
Fig. 4.

CA-induced LC3 expression in autophagosomes. SH-SY5Y cells were treated with 25 μM of CA, 10 μM of rapmycin and 5 mM of 3MA for 24 h and then, were labeled with 40, 6-diamidino-2-phenylindole (DAPI, blue), and Alexa 488 secondary antibody against LC3 (green). LC3 fluorescent puncta were observed in cells by using the confocal microscope.
CA showed protective effect against MPP+-induced cytotoxicity in SH-SY5Y cells
SH-SY5Y cells were treated with CA at the concentrations of 1.56, 3.13, 6.25, 12.5 and 25 μM for 2 h. Then MPP+ (600 μM) was added and cells were incubated for another 48 h. As shown in Fig. 5, MPP+ caused a significant cytotoxicity on SHSY5Y cells. However, this cytotoxicity was attenuated by CA treatment in a dose-dependent manner.
Fig. 5.
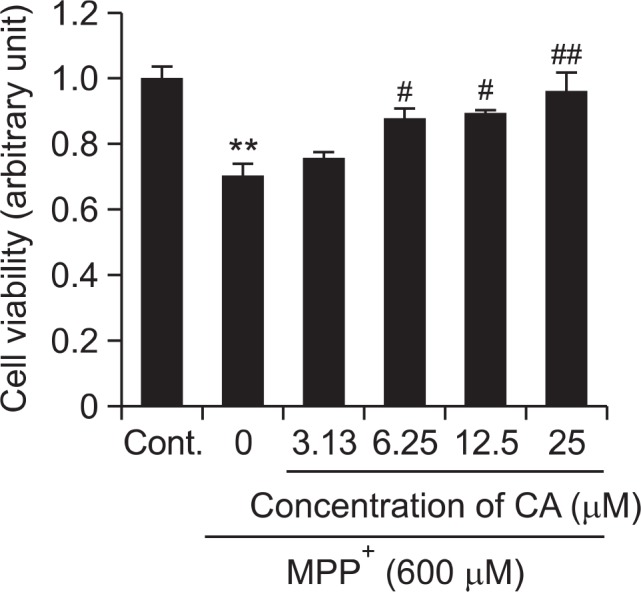
Neuroprotective effect of CA on MPP+-induced cytotoxicity in SH-SY5Y cells. Neuroprotective effect of CA on MPP+-induced cytotoxicity was determined by MTT assay. Cells were treated with MPP+ 600 μM alone, or MPP+ and CA for 48 h. SH-SY5Y cells were pre-treated with CA at the concentrations of 3.13, 6.25, 12.5 and 25 μM for 2 h followed by addition of MPP+ 600 μM for 48 hr. The cells viability were measured by MTT assay. Each data represents the mean ± SD (n=3). Significantly different (**p<0.01) from control. Significantly different (#p<0.05, ##p<0.01) from MPP+ treated group.
DISCUSSION
Basal expression of autophagy acts as one of the cytoprotective mechanisms and participates in maintaining homeostasis (Hara et al., 2006; Dadakhujaev et al., 2010). Also in diseased conditions, autophagy enhancement is known to play a role as protective mechanism. The results are consistent with previous studies supporting the putative neuroprotective effects of rapamycin, an autophagy enhancing agent, in various kinds of neurodegenerative disorders including PD (Ravikumar et al., 2006; Pan et al., 2008a; Pan et al., 2009; Underwood et al., 2010). Autophagy is the bulk degradation of proteins and organelles which is, essential for cellular maintenance and cell survival. Recently, LC3, DOR/TP53INP2 and GATE16 have been reported as new regulators involved in mammalian autophagy (Nowak et al., 2009; Spowart and Lum, 2010). LC3, GATE 16 and DOR co-localize during autophagosome formation and DOR/TP53INP2 has a short life cycle and degraded during fusion process of autophagosome and lysosome (Kabeya et al., 2004; Tanida et al., 2004; Nowak and Iovanna, 2009; Mauvezin et al., 2010; Weidberg et al., 2010). In this study, we demonstrated CA can induce LC3, GATE 16 and DOR/TP53INP2 protein expressions without any cytotoxicity (Fig. 1). These results indicate that CA can effectively increase the formation of autophagosomes which can dispose aggregated or misfolded proteins by fusing with lysosomes.
To elucidate the specific action mechanism of CA on enhancing the autophagy, we investigated some kinds of the most important proteins related with autophagy (Fig. 2). Beclin-1 was originally isolated from a yeast two-hybrid screen for Bcl-2 interacting proteins and has been shown to interact with anti-apoptosis protein such as Bcl-2 and Bcl-xL, and is also a principal regulator in autophagosome formation which initiates autophagy through class III PI3K pathway (Liu et al., 2007; Ko et al., 2011). mTOR, a mammalian target of rapamycin, acts as an inhibitor protein of autophagy mechanism (Gurusamy et al., 2010; Han et al., 2012). In this study, CA significantly induced Beclin-1 protein expression in dose dependent manner. Also, CA significantly attenuated protein expression of mTOR in dose dependent manner. Next, our study expanded to the upstream regulator of mTOR, AMPK protein. AMPK, AMP activated protein kinase, is a key energy sensor and can regulate metabolism (Williams et al., 2009; Wu et al., 2011). AMPK demonstrated conflicting roles for mTOR in autophagy. Several studies indicated that AMPK is an inducer of autophagy and many studies focus on it as a pharmacological agent (Melendez et al., 2003; Patschan et al., 2008; Williams et al., 2009; Wu et al., 2011). Our results showed that CA can increase the phosphorylated form of AMPK in a dose dependent manner. Taken together, CA revealed that it enhances autophagy through modulation of various kinds of proteins not only in lower stream of autophagy mechanism but also the proteins working in upper stream of the mechanism, too.
Furthermore, we investigated whether enhancement of autophagy by CA will be inhibited by 3MA treatment, the most commonly used specific inhibitor of autophagic removal. 3MA has an inhibitory effect on autophagy in the early stage of autophagosome formation and is thought to induce inhibition of PI3 kinase III (Bae et al., 2011). In this study, LC3II and beclin-1 were upregulated in the rapamycin and CA treated cells, and the protein expression levels of LC3 and beclin-1 were suppressed by the co-treatment of CA and 3MA. Recent reports have shown 3MA inhibits class III PI3K-mediated mTOR suppression, resulting in autophagy induction (Bae et al., 2011; Ko et al., 2011; Bao et al., 2012). In this study, mTOR was suppressed in the CA and rapamycin treated cells, and the protein expression level of mTOR was increased by the co-treatment of CA and 3MA. Therefore, CA acts similarly as rapamycin, a well established autophagy inducer via suppression of mTOR function (Fig. 3, 4).
Currently in clinics, Levo-dopa is used for PD patients to increase dopamine concentration in the brain and other neurodegenerative diseases (Chen et al., 2003; Bollimuntha et al., 2005; Bae et al., 2011). But, several studies showed that Levo-dopa has side effects such as wearing-off and dyskinesia (Chen et al., 2003; Choi et al., 2005; Bae et al., 2011). Recently, the PD etiology and mechanisms related to dopaminergic neuronal death became focused for prevention and alleviating the progress of PD (Chen et al., 2003; Bollimuntha et al., 2005). Research on PD models were created by using toxic materials such as 6-OHDA and MPTP which were known to selectively destroy dopaminergic neurons. MPP+, the active neurotoxic metabolite converted from MPTP by monoamine oxidase-B in astrocytes, is the proximal neurotoxin destroying the nigrostriatal pathway in human and mouse (Mathiasen et al., 2004; Chen et al., 2010). Therefore, MPTP and MPP+ have been widely employed to mimic in vivo and in vitro models of PD (Virmani et al., 2004). In this study of MPP+ in vitro model, CA significantly rescued neuronal cells from the cytotoxicity induced by MPP+ (Fig. 5) which may suggest a possible role of CA in protection of PD-related neuronal damages.
CONCLUSION
In conclusion, autophagy inducing effect of CA was confirmed for the first time in SH-SY5Y cells through the main control factors of AMPK, mTOR, Beclin-1 and LC3 as shown in Fig. 6. In addition, CA showed significant neuroprotective effect against MPP+-induced cellular damages and it may possesses protective function against PD-related neuronal damages which needs to be elucidated in future studies.
Fig. 6.

Regulation of autophagy key regulators, AMPK, mTOR, Beclin-1 and LC3, by CA treatment. CA causes activation of AMPK, Beclin-1 and LC3, and inhibition of mTOR followed by the induction of autophagy.
Acknowledgments
This work was supported by the institutional program of the Korea Institute of Science and Technology (Grant No. 2Z04210), The Business Promotion of the “Baekdudaegan Gr eenmine” Resource program (Grant No. R0000487), and Bio-Synergy Research Project (NRF-2012M3A9C4048793) of the Ministry of Science, ICT and Future Planning through the National Research Foundation, Republic of Korea.
REFERENCES
- Bae NY, Ahn TK, Chung SK, Oh MS, Ko HS, Oh HG, Park GH, Yang HO. The neuroprotective effect of modified Yeoldahanso-tang via autophagy enhancement in models of Parkinson’s disease. J Ethnopharmacol. 2011;134:313–322. doi: 10.1016/j.jep.2010.12.016. [DOI] [PubMed] [Google Scholar]
- Baliga MS, Dsouza JJ. Amla(Emblica officinalis Gaertn), a wonder berry in the treatment and prevention of cancer. Eur J Cancer Prev. 2011;20:225–239. doi: 10.1097/CEJ.0b013e32834473f4. [DOI] [PubMed] [Google Scholar]
- Bao XX, Xie BS, Li Q, Li XP, Wei LH, Wang JL. Nifedipine induced autophagy through Beclin1 and mTOR pathway in endometrial carcinoma cells. Chin Med J. 2012;125:3120–3126. [PubMed] [Google Scholar]
- Bollimuntha S, Singh BB, Shavali S. TRPC1-mediated inhibition of 1-methyl-4-phenylpyridinium ion neurotoxicity in human SH-SY5Y neuroblastoma cells. J Biol Chem. 2005;280:2132–2140. doi: 10.1074/jbc.M407384200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CL, Lin CS. Phytochemical composition, anti-oxidant activity, and neuroprotective effect of Terminalia chebula Retzius extracts. Evid Based Complement Alternat Med. 2012;2012:125247. doi: 10.1155/2012/125247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XC, Fang F, Zhu YG, Chen LM, Zhou YC, Chen Y. Protective effect of ginsenoside Rg1 on MPP+-induced apoptosis in SH-SY5Y cells. J Neural Transm. 2003;110:835–845. doi: 10.1007/s00702-003-0005-y. [DOI] [PubMed] [Google Scholar]
- Chen Z, Lu T, Yue X, Wei N, Jiang Y, Chen M, Ni G, Liu X, Xu G. Neuroprotective effect of ginsenoside Rb1 on glutamate-induced neurotoicity : With emphasis on autophagy. Neurosci Lett. 2010;482:264–268. doi: 10.1016/j.neulet.2010.07.052. [DOI] [PubMed] [Google Scholar]
- Choi JY, Jang EH, Park CS, Kang JH. Enhanced susceptibility to 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine neurotoxicity in high-fat diet induced obesity. Free Radic Biol Med. 2005;38:806–816. doi: 10.1016/j.freeradbiomed.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Dadakhujaev S, Noh HS, Jung EJ, Cha JY, Baek SM, Ha JH, Kim DR. Autophagy protects the rotenone-induced cell death in alpha-synuclein overexpressing SH-SY5Y cells. Neurosci Lett. 2010;472:47–52. doi: 10.1016/j.neulet.2010.01.053. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan K, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–112. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Ichiro S, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L, Pan Y, Li XJ. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy. 2012;8:812–825. doi: 10.4161/auto.19471. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Satsuki OO, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi: 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Ko HS, Kim YJ, Amor EC, Lee JW, Kim HC, Kim HJ, Yang HO. Induction of autophagy by dimethyl cardamonin is associated with proliferative arrest in human colorectal carcinoma HCT116 and LOVO cells. J Cell Biochem. 2011;112:2471–2479. doi: 10.1002/jcb.23171. [DOI] [PubMed] [Google Scholar]
- Larsen KE, Sulzer D. Autophagy in neurons: a review. Histol Histopathol. 2002;17:897–908. doi: 10.14670/HH-17.897. [DOI] [PubMed] [Google Scholar]
- Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slinger-land JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Liu D, Si H, Reynolds KA, Zhen W, Jia Z, Dillon JS. Dehydroepiandrosterone protects vascular endothelial cells against apoptosis through a Galphai protein-dependent activation of phosphatidylinositol 3-kinase/Akt and regulation of antiapoptotic Bcl-2 expression. Endocrinology. 2007;148:3068–3076. doi: 10.1210/en.2006-1378. [DOI] [PubMed] [Google Scholar]
- Mathiasen JR, McKenna BA, Saporito MS, Ghadge GD, Roos RP, Holskin BP, Wu ZL, Trusko SP, Connors TC, Maroney AC, Thomas BA, Thomas JC, Bozyczko-Coyne D. Inhibition of mixed lineage kinase 3 attenuates MPP+-induced neurotoxicity in SH-SY5Y cells. Brain Res. 2004;1003:86–97. doi: 10.1016/j.brainres.2003.11.073. [DOI] [PubMed] [Google Scholar]
- Mauvezin C, Orpinell M, Francis VA, Mansilla F, Duran J, Ribas V, Palacín M, Boya P, Teleman AA, Zorzano A. The nuclear cofactor DOR regulates autophagy in mammalian and Drosophila cells. EMBO Rep. 2010;11:37–44. doi: 10.1038/embor.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science. 2003;301:1387–1391. doi: 10.1126/science.1087782. [DOI] [PubMed] [Google Scholar]
- Nowak J, Archange C, Joel TL, Pontarotti P, Pe´busque M, Vaccaro MI, Velasco G, Dagorn JC, Iovanna JL. The TP53INP2 protein is required for autophagy in mammalian cells. Mol. Biol. Cell. 2009;20:870–881. doi: 10.1091/mbc.E08-07-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak J, Iovanna JL. TP53INP2 is the new guest at the table of self-eating. Autophagy. 2009;5:383–384. doi: 10.4161/auto.5.3.7698. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Le W, Jankovic J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain. 2008a;131:1969–1978. doi: 10.1093/brain/awm318. [DOI] [PubMed] [Google Scholar]
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis. 2008b;32:16–25. doi: 10.1016/j.nbd.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Pan T, Rawal P, Wu Y, Xie W, Jankovic J, Le W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience. 2009;164:541–551. doi: 10.1016/j.neuroscience.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Park JH, Joo HS, Yoo KY, Shin BN, Kim IH, Lee CH, Choi JH, Byun K, Lee B, Lim SS, Kim MJ, Won MH. Extract from Terminalia chebula seeds protect against experimental ischemic neuronal damage via maintaining SODs and BDNF levels. Neurochem Res. 2011;36:2043–2050. doi: 10.1007/s11064-011-0528-9. [DOI] [PubMed] [Google Scholar]
- Patschan S, Chen J, Polotskaia A. Lipid mediators of autophagy in stressinduced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol. 2008;294:1119–1129. doi: 10.1152/ajpheart.00713.2007. [DOI] [PubMed] [Google Scholar]
- Per OS, Paul BG. 3-methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Nati Acad Sci USA. 1982;79:1889–1892. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC. Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet. 2006;15:1209–1216. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- Shin HY, Chu SH, Lee HK, Lee JW. mTOR inhibitor as a potential drug of age-related disease. Korean J Clin Geri. 2011;12:149–159. [Google Scholar]
- Spowart J, Lum JJ. Opening a new DOR to autophagy. EMBO Rep. 2010;11:4–5. doi: 10.1038/embor.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36:2503–2518. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S, García-Arencibia M, O’Kane CJ, Murphy MP, Rubin-sztein DC. Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet. 2010;19:3413–3429. doi: 10.1093/hmg/ddq253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani A, Gaetani F, Binienda Z, Xu A, Duhart H, Ali SF. Role of mitochondrial dysfunction in neurotoxicity of MPP+: partial protection of PC12 cells by acetyl-L-carnitine. Ann N Y Acad Sci. 2004;1025:267–273. doi: 10.1196/annals.1316.033. [DOI] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T, Forsberg LJ, Viollet B, Brenman JE. Basal autophagy induction without AMP-activated protein kinase under low glucose conditions. Autophagy. 2009;5:1155–1165. doi: 10.4161/auto.5.8.10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z, Jankovic J, Pan T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals. 2011;19:163–174. doi: 10.1159/000328516. [DOI] [PMC free article] [PubMed] [Google Scholar]