

Abstract
Background
The mechanistic target of rapamycin (mTOR) comprises 2 structurally distinct multiprotein complexes, mTOR complexes 1 and 2 (mTORC1 and mTORC2). Deregulation of mTOR signaling occurs during and contributes to the severity of myocardial damage from ischemic heart disease. However, the relative roles of mTORC1 versus mTORC2 in the pathogenesis of ischemic damage are unknown.
Methods and Results
Combined pharmacological and molecular approaches were used to alter the balance of mTORC1 and mTORC2 signaling in cultured cardiac myocytes and in mouse hearts subjected to conditions that mimic ischemic heart disease. The importance of mTOR signaling in cardiac protection was demonstrated by pharmacological inhibition of both mTORC1 and mTORC2 with Torin1, which led to increased cardiomyocyte apoptosis and tissue damage after myocardial infarction. Predominant mTORC1 signaling mediated by suppression of mTORC2 with Rictor similarly increased cardiomyocyte apoptosis and tissue damage after myocardial infarction. In comparison, preferentially shifting toward mTORC2 signaling by inhibition of mTORC1 with PRAS40 led to decreased cardiomyocyte apoptosis and tissue damage after myocardial infarction.
Conclusions
These results suggest that selectively increasing mTORC2 while concurrently inhibiting mTORC1 signaling is a novel therapeutic approach for the treatment of ischemic heart disease.
Keywords: AKT1S1 protein, human, RICTOR protein, human, TOR serine-threonine kinases
Ischemic damage affects cardiac function through multiple mechanisms, including infarction injury with cardiac myocyte death by apoptosis. The mechanistic target of rapamycin (mTOR), an atypical serine/threonine protein kinase and central regulator of cell function, determines cell viability. mTOR signaling deregulation in cardiac disease contributes to cardiomyocyte apoptosis with associated impairment of cardiac function. mTOR resides in 2 distinct complexes, mTOR complexes 1 and 2 (mTORC1 and mTORC2).1,2 Activation of mTORC1 phosphorylates several downstream targets, including eukaryotic translation initiation factor 4E-binding protein and ribosomal S6Kinase, which in turn phosphorylate ribosomal S6 protein (RibS6). In comparison, mTORC2 activation phosphorylates the prosurvival kinase Akt on Ser-473 required for Akt activation.3 Although the mechanisms that regulate mTORC1 are well understood, the regulation of mTORC2 is relatively poorly characterized.
mTOR signaling in cardiac physiology and disease has been examined by manipulating the level or activity of mTOR kinase in hearts of mice. Conditional genetic deletion of mTOR kinase induced heart failure,4 whereas pharmacological inhibition of mTORC1 with rapamycin improved cardiac function in models of pressure overload, myocardial infarction (MI), and hypertrophic cardiomyopathy.5-7 Moreover, cardiomyocyte-specific mTOR overexpression protected hearts from challenge with pressure overload or ischemia.8,9 Because mTOR kinase participates in signaling through mTORC1 and mTORC2, neither genetic nor pharmacological manipulation of mTOR kinase is sufficient to distinguish the roles of these 2 complexes in cardiac pathology. For example, although short-term inhibition of mTORC1 with rapamycin stimulates mTORC2 in cancer cells and cardiac tissue,10,11 long-term treatment with rapamycin decreases the assembly and function of mTORC2 in cardiac tissue.10,12,13 Moreover, rapamycin does not inhibit all mTORC1 functions.14 In addition, pharmacological inhibition of mTORC1 by rapamycin affects resident and infiltrating cells in the heart. Therefore, studies using systemic administration of rapamycin have been of limited value in delineating the relative importance of mTORC1 and mTORC2 in the heart. Thus, tissue- and cell-specific analyses of mTOR signaling are required to reveal the multifaceted role of mTOR.
Knockdown of Rictor, a component essential for mTORC2 function, decreases mTORC2 activity and Akt activation and increases cell death with impaired proliferation in cancer cells.15,16 Effects of mTOR2 inhibition in the noncardiac context imply a role in the pathogenesis of ischemic heart disease, but cardiac-related studies of mTOR2 have not specifically addressed this issue. Therefore, acute MI was used as the setting to assess the impact of mTORC2 signaling in amelioration of pathological injury, with implications for mTORC2 as novel therapeutic target for cardiac ischemia.
Methods
Mice, Surgery, and Cardiac Function Analysis
Unless otherwise indicated, all experiments were performed in 7-week-old male C57BL/6 mice purchased from Jackson Laboratories. PI3K γ-/- mice and Akt1 knockout-/- have been described previously.17 Male mice underwent permanent ligation (MI) of the left anterior descending coronary artery or a sham operation, as previously described.18 For echocardiography, mice were anesthetized with 2% isoflurane and scanned with a Vevo770 imaging system (Visual Sonics, Toronto, ON, Canada), as previously described.19 Closed-chest hemodynamic assessment was performed after insertion of a microtip pressure transducer (FT111B, Scisense) into the right carotid artery and advancement into left ventricle.19 For in vivo injection of insulin, mice were injected intraperitoneally with PBS or insulin (1 U/kg body weight for 1 hour) after an overnight fast. Institutional Animal Care and Use Committee approval was obtained for all animal studies.
Isolation and Primary Cultures of Neonatal and Adult Ventricular Cardiomyocytes
Isolation and primary cultures of neonatal (NRCMs) and adult ventricular cardiomyocytes were prepared by standard procedures. Cells were treated with insulin (100 nmol/L) or H2O2 for multiple individual time points as indicated in the figures.
Adenoviral Constructs and siRNA
Recombinant adenoviruses were generated with human and mutated human PRAS40 (proline-rich Akt substrate of 40 kDa) cDNAs sub-cloned into the pShuttle-cytomegalovirus vector using the AdEasy XL Adenoviral Vector System (Stratagene), as previously described.18 The human full-length PRAS40 contained a C-terminal FLAG tag. NRCMs and adult cardiomyocytes were infected with adenoviruses, as described previously, at a multiplicity of infection of 20.20
Adeno-Associated Virus Serotype 9 Generation and Systemic In Vivo Adeno-Associated Virus Serotype 9 Cardiac-Targeted Gene Transfer Protocol
In vivo cardiac-targeted PRAS40 expression in normal mouse hearts was obtained by using tail vein injection of an adeno-associated virus serotype 9 (AAV9) harboring the PRAS40 gene or shRNA targeting Rictor (AAV–sh-Rictor) driven by a cardiomyocyte-specific cytomegalovirus–myosin light chain-promoter or U6 promoter, respectively. Recombinant AAV9 vector carrying the same promoters without a downstream encoded transgene product served as control. Ringer lactate (100 _L) heated to 37°C containing 1×1011 total viral particles of AAV9-control, AAV9-PRAS40, or AAV9-sh-Rictor was injected into the tail vein, as previously described.21
Sample Preparation, Immunoblotting, and Real-Time Quantitative Polymerase Chain Reaction
Whole hearts and isolated myocytes were prepared as described previously.18 Immunoblots from isolated cells or tissue were conducted as previously described. RNA was isolated with the Quick RNA MiniPrep kit (ZymoResearch) according to the manufacturer’s protocol. cDNA was generated, and real-time quantitative polymerase chain reaction was carried out with the cDNA preparation kit and SYBR real-time polymerase chain reaction (Biorad) according to the manufacturer’s protocol. Differences were calculated with the ΔΔC(T) method. A full list of primers and antibodies is provided in the Tables I and II in the online-only Data Supplement.
Histology and Staining
Immunostaining of isolated myocytes was performed on cells grown on Permanox chamber slides. Sections for Masson trichrome, hematoxylin and eosin, and immunohistochemistry were generated from paraffin-embedded hearts. Hearts were perfused in situ with formalin for 15 minutes, excised, and fixed in formalin for 24 hours at room temperature. Sections were deparaffinized with standard procedures. Immunostaining of paraffin-embedded hearts was performed as described previously in detail.22 Sections were also used to visualize the sarcolemma by staining with tetramethyl rhodamine isothiocyanate–conjugated wheat germ agglutinin (Sigma-Aldrich). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining was performed using the In Situ Cell Death Detection Kit, TMR red (Roche Applied Science) according to the manufacturer’s directions.
Flow Cytometry
Cell death was measured with an Annexin V kit (BD Biosciences) according to the manufacturer’s instructions. To induce cell death, NRCMs were treated with 50 μmol/L H2O2 for 4 hours.
Statistical Analysis
Kaplan–Meier curves were generated to illustrate survival after MI; statistical assessment was carried out by the log-rank test. Statistical analysis was performed with GraphPad Prism 5.0 (Graphpad Software Inc; www.graphpad.com). Values of P<0.05 were considered significant. All the data sets were tested for normality of distribution with the Shapiro-Wilk test. To compare 2 groups with normal distribution, the Student t test was applied; otherwise, a nonparametric test was used. Nonparametric tests were used when n<5 per group. For comparison of >2 groups, 1-way ANOVA was applied; for the echo-cardiographic time course analysis, repeated measures ANOVA was used. Bonferroni post hoc tests were included in both cases..
Results
mTOR Activation After MI
mTORC1 and mTORC2 signaling (Figure 1A) was assessed by phosphorylation of RibS6 (for mTORC1) and Akt (for mTORC2) after permanent occlusion of the left anterior descending coronary artery at 2 days after challenge. Phosphorylation of both proteins was increased in the infarcted mouse heart, indicating activation of both mTORC1 and mTORC2 (Figure IA in the online-only Data Supplement). Activation of mTORC1 and mTORC2 in cardiomyocytes was confirmed by confocal immunolocalization of RibS6 and AktS473 phosphorylation after the infarction challenge (Figure IA in the online-only Data Supplement).
Figure 1.
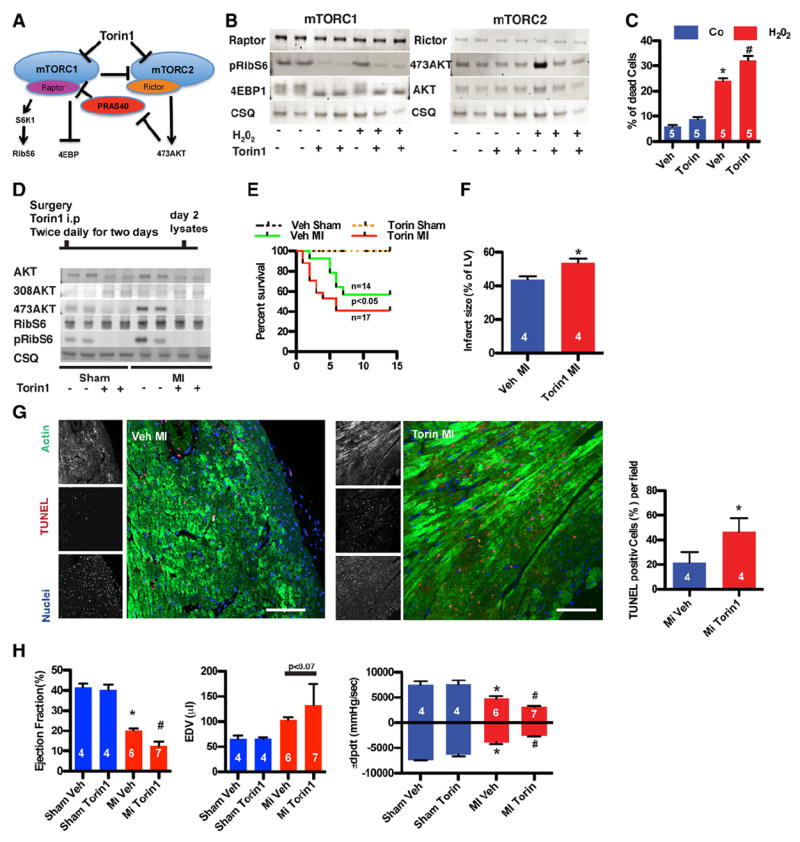
Decreasing mechanistic target of rapamycin complex 1 (mTORC1) and 2 (mTORC2) activity increases damage after stress. A, Schematic overview of mTOR signaling. B, mTORC1 and mTORC2 are inactivated after treatment with Torin1, as shown by immunoblots. C, Cell death in neonatal rat cardiomyocytes. Challenge with H2O2 (50 μmol/L for 4 hours) after mTOR kinase inhibition with Torin1 (50 nmol/L). Torin1 exposure increases apoptosis in response to H2O2. *P<0.05 vs vehicle (Veh); #P<0.05 vs H2O2 vehicle. D, Torin1 inhibits mTOR kinase activity in vivo, as shown by immunoblots. E, Kaplan–Meier survival curve of vehicle- and Torin1- (5 mg/kg body weight) injected mice. Mortality early after infarction is increased after injection of Torin1. n=4 in the sham groups. F, Torin1 increases infarct size. P<0.05 vs control myocardial infarction (MI). G, Representative confocal scans are shown for terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL), actin, and nuclei (red, green and blue, respectively, in overlays). Bar, 150 μm. Percentage of TUNEL-labeled cells in the left ventricle (LV) of remote area 1 day after MI. *P<0.05 vs control MI. H, Echocardiographic assessment of control or Torin1-injected mice for ejection fraction and LV end-diastolic volume (EDV). *P<0.05 vs sham; #P<0.05 vs MI vehicle. Hemodynamic measurements of ±dP/dt 2 weeks after surgery. *P<0.05 vs sham; #P<0.05 vs MI vehicle. Numbers of mice or of independent experiments per group are indicated in the bar graphs.
mTOR Kinase Inhibition With Torin1 Increases Cardiomyocyte Death In Vitro
Kinase activity of both mTORC1 and mTORC2 was inhibited with Torin1, a second- generation mTOR inhibitor (Figure 1A). mTORC1 and mTORC2 were inhibited by Torin1, as shown by decreased insulin-dependent phosphorylation of S6Kinase and AktS473, respectively, in a dose-dependent fashion, whereas mitogen-activated protein kinase phosphorylation was unchanged (Figure IB in the online-only Data Supplement). mTOR activity mediates survival in response to oxidative stress, as demonstrated with Torin1 treatment (50 nmol/L) of NRCMs, which blocks mTORC1 and mTORC2 activation in response to H2O2 (Figure 1B), consequently leading to increased cell death (Figure 1C). The increased cell death after Torin1 treatment was mainly apoptotic, as indicated by increased annexin V binding (Figure IC in the online-only Data Supplement).
mTOR Kinase Inhibition With Torin1 Increases Damage After MI
Acute mTOR inhibition in vivo was performed with Torin1 injected immediately after MI with a short 2-day follow-up treatment to minimize systemic effects of the inhibitor. Pharmacokinetic properties of Torin1 render the compound short-lived23; therefore, Torin1 was injected twice per day for 2 days (Figure ID in the online-only Data Supplement). mTOR signaling assessment in mice receiving Torin1 treatment revealed decreased phosphorylation for the downstream targets RibS6 and phosphorylation of AktS473, whereas AktT308 phosphorylation was increased after MI (Figure 1D). Increased mortality was observed in Torin1-injected mice by 10 days after MI (Figure 1E), and infarct size was increased by 23% after Torin1 treatment compared with controls (Figure 1F). Apoptotic cell death contributed to increased infarct size and mortality after Torin1 administration, as observed in myocardial sections where TUNEL labeling was significantly increased (Figure 1G). Ejection fraction was significantly impaired with a trend toward enlarged left ventricular volume as a consequence of Torin1 treatment compared with control mice 2 weeks after MI, indicating that Torin1 treatment accelerated decompensation (Figure 1H and Table III in the online-only Data Supplement). Hemodynamic performance was also impaired in Torin1-treated animals relative to nontreated control MI groups (Figure 1H). Heart rates were indistinguishable between the control and treated groups (Figure IE in the online-only Data Supplement). Collectively, these findings indicate that inactivation of mTOR kinase activity is detrimental in the adaptive response to MI.
mTORC2 Inhibition Increases Cardiomyocyte Death In Vitro
Functional activity for either mTORC1 or mTORC2 was selectively inhibited by targeted downregulation of either Rictor or Raptor expression with siRNA (see Figure 1A).1 mTORC1 signaling was inhibited by siRNA-induced depletion of Raptor with no discernible effect on mTORC2 activation, expression of Rictor (Figure 2A-2C), or NRCM death (Figure 2D). mTOR protein levels were slightly reduced after knockdown of Raptor, consistent with prior observations in Raptor knockout mice (Figure 2C).24 mTORC2 signaling was inhibited by Rictor depletion with siRNA, which was associated with increased cell death (Figure 2D) without an apparent impact on mTORC1 activation in response to H2O2 (Figure 2E-2G). Decreased NRCM survival was also observed by overexpression of AktS473A, a point mutant of the kinase that cannot be phosphorylated on AktS473 (Figure IIA in the online-only Data Supplement), showing that AktS473 must be phosphorylated mTORC2 to mediate protection from oxidative stress. FoxO phosphorylation downstream of Akt was decreased after knockdown of Rictor, whereas glycogen synthase kinase 3 phosphorylation remained unchanged (Figure IIB in the online-only Data Supplement), consistent with AKTS473-mediated regulation of target molecules that influence cellular survival.25 Increased cell death after Rictor knockdown was primarily apoptotic, as indicated by annexin V–positive cells, consistent with the effect of Torin1 inhibition (Figure IIC in the online-only Data Supplement).
Figure 2.
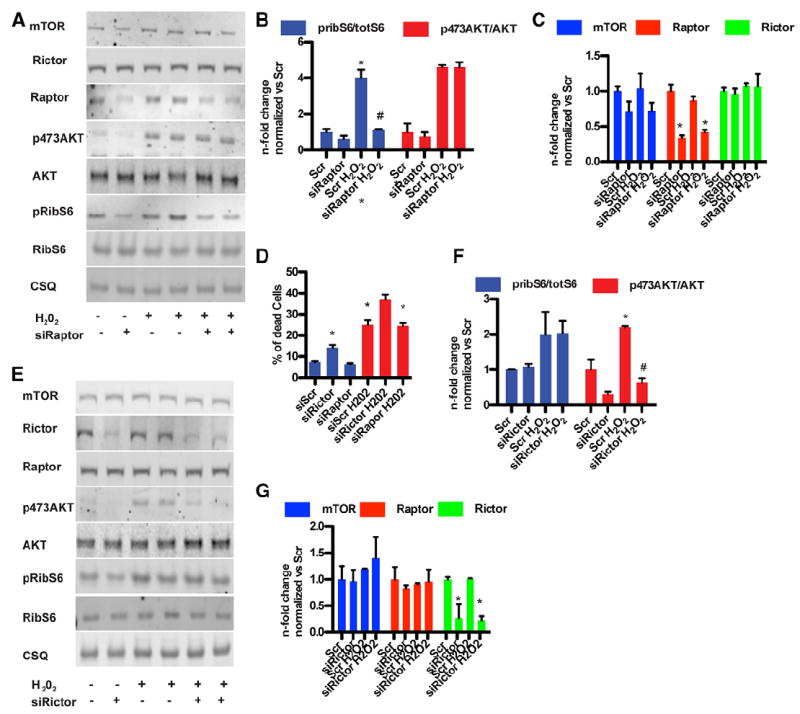
Loss of mechanistic target of rapamycin complex 2 (mTORC2) signaling increases cardiomyocyte death in vitro. A, Silencing of Raptor confirmed by immunoblot. B, Bar graphs depicting quantification of Akt phosphorylation and ribosomal S6 protein (RibS6) phosphorylation after Raptor silencing. *P<0.05 vs scramble (Scr); #P<0.05 vs Scr H2O2 vehicle (Veh). C, Bar graphs depicting quantitation of Rictor, mTOR, and Raptor expression after Raptor silencing. n=4 independent experiments. D, Cell death in neonatal rat cardiomyocytes. Rictor silencing but not Raptor silencing increases cell death after challenge with H2O2 (50 μmol/L for 4 hours). *P<0.05 vs control; #P<0.05 vs Scr H2O2. n=5 independent experiments. E, Silencing of Rictor confirmed by immunoblot. n=4 independent experiments. F, Bar graphs depicting quantification of Akt phosphorylation and RibS6 phosphorylation after Rictor silencing. *P<0.05 vs Scr; #P<0.05 vs Scr H2O2 vehicle. n=4 independent experiments. G, Histogram depicting quantification of Raptor, mTOR, and Rictor expression after Rictor silencing. n=4 independent experiments.
mTORC2 Inhibition Increases Damage After MI
mTORC2-selective inhibition by knocking down Rictor in vivo was accomplished using an shRNA targeted to mouse Rictor delivered via recombinant cardiotropic AAV9. Green fluorescent protein reporter virus was used to test the efficiency of the AAV9-mediated gene delivery. AAV9-mediated gene delivery resulted in 80% of the cardiomyocytes being positive for the green fluorescent protein transgene measured by fluorescence-activated cell sorter (Figure IIIA in the online-only Data Supplement). Decreases in expression of Rictor (80% reduction) and mTORC2 signaling were confirmed in whole-heart lysates 2 days after infarction (Figure 3A), associated with diminished AktS473 phosphorylation. Rictor knockdown did not affect either Raptor expression or mTORC1 activity compared with control mice, confirming the specificity of decreased mTORC2 signaling (Figure 3B and 3C), although total mTOR protein level was slightly decreased after Rictor knockdown (Figure 3C). Increased mortality was associated with knockdown of Rictor during the first 10 days after MI (Figure 3D), similar to the results obtained with Torin1 (Figure 1D). Apoptotic cell death contributed to increased mortality, with TUNEL-positive cells increased after Rictor knockdown in myocardial sections (Figure 3E). Impaired systolic function with enlarged left ventricular chamber size was evident after Rictor knockdown compared with control animals (Figure 3F and Table IV in the online-only Data Supplement). Successful knockdown of Rictor in cardiomyocytes was confirmed by immunocytofluorescence (Figure 3G). Increased fibrotic area resulted from knockdown of Rictor associated with pathological remodeling (Figure 3H). Therefore, mTORC2 inhibition decreases cardiomyocyte survival in response to MI challenge in vivo.
Figure 3.
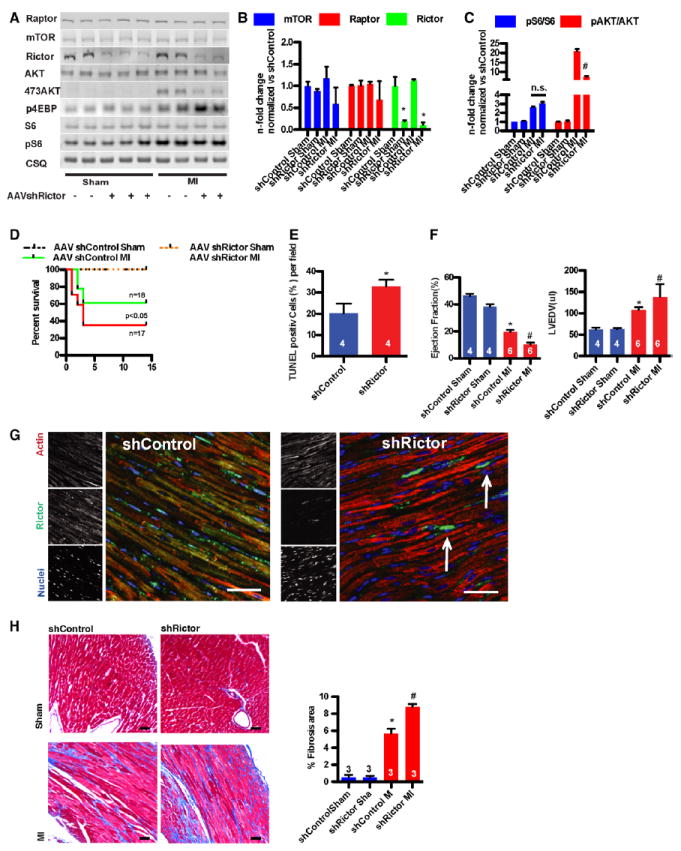
Loss of mechanistic target of rapamycin complex 2 (mTORC2) signaling increases cardiomyocyte damage after myocardial infarction (MI). A, Silencing of Rictor confirmed by immunoblot. Adeno-associated virus serotype 9 (AAV9)–sh-Rictor hearts exhibit decreased Akt phosphorylation without altered mTORC1 activation. B, Bar graphs depicting quantification of Raptor/mTOR/ Rictor expression. *P<0.05 vs control sham. n=3 per group. C, Bar graphs depicting quantification of ribosomal S6 protein (pRibS6) phosphorylation. n=3 per group. D, Kaplan–Meier survival curve of AAV–sh-control and AAV–sh-Rictor mice. AAV–sh-Rictor increases mortality early after infarction. n=6 in the sham groups. E, Percentage of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)–labeled cells in the left ventricle of the remote area 1 day after myocardial infarction (MI).*P<0.05 vs control MI. F, Echocardiographic assessment of AAV–sh-control or AAV–sh-Rictor mice for ejection fraction (EF) and left ventricular end-diastolic volume (LVEDV). *P<0.05 vs sh-control sham; #P<0.05 vs MI sh-Control. G, Silencing of Rictor in myocytes confirmed by confocal microscopy. Representative confocal scans for Rictor, actin, and nuclei (green, red, and blue, respectively, in overlays). Interstitial nonmyocytes express Rictor in AAV–sh-Rictor hearts (arrow), Bar, 150 μm. H, Masson trichrome staining from control and sh-Rictor– treated hearts. Bar graphs depicting quantification of fibrotic area. *P<0.05 vs control sham; #P<0.05 vs MI. Bar, 1mm. Numbers of mice per group are indicated in the bar graphs.
Decreased mTORC2 Activity Resulting From Prolonged mTORC1 Inhibition With Rapamycin
Genetic deletion of Raptor inhibits mTORC1 signaling and increases mTORC2 function by suppressing a negative feedback loop, but it also leads to severe cardiomyopathy.24 Longterm pharmacological inhibition of mTORC1 with rapamycin was used to determine the impact on TORC2 signaling. Indeed, mTORC2 signaling was disrupted after prolonged treatment of NRCMs with rapamycin, shown by diminished phosphorylation of the mTOR2 target AktS473 (Figure IIIB in the onlineonly Data Supplement). Cell death inhibition after short-term treatment with rapamycin in response to H2O2 was abrogated on long-term treatment (Figure IIIB in the online-only Data Supplement). The consequences of long-term inhibition of mTORC1 on mTORC2 signaling in vivo were assessed by intraperitoneal injection of rapamycin daily for 2 weeks. mTORC2 signaling was unaffected by short-term treatment with rapamycin (2 mg/kg, with the first dose on the day of MI), but prolonged treatment blocked AktS473 after MI (Figure IIIC in the online-only Data Supplement). In conclusion, prolonged treatment with rapamycin also impairs mTORC2 function, similar to the effects of Torin1 in cardiomyocytes.
Increasing mTORC2 Signaling While Inhibiting mTORC1 Decreases Cardiomyocyte Death
An endogenous molecular mechanism exists that blocks mTORC1 activity to regulate growth by maintaining the appropriate balance between anabolic processes and catabolic processes. PRAS40, a specific component of mTORC1 that interacts with RAPTOR to inhibit mTORC1 kinase activity, 26,27 has never been studied in the cardiac context. PRAS40 expression and regulation in myocytes were confirmed, with Akt-mediated phosphorylation of PRAS40T246 that was absent in Akt1 knockout mice (Figure IVA in the online-only Data Supplement). Stimulation of Akt with insulin induced PRAS40 phosphorylation in isolated adult wild-type myocytes (Figure IVB in the online-only Data Supplement), thereby releasing the inhibitory function of PRAS40 on mTORC1, as previously described.26 PRAS40-mediated inhibition of mTORC1 signaling after H2O2 stimulation was mediated by adenoviral overexpression of PRAS40 (Figure IVC in the online-only Data Supplement). mTORC2 signaling was increased by PRAS40 expression, as evidenced by higher levels of AktS473 compared with controls after H2O2 stimulation (Figure IVD in the online-only Data Supplement), consequently inhibiting apoptotic cell death (Figure IVE in the online-only Data Supplement). Protective effects of PRAS40 against oxidative stress were abrogated by pharmacological inhibition of Akt or after overexpression of a phosphorylation-impaired AktS473A that is point mutated at a critical activation site of the kinase (Figure IVF in the online-only Data Supplement). Increased PRAS40 phosphorylation is also observed in cardiomyocytes after MI (Figure VA and VB in the online-only Data Supplement). Thus, PRAS40 overexpression decreases mTORC1 and increases mTORC2 signaling, decreasing oxidative stress–induced cardiomyocyte death in vitro.
Loss of PRAS40-mediated cytoprotection on inhibition of Akt prompted further studies to delineate the involvement of mTORC2 using pharmacological and genetic approaches. Cytoprotective effects of PRAS40 were lost on mTOR kinase inhibition with Torin1 (Figure 4A), which effectively inhibited both mTORC1 and mTORC2 (Figure 4B). Silencing of Akt expression increased cell death in NRCMs in all groups (Figure 4C), and PRAS40 was not protective when Akt was absent, regardless of mTORC1 inhibition (Figure 4D). mTORC2-mediated Akt activation was diminished after silencing of Rictor, abrogating the cytoprotective effect of PRAS40 overexpression. In contrast, silencing of Raptor expression that inhibits mTORC1 did not affect PRAS40- mediated protection (Figure 4E). Efficiency of gene silencing in all studies involving either Raptor or Rictor, loss of mTORC1 function in Raptor-deficient cells, or loss of mTORC2 in Rictor-deficient cells was confirmed by immunoblots (Figure 4F). Protective effects of PRAS40 were not attributable to increased autophagy in our samples (data not shown). Taken together, these data show that mTORC2 signaling is required for PRAS40-mediated protection and that Akt activation is integral to cytoprotection in response to oxidative stress in vitro.
Figure 4.
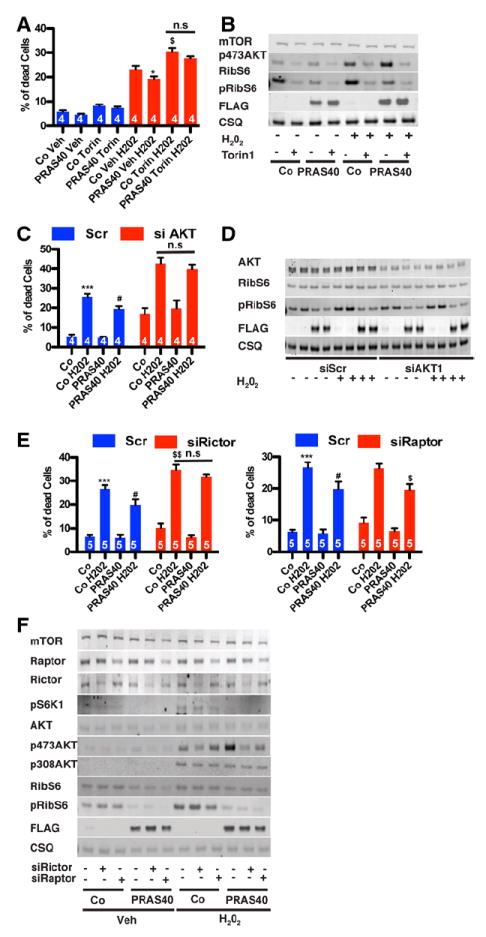
Mechanistic target of rapamycin complex 2 (mTORC2) function is necessary for PRAS40-mediated protection. A, Cell death quantified by flow cytometric detection on H2O2 treatment. Torin1 blunts the protective effects of PRAS40. *P<0.05 vs control; $P<0.05 vs control H2O2. B, mTOR kinase inhibition after Torin1 (50 nmol/L) confirmed by immunoblot. C, siRNA Akt silencing blunts PRAS40-induced protection. Data are depicted as percentage of dead cells. ***P<0.001 vs control; #P<0.05 vs control H2O2. D, Akt1 silencing confirmed by immunoblot. E, siRNA Rictor silencing, but not Raptor silencing, blunts PRA40-induced protection. Data are depicted as percentage of dead cells. ***P<0.001 vs control; #P<0.05 vs control H2O2; $$P<0.01 vs control H2O2. F, Rictor or Raptor silencing and blunting of mTOR downstream target phosphorylation by immunoblot. Numbers of independent experiments per group are indicated in the bar graphs.
Increasing mTORC2 Signaling While Inhibiting mTORC1 Decreases Damage After MI
Selective mTORC1 inhibition was achieved with PRAS40 delivered via recombinant cardiotropic AAV9 with a cardiomyocyte- specific myosin light chain promoter construct (Figure VIA and VIB in the online-only Data Supplement). Strong transgene delivery of AAV9 myosin light chain-2 PRAS40 (80% positive cardiomyocytes) by tail vein injection was confirmed by counting FLAG-tag–positive cardiomyocytes (Figure VIC in the online-only Data Supplement), similar to the results obtained with the green fluorescent protein reporter virus (Figure IIIA in the online-only Data Supplement). PRAS40 expression was increased ≈2-fold over endogenous levels in the heart 3 weeks after tail vein injection of AAV-PRAS40 without apparent changes in cardiac function.
Increased mortality during the first 10 days after MI in the AAV-control mice was significantly reduced by administration of AAV-PRAS40 (Figure 5A). Infarct size was reduced by 30% in AAV-PRAS40–treated mice compared with control mice (Figure 5B), associated with decreased cell death in the infarct border zone (2.8% versus 1.6% TUNEL-positive cardiomyocytes in AAV-control and AAV-PRAS40, respectively; Figure 5C). AAV-PRAS40–treated mice showed significantly higher ejection fraction relative to the AAV-control group within 1 week after infarction. End-diastolic and end-systolic dimensions were reduced in AAV-PRAS40 mice relative to the AAV-control group, indicative of blunted remodeling with AAV-PRAS40 (Figure 5D and Table V in the online-only Data Supplement), which was maintained over the 6-week time course for assessment. Hemodynamic performance was improved and end-diastolic pressure was reduced in AAVPRAS40 relative to AAV-control mice at the termination of the study 6 weeks after MI (Figure 5E). Heart rate was comparable between the groups (Figure XID in the online-only Data Supplement). A hypertrophic increase in the ratio of heart weight to body weight was blocked in PRAS40-overexpressing mice (Figure 5F), consistent with blunted increases in cardiomyocyte cross-sectional area and hypertrophic gene signature in mice receiving AAV-PRAS40 relative to the AAV-control group (Figure 5G and 5H). Decreased pathological remodeling resulted in less fibrosis in PRAS40-overexpressing mice measured by Mason trichrome staining and collagen1 transcription 6 weeks after MI (Figure 5I and 5J). Overall, PRAS40 selectively inhibited mTORC1 in cardiomyocytes, thereby protecting against MI damage and characteristic sequelae in vivo.
Figure 5.
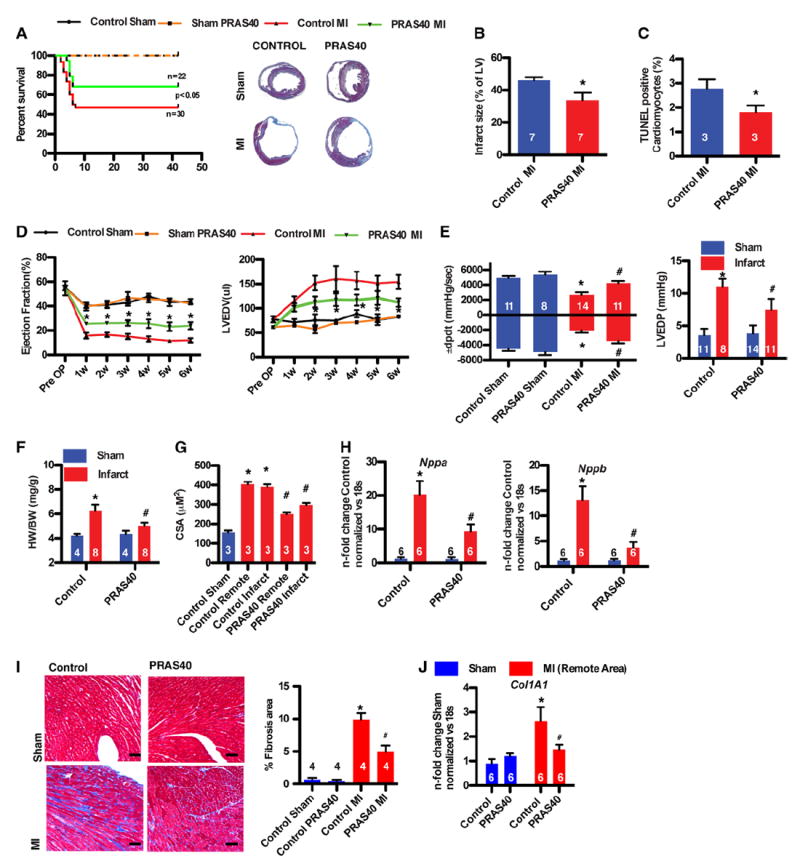
PRAS40 protects against ischemic injury. A, Kaplan–Meier survival analysis of control and PRAS40 animals after myocardial infarction (MI) or sham operation. Masson trichrome staining of control and PRAS40-treated hearts 6 weeks after surgery. n=5 for the sham groups. B, Infarct size measurements 6 weeks after MI. *P<0.05 vs control MI. C, Percentage of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)–labeled myocytes in the left ventricle (LV) in the remote area 2 days after MI. *P<0.05 vs control MI. D, Weekly echocardiographic assessment of control or PRAS40 sham and MI hearts for ejection fraction and LV enddiastolic volume (LVEDV). *P<0.05 vs control MI; n=14 to 15 for the MI group and n=5 to 6 for the sham group. E, In vivo hemodynamic measurements of ±dP/dt and LV end-diastolic pressure (LVEDP) 6 weeks after surgery. F, Ratio of heart weight to body weight (HW/BW) in control and PRAS40 mice 6 weeks after sham and MI surgery. G, Cardiomyocyte area (CSA) in control and PRAS40 mice 6 weeks after sham or MI surgery (*P<0.01 vs control sham; #P<0.05 vs control MI. H, Nppa and Nppb transcription in hearts of mice of the indicated group 6 weeks after sham or MI (*P<0.01 vs control sham; #P<0.05 vs control MI). Error bars indicate mean±SEM. I, Masson trichrome staining from control and PRAS40-treated hearts. Bar graphs depicting quantification of fibrotic area. *P<0.05 vs control sham. #P<0.05 vs MI. Bar, 1 mm. J, Collagen1 transcription in hearts of mice of the indicated group 6 weeks after sham or MI. *P<0.01 vs control sham; #P<0.05 vs control MI. Error bars indicate mean±SEM. Number of mice per group is indicated in the bar.
Mechanism of PRAS40-Mediated Protection
Molecular mechanisms of PRAS40-mediated reduction in ischemic injury were correlated with assessments of mTOR signaling in the acute phase after MI. AKTS473 phosphorylation in AAV-PRAS40–treated hearts displayed a 9.7-fold increase in AKTS473 in the infarct zone, together with increased PRAS40 phosphorylation relative to a 6.3-fold increase in AKTS473 phosphorylation for AAV-control hearts at 2 days after MI (Figure 6A and Figure VIE in the online-only Data Supplement). AktT308 phosphorylation was also increased in AAV-PRAS40–treated hearts compared with AAV-controls. As intended, mTORC1 activation was decreased by 39% by AAV-PRAS40 after MI, along with 5.7-fold higher RibS6 phosphorylation in AAV-control mice compared with shamoperated animals (Figure 6B). Enhanced AKTS473 phosphorylation (Figure 6C) and reduced mTORC1 activation (Figure 6D) resulting from AAV-PRAS40 administration were confirmed by immunohistochemistry. AKTS473 phosphorylation increases persist in AAV-PRAS40–treated hearts up to 6 weeks after MI surgery (Figure 6E and 6F) without evidence of pathological hypertrophy (Figure 5F) or RibS6 phosphorylation in the border zone (Figure VIF in the onlineonly Data Supplement). Preservation of insulin receptor substrate-1 expression in hearts of the AAV-PRAS40 group relative to the AAV-control group at 6 weeks (Figure 6E) suggests that decreased mTORC1 signaling slows degradation of insulin receptor substrate-1, consequently improving insulin signaling and mTORC2 function. Expression of the sarcoplasmic reticulum ATPase was decreased and sodium-calcium exchanger was increased in AAV-control hearts in the remote area at 6 weeks but remained essentially unchanged in AAV-PRAS40–treated hearts compared with sham-operated animals (Figure 6E and Figure VIG in the online-only Data Supplement).
Figure 6.
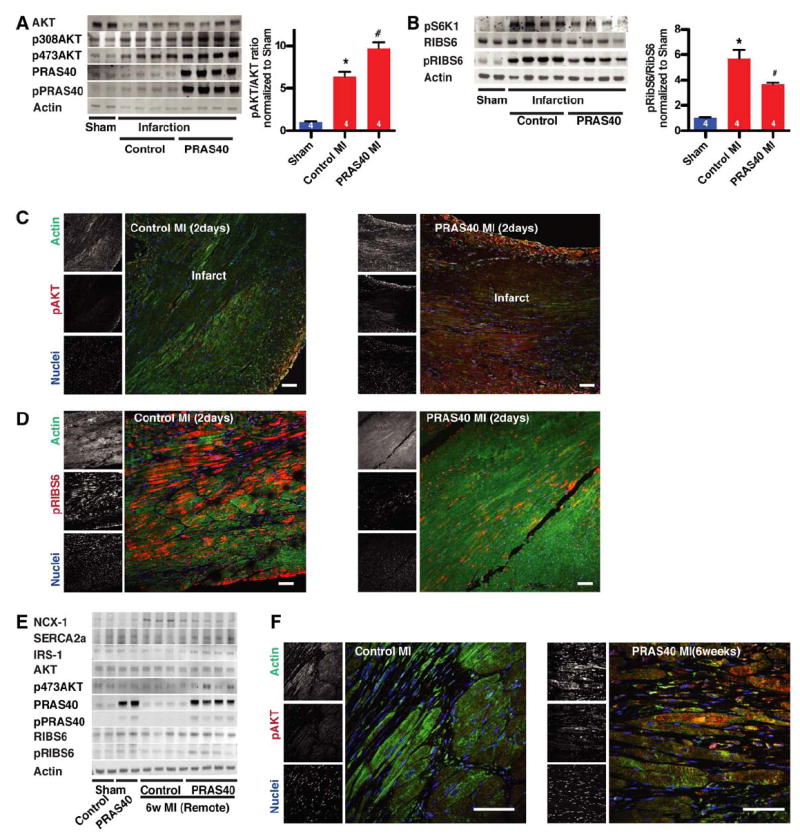
PRAS40 promotes protective mechanistic target of rapamycin complex 2 (mTORC2) signaling in vivo. A, Heart lysates for indicated proteins 2 days after myocardial infarction (MI) assessed by immunoblot. Akt phosphorylation quantification histogram. P<0.01 vs control sham; #P<0.05 vs control MI. B, Heart lysates for indicated proteins 2 days after MI assessed by immunoblot. Ribosomal S6 protein (RibS6) phosphorylation quantification by histogram. *P<0.01 vs control sham; #P<0.05 vs control MI. C, Paraffin-embedded sections from control hearts and PRAS40-treated hearts 2 days after MI stained for pAkt473 (red), actin (green), and nuclei (blue) and assessed by confocal microscopy. D, Sections of control hearts and PRAS40-treated hearts 2 days after MI stained for pRibS6 (red), actin (green), and nuclei (blue) and assessed by confocal microscopy. Bar, 150 μm. E, Heart lysates assessed for indicated protein expression 6 weeks after surgery by immunoblot. F, PRAS40 overexpression increases Akt phosphorylation in myocytes by confocal microscopy of myocardial sections at 6 weeks after surgery. Bar, 50 μm. Number of mice per group is indicated in the bar.
mTORC2 Deletion Diminishes PRAS40-Mediated Protection From Infarction Injury
Participation of the phosphatidylinositol-2-kinase (PI3K)-mTORC2-Akt pathway in PRAS40-mediated cardioprotection in vivo was established with genetically engineered mouse strains lacking PI3K or AKT. Global deletion of PI3K−/− results in impaired Akt activation in response to MI.28 PI3K−/− mice treated with AAV-PRAS40 or AAV-control show comparable cardiac function at 8 weeks of age (Figure 7A), but cardioprotection conferred by AAVPRAS40 was absent in PI3K−/− mice relative to wild-type control mice at 1 week after MI. AAV-PRAS40 also fails to provide protective effects in Akt1-null mice with a comparable decline in cardiac function between AAV-control and AAV-PRAS40 after MI (Figure 7B). Comparable PRAS40 overexpression between all groups of mice was confirmed by immunoblots (Figure VIIA and VIIB in the online-only Data Supplement). Causality of mTORC2 function for PRAS40-mediated cardioprotection was established with Rictor knockdown with AAV (AAV–sh-Rictor). Experimental groups consisted of mice receiving AAV–sh-Rictor alone or in conjunction with AAV-PRAS40 and control groups receiving AAV–sh-control alone or in combination with AAV-PRAS40 injected at 7 weeks of age followed by MI 3 weeks later. AAV–sh-Rictor treatment exacerbated the decline in cardiac function, whereas AAVPRAS40 improved ejection fraction measured 2 weeks after MI challenge (Figure 7C). Moreover, AAV–sh-Rictor decreased cardiac function and increased left ventricular dimension in mice receiving the otherwise cardioprotective AAV-PRAS40 treatment (Figure 7C). mTORC1 function was impaired in mice receiving AAV-PRAS40, as evidenced by blunted hypertrophic remodeling (Figure 7D). Apoptotic cell death increased with AAV–sh-Rictor treatment and decreased with AAV-PRAS40 treatment, but cytoprotection from AAV-PRAS40 was overridden by AAV–sh-Rictor (Figure 7E). In summary, mTORC2 activity is responsible for the protective effects of PRAS40 in vivo (Figure 8).
Figure 7.
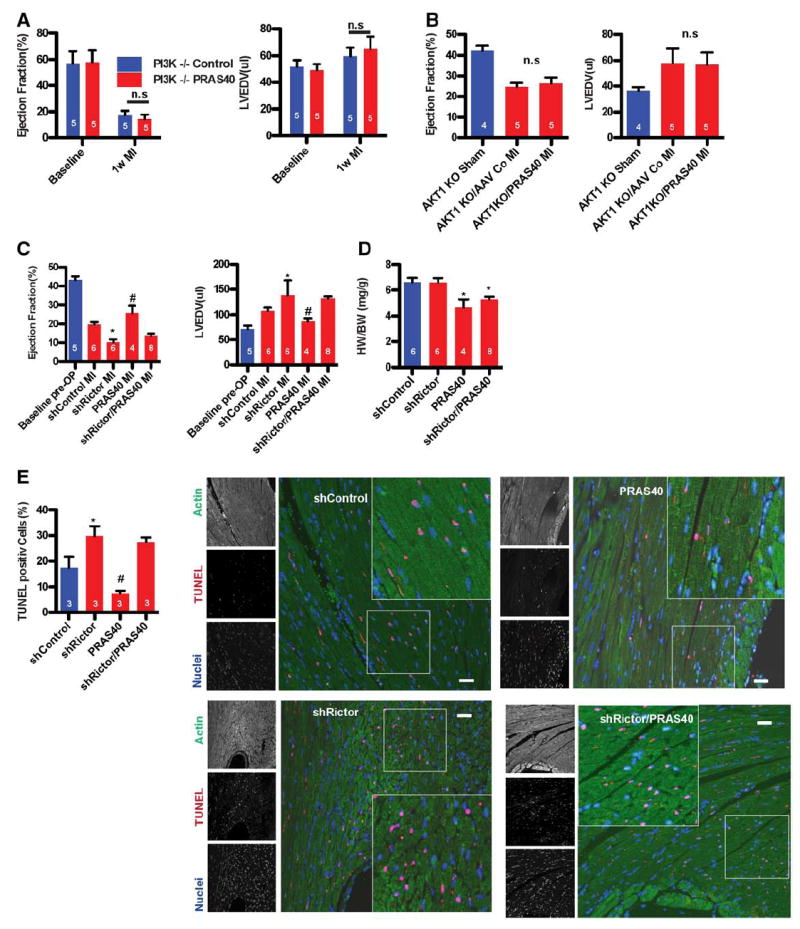
Cardioprotection by PRAS40 is mechanistic target of rapamycin complex 2 (mTORC2) dependent in vivo. A, Ejection fraction (EF) and left ventricular end-diastolic volume (LVEDV) in PI3K−/− mice before and after myocardial infarction (MI) assessed by echocardiography. B, EF and LVEDV in Akt1−/− mice before and after MI assessed by echocardiography. C, EF and LVEDV in mice injected with adeno-associated virus serotype 9 (AAV9)–sh-control, AAV–sh-Rictor, AAV-PRAS40, or AAV–sh-Rictor and AAV-PRAS40 and assessed by echocardiography. *P<0.05 vs sh-control; #P<0.05 vs sh-control. D, Ratio of heart weight to body weight (HW/BW) in mice 2 weeks after MI surgery. *P<0.05 vs sh-control. E, Percentage of terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL)– labeled myocytes in the LV in the remote area 1 day after MI. n=3 per group. *P<0.05 vs control MI; #P<0.05 vs sh-control. Bar, 150 μm. Number of mice per group is indicated in the bar.
Figure 8.
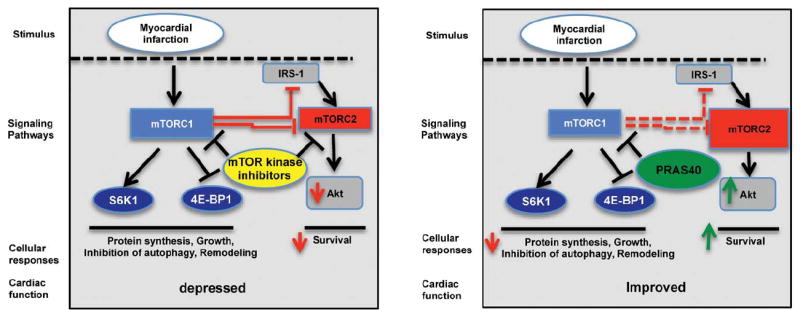
Model for PRAS40-mediated cardioprotection. mechanistic target of rapamycin (mTOR) kinase inhibitors or impairment of mTOR complex 2 (mTORC2) function worsens cardiac function after myocardial infarction. PRAS40 blocks mTORC1 in myocytes and diverts toward mTORC2 function, increasing Akt activation and leading to increased cellular survival after infarction. IRS-1 indicates insulin receptor substrate-1.
Discussion
Loss of viable myocardium after MI is the leading cause of cardiac dysfunction resulting in chronic heart failure. A cardioprotective role for mTORC2 activation in cardiomyocytes after MI is supported by findings presented in this study. Pharmacological inhibition with second-generation mTOR inhibitors and genetic silencing of Rictor result in increased ischemic damage and are associated with increased mortality after MI, consistent with a beneficial role for mTORC2.
Role of mTORC2 Signaling in Ischemic Damage
Surprisingly little is known about the specific role of mTORC2 in cell survival other than impairment of mTORC2 function by Rictor knockdown worsens cell survival in vitro.29 Moreover, evidence for a direct role of mTORC2 in regulating survival after ischemic damage in vivo has, to the best of our knowledge, never been reported. Genetic and pharmacologic evidence shows that mTORC1 inhibition is beneficial after MI.5-7,30-32 However, feedback occurring in mTORC1 and mTORC2 signaling on rapamycin treatment intertwines the relative roles of these 2 distinct complexes and renders the system inadequate to dissect the specific role of mTORC2 in cell survival and disease progression.
Pharmacological inhibition of mTOR kinase function with Torin1 is detrimental to cardiomyocytes both in vitro and in vivo after MI (Figure 1), consistent with genetic evidence that mTOR deletion results in lethal, fully penetrant dilated cardiomyopathy4 and highlighting the importance of intact mTOR signaling in cell survival and function. Moreover, the use of novel mTOR kinase inhibitors to treat cancer may carry inherent cardiotoxic side effects in patients, especially those with comorbidities of heart failure or infarction injury. With next-generation mTOR inhibitors entering clinical trials, it is imperative to further characterize their cardiotoxic potential.33
Genetic deletion of mTORC2 is associated with increased ischemic damage (Figure 3). The findings presented here confirm the important role of mTORC2, Rictor, and AktS473 phosphorylation in cardiomyocytes in controlling cellular survival. Previous studies have shown that Rictor plays a critical role in maintaining tissue metabolism, T-cell development, and neuronal function, but the role of Rictor in response to ischemic tissue injury has not been explored.15,34,35 Silencing of Rictor exacerbates negative consequences after MI (Figure 3), supporting the premise that Rictor plays an important role in the regulation of cellular survival after ischemic damage. Reduced AktS473 phosphorylation after Rictor silencing cascades into decreased phosphorylation of downstream targets for Akt, including FoxO, that presumably could contribute to cell death.36,37 Further confirmation of an essential role for AktS473 phosphorylation was evident from the use of a phosphorylation- deficient inactive Akt mutant (S473→A) unable to block cell death in response to oxidative stress (Figure II in the online-only Data Supplement). Indeed, prior studies show that mTORC2 regulates Akt substrate specificity for downstream phosphorylation targets, including FoxO, glycogen synthase kinase 3, and tuberous sclerosis 2.25 Importantly, long-term use of rapamycin disrupts mTORC2 function in cardiomyocytes and compromises cell survival (Figure III in the online-only Data Supplement). Detrimental side effects of chronic rapamycin administration in the clinical setting demonstrate a pressing need for studies to elucidate cardiotoxicity risk, especially in combination with ischemic injury.
PRAS40 Is Cardioprotective via Increased mTORC2 Signaling
Although short-term inhibition of mTORC1 with rapamycin is beneficial after MI, long-term treatment disrupts mTORC2 function. Therefore, approaches to block mTORC1 in myocytes using selective endogenous molecular inhibitors can provide the beneficial effect of blocking pathological responses without compromising cell survival. PRAS40 was initially identified as a 14-3-3 binding protein38 and was subsequently found to be an mTORC1 inhibitor and substrate. 26,27,39,40 PRAS40 contains 2 proline-enriched stretches at the amino-terminus and an Akt consensus phosphorylation site (RXRXXS/T) located at Thr246. Phosphorylated PRAS40 dissociates from mTORC1, resulting in mTORC activation in response to growth factors, insulin, glucose, and nutrients.39-41 PRAS40 is expressed and regulated by Akt in cardiomyocytes (Figure 4). Furthermore, findings presented here demonstrate that clinically relevant AAV gene therapy with PRAS40 is protective in response to infarction injury. Importantly, PRAS40 overexpression reduced mortality after MI and improved cardiac function (Figure 6). PRAS40 reduced ischemic injury in myocytes in part via increased mTORC2 signaling because genetic interference with the PI3K-Akt-Rictor pathway resulted in loss of protection (Figure 8). Prevention of pathological remodeling with decreased fibrosis in the remote area and prevention of the deregulation of sarcoplasmic reticulum calcium handling proteins likely contribute to the improved contractile function after PRAS40 overexpression after MI. Overall, our findings concur with previous reports of PRAS40 involvement in the regulation of Akt-dependent cell survival and apoptosis.42-44 The importance of PRAS40/mTORC2 is also supported by recent reports that PRAS40 protects neurons from death after spinal cord injury.43,44 In addition to regulation of mTOR kinase activity, unbound PRAS40 may have effects on complexes formed with 14-3-3 and FoxO, resulting in cytoplasmic retention of FoxO, downregulation of proapoptotic genes, and resistance to cell death.45
Recent advancements in the development of AAVs have enabled the first clinical trial with AAV-based cardiac-specific gene transfer.46,47 Thus, cardioprotection by PRAS40 could represent a future therapeutic option for treatment of myocardial injury. Traditional molecular interventions have focused on inhibition of pathological remodeling and potentiation of cell survival or growth, 2 goals that are often at odds with each another. The unique capacity of PRAS40 to block hypertrophic growth while concurrently promoting TORC2-mediated cell survival without affecting cardiac function represents a previously unrecognized constellation of features to prevent cardiac dysfunction. Correctly timed inhibition of mTORC1 in conjunction with mTORC2 potentiation could be a potent combinatorial approach to blunt cellular losses and to ameliorate the progression of degenerative changes accompanying ischemic damage.
Supplementary Material
CLINICAL PERSPECTIVE.
The mechanistic target of rapamycin (mTOR), an atypical Ser/Thr protein kinase and central regulator of cell function, is deregulated after myocardial infarction. mTOR comprises 2 structurally distinct multiprotein complexes, mTOR complexes 1 and 2 (mTORC1 and mTORC2). mTORC1 controls cell growth; mTORC2 controls cellular survival. Inhibiting mTORC1 with rapamycin is already an established clinical application to prevent restenosis after percutaneous coronary stent implantation, and rapamycin is an inherent component of pharmacological cancer therapeutic regimens. Although oral administration of rapamycin to inhibit mTORC1 has been successfully used in rodent models of heart failure, clinical administration raises concerns about potential side effects of systemic mTOR inhibition in other organs with concomitant risk of immunosuppression. This study shows that inhibition of mTORC1 specifically in cardiomyocytes with PRAS40, an endogenous mTORC1 inhibitor, enhances cardioprotection after myocardial infarction. A unique molecular feature of PRAS40 is the inhibition of mTORC1 while simultaneously increasing mTORC2 activation, which increases cellular survival. The findings presented in this study demonstrate that a clinically relevant adeno-associated virus serotype 9 gene therapy with PRAS40 is protective in response to infarction injury and reduced mortality after infarction. Most existing therapies target outside-in signaling in cardiac cells but are limited in effectiveness in preventing cardiac remodeling. Targeting intracellular mTORC1 signaling in cardiomyocytes with PRAS40 using the recent advancements in the development of adeno-associated virus serotype 9 vectors might have better therapeutic potential than existing therapies to blunt remodeling and to potentiate cell survival and is unlike rapamycin without systemic side effects.
Acknowledgments
We thank all members of Dr Sussman’s laboratory for helpful discussions and comments.
Sources of Funding
This study was supported by grants from the National Institutes of Health to Drs Sussman (R37 HL091102-06, R01 HL105759-03, R01 HL067245-12, R01 HL113656-02, R01 HL117163-01, R01 HL113647-01, and 2P01HL085577) and Glembotski ((RO1 HL75573, RO1 HL104535, RO3 EB011698, and PO1 HL085577): the Deutsche Forschungsgemeinschaft (1659/1-1 to Dr Völkers and 3900/1-1 to Dr Konstandin); the Rees-Stealy Research Foundation to S. Din, P. Quijada, and Dr Doroudgar; and the San Diego Chapter of the Achievement Rewards for College Scientists Foundation, the American Heart Association (Predoctoral Fellowship 10PRE3410005), and the Inamori Foundation to Dr Doroudgar.
Footnotes
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.113.003638/-/DC1.
Disclosures
None.
References
- 1.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sussman MA, Völkers M, Fischer K, Bailey B, Cottage CT, Din S, Gude N, Avitabile D, Alvarez R, Sundararaman B, Quijada P, Mason M, Konstandin MH, Malhowski A, Cheng Z, Khan M, McGregor M. Myocardial AKT: the omnipresent nexus. Physiol Rev. 2011;91:1023–1070. doi: 10.1152/physrev.00024.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang D, Contu R, Latronico MV, Zhang J, Zhang JL, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 6.Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, Katus HA, Hardt SE. Beneficial effects of mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol. 2009;54:2435–2446. doi: 10.1016/j.jacc.2009.08.031. [DOI] [PubMed] [Google Scholar]
- 7.Marin TM, Keith K, Davies B, Conner DA, Guha P, Kalaitzidis D, Wu X, Lauriol J, Wang B, Bauer M, Bronson R, Franchini KG, Neel BG, Kontaridis MI. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J Clin Invest. 2011;121:1026–1043. doi: 10.1172/JCI44972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M, Hara K, Rosenzweig A, Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol, Cell Physiol. 2010;299:C1256–C1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aoyagi T, Kusakari Y, Xiao CY, Inouye BT, Takahashi M, Scherrer-Crosbie M, Rosenzweig A, Hara K, Matsui T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2012;303:H75–H85. doi: 10.1152/ajpheart.00241.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 11.Harston RK, McKillop JC, Moschella PC, Van Laer A, Quinones LS, Baicu CF, Balasubramanian S, Zile MR, Kuppuswamy D. Rapamycin treatment augments both protein ubiquitination and Akt activation in pressure-overloaded rat myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1696–H1706. doi: 10.1152/ajpheart.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzman JA, O’Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148:3477–3484. doi: 10.1210/en.2007-0099. [DOI] [PubMed] [Google Scholar]
- 13.Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, Abel ED, Symons JD, Jalili T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327. doi: 10.1161/HYPERTENSIONAHA.109.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huo Y, Iadevaia V, Proud CG. Differing effects of rapamycin and mTOR kinase inhibitors on protein synthesis. Biochem Soc Trans. 2011;39:446–450. doi: 10.1042/BST0390446. [DOI] [PubMed] [Google Scholar]
- 15.Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305–2316. doi: 10.4161/cc.10.14.16586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goncharova EA, Goncharov DA, Li H, Pimtong W, Lu S, Khavin I, Krymskaya VP. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol Cell Biol. 2011;31:2484–2498. doi: 10.1128/MCB.01061-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-dos-Santos AJ, Stanford WL, Bolon B, Wakeham A, Itie A, Bouchard D, Kozieradzki I, Joza N, Mak TW, Ohashi PS, Suzuki A, Penninger JM. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 2000;287:1040–1046. doi: 10.1126/science.287.5455.1040. [DOI] [PubMed] [Google Scholar]
- 18.Muraski JA, Rota M, Misao Y, Fransioli J, Cottage C, Gude N, Esposito G, Delucchi F, Arcarese M, Alvarez R, Siddiqi S, Emmanuel GN, Wu W, Fischer K, Martindale JJ, Glembotski CC, Leri A, Kajstura J, Magnuson N, Berns A, Beretta RM, Houser SR, Schaefer EM, Anversa P, Sussman MA. Pim-1 regulates cardiomyocyte survival downstream of Akt. Nat Med. 2007;13:1467–1475. doi: 10.1038/nm1671. [DOI] [PubMed] [Google Scholar]
- 19.Quijada P, Toko H, Fischer KM, Bailey B, Reilly P, Hunt KD, Gude NA, Avitabile D, Sussman MA. Preservation of myocardial structure is enhanced by pim-1 engineering of bone marrow cells. Circ Res. 2012;111:77–86. doi: 10.1161/CIRCRESAHA.112.265207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng Z, Völkers M, Din S, Avitabile D, Khan M, Gude N, Mohsin S, Bo T, Truffa S, Alvarez R, Mason M, Fischer KM, Konstandin MH, Zhang XK, Heller Brown J, Sussman MA. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J. 2011;32:2179–2188. doi: 10.1093/eurheartj/ehq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Völkers M, Weidenhammer C, Herzog N, Qiu G, Spaich K, von Wegner F, Peppel K, Müller OJ, Schinkel S, Rabinowitz JE, Hippe HJ, Brinks H, Katus HA, Koch WJ, Eckhart AD, Friedrich O, Most P. The inotropic peptide ßARKct improves ßAR responsiveness in normal and failing cardiomyocytes through G(βγ)-mediated L-type calcium current disinhibition. Circ Res. 2011;108:27–39. doi: 10.1161/CIRCRESAHA.110.225201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Avitabile D, Bailey B, Cottage CT, Sundararaman B, Joyo A, McGregor M, Gude N, Truffa S, Zarrabi A, Konstandin M, Khan M, Mohsin S, Völkers M, Toko H, Mason M, Cheng Z, Din S, Alvarez R, Jr, Fischer K, Sussman MA. Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin. Proc Natl Acad Sci U S A. 2011;108:6145–6150. doi: 10.1073/pnas.1017935108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Chang JW, Wang J, Kang SA, Thoreen CC, Markhard A, Hur W, Zhang J, Sim T, Sabatini DM, Gray NS. Discovery of 1-(4-(4-Propionylpiperazin-1-yl)-3-(trifluoromethyl)phenyl)-9-(quinolin- 3-yl)benzo[h][1,6]naphthyridin-2(1 H)-one as a highly potent, selective mammalian target of rapamycin (mTOR) inhibitor for the treatment of cancer. J Med Chem. 2010;53:7146–7155. doi: 10.1021/jm101144f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Rüegg MA, Pedrazzini T, Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 25.Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 26.Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 28.Siragusa M, Katare R, Meloni M, Damilano F, Hirsch E, Emanueli C, Madeddu P. Involvement of phosphoinositide 3-kinase gamma in angiogenesis and healing of experimental myocardial infarction in mice. Circ Res. 2010;106:757–768. doi: 10.1161/CIRCRESAHA.109.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hung CM, Garcia-Haro L, Sparks CA, Guertin DA. mTOR-dependent cell survival mechanisms. Cold Spring Harb Perspect Biol. 2012:4a008771. doi: 10.1101/cshperspect.a008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koitabashi N, Kass DA. Reverse remodeling in heart failure: mechanisms and therapeutic opportunities. Nat Rev Cardiol. 2012;9:147–157. doi: 10.1038/nrcardio.2011.172. [DOI] [PubMed] [Google Scholar]
- 31.McKinsey TA, Kass DA. Small-molecule therapies for cardiac hypertrophy: moving beneath the cell surface. Nat Rev Drug Discov. 2007;6:617–635. doi: 10.1038/nrd2193. [DOI] [PubMed] [Google Scholar]
- 32.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagiwara A, Cornu M, Cybulski N, Polak P, Betz C, Trapani F, Terracciano L, Heim MH, Rüegg MA, Hall MN. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 35.Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121:4477–4490. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta. 2011;1813:1978–1986. doi: 10.1016/j.bbamcr.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 37.Ronnebaum SM, Patterson C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol. 2010;72:81–94. doi: 10.1146/annurev-physiol-021909-135931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovacina KS, Park GY, Bae SS, Guzzetta AW, Schaefer E, Birnbaum MJ, Roth RA. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem. 2003;278:10189–10194. doi: 10.1074/jbc.M210837200. [DOI] [PubMed] [Google Scholar]
- 39.Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007;282:20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiza C, Nascimento EB, Ouwens DM. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab. 2012;302:E1453–E1460. doi: 10.1152/ajpendo.00660.2011. [DOI] [PubMed] [Google Scholar]
- 41.Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- 42.Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Modulation of prolinerich Akt substrate survival signaling pathways by oxidative stress in mouse brains after transient focal cerebral ischemia. Stroke. 2006;37:513–517. doi: 10.1161/01.STR.0000198826.56611.a2. [DOI] [PubMed] [Google Scholar]
- 43.Saito A, Narasimhan P, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci. 2004;24:1584–1593. doi: 10.1523/JNEUROSCI.5209-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu F, Narasimhan P, Saito A, Liu J, Chan PH. Increased expression of a proline-rich Akt substrate (PRAS40) in human copper/zinc-superoxide dismutase transgenic rats protects motor neurons from death after spinal cord injury. J Cereb Blood Flow Metab. 2008;28:44–52. doi: 10.1038/sj.jcbfm.9600501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim W, Youn H, Seong KM, Yang HJ, Yun YJ, Kwon T, Kim YH, Lee JY, Jin YW, Youn B. PIM1-activated PRAS40 regulates radioresistance in non-small cell lung cancer cells through interplay with FOXO3a, 14-3-3 and protein phosphatases. Radiat Res. 2011;176:539–552. doi: 10.1667/rr2609.1. [DOI] [PubMed] [Google Scholar]
- 46.Pacak CA, Byrne BJ. AAV vectors for cardiac gene transfer: experimental tools and clinical opportunities. Mol Ther. 2011;19:1582–1590. doi: 10.1038/mt.2011.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium Up-Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-inhuman phase 1/2 clinical trial. J Card Fail. 2009;15:171–181. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.