

Abstract
Cherubism is a genetic disorder of the craniofacial skeleton caused by gain-of-function mutations in the signaling adaptor protein, SH3-domain binding protein 2 (SH3BP2). In a knock-in mouse model for cherubism, we previously demonstrated that homozygous mutant mice develop T/B cell-independent systemic macrophage inflammation leading to bone erosion and joint destruction. Homozygous mice develop multiostotic bone lesions while cherubism lesions in humans are limited to jawbones. We identified a critical role of TNF-α in the development of autoinflammation by creating homozygous TNF-α-deficient cherubism mutants, where systemic inflammation and bone destruction were rescued. In the current study, we examined whether postnatal administration of an anti-TNF-α antagonist can prevent or ameliorate the disease progression in cherubism mice. Neonatal homozygous mutants, where active inflammation has not yet developed, were treated with a high dose of etanercept (25 mg/kg, twice/week) for 7 weeks. Etanercept-treated neonatal mice showed strong rescue of facial swelling and bone loss in jaws and calvariae. Destruction of joints was fully rescued in the high dose group. Moreover, the high dose treatment group showed a significant decrease in lung and liver inflammatory lesions. However, inflammation and bone loss, which were successfully treated by etanercept administration recurred after etanercept discontinuation. No significant effect was observed in low dose- (0.5 mg/kg, twice/week) and vehicle-treated groups. In contrast, when 10-week-old cherubism mice with fully active inflammation were treated with etanercept for 7 weeks, even the high dose administration did not decrease bone loss, lung or liver inflammation. Taken together, the results suggest that anti-TNF-α therapy may be effective in young cherubism patients, if treated before the inflammatory phase or bone resorption occurs. Therefore, early genetic diagnosis and early treatment with anti-TNF-α antagonists may be able to prevent or ameliorate cherubism, especially in patients with a mutation in SH3BP2.
Keywords: cherubism, etanercept, TNF-α, inflammation, bone loss
Introduction
Cherubism (OMIM#118400) is a craniofacial disorder in children characterized by bilateral swelling of the lower face due to excessive development of expansile fibrous lesions, which also results in the destruction of maxillary and mandibular bones. Cherubism lesions also lead to tooth defects and a characteristic disfiguring facial appearance. Lesions contain a large number of tartrate-resistant acid phosphatase (TRAP)-positive multinucleated giant cells and spindle-shaped fibroblastoid cells. Typically, expansion of the lesions ceases and they regress after puberty, but the multilocular radiolucent cavities fill with bone later in life.(1,2) Previously, linkage analysis and subsequent sequencing of candidate genes have successfully identified heterozygous mutations responsible for autosomal dominant cherubism in the SH3 domain binding protein 2 (SH3BP2).(3,4) SH3BP2 is a signaling adaptor protein originally discovered as one of the proteins that bind to the SH3 domain of the protein tyrosine kinase ABL1.(5) Subsequently, SH3BP2 turned out to interact with a variety of proteins including SYK,(6) 14-3-3,(7) VAV,(8) LYN,(9) CIN85,(10) HIP-55,(10) PLCγ1 and PLCγ2,(6,11) SHP-1,(12,13) BLNK,(14) and SRC(15) in various hematopoietic cell types including T cells, B cells, mast cells, neutrophils and macrophages as well as in osteoblasts and osteoclasts, suggesting that SH3BP2 is involved in signaling pathways that control both the immune and skeletal system.
We generated a knock-in (KI) mouse model of cherubism and discovered that while systemic bone loss due to increased RANKL-induced osteoclast differentiation was seen in heterozygous KI mutants (Sh3bp2KI/+), homozygous mutants (Sh3bp2KI/KI) spontaneously developed systemic inflammatory infiltrates rich in macrophages in skin, lung, liver, stomach, lymph nodes, joints as well as in jawbones with serum TNF-α elevation (200 to 650 pg/ml).(16) Infiltrates surrounding jawbones caused bone erosion similar to cherubism patients.(16) This inflammation is regarded as autoinflammatory since T and B cell are not required for the pathology.(17,18) The key role of TNF-α in the development of inflammation in cherubism mice has been demonstrated in homozygous cherubism mutants deficient in TNF-α, which showed significant rescue of systemic inflammation as well as of inflammatory bone loss and erosion, suggesting that cherubism patients may benefit from anti-TNF-α treatment.(16) Recently, it was discovered that SH3BP2 is a substrate of TANKYRASE1 and TANKYRASE2 (TNKS and TNKS2, respectively), members of the poly (ADP-ribose) polymerase (PARP) superfamily. Reduced ADP-ribosylation of mutant SH3BP2 protein results in decreased proteasomal degradation and is the key mechanism that leads to the increased TNF-α production in macrophages, hence resulting in inflammation.(19,20)
Standard management of cherubism symptoms are generally limited to supportive care including observation of the disease course with the expectation that lesions regress after puberty.(2) Surgical intervention is considered only when functional or aesthetic concerns arise, including nasal obstruction, proptosis or severe facial deformity.(2) Currently, there is no established pharmacological treatment for cherubism.
Homozygous cherubism mice lacking TNF-α exhibit dramatically reduced inflammation as well as increased bone mass, suggesting that pharmacological neutralization of TNF-α can be a potential therapeutic approach to treat human cherubism patients.(16) In Sh3bp2KI/KI/Tnf-α-/- double mutants, TNF-α protein is completely deficient throughout all embryonic stages due to global deletion of the Tnf-α gene.(21) However, human cherubism patients are usually diagnosed at 2-5 years of age after manifesting facial or submandibular lymph node swelling. Therefore, in this study, we examined whether postnatal pharmacological treatment of our cherubism mice with an anti-TNF-α drug is effective to reduce inflammation.
Etanercept (Enbrel®) is a dimeric fusion protein consisting of human type II TNF-α receptor linked to the Fc portion of human IgG1. Etanercept is one of the widely used anti-TNF-α drugs which is approved for the treatment of a variety of inflammatory diseases including rheumatoid arthritis, ankylosing spondylitis, psoriasis, and psoriatic arthropathies.(22,23) While other anti-TNF-α inhibitors such as infliximab, adalimumab, golimumab, certolizumab do not effectively inhibit mouse TNF-α, there are many reports that etanercept blocks mouse TNF-α and reduces TNF-α mediated inflammatory reactions in various disease models in rodents.(24-28)
First, we demonstrate that neonatal homozygous mice treated with etanercept develop significantly reduced systemic inflammation and bone loss. Second, we show that etanercept treatment of adult homozygous mutants with fully active inflammation does not result in a reduction of inflammation and bone loss. These outcomes suggest that anti-TNF-α drugs might be suitable as a therapeutic agent for cherubism when administered at the early stage of the disease before the onset of inflammation and lesion formation and might be able to prevent the future development of lesions in jawbones. Our study also indicates the importance and usefulness of early genetic diagnosis of SH3BP2 mutations in children born to families affected with cherubism, allowing the patients to undergo early anti-TNF-α treatments.
Materials and Methods
Mice
A cherubism mouse model was created by introducing the most common mutation in cherubism patients (P418R) into the mouse Sh3bp2 gene (P416R in mouse) by homologous recombination.(16) Homozygous cherubism mutant mice (Sh3bp2KI/KI) and wild-type littermates (Sh3bp2+/+) were created by crossing heterozygous mutants that have been backcrossed into a C57BL/6 genetic background for at least 12 generations.
Etanercept administration in cherubism mice
Etanercept (Enbrel®, Pfizer,Inc., New York, NY, USA) was chosen for our experiments, because among five anti-TNF-α inhibitors (etanercept, infliximab, adalimumab, golimumab, certolizumab), which have been approved for human use, only etanercept is reported to effectively block mouse TNF-α (European Medicines Agency (http://www.ema.europa.eu/ema/), Scientific Discussion in initial marketing authorization documents). The risk of tuberculosis reactivation in patients receiving etanercept is lower than in those receiving TNF antibodies, suggesting that soluble TNFR (Etanercept) may be safer and etanercept might have advantages in human clinical use compared to other TNF inhibitors. (29-31) The effective dose of etanercept in mice is dependent on the model of inflammatory disease, ranging from 0.06 mg/kg/week for 60 weeks in degenerative/inflammatory changes in skeletal muscle in aged SJL/J mice (27) to 100 mg/kg/week for 8 weeks in GGTase-I deficient arthritis model. (32) Generally, the effective dose in mice is higher compared to the standard dose for the treatment of adult rheumatoid arthritis, ankylosing spondylitis, psoriasis, and psoriatic arthropathies in humans (50 mg/week: 0.8mg/kg/week at a body weight of 62.5 kg).(22) Therefore, we decided to use 1 mg/kg/week (0.5 mg/kg twice per week; low dose in this study) as an equivalent dose for human use and 50 mg/kg/week (25 mg/kg twice per week; high dose in this study) as an intermediate dose for mice.
Etanercept diluted with phosphate buffered saline (PBS) was subcutaneously injected into homozygous mutants and wild-type controls twice a week from either 1 or 10 weeks of age for 7 weeks. For the administration of the 1-week-old mice, Sh3bp2KI/KI mice were divided into 3 groups: low (0.5 mg/kg) and high (25 mg/kg) dose of etanercept and a PBS vehicle control group. For 10-week-old mice, Sh3bp2KI/KI mice were divided into 2 groups: high dose of etanercept (25 mg/kg) and PBS vehicle control group. At age of 8 or 17 weeks, respectively, mice were sacrificed and analyzed. Etanercept administration and all other animal studies were approved by the Institutional Animal Care and Use Committee at the University of Missouri-Kansas City.
Tissue preparation for histological analysis
Postmortem tissues were harvested and fixed with Bouin's fixative solution or PBS solution containing 4% paraformaldehyde for 2-3 days, then embedded in paraffin. Sections (6μm) were stained with hematoxylin and eosin (H&E).
RNA and quantitative PCR analysis
Total RNA from liver tissue was extracted using TRIzol and converted to cDNA with Superscript III First Strand Synthesis System (Life technologies, Grand Island, NY, USA). Quantitative real-time PCR analysis was performed using a StepOnePlus system (Applied Biosystems, Carlsbad, CA, USA) to compare TNF-α mRNA expression levels in whole liver tissue by TaqMan® probes (Mm00443260_g1) and normalized against β-actin (Mm00607939_s1). To compare IL-1α and IL-1β mRNA expression levels in bone marrow-derived M-CSF dependent macrophages, TaqMan probes, Mm00439620_m1 and Mm00434228_m1 were used, respectively. TNF-α, IL-1α, and IL-1β mRNA expression levels relative to β-actin were calculated with the Δ-Δ Ct method.
MicroCT analysis
Paraformaldehyde-fixed mandibulae, calvariae, and elbow joints were subjected to microCT (vivaCT 40, Scanco Medical, PA, USA) analysis. Resulting images were used to quantitate inflammatory bone loss in mandibular and calvarial bone as well as to evaluate the rescue of elbow joint destruction. Two-dimensional (2D) images were taken with a threshold of 300 for mineralized tissue and further processed for the three-dimensional (3D) image reconstruction with a spatial resolution of 10μm. Distance between cemento-enamel junction (CEJ) and alveolar bone crest (ABC) at the distal lingual surface of mandibular first molar (M1) was measured in reconstructed 3D images using modified procedures published by Park and colleagues.(33) Calvarial microCT images (6 mm × 6 mm) were used to calculate the proportion of erosion area (%) of calvarial bone. The intersections of the coronal and sagittal sutures were set as reference positions of the square center. Total area of bone erosions including suture areas in 6 mm × 6 mm calvarial bone images were measured as pixels with ImageJ (NIH) and divided by the total number of pixels.
Assessment of facial swelling and analysis of elbow joint destruction
Facial swelling was assessed by whether the cornea right above pupil is located underneath the eyelid margin by two blinded independent observers. Elbow joints were given semi-quantitative scores (0 to 3) for joint destruction by two independent observers using 3D microCT images of right elbow joint, according to the following criteria: 0 = normal; 1 = mild coronoid fossa erosion with or without resorption pits at lateral epicondyle; 2 = moderate coronoid fossa erosion with resorption pits at lateral epicondyle; 3 = severe coronoid fossa erosion with resorption pits at lateral epicondyle.
Histomorphometry
Total inflammatory area in liver and lung stained with H&E was measured as pixels with ImageJ (NIH), which was divided by the pixels of the entire tissue to calculate the proportion of inflammatory area (%). With regard to liver inflammation, areas of vasculature were excluded from entire tissue areas. Lung and liver images were taken with 1× or 4× objectives, respectively (Nikon E800 microscope, NIKON Instruments Inc., Melville, NY, USA). Results from four to five images from different areas of each tissue section were averaged.
Bone marrow-derived M-CSF-dependent macrophages (BMMs) culture
Bone marrow cells were harvested from tibia and femur of 8- to 10-week-old of Sh3bp2KI/KI and Sh3bp2+/+ mice. After red blood cell lysis, non-adherent cells were collected and cultured for 3 days with α–MEM supplemented with 10% FBS, penicillin/streptomycin, and 30 ng/ml of M-CSF (Peprotech, Rocky Hill, NJ, USA). Adherent macrophages were harvested and 5×104 cells/well were plated in 96-well plates without M-CSF. After 6 to 8 hours, macrophages were stimulated with TNF-α (10 ng/ml), IL-1α (20 ng/ml), or IL-1β (20 ng/ml) (Peprotech).
ELISA assay
After 24 hours of BMMs stimulation with IL-1α (20 ng/ml) or IL-1β (20 ng/ml) culture supernatants were collected and TNF-α levels were measured with mouse TNF-α DuoSet (R&D Systems Inc., Minneapolis, MN, USA) according to manufacturer's protocol.
Osteoclast culture
Non-adherent bone marrow cells were plated in 48-well plates at the density of 2.1×105 cells/well with α–MEM supplemented with 10% FBS, penicillin/streptomycin, and 25 ng/ml of M-CSF. After 48 hours, BMMs were stimulated with RANKL for 3 days with or without etanercept. Osteoclasts were visualized by tartrate-resistant acid phosphatase (TRAP) staining kit (Sigma, St. Louis, MO, USA) and TRAP-positive cells with more than 3 nuclei were counted as osteoclasts.
Statistics
Power analysis (G*Power 3.1.3)(34) with parameters obtained from preliminary studies suggested that sample sizes of six would sufficiently detect a statistical difference of therapeutic effects of high dose (25 mg/kg) etanercept compared to the PBS-treated group. Statistical analyses were performed with SPSS statistics software (IBM, Armonk, New York, USA). Mean value comparison between two groups was performed with Student's t-test (two-tailed distribution, equal variance). One-way ANOVA was used to compare means among three or more groups. If the ANOVA showed a significant difference, the Tukey-Kramer test was used as a post-hoc test. p < 0.05 was considered as statistically significant. The numbers after ± represent SD except for joint destruction scores in Fig. 3E and 6E in which the numbers after ± represent SEM.
Fig. 3. Rescued craniofacial bone erosion and elbow joint destruction in etanercept-treated Sh3bp2KI/KI mice.
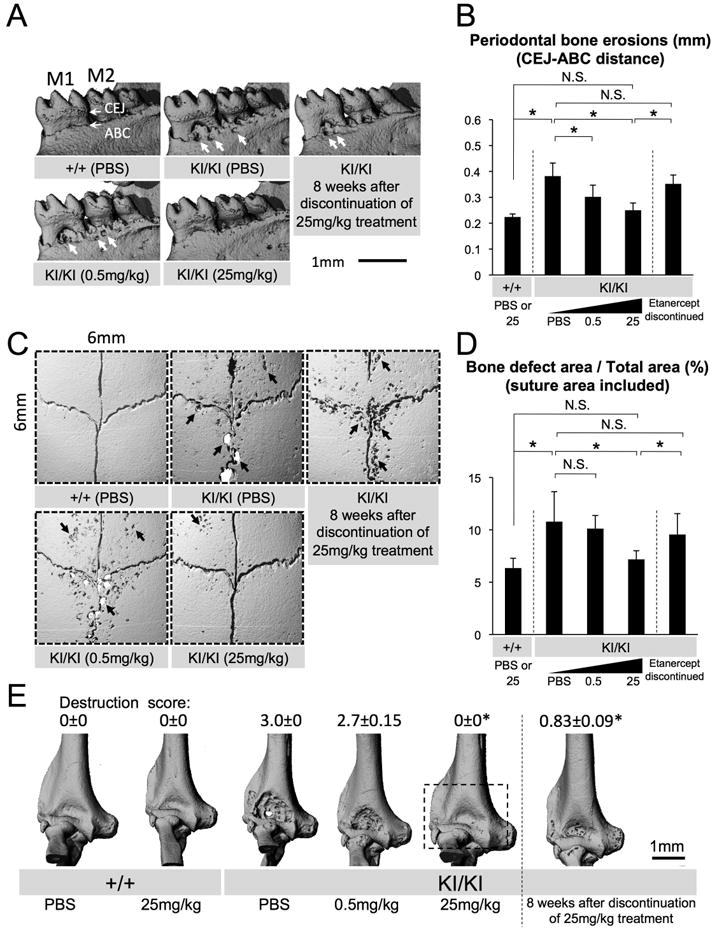
(A) Reconstructed microCT images of the mandibular bone from 8-week-old PBS- or etanercept-treated Sh3bp2+/+ and Sh3bp2KI/KI mice. Thick arrows indicate erosion pits due to inflammation (M1: first molar, M2: second molar, CEJ: cemento-enamel junction, ABC: alveolar bone crest). (B) Quantitative measurement of the distance between CEJ and ABC at the lingual distal end of the mandibular first molar. (C) Reconstructed microCT images of the calvarial bone from PBS- or etanercept-treated 8-week-old Sh3bp2+/+ and Sh3bp2KI/KI mice. Arrows indicate inflammatory erosion pits on the surface. (D) Quantitative analysis of calvarial bone erosion. Proportion (%) of bone erosion area including suture to total calvarial bone area (6 mm × 6 mm) was calculated. (E) Reconstructed microCT images of elbow joint from PBS- or etanercept-administered Sh3bp2+/+ and Sh3bp2KI/KI mice. Dotted box on Sh3bp2KI/KI image treated with 25 mg/kg etanercept indicates the prevented elbow joint destruction by etanercept treatment. Numbers represent average joint destruction score with ± SEM. Joint destruction score in Sh3bp2KI/KI mice treated with 25 mg/kg etanercept or in mice which underwent 8-week etanercept discontinuation (asterisks) is lower than in those with 0.5 mg/kg etanercept and PBS treatment (p < 0.05). Error bars represent SD. Asterisks represent significant difference (p < 0.05). N.S.: not significant. n=6-7 except for n=5-9 in Fig. 3E.
Fig. 6. No significant improvement in bone loss when etanercept is administered to adult Sh3bp2KI/KI mice with active inflammation.
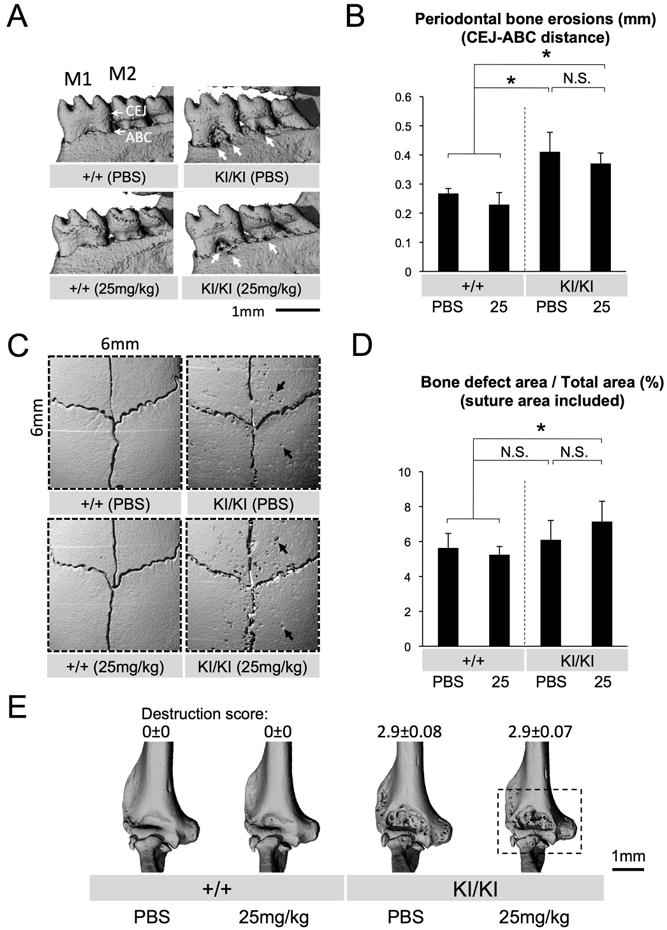
(A) Reconstructed microCT images of the mandibular bone from 17-week-old Sh3bp2+/+ and Sh3bp2KI/KI mice treated with PBS or etanercept for 7 weeks. Thick arrows indicate erosion pits due to inflammation (M1: first molar, M2: second molar, CEJ: cemento-enamel junction, ABC: alveolar bone crest). (B) Quantitative measurement of the CEJ-ABC distance at the lingual distal end of the mandibular first molar. (C) Reconstructed microCT images of the calvarial bone from PBS- or etanercept-treated 17-week-old Sh3bp2+/+ and Sh3bp2KI/KI mice. Arrows indicate inflammatory erosion pits. Note that overall numbers and areas of erosion pits in PBS-treated Sh3bp2KI/KI mutant are decreased compared to PBS-treated 8-week-old Sh3bp2KI/KI mice (Fig. 3C). (D) Quantitative measurement of calvarial bone erosion. Proportion (%) of bone erosion area including suture to total calvarial bone area (6 mm × 6 mm) was calculated. (E) MicroCT images of elbow joint from PBS- or etanercept-administered 17-week-old Sh3bp2+/+ and Sh3bp2KI/KI mice. Etanercept-treatment of Sh3bp2KI/KI mice did not improve destruction of the elbow joint. Dotted box on Sh3bp2KI/KI image treated with 25 mg/kg etanercept indicates the unchanged elbow joint destruction after etanercept treatment. Numbers represent average joint destruction score with ± SEM. Joint destruction score in Sh3bp2KI/KI mice treated with 25 mg/kg etanercept is comparable to that in PBS-treated Sh3bp2KI/KI mice. Error bars represent SD. Asterisks represent significant difference (p < 0.05). N.S.: not significant. n=5-7 except for Fig. 6E (n=6-7).
Results
Postnatal development of macrophage inflammation in homozygous cherubism knock-in mice
Previously we have shown that homozygous Sh3bp2KI/KI mutants develop severe systemic inflammatory infiltrates rich in macrophages by 10 weeks of age.(16) Before starting etanercept administration, we first examined when macrophage infiltrates start to develop in homozygous mutants. We performed H&E staining and histologically inspected the inflammation in the soft tissue of oral cavity, lung, and liver from various postnatal ages of homozygous mutants compared with wild-type control mice. In the oral soft tissues, no obvious inflammatory lesions were observed in 1-week-old mice, but at 2 weeks of age, mild inflammatory cell accumulation rich in macrophages became evident and the accumulation continued to increase thereafter (Fig. 1A). In the lung, initial accumulation of macrophages was seen at 1 week of age in Sh3bp2KI/KI mutants, followed by the development of typical inflammatory nodules containing a large number of macrophages and lymphocytes at 2 weeks of age (Fig. 1B). In contrast, inflammatory lesions in liver started to appear at 2 weeks of age (Fig. 1C). We did not see any histological signs of inflammation at embryonic day 18.5 (data not shown). These findings indicate that inflammation in homozygous Sh3bp2KI/KI mice develops postnatally approximately at 1 week of age with tissue-dependent age of onset.
Fig. 1. Postnatal development of inflammation in homozygous Sh3bp2KI/KI knock-in mice.
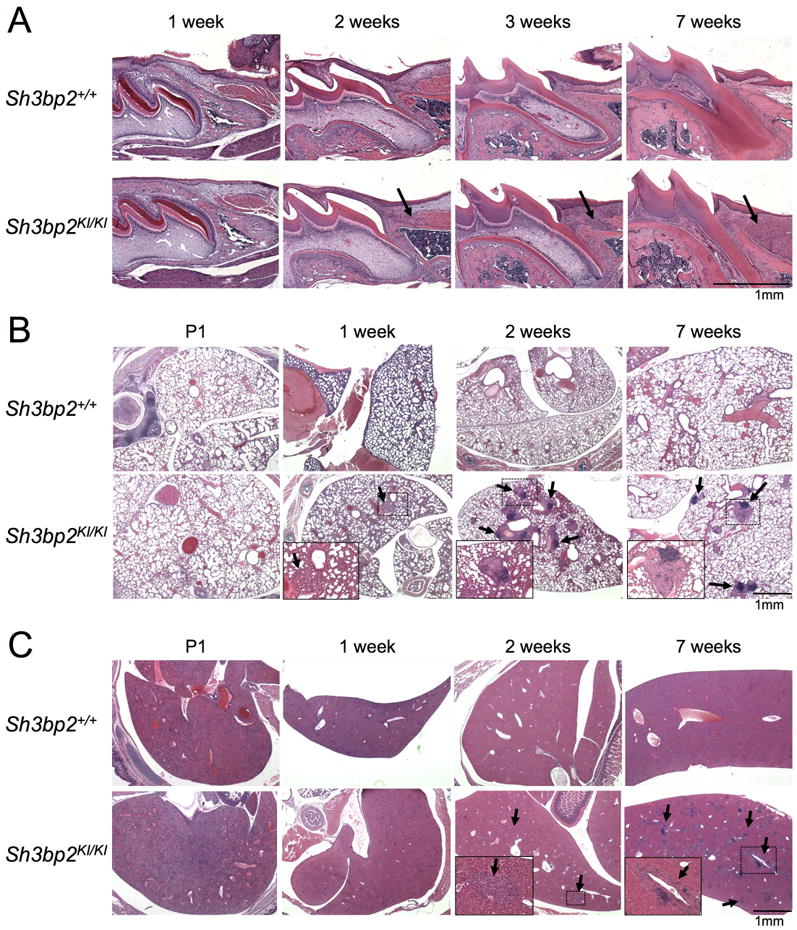
H&E staining of (A) first molar in maxilla and surrounding oral soft tissues, (B) lung, and (C) liver tissue section from wild-type (Sh3bp2+/+) and homozygous knock-in (KI) cherubism mutant mice (Sh3bp2KI/KI). Tissues were harvested at various postnatal ages from P1 (postnatal 1 day) to 7 weeks of age. Arrows indicate inflammatory lesions. Insets are magnifications of the dotted areas. Inflammatory nodular lesions in lung (B) develop at 1 week of age, and inflammatory infiltrates in oral (A) and liver (C) tissues are observed at 2 weeks of age.
Etanercept administration to neonatal Sh3bp2KI/KI mutants prevents facial swelling and rescues loss of body weight
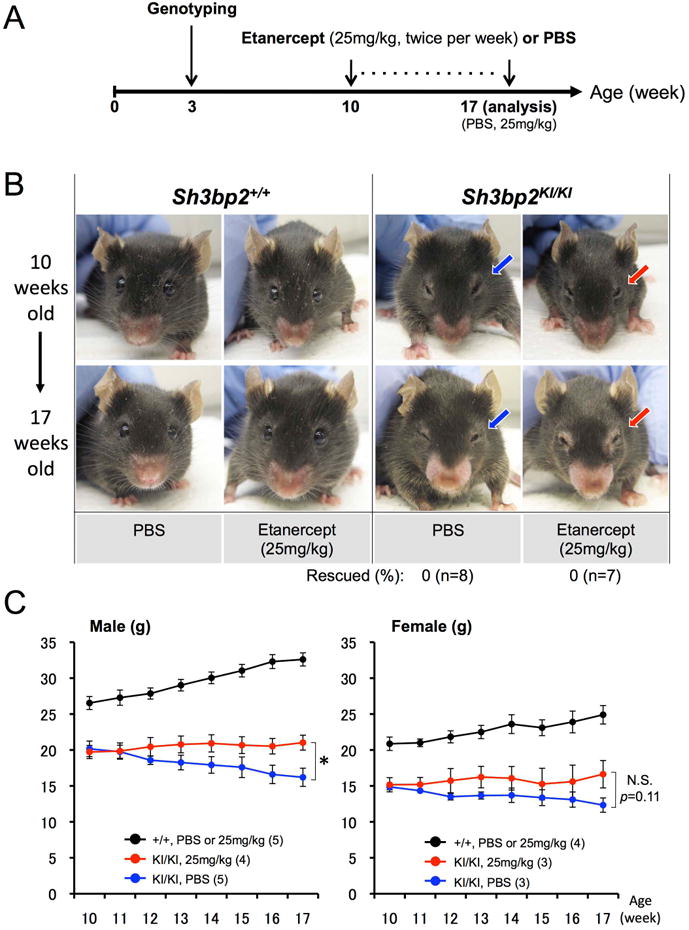
One-week-old Sh3bp2KI/KI mutants were subcutaneously injected with high (25 mg/kg) or low (0.5 mg/kg) dose etanercept twice per week for 7 weeks. Results of TNF-α inhibition were compared with vehicle (PBS)-injected Sh3bp2KI/KI mutants at the age of 8 weeks (Fig. 2A). Strikingly, high dose etanercept treatment rescued the closure of eyelids associated with facial skin inflammation in all Sh3bp2KI/KI mutants examined (n=7) (Fig. 2B: red arrow), which is typically seen in the all inflamed homozygous mutant mice(16) (n=9) (Fig. 2B: blue arrow), while low dose etanercept administration failed to rescue in all cases (n=6) (Fig. 2B: green arrow). Consistent with this observation, loss of body weight was improved in the homozygous mutants treated with 25 mg/kg etanercept in both males and females compared with PBS-treated Sh3bp2KI/KI mutants (Fig. 2C). Next we investigated whether facial inflammation recurs in the Sh3bp2KI/KI mutants after etanercept treatment was discontinued. The Sh3bp2KI/KI mice that were successfully treated for 7 weeks with 25 mg/kg etanercept administration showed closure of eyelids after 8 weeks discontinuation of etanercept administration in all cases (n=9) (Fig. 2D), suggesting a relapse of inflammation. Recurrence of inflammation was concurrent with the arrest of body weight gain in etanercept-treated Sh3bp2KI/KI mutants after etanercept discontinuation (Fig. 2E).
Fig. 2. Etanercept administration to neonatal Sh3bp2KI/KI mutants improves facial swelling and loss of body weight.
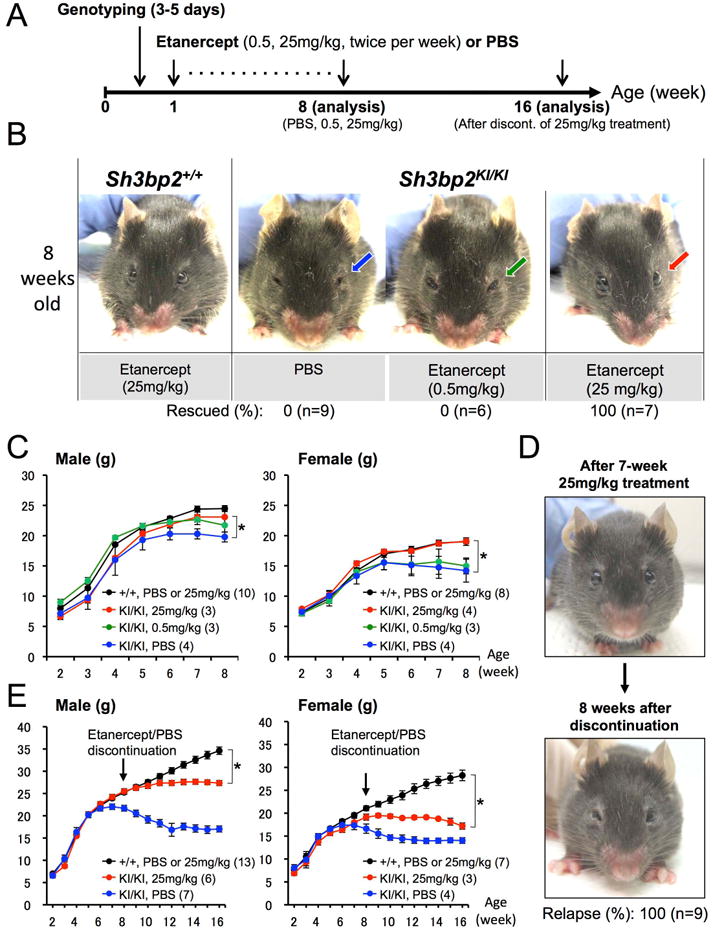
(A) Overall experimental procedure of the etanercept administration to neonatal mice. One-week-old wild-type (Sh3bp2+/+) and homozygous cherubism mutant (Sh3bp2KI/KI) mice were injected with PBS or etanercept (0.5 mg/kg or 25 mg/kg twice per week) for 7 weeks following genotyping at 3-5 days of age. Mice were sacrificed at 8 weeks of age for analysis. Some of the Sh3bp2KI/KI mice treated with 25 mg/kg etanercept were analyzed 8 weeks after discontinuation of etanercept treatment (16 weeks old). (B) Facial appearance of 8-week-old PBS-administered Sh3bp2KI/KI mouse and etanercept-treated Sh3bp2+/+ and Sh3bp2KI/KI mouse after 7 weeks of treatment. Blue or green arrow indicates the closed eyelids caused by skin inflammation, which is typically seen in Sh3bp2KI/KI mice around 6 weeks after birth. Red arrow points at open eyelids of a Sh3bp2KI/KI mouse treated with 25 mg/kg etanercept, demonstrating rescue of facial skin inflammation. Numbers represent percentages of Sh3bp2KI/KI mice with rescue of facial swelling. (C) Body weight changes in PBS- or etanercept-administered Sh3bp2+/+ and Sh3bp2KI/KI mice. Weights are significantly higher in etanercept-administered Sh3bp2KI/KI mice (red line) compared to PBS-administered Sh3bp2KI/KI mice (blue line) after 7-week treatment. Numbers in parentheses represent the number of the mice weighed. (D) Facial appearance of 8-week-old etanercept (25 mg/kg)-treated Sh3bp2KI/KI mouse (top) and the same mouse after 8 weeks of etanercept discontinuation (bottom). (E) Body weight changes in PBS- or etanercept-treated Sh3bp2+/+ and Sh3bp2KI/KI mice. Weight gain is arrested in etanercept-discontinued Sh3bp2KI/KI mice (red line) compared to PBS or etanercept-treated Sh3bp2+/+ mice (black line). Arrows indicate the age at etanercept discontinuation. Numbers in parentheses represent the number of the mice weighed. Error bars represent ± SEM. Asterisks represent the significant difference (p < 0.05).
Etanercept administration to neonatal Sh3bp2KI/KI mutants prevents bone loss in jaws and calvariae and inflammation-induced joint destruction
We examined whether jaw and calvarial bone loss in Sh3bp2KI/KI mutants can be prevented by etanercept administration. Remarkably, microCT images of mandibular bone of Sh3bp2KI/KI mutants treated with high dose etanercept were indistinguishable from those of PBS-treated Sh3bp2+/+ mice (Fig. 3A). To quantitate the jawbone loss, distance between cemento-enamel junction (CEJ) and alveolar bone crest (ABC) at the distal lingual surface of the mandibular first molar (M1) was measured using reconstructed three-dimensional microCT images from etanercept-treated Sh3bp2KI/KI mutants, compared with those from the PBS-treated Sh3bp2KI/KI control group. Both low- (0.5 mg/kg) and high- (25 mg/kg) dose etanercept administration significantly contributed to the reduction in the average CEJ-ABC distance (0.31 ± 0.045 mm and 0.25 ± 0.028 mm, respectively) compared to those of PBS-treated Sh3bp2KI/KI mutants (0.38 ± 0.050 mm) (Fig. 3B). Next, we investigated the erosion pits on calvarial bone surface in etanercept-treated Sh3bp2KI/KI mutants and compared with those from PBS-treated Sh3bp2KI/KI mutants. Consistent with the reduction in the number of erosion pits (Fig. 3C), the total area of bone erosion was significantly reduced in Sh3bp2KI/KI mutants treated with 25 mg/kg etanercept (7.2 ± 2.6%), compared with PBS-treated Sh3bp2KI/KI mutants (10.8 ± 2.9%) (Fig. 3D).
We further examined whether etanercept administration ameliorates inflammatory joint destruction because our previous data showed that Sh3bp2KI/KI mutants develop bone-destructive synovial tissue inflammation in elbow and knee joints.(16) MicroCT analysis of elbow joints from Sh3bp2KI/KI mutants treated with high dose etanercept revealed protection from bone destruction and intact coronoid fossa in all elbow joints examined (n=6), which were indistinguishable from wild-type controls (Fig. 3E). However, after discontinuation of etanercept administration, Sh3bp2KI/KI mutants, which were successfully treated with 25 mg/kg etanercept, showed a relapse of jaw and calvarial bone loss at levels comparable to those of PBS-treated 8-week-old Sh3bp2KI/KI mice (Fig. 3A, B, C, D). Elbow joint destruction redeveloped as well, but less severe compared to PBS-treated Sh3bp2KI/KI mice (Fig. 3E). Taken together, these results demonstrate that etanercept treatment effectively prevents the bone loss associated with inflammation and discontinuation of etanercept treatment results in the relapse into inflammatory bone loss in Sh3bp2KI/KI mutants.
Etanercept administration to neonatal Sh3bp2KI/KI mutants reduces the development of inflammation in lung and liver
In Sh3bp2KI/KI mutants, accumulation of inflammatory infiltrates is not only seen in craniofacial tissues but also in various internal organs including lung and liver tissues.(16) Therefore, we examined if etanercept treatment can effectively reduce the development of inflammation in lung and liver. In lung, a large number of inflammatory nodules, which look whitish on the surface of lung tissue after fixation with Bouin's solution, are typically seen in 8-week-old Sh3bp2KI/KI mice (Fig. 4A: white arrows, top panel). However, Sh3bp2KI/KI mutants administered with 25 mg/kg etanercept exhibited significantly decreased superficial inflammatory nodules compared to the PBS-treated Sh3bp2KI/KI mice. Homozygous mutants injected with 0.5 mg/kg etanercept did not exhibit the reduction in the number of superficial nodules (Fig. 4A, top panel). This reduction was confirmed by histomorphometric analysis of lung tissue showing the proportion of total inflammatory area to the total lung tissue area in lung sections (average lesion area: 9.1 ± 4.0% in PBS treatment, 9.6 ± 5.7% in 0.5 mg/kg treatment vs. 2.7 ± 2.3% in 25 mg/kg treatment) (Fig. 4A, bottom panel, 4B). In liver, 25 mg/kg etanercept administration significantly reduced the total area of inflammation, compared to 0.5 mg/kg etanercept- and PBS-treated Sh3bp2KI/KI mutants (Fig. 4C). This striking reduction was also confirmed by histomorphometric analysis of the total inflammatory area in liver tissue (average lesion area: 10.6 ± 5.0% in PBS treatment, 9.9 ± 7.2% in 0.5 mg/kg treatment vs. 0.1 ± 0.08% in 25 mg/kg treatment) (Fig. 4D). Consistent with a decreased area of the inflamed lesion, lower expression levels of TNF-α mRNA in liver tissue were seen in 25 mg/kg etanercept-treated Sh3bp2KI/KI mice as quantitated by real-time PCR (Fig. 4E). After discontinuation of etanercept treatment, lung lesions which were reduced by high dose 25 mg/kg etanercept showed a trend of regrowth (p = 0.18) to comparable levels of PBS-treated 8-week-old Sh3bp2KI/KI mice (Fig. 4A, B). Liver lesions redeveloped in Sh3bp2KI/KI mice after etanercept discontinuation, which was also equivalent to PBS-treated Sh3bp2KI/KI mice (Fig. 4C, D). However, liver TNF-α mRNA expression was still comparable to 25 mg/kg etanercept-treated Sh3bp2KI/KI mice (before discontinuation) (Fig. 4E), suggesting that TNF-α produced outside the liver is involved in the redevelopment of liver lesions. Collectively, these results demonstrate that etanercept treatment effectively prevents lung and liver inflammation, but discontinuation of etanercept results in the relapse of inflammation in lung and liver in Sh3bp2KI/KI mutants.
Fig. 4. Reduced lung and liver inflammatory infiltrates in etanercept-treated Sh3bp2KI/KI mice.
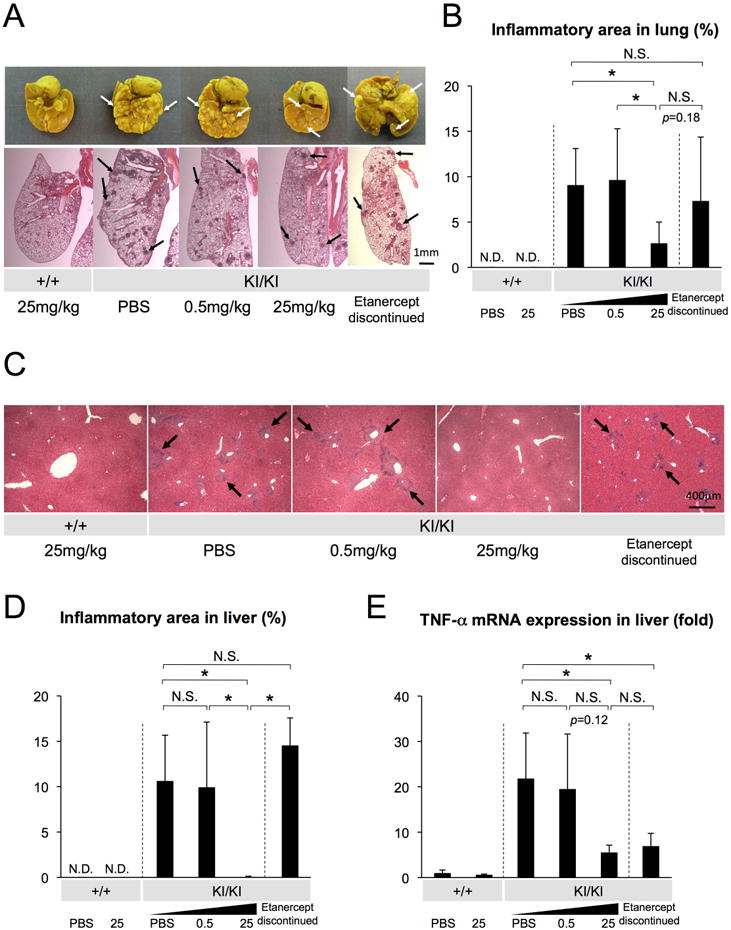
(A) Lung tissues fixed with Bouin's solution (top), and H&E-stained lung tissue sections (bottom). Arrows indicate inflammatory nodules on the lung surface (white) and in sections (black). (B) Quantitative analysis of total inflamed areas in lung. (C) Liver sections from PBS- or etanercept-treated Sh3bp2+/+ and Sh3bp2KI/KI mice (H&E). Arrows indicate inflammatory infiltrates surrounding vessels. (D) Quantitative measurement of total area of the inflammatory infiltrates in liver. (E) Quantitative-PCR analysis of TNF-α mRNA expression in liver. Values are the average of 2-9 independent RNA samples from each group. Average expression level of two Sh3bp2+/+ mice treated with PBS was set as 1. Error bars represent SD. Asterisks represent significant difference (p < 0.05). N.S.: not significant. N.D.: not detected. n=6-7. In Fig. 4E, n=3-9 per group except for n=2 in each Sh3bp2+/+ group.
Etanercept administration to adult Sh3bp2KI/KI mutants with active inflammation does not improve facial swelling
A desirable therapeutic strategy for patients already diagnosed with cherubism would be to stabilize or shrink actively growing lesions. We therefore raised the question whether etanercept treatment is able to improve fully active inflammatory lesions in Sh3bp2KI/KI mice. We administered etanercept to 10-week-old Sh3bp2KI/KI mice, which have already developed active inflammatory lesions. Sh3bp2KI/KI mice were treated for 7 weeks with 25 mg/kg etanercept (twice per week) (Fig. 5A). This same dose that successfully prevented facial swelling in neonatal Sh3bp2KI/KI mice (Fig. 2B: red arrow) failed to rescue facial swelling associated with eyelid closure in all 17-week-old Sh3bp2KI/KI mice after 7-week etanercept treatment (n=7) (Fig. 5B: red arrow, bottom panel). However, the progressive loss of body weight with age was ameliorated by etanercept injection (Fig. 5C), suggesting that TNF-α inhibition can substantially improve the systemic condition of Sh3bp2KI/KI mice.
Fig. 5. Etanercept administration to actively inflamed 10-week-old Sh3bp2KI/KI mice does not rescue facial swelling.
(A) Experimental procedure of etanercept administration to the 10-week-old Sh3bp2KI/KI mice with active inflammatory lesions. Ten-week-old Sh3bp2+/+ and homozygous Sh3bp2KI/KI mutant mice were treated with PBS or 25 mg/kg of etanercept twice per week for 7 weeks. Mice were sacrificed at 17 weeks of age for analysis. (B) Facial appearance of PBS- or etanercept-treated mice (top: before treatment at 10 weeks of age; bottom: after treatment at 17 weeks of age). Blue arrows indicate closed eyelids due to facial skin inflammation. Etanercept treatment failed to rescue eyelid closure in Sh3bp2KI/KI mice after etanercept administration (red arrows). Numbers represent the percentages of Sh3bp2KI/KI mice rescued from facial swelling. (C) Body weight changes in PBS- or etanercept-administered Sh3bp2+/+ and Sh3bp2KI/KI mice. Body weight in etanercept-administered Sh3bp2KI/KI mice (red line) was maintained, whereas weight of PBS-administered Sh3bp2KI/KI mice (blue line) continued to decrease. Numbers in parentheses represent the number of the mice weighed. Error bars represent ± SEM. Asterisk represents the significant difference (p < 0.05). N.S.: not significant.
Etanercept administration to adult Sh3bp2KI/KI mutants with active inflammation does not improve jaw and calvarial bone loss and joint destruction
Consistent with the unsuccessful rescue of facial swelling in adult Sh3bp2KI/KI mice (Fig. 5B), microCT analysis followed by the quantitative measurement of the distance between the CEJ and ABC revealed that 25 mg/kg etanercept treatment for 7 weeks does not reduce mandibular pitting (Fig. 6A: arrows) and bone loss (average distance: 0.41 ± 0.07 mm in PBS-treated group vs. 0.37 ± 0.04 mm in etanercept-treated group) (Fig. 6B). Interestingly, the average area of calvarial bone erosion was decreased in 17-week-old Sh3bp2KI/KI mice treated with PBS (6.1 ± 1.1%) (Fig. 6D) compared to 8-week-old Sh3bp2KI/KI mice treated with PBS (10.8 ± 2.9%) (p = 0.004) (Fig. 3D). The improved calvarial bone formation in the adult 17-week-old Sh3bp2KI/KI mice suggests that calvarial bone erosion ameliorates with age, which may be comparable to the regression of bone lesions after puberty in human cherubism patients. Further microCT analysis revealed that the elbow joint destruction continued after etanercept treatment in 17-week-old Sh3bp2KI/KI mice in all cases examined (n=7) (Fig. 6E). Taken together, these results indicate that inhibition of TNF-α in adult cherubism mice with active inflammation is less effective in improving bone loss.
Etanercept administration to adult Sh3bp2KI/KI mutants with active inflammation does not improve lung and liver inflammation
Next, we quantitated the inflamed area in lung and liver from 17-week-old Sh3bp2KI/KI mice, which had received etanercept treatment for 7 weeks. Etanercept treatment failed to reduce inflammatory lesions in lung (Fig. 7A, B). Interestingly, similar to the age-associated resolution of calvarial bone erosion, the area of inflamed lung tissues showed a decreasing trend with age in PBS-treated Sh3bp2KI/KI mice from 9.1 ± 4.0% at 8 weeks (Fig. 4B) to 5.2 ± 2.7% at 17 weeks (p = 0.079) (Fig. 7B). However, we even saw a non-significant increase (p = 0.31) in inflamed lung tissue area in the etanercept-treated group (7.4 ± 4.3%) compared to the PBS-treated controls (5.2 ± 2.7%) (Fig. 7B). Therefore, it can be assumed that etanercept does not contribute to the age-related resolution of lung lesions. In liver, the proportion of inflammation area is quite variable at 17 weeks of age and the average inflammation area in the PBS-treated Sh3bp2KI/KI group (10.6 ± 6.2%) (Fig. 7D) did not change with age compared to the PBS-treated 8-week-old Sh3bp2KI/KI mice (10.6 ± 5.0%) (Fig. 4D). However, etanercept treatment effectively reduced the total area of inflammation in the 17-week-old treated cohort (4.1 ± 3.50%) compared to the PBS-treated group (10.6 ± 6.2%) (Fig. 7C, D). In contrast, TNF-α mRNA expression in liver was comparable between etanercept- and PBS- treated groups (p = 0.13) (Fig. 7E). Interestingly, TNF-α stimulation of bone marrow derived M-CSF-dependent macrophages (BMMs) from Sh3bp2KI/KI mice induced more IL-1α and IL-1β mRNA expression compared to wild-type macrophages (Fig. 7F). Furthermore, TNF-α secretion in response to IL-1α and IL-1β was increased in Sh3bp2KI/KI BMMs (Fig. 7G), suggesting that a (TNF-α) ➜ (IL-1α/β) ➜ (TNF-α) inflammatory circuit is involved in the development of inflammatory lesions in cherubism mice and that IL-1α/β play a potential role in the maintenance of active lesions. Collectively, these results demonstrate that etanercept treatment of adult cherubism mice with fully active inflammation has limited efficacy only for the reduction of liver inflammation.
Fig. 7. Limited effect of etanercept treatment on active lung and liver lesions and IL-1α/β-mediated TNF-α positive feedback loop in Sh3bp2KI/KI macrophages.
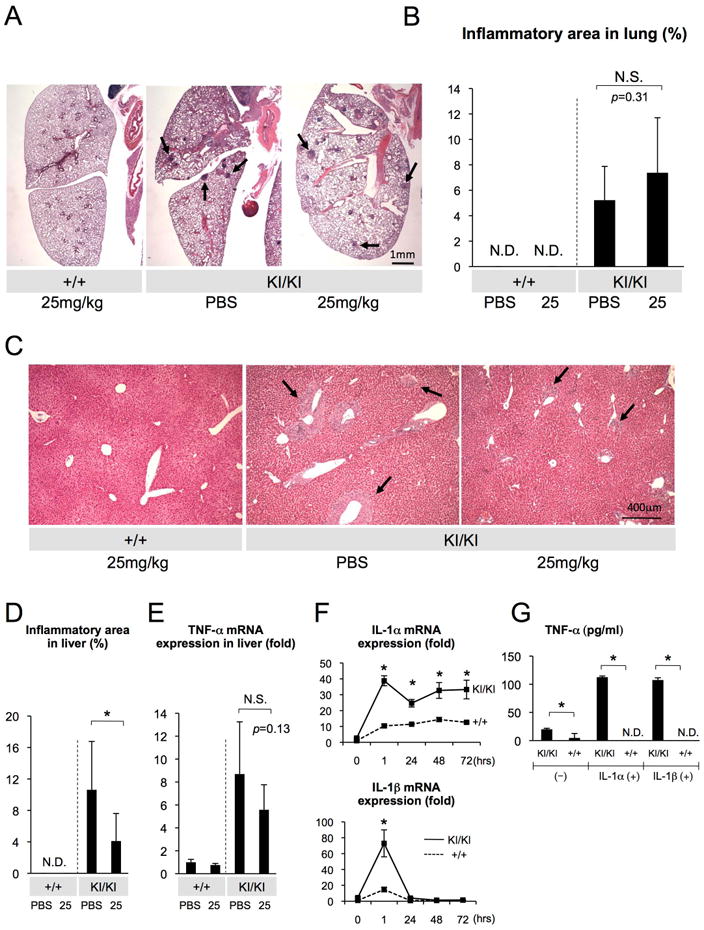
(A) Lung tissue sections from PBS- or etanercept-treated 17-week-old Sh3bp2+/+ and Sh3bp2KI/KI mice stained with H&E. Arrows indicate inflammatory nodules. (B) Quantitative analysis of total inflamed nodule area in lung sections. (C) H&E staining of liver sections from PBS- or etanercept-treated mice. Arrows indicate inflammatory infiltrates. (D) Quantitative measurement of the total area of inflammatory infiltrates in liver. (E) Quantitative-PCR analysis of the TNF-α mRNA expression in liver. Average expression level of two PBS-treated Sh3bp2+/+ mice was set as 1. (F) Quantitative-PCR analysis of the IL-1α and IL-1β mRNA expression in BMMs stimulated with TNF-α. Values are the average of three independent RNA samples from each genotype. Average expression level in Sh3bp2+/+ macrophages was set as 1. (G) TNF-α level in the culture supernatants of BMMs stimulated with IL-1α or IL-1β. Error bars represent SD. Asterisks represent significant difference (p < 0.05). N.S.: not significant. N.D.: not detected. n=5-7. In Fig. 7E, n=2 in each Sh3bp2+/+ group.
Discussion
Cherubism is a human craniofacial bone disorder developing in childhood with characteristic bilateral jaw swelling and jawbone destruction caused by excessive osteoclast activity and progressive proliferation of fibrous tissues. Because the growing lesion is a non-malignant tumor and jaw swelling typically regresses after puberty, current standard management of cherubism is conservative and includes close observation of disease progression unless aesthetic or functional concerns such as nasal obstruction, proptosis or severe facial deformity develop. In such cases surgical resection is considered.(2) At present, there is no promising pharmacological treatment for cherubism.
In this study, we demonstrate that administration of etanercept, an FDA-approved TNF-α blocker for the treatment of inflammatory diseases such as rheumatoid arthritis, to inflammation-free neonatal Sh3bp2KI/KI mice is effective in preventing the development of inflammation. Therefore, our study presents a new pharmacological therapeutic approach for cherubism treatment, which can potentially prevent any future facial swelling and jawbone destruction. In contrast, we also demonstrate that the same etanercept treatment of adult Sh3bp2KI/KI mice with fully active inflammation does not necessarily contribute to the reduction and regression of inflammation.
It is reported that mice treated with etanercept generate antibodies against etanercept.(35) Although generation of anti-etanercept antibodies in Sh3bp2KI/KI mice during treatment could be one of the causes of failed etanercept treatment in adult Sh3bp2KI/KI mice, the unsuccessful therapeutic effect of TNF-α antagonist on adult Sh3bp2KI/KI mutants with active inflammation is consistent with a recent report by Hero et al.(36) In this report, the authors treated two juvenile cherubism patients with actively growing lesions due to a mutation in SH3BP2 with the human anti-human TNF-α antibody adalimumab over 2-years and failed to find regression or prevention of lesion growth, although the number of giant cells decreased substantially.(37) The fact that there is only limited therapeutic efficacy of the TNF-α antagonist in both adult cherubism mice and human cherubism patients with active lesions (36), but high efficiency in preventing lesions in young mice suggests that anti-TNF-α treatment is more effective when started prior to the development of cherubism lesions. In other words, anti-TNF-α treatment is not likely to suppress the growth of active lesions or enhance their regression. This age-associated effectiveness further suggests that TNF-α plays a role exclusively in the initiation of lesion development. Hence, other inflammatory cytokines following initial TNF-α expression contribute most likely to the maintenance and progression of the cherubism lesions. IL-1α or IL-1β may play such a role, since both cytokines mediate TNF-α–driven inflammatory positive feedback loop in Sh3bp2KI/KI macrophages (Fig. 7F, G). Therefore, treatment with anti-TNF-α drugs in combination with IL-1α or IL-1β inhibitors might show increased therapeutic efficacy in cherubism patients with active lesions. Interestingly, a clinical study reported that continued treatment with denosumab, a human monoclonal antibody against human RANKL, could be useful as a therapeutic agent for giant cell tumors of bone (GCTB) by eliminating osteolytic giant cells and inhibiting tumor progression.(38) This drug has recently been approved by FDA for the treatment of GCTB. Because there is an indistinguishable histological similarity between GCTB and cherubism,(39) RANKL could be an additional potential target to inhibit osteoclast-mediated bone loss in cherubism.
Since most cherubism patients are diagnosed after showing active signs like submandibular lymph node swelling and bilateral jaw expansion around 2-5 years of age,(40) starting anti-TNF-α treatment for pre-symptomatic cherubism patients is not realistic. However, anti-TNF-α treatment could be started early if genetic testing of children in cherubism family showed positive for a mutation in SH3BP2 shortly after birth. Given the fact that long-term TNF-α suppression increases the risk for serious infectious diseases such as tuberculosis,(41-46) anti-TNF-α therapy needs to be cautiously applied and long-term administration, most likely until puberty, should be carefully monitored.
The 25 mg/kg administration twice per week, which prevented the inflammation and bone loss in Sh3bp2KI/KI mice, is about 60-fold higher compared to the standard dose in humans.(22) The requirement of such a high dose of etanercept in cherubism mice may be due to the circumstances that mice generate anti-etanercept antibodies,(35) inflammation in cherubism mice is more systemic, and etanercept is designed to neutralize human TNF-α. We have also tested a 5 mg/kg dose, but the prevention was not as successful as with 25 mg/kg (Supplemental Fig. 1). Therefore, the effective dose of TNF-α inhibitor for cherubism patients should be carefully determined, since the severity of cherubism lesions and immune reactions differ between human and mouse.
TNF-α inhibits osteoblast differentiation in vitro(47,48) and in vivo,(49) where Li et al. showed that the increased bone mass in TNF-α knock-out mice is associated with the elevation of serum osteocalcin with no change in the serum type I collagen C-terminal telopeptide (CTX). This suggests that endogenous TNF-α controls bone formation through osteoblasts. On the other hand, TNF-α stimulates osteoclast differentiation.(48,50) Taken together, both promotion of bone formation and inhibition of bone resorption due to TNF-α inhibition by etanercept are likely to contribute to the prevention of bone loss and joint destruction in etanercept-treated Sh3bp2KI/KI mice. However, the effect of TNF-α inhibition on osteocyte-dependent regulation of osteoclastogenesis through RANKL expression(51,52) or via apoptotic osteocytes(53,54) still needs to be investigated to fully understand the mechanisms by which TNF-α inhibition prevents bone loss in Sh3bp2KI/KI mice. Our osteoclast cultures with RANKL-stimulated BMMs treated with high dose etanercept showed a small, but statistically significant, reduction in the osteoclast formation (Supplemental Fig. 2), suggesting that the prevention of bone loss is, at least in part, due to the direct inhibition of osteoclastogenesis by etanercept.
We found that the effectiveness of etanercept in cherubism mice is organ-dependent because the treatment of neonatal mutants with 25 mg/kg etanercept failed to prevent inflammation in stomach and lymph nodes in all examined cases (n=7) (data not shown). The organ-specific efficacy of etanercept in cherubism mice is consistent with previous reports that each TNF inhibitor has a distinct efficacy profile in human inflammatory diseases(55,56) and that etanercept treatment failed to show clinical response in Crohn's disease,(57) whereas other TNF-α antagonist such as infliximab and adalimumab were effective.(58,59) These studies suggest that each TNF-α antagonist has specific disease targets and TNF-α antagonists other than adalimumab (administered by Hero et al. (36)) may have better clinical efficacy for the treatment of cherubism patients.
In cherubism patients, regression of the jaw swelling after puberty is a characteristic clinical feature. Age-related spontaneous regression of resorptive areas in calvariae and inflammatory lesions in lung were also found in Sh3bp2KI/KI mice. But bone loss in mandibulae, hepatic lesions, and facial swellings were unchanged with age in Sh3bp2KI/KI mice, suggesting that spontaneous regression occurs in a tissue-specific manner. However, etanercept administration did not seem to contribute to age-associated regression of lesions in Sh3bp2KI/KI mice. Given the fact that Sh3bp2KI/KI mice that were successfully treated with etanercept develop inflammation after termination of etanercept administration, cherubism patients will likely need to undergo anti-TNF-α treatment until they reach puberty at which time cherubism lesions normally start to regress. The mechanism of this age-associated regression remains unknown and needs to be investigated to fully understand the pathogenesis of cherubism and to establish more effective therapeutic strategies.
In summary, this study provides evidence that anti-TNF-α therapy might be effective for cherubism patients, if it can be started at an age when active lesions are not fully developed. Early treatment could prevent future development of cherubism lesions. To maximize the effectiveness of anti-TNF-α therapy, early genetic testing for SH3BP2 mutations will be required and a combination therapy with inhibitors that target inflammatory cytokines other than TNF-α or anti-bone resorptive agents should be considered for improved treatment efficacy.
Supplementary Material
Supplemental Fig. 1. 5 mg/kg etanercept treatment does not effectively prevent body weight loss, inflammatory jaw bone and elbow joint destruction, and liver inflammation in Sh3bp2KI/KI mice compared to 25 mg/kg treatment. (A) Body weight changes in PBS- or etanercept-administered Sh3bp2+/+ and Sh3bp2KI/KI mice. 5 mg/kg etanercept administration (light-blue line) prevented weight loss in male Sh3bp2KI/KI mice compared to PBS-administered male Sh3bp2KI/KI mice (dark-blue line), but not in female Sh3bp2KI/KI mice. Numbers in parentheses represent the number of the mice weighed. (B) Reconstructed microCT images of the mandibular bone from 8-week-old Sh3bp2KI/KI mice treated with 5 mg/kg etanercept. Arrows indicate erosion pits due to inflammation. (C) Reconstructed microCT images of elbow joint from 5 mg/kg etanercept-administered Sh3bp2KI/KI mice, demonstrating the elbow joint destruction. (D) Representative liver tissue (H&E staining) from Sh3bp2KI/KI mice treated with 5mg/kg etanercept and quantitative measurement of total area of the inflammatory infiltrates in liver, showing less effectiveness of 5 mg/kg administration compared to 25 mg/kg. Arrows indicate inflammatory lesions. Error bars represent SEM in (A) and SD in (D). Asterisks represent significant difference (p < 0.05). N.S.: not significant. N.D.: not detected, n=6-9.
Supplemental Fig 2. Etanercept treatment decreases the number of RANKL-induced osteoclast formation in Sh3bp2KI/KI BMMs cultures. (A) TRAP staining of osteoclasts differentiated from Sh3bp2KI/KI BMMs stimulated with RANKL in the presence or absence of etanercept. (B) Quantitative analysis of the number of TRAP-positive multinucleated cells. n= 4/group. Asterisks represent significant difference (p < 0.001) compared to 0 μg/ml etanercept culture (one-way ANOVA, Tukey post-hoc analysis). N.S.: not significant.
Acknowledgments
Grants: This work was supported by a grant from the National Institute of Health (R01DE020835) to Y.U.
We would like to thank Drs. L. Bonewald, M. Johnson, J. Gorski, S. Dallas, and all members of Bone Biology Research Program in the Department of Oral and Craniofacial Sciences at the University of Missouri-Kansas City, School of Dentistry for critical discussion and helpful suggestions. We are also grateful to M. Ueki, S. Ueki, R. Ishikawa, and N. Mizuno for technical assistance. This work was supported by a grant from the National Institute of Health (R01DE020835) to YU.
Authors' roles: Study design: TY, TM, ER, and YU. Study conduct and data collection: TY, SI, TM, and MK. Data analysis: TY, SI, TM, MK, and YU. Data interpretation: TY, TM, and YU. Drafting manuscript: TY and YU. Revising manuscript content: TY, ER, and YU. YU supervised the overall study and wrote the final manuscript. TY and YU take responsibility for the integrity of data analysis.
Footnotes
Disclosures: All authors state that they have no conflict of interest.
References
- 1.Von Wowern N. Cherubism: a 36-year long-term follow-up of 2 generations in different families and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2000;90(6):765–72. doi: 10.1067/moe.2000.108438. [DOI] [PubMed] [Google Scholar]
- 2.Papadaki ME, Lietman SA, Levine MA, Olsen BR, Kaban LB, Reichenberger EJ. Cherubism: best clinical practice. Orphanet J Rare Dis. 2012;7(Suppl 1):S6. doi: 10.1186/1750-1172-7-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiziani V, Reichenberger E, Buzzo CL, Niazi S, Fukai N, Stiller M, et al. The gene for cherubism maps to chromosome 4p16. Am J Hum Genet. 1999;65(1):158–66. doi: 10.1086/302456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueki Y, Tiziani V, Santanna C, Fukai N, Maulik C, Garfinkle J, et al. Mutations in the gene encoding c-Abl-binding protein SH3BP2 cause cherubism. Nat Genet. 2001;28(2):125–6. doi: 10.1038/88832. [DOI] [PubMed] [Google Scholar]
- 5.Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259(5098):1157–61. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- 6.Deckert M, Tartare-Deckert S, Hernandez J, Rottapel R, Altman A. Adaptor function for the Syk kinases-interacting protein 3BP2 in IL-2 gene activation. Immunity. 1998;9(5):595–605. doi: 10.1016/s1074-7613(00)80657-3. [DOI] [PubMed] [Google Scholar]
- 7.Foucault I, Liu YC, Bernard A, Deckert M. The chaperone protein 14-3-3 interacts with 3BP2/SH3BP2 and regulates its adapter function. J Biol Chem. 2003;278(9):7146–53. doi: 10.1074/jbc.M209509200. [DOI] [PubMed] [Google Scholar]
- 8.Foucault I, Le Bras S, Charvet C, Moon C, Altman A, Deckert M. The adaptor protein 3BP2 associates with VAV guanine nucleotide exchange factors to regulate NFAT activation by the B-cell antigen receptor. Blood. 2005;105(3):1106–13. doi: 10.1182/blood-2003-08-2965. [DOI] [PubMed] [Google Scholar]
- 9.Maeno K, Sada K, Kyo S, Miah SM, Kawauchi-Kamata K, Qu X, et al. Adaptor protein 3BP2 is a potential ligand of Src homology 2 and 3 domains of Lyn protein-tyrosine kinase. J Biol Chem. 2003;278(27):24912–20. doi: 10.1074/jbc.M301201200. [DOI] [PubMed] [Google Scholar]
- 10.Le Bras S, Moon C, Foucault I, Breittmayer JP, Deckert M. Abl-SH3 binding protein 2, 3BP2, interacts with CIN85 and HIP-55. FEBS Lett. 2007;581(5):967–74. doi: 10.1016/j.febslet.2007.01.084. [DOI] [PubMed] [Google Scholar]
- 11.Jevremovic D, Billadeau DD, Schoon RA, Dick CJ, Leibson PJ. Regulation of NK cell-mediated cytotoxicity by the adaptor protein 3BP2. J Immunol. 2001;166(12):7219–28. doi: 10.4049/jimmunol.166.12.7219. [DOI] [PubMed] [Google Scholar]
- 12.Yu Z, Maoui M, Zhao ZJ, Li Y, Shen SH. SHP-1 dephosphorylates 3BP2 and potentially downregulates 3BP2-mediated T cell antigen receptor signaling. FEBS J. 2006;273(10):2195–205. doi: 10.1111/j.1742-4658.2006.05233.x. [DOI] [PubMed] [Google Scholar]
- 13.Chihara K, Nakashima K, Takeuchi K, Sada K. Association of 3BP2 with SHP-1 regulates SHP-1-mediated production of TNF-alpha in RBL-2H3 cells. Genes Cells. 2011;16(12):1133–45. doi: 10.1111/j.1365-2443.2011.01557.x. [DOI] [PubMed] [Google Scholar]
- 14.Shukla U, Hatani T, Nakashima K, Ogi K, Sada K. Tyrosine phosphorylation of 3BP2 regulates B cell receptor-mediated activation of NFAT. J Biol Chem. 2009;284(49):33719–28. doi: 10.1074/jbc.M109.049999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levaot N, Simoncic PD, Dimitriou ID, Scotter A, La Rose J, Ng AH, et al. 3BP2-deficient mice are osteoporotic with impaired osteoblast and osteoclast functions. J Clin Invest. 2011;121(8):3244–57. doi: 10.1172/JCI45843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ueki Y, Lin CY, Senoo M, Ebihara T, Agata N, Onji M, et al. Increased myeloid cell responses to M-CSF and RANKL cause bone loss and inflammation in SH3BP2 “cherubism” mice. Cell. 2007;128(1):71–83. doi: 10.1016/j.cell.2006.10.047. [DOI] [PubMed] [Google Scholar]
- 17.Ferguson PJ, El-Shanti HI. Autoinflammatory bone disorders. Curr Opin Rheumatol. 2007;19(5):492–8. doi: 10.1097/BOR.0b013e32825f5492. [DOI] [PubMed] [Google Scholar]
- 18.Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*) Annu Rev Immunol. 2009;27:621–68. doi: 10.1146/annurev.immunol.25.022106.141627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levaot N, Voytyuk O, Dimitriou I, Sircoulomb F, Chandrakumar A, Deckert M, et al. Loss of Tankyrase-Mediated Destruction of 3BP2 Is the Underlying Pathogenic Mechanism of Cherubism. Cell. 2011;147(6):1324–39. doi: 10.1016/j.cell.2011.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guettler S, LaRose J, Petsalaki E, Gish G, Scotter A, Pawson T, et al. Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell. 2011;147(6):1340–54. doi: 10.1016/j.cell.2011.10.046. [DOI] [PubMed] [Google Scholar]
- 21.Pasparakis M, Alexopoulou L, Episkopou V, Kollias G. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J Exp Med. 1996;184(4):1397–411. doi: 10.1084/jem.184.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caporali R, Pallavicini FB, Filippini M, Gorla R, Marchesoni A, Favalli EG, et al. Treatment of rheumatoid arthritis with anti-TNF-alpha agents: a reappraisal. Autoimmun Rev. 2009;8(3):274–80. doi: 10.1016/j.autrev.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Kerensky TA, Gottlieb AB, Yaniv S, Au SC. Etanercept: efficacy and safety for approved indications. Expert Opin Drug Saf. 2012;11(1):121–39. doi: 10.1517/14740338.2012.633509. [DOI] [PubMed] [Google Scholar]
- 24.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, et al. Tumor necrosis factor-alpha antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension. 2010;56(4):643–9. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R, Bal HS, Desta T, Behl Y, Graves DT. Tumor necrosis factor-alpha mediates diabetes-enhanced apoptosis of matrix-producing cells and impairs diabetic healing. Am J Pathol. 2006;168(3):757–64. doi: 10.2353/ajpath.2006.050907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muia C, Bramanti P, et al. Immunomodulatory effects of etanercept in an experimental model of spinal cord injury. J Pharmacol Exp Ther. 2006;316(3):1006–16. doi: 10.1124/jpet.105.097188. [DOI] [PubMed] [Google Scholar]
- 27.Nemoto H, Konno S, Sugimoto H, Nakazora H, Nomoto N, Murata M, et al. Anti-TNF therapy using etanercept suppresses degenerative and inflammatory changes in skeletal muscle of older SJL/J mice. Exp Mol Pathol. 2011;90(3):264–70. doi: 10.1016/j.yexmp.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Rose S, Eren M, Murphy S, Zhang H, Thaxton CS, Chowaniec J, et al. A novel mouse model that develops spontaneous arthritis and is predisposed towards atherosclerosis. Ann Rheum Dis. 2013;72(1):89–95. doi: 10.1136/annrheumdis-2012-201431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wallis RS, Broder M, Wong J, Beenhouwer D. Granulomatous infections due to tumor necrosis factor blockade: correction. Clin Infect Dis. 2004;39(8):1254–5. doi: 10.1086/424455. [DOI] [PubMed] [Google Scholar]
- 30.Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis. 2004;38(9):1261–5. doi: 10.1086/383317. [DOI] [PubMed] [Google Scholar]
- 31.Plessner HL, Lin PL, Kohno T, Louie JS, Kirschner D, Chan J, et al. Neutralization of tumor necrosis factor (TNF) by antibody but not TNF receptor fusion molecule exacerbates chronic murine tuberculosis. J Infect Dis. 2007;195(11):1643–50. doi: 10.1086/517519. [DOI] [PubMed] [Google Scholar]
- 32.Khan OM, Ibrahim MX, Jonsson IM, Karlsson C, Liu M, Sjogren AK, et al. Geranylgeranyltransferase type I (GGTase-I) deficiency hyperactivates macrophages and induces erosive arthritis in mice. J Clin Invest. 2011;121(2):628–39. doi: 10.1172/JCI43758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park CH, Abramson ZR, Taba M, Jr, Jin Q, Chang J, Kreider JM, et al. Three-dimensional micro-computed tomographic imaging of alveolar bone in experimental bone loss or repair. J Periodontol. 2007;78(2):273–81. doi: 10.1902/jop.2007.060252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39(2):175–91. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 35.Lories RJ, Derese I, de Bari C, Luyten FP. Evidence for uncoupling of inflammation and joint remodeling in a mouse model of spondylarthritis. Arthritis Rheum. 2007;56(2):489–97. doi: 10.1002/art.22372. [DOI] [PubMed] [Google Scholar]
- 36.Hero M, Suomalainen A, Hagstrom J, Stoor P, Kontio R, Alapulli H, et al. Anti-tumor necrosis factor treatment in cherubism - Clinical, radiological and histological findings in two children. Bone. 2013;52(1):347–53. doi: 10.1016/j.bone.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 37.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov. 2003;2(9):736–46. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 38.Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275–80. doi: 10.1016/S1470-2045(10)70010-3. [DOI] [PubMed] [Google Scholar]
- 39.Kaugars GE, Niamtu J, 3rd, Svirsky JA. Cherubism: diagnosis, treatment, and comparison with central giant cell granulomas and giant cell tumors. Oral Surg Oral Med Oral Pathol. 1992;73(3):369–74. doi: 10.1016/0030-4220(92)90137-f. [DOI] [PubMed] [Google Scholar]
- 40.Reichenberger EJ, Levine MA, Olsen BR, Papadaki ME, Lietman SA. The role of SH3BP2 in the pathophysiology of cherubism. Orphanet J Rare Dis. 2012;7(Suppl 1):S5. doi: 10.1186/1750-1172-7-S1-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl J Med. 2006;355(7):704–12. doi: 10.1056/NEJMct055183. [DOI] [PubMed] [Google Scholar]
- 42.Listing J, Strangfeld A, Kary S, Rau R, von Hinueber U, Stoyanova-Scholz M, et al. Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum. 2005;52(11):3403–12. doi: 10.1002/art.21386. [DOI] [PubMed] [Google Scholar]
- 43.Dixon WG, Symmons DP, Lunt M, Watson KD, Hyrich KL, Silman AJ. Serious infection following anti-tumor necrosis factor alpha therapy in patients with rheumatoid arthritis: lessons from interpreting data from observational studies. Arthritis Rheum. 2007;56(9):2896–904. doi: 10.1002/art.22808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dixon WG, Watson K, Lunt M, Hyrich KL, Silman AJ, Symmons DP. Rates of serious infection, including site-specific and bacterial intracellular infection, in rheumatoid arthritis patients receiving anti-tumor necrosis factor therapy: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006;54(8):2368–76. doi: 10.1002/art.21978. [DOI] [PubMed] [Google Scholar]
- 45.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA. 2006;295(19):2275–85. doi: 10.1001/jama.295.19.2275. [DOI] [PubMed] [Google Scholar]
- 46.Rosenblum H, Amital H. Anti-TNF therapy: safety aspects of taking the risk. Autoimmun Rev. 2011;10(9):563–8. doi: 10.1016/j.autrev.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 47.Gilbert L, He X, Farmer P, Boden S, Kozlowski M, Rubin J, et al. Inhibition of osteoblast differentiation by tumor necrosis factor-alpha. Endocrinology. 2000;141(11):3956–64. doi: 10.1210/endo.141.11.7739. [DOI] [PubMed] [Google Scholar]
- 48.Nanes MS. Tumor necrosis factor-alpha: molecular and cellular mechanisms in skeletal pathology. Gene. 2003;321:1–15. doi: 10.1016/s0378-1119(03)00841-2. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Li A, Strait K, Zhang H, Nanes MS, Weitzmann MN. Endogenous TNFalpha lowers maximum peak bone mass and inhibits osteoblastic Smad activation through NF-kappaB. J Bone Miner Res. 2007;22(5):646–55. doi: 10.1359/jbmr.070121. [DOI] [PubMed] [Google Scholar]
- 50.Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191(2):275–86. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–4. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 52.Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O'Brien CA. Matrix-embedded cells control osteoclast formation. Nat Med. 2011;17(10):1235–41. doi: 10.1038/nm.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheung WY, Simmons CA, You L. Osteocyte apoptosis regulates osteoclast precursor adhesion via osteocytic IL-6 secretion and endothelial ICAM-1 expression. Bone. 2012;50(1):104–10. doi: 10.1016/j.bone.2011.09.052. [DOI] [PubMed] [Google Scholar]
- 54.Al-Dujaili SA, Lau E, Al-Dujaili H, Tsang K, Guenther A, You L. Apoptotic osteocytes regulate osteoclast precursor recruitment and differentiation in vitro. J Cell Biochem. 2011;112(9):2412–23. doi: 10.1002/jcb.23164. [DOI] [PubMed] [Google Scholar]
- 55.Haraoui B. Differentiating the efficacy of the tumor necrosis factor inhibitors. Semin Arthritis Rheum. 2005;34(5 Suppl1):7–11. doi: 10.1016/j.semarthrit.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 56.Rigby WF. Drug insight: different mechanisms of action of tumor necrosis factor antagonists-passive-aggressive behavior? Nat Clin Pract Rheumatol. 2007;3(4):227–33. doi: 10.1038/ncprheum0438. [DOI] [PubMed] [Google Scholar]
- 57.Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, et al. Etanercept for active Crohn's disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121(5):1088–94. doi: 10.1053/gast.2001.28674. [DOI] [PubMed] [Google Scholar]
- 58.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50(2):206–11. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papadakis KA, Shaye OA, Vasiliauskas EA, Ippoliti A, Dubinsky MC, Birt J, et al. Safety and efficacy of adalimumab (D2E7) in Crohn's disease patients with an attenuated response to infliximab. Am J Gastroenterol. 2005;100(1):75–9. doi: 10.1111/j.1572-0241.2005.40647.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. 5 mg/kg etanercept treatment does not effectively prevent body weight loss, inflammatory jaw bone and elbow joint destruction, and liver inflammation in Sh3bp2KI/KI mice compared to 25 mg/kg treatment. (A) Body weight changes in PBS- or etanercept-administered Sh3bp2+/+ and Sh3bp2KI/KI mice. 5 mg/kg etanercept administration (light-blue line) prevented weight loss in male Sh3bp2KI/KI mice compared to PBS-administered male Sh3bp2KI/KI mice (dark-blue line), but not in female Sh3bp2KI/KI mice. Numbers in parentheses represent the number of the mice weighed. (B) Reconstructed microCT images of the mandibular bone from 8-week-old Sh3bp2KI/KI mice treated with 5 mg/kg etanercept. Arrows indicate erosion pits due to inflammation. (C) Reconstructed microCT images of elbow joint from 5 mg/kg etanercept-administered Sh3bp2KI/KI mice, demonstrating the elbow joint destruction. (D) Representative liver tissue (H&E staining) from Sh3bp2KI/KI mice treated with 5mg/kg etanercept and quantitative measurement of total area of the inflammatory infiltrates in liver, showing less effectiveness of 5 mg/kg administration compared to 25 mg/kg. Arrows indicate inflammatory lesions. Error bars represent SEM in (A) and SD in (D). Asterisks represent significant difference (p < 0.05). N.S.: not significant. N.D.: not detected, n=6-9.
Supplemental Fig 2. Etanercept treatment decreases the number of RANKL-induced osteoclast formation in Sh3bp2KI/KI BMMs cultures. (A) TRAP staining of osteoclasts differentiated from Sh3bp2KI/KI BMMs stimulated with RANKL in the presence or absence of etanercept. (B) Quantitative analysis of the number of TRAP-positive multinucleated cells. n= 4/group. Asterisks represent significant difference (p < 0.001) compared to 0 μg/ml etanercept culture (one-way ANOVA, Tukey post-hoc analysis). N.S.: not significant.