

Abstract
Nanoscale vehicles for delivery have been of interest and extensively studied for two decades. However, the encapsulation stability of hydrophobic drug molecules in delivery vehicles and selective delivery into targeting disease cells is a potential hurdle for efficient delivery systems. Here, we demonstrate a simple and fast synthetic protocol of nanogels that shows high encapsulation stabilities. These nanogels can also be modified various targeting ligands for active targeting. We show that the targeting nanogels (T-NGs), which are prepared within 2 hours by a one-pot synthesis, exhibit very narrow size distributions and have the versatility of surface modification with cysteine-modified ligands including folic acid, cyclic arginine-glycine-aspartic acid (cRGD) peptide, and cell-penetrating peptide. T-NGs hold their payloads, undergo facilitated cell internalization by receptor-mediated uptake, and release their drug content inside cells due to the reducing intracellular environment. Selective cytotoxicity to cells, which have complementary receptors, is also demonstrated.
INTRODUCTION
The decoration of nanoscopic objects with ligands is of great interest due to implications in a variety of areas, including drug delivery and sensing.1-3 When such nanoscale platforms also exhibit the propensity to act as carriers for non-covalently loaded cargo, these implications are even greater. For example, ligand-modified nanocarriers that are capable of targeting specific cells hold great promise in therapeutic applications, such as cancer chemotherapy.4-5 Targeting mechanisms for nanocarriers in cancer chemotherapy can be broadly classified into two categories, active and passive. Passive targeting is based on the propensity of nanoscopic objects, with 10-200 nm sizes, to selectively accumulate in solid tumor tissue due to the increased permeability of the tumor vasculature and ineffective lymphatic drainage, which is known as the enhanced permeation retention (EPR) effect.6-7 Active targeting is achieved by coating the nanocarrier with ligands that exhibit high affinity for receptors that are overexpressed on the cancer cell surface.8-9 While either of these targeting approaches can be effective, the most successful approach will likely be achieved by combining the key features of both strategies.
Targeting ligands, such as folic acid, are widely studied as components of active targeting systems in the field of drug delivery.10-13 In many cases, these ligands are attached to the hydrophilic ends of assembly-forming amphiphilic molecules (e.g. block copolymers).14-16 However, the installation of such ligands onto these molecules requires complicated synthetic steps, limiting the versatility of ligand functionalization to tailor carriers for targeting specific cell types. Additionally, modification of the self-assembling molecules by this method could alter their hydrophilic-lipophilic balance (HLB) and introduce undesirable variations in assembly behavior, thus requiring optimization on a case-by-case basis. A class of drug delivery vehicle, which affords surface modification of chemically-crosslinked nanocarriers with reactive functional groups in a post-assembly step, can address these complications. In such a system, the nanocarrier morphology is maintained during modification, providing versatility in surface functionalization without compromising the structural integrity of assembly properties of the nanoscopic scaffold. It is additionally essential that the nanocarrier hosts stably encapsulate guest/drug molecules until they reach the site of the targeted cells.
We have recently reported on a crosslinked nanogel system that can preferentially address these needs.17-18 Briefly, this system is generated from self-crosslinking polymers that exploit disulfide exchange reactions to trap non-covalently bound guest molecules by a lock-in strategy. The template polymers, which form nanoscale aggregates in the aqueous phase, contain pyridyl disulfide (PDS) units to serve as both the lipophilic moieties and crosslinking functionalities. Initial sequestration of hydrophobic guest molecules is achieved within the lipophilic cores of these micelle-type polymer aggregates. However, the aggregates themselves are very leaky, permitting transient guest leakage into the solvent exterior and transfer to surrounding hydrophobic compartments.19 Intra/interchain disulfide crosslinking of the PDS functionalities that are buried in the lipophilic interiors of these amphiphilic nanoassemblies lock the drug molecules in the core, providing enhanced noncovalent encapsulation stability. While crosslinking is observed to improve encapsulation stability over the leaky amphiphilic polymer aggregates, it is noteworthy that the cellular uptake efficiencies of the crosslinked polymer nanogels are rather poor. However, this feature could be advantageous, providing an opportunity to tailor the nanogels with surface ligands that facilitate selective uptake by specific cells (Figure 1). In this paper, we report on a fast, one-pot synthesis of ligand-decorated nanogels. The resulting delivery vehicles exhibit high encapsulation stability to exploit passive targeting mechanisms and can be easily modified at their surfaces with various ligands for the active targeting of cell surface receptors. We show that the targeted nanogels (T-NGs), which are prepared within 2 hours by a one-pot synthesis, exhibit very narrow size distributions and can be simply prepared with cysteine-modified ligands including folic acid, cyclic arginine-glycine-aspartic acid (RGD) peptide, and cell-penetrating peptide. We demonstrate that the T-NGs hold their payloads, undergo facilitated cell internalization by receptor-mediated uptake, and release their drug content inside cells due to cleavage of crosslinked disulfide bonds by the reducing intracellular environment.20-23
Figure 1.
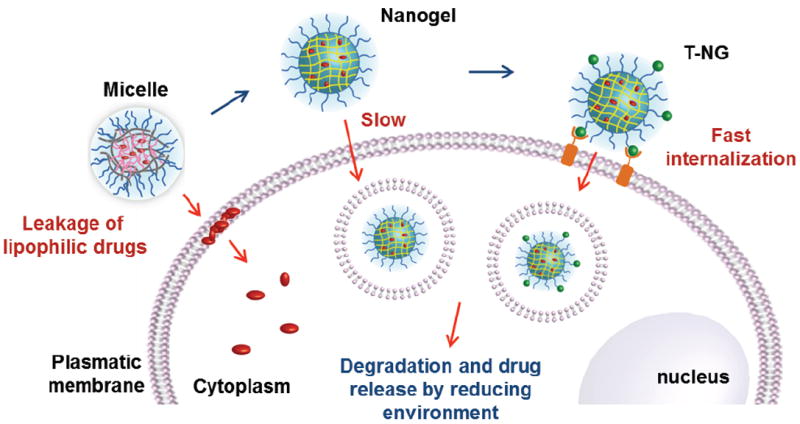
Nanogels are non-leaky, but exhibit slow celluar internalization; polymeric micelles exhibit rapid internalizations of lipophilic drug molecules due to leakage of free drug from the assembly interior; T-NGs exhibit high encapsulation stability and fast cellular internalization, which has a great potential for passive and active targeting drug delivery.
EXPERIMENTAL SECTION
General
2,2’-Dithiodipyridine, 2-mercaptoethanol, polyethylene glycol monomethyl ether methacrylate (MW 450), d,l-dithiothreitol (DTT), 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbo-cyanine perchlorate (DiI) and 3,3’-dioctadecyloxacarbocyanine perchlorate (DiO), folic acid, cyclo(Arg-Gly-Asp-d-Phe-Cys) peptide, 2-cyano-2-propyl benzodithioate and other conventional reagents were obtained from commercial sources and were used as received, unless otherwise mentioned. Polymers were synthesized using RAFT polymerization and then purified by precipitation. Pyridyl disulfide ethyl methacrylate (PDSEMA) was prepared using a previously reported procedure.24 1H-NMR spectra were recorded on a 400 MHz Bruker NMR spectrometer using the residual proton resonance of the solvent as the internal standard. Molecular weights of the polymers were estimated by gel permeation chromatography (GPC) using PMMA standard with a refractive index detector. Dynamic light scattering (DLS) measurements were performed using a Malvern Nanozetasizer. The fluorescence spectra were obtained from a JASCO FP-6500 spectrofluorimeter. Transmission electron microscopy (TEM) images were taken from JEOL 100CX at 100 KV.
Random copolymer
A mixture of 2-cyano-2-propyl benzodithioate (21.0 mg, 0.095 mmol), PDSEMA (860 mg, 3.37 mmol), polyethylene glycol monomethyl ether methacrylate (620 mg, 1.31 mmol) and AIBN (5.0 mg, 0.031 mmol) was dissolved in THF (3 mL) and degassed by performing three freeze-pump-thaw cycles. This reaction vessel was sealed and then put into a pre-heated oil bath at 60 °C for 12 h. To remove unreactive monomers, the resultant mixture was precipitated in cold diethylether (20 mL) to yield the random copolymer as a waxy liquid. GPC (THF) Mn: 23 K. PDI: 1.38. 1H NMR (400 MHz, CDCl3) δ: 8.45, 7.66, 7.09, 4.20-4.06, 3.90-3.36, 3.01, 2.15-1.62, 1.43-0.86. The molar ratio between two blocks was determined by integrating the methoxy proton in the polyethylene glycol unit and the aromatic proton in the pyridine and found to be 29%:71% (PEO:PDSEMA).
Cell penetrating peptide CRRR
Peptide was synthesized on 2-chloro-trityl chloride resin by solid phase synthesis using standard Fmoc methodology. Briefly, coupling reaction was achieved by 3 equiv. Fmoc protected amino acid, 3 equiv. HATU, and 3 equiv DIPEA in DMF and monitored by Kaiser Test. Fmoc protection group was removed by 20% piperidine in DMF. Cleavage of peptide from resin was performed in the presence of TFA/TIS/H2O/EDT mixture. Then cleavage mixture was precipitated in cold ether 5 times to afford crude peptide. Peptide was used without further purification. Yield: 80%. Peptide was characterized by 1H-NMR and mass spectrometry (see Supporting Information).
Cysteine-folic acid
The ligand was prepared on cysteine preloaded 2-chlorotrityl resin by solid phase synthesis. The resin was treated with 1.2 eq folic acid and 1.2 eq DIPEA in DMSO for 24 hours. The coupling reaction was repeated twice. Cleavage of cysteine-folic acid was done by treating the resin with TFA/TIS/H2O/EDT mixture. Crude product was obtained by precipitation in cold ether 5 times. Yield: 78%. Characterization was followed by 1H-NMR and mass spectrometry (see Supporting Information).
T-NGs preparation
The polymer (10 mg) was dissolved in water (1 mL) and hydrophobic dyes (0.1 mg of DiO or DiI) or 0.5 mg paclitaxel in acetone (100 μL) was added and the mixture was stirred for 6 h at room temperature, open to the atmosphere allowing the organic solvent to evaporate. It was placed in a vessel pre-heated at 60 °C for 10 min and then a measured amount of DTT was added. Then the mixture was stirred for 30 min to allow for crosslinking. To modify the nanogels surface, the ligand (5 mg), CRRR, cRGD, or cyteine-modified folic acid, was added and then stirred for another 1 h. The resulting nanogels were purified by filtration and unattached excess ligand was removed from the nanogel solution by ultrafiltration (triplicate) using a membrane with a molecular weight cutoff of 10,000 g/mol (Amicon Ultra cell-10K).
Crosslinking density and surface modification
In order to determine the crosslinking density, we first treated the polymer in water with the requisite amount of DTT (20 mol% compared to PDS groups), and stir the mixture at 60 °C. UV-visible spectroscopic measurements were performed with samples of this solution diluted ten times. Once this was measured, we calculated the amount of pyridothione based on its known molar extinction coefficient (8.08 × 103 M-1cm-1 at 343 nm).25 The percentage of cross-linking was calculated by assuming that formation of a single, crosslinking disulfide bond would require cleavage of two PDS units and produce two pyridothione molecules. The attachment of the ligand was evaluated by further increase in pyridothione absorption.
Cell culture
The cell viabilities of the nanogels were tested with 293T cells. 293T cells were cultured in T75 cell culture flasks using Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12) with 10% fetal bovine serum (FBS) supplement. The cells were seeded at 10,000 cells/well/200 μL in a 96 well plate and allowed to grow for 24 hours under incubation at 37 °C and 5% CO2. These cells were then treated with nanogels of different concentrations and were incubated for another 24 hours. Cell viability was measured using the Alamar Blue assay with each data point measured in triplicate. Fluorescence measurements were made using the plate reader SpectraMax M5 by setting the excitation wavelength at 560 nm and monitoring emission at 590 nm on a black well plate. The toxicity of paclitaxel (PTX)-encapsulated polymer aggregates, NG, and NG-RGD were tested against the MCF7 and HeLa cell line. The cells were treated with PTX-encapsulated polymer nanogels of 10 μM concentrations and were incubated for 72 hours. Cell death was measured by the Alamar Blue assay in triplicate.
Laser scanning confocal microscopy
The laser confocal experiment was performed with different cell lines. Each cell line was cultured in T75 cell culture flask containing DMEM/F12 with 10% FBS supplement. The cells were seeded at 10,000 cells/100 μL in cover slip-bottomed petri dishes and allowed to grow for 1 day at 37 °C in a 5% CO2 incubator. The cells in 2 mL of culture medium were treated with polymer aggregates, nanogels, or T-NGs (0.1 mg/mL) containing dyes and incubated at 37 °C for different time intervals before monitoring the cells by confocal microscopy.
RESULT AND DISCUSSION
Preparation of T-NG
T-NGs were prepared through the lock-in strategy using a random copolymer that contains oligoethylene glycol (OEG) units (29%) and pyridyldisulfide (PDS) moieties (71%) as side chain functionalities (Scheme 1). At elevated temperatures, the polymer formed larger aggregates (42 nm in diameter) than at room temperature (24 nm in diameter), likely due to intermolecular associations between the polymer chains caused by the lower critical solution temperature (LCST) behaviour of the OEG units (Figure 2a). We hypothesized that preparation at high temperatures would facilitate the crosslinking reaction and that subsequent addition of cysteine-containing ligands would enable a fast, one-pot T-NG synthesis. The crosslinking and surface modification reactions were observed to finish within a very short time period. Figure 2b, which traces production of the pyridothione byproduct of disulfide bond formation during nanogel synthesis, plateaus within 20 min. A deficient amount of dithiothreitol (DTT) (20 mol% against the precursor PDS groups) cleaved a corresponding amount of polymer PDS functionalities, generating free thiols that can then react with remaining PDS functionalities in the polymer chain (both intra- and inter-chain) to provide the crosslinked polymer nanogel. Based on pyridothione release, the actual crosslinking density was found to be 13-14% (39 mol% of PDS is consumed), which is very close to the theoretically calculated crosslinking density of 14%.
Scheme 1.

a) Schematic representation of the one-pot synthesis of T-NGs, b) chemical structure of polymer and T-NGs, and c) cysteine-containing targeting ligands.
Figure 2.
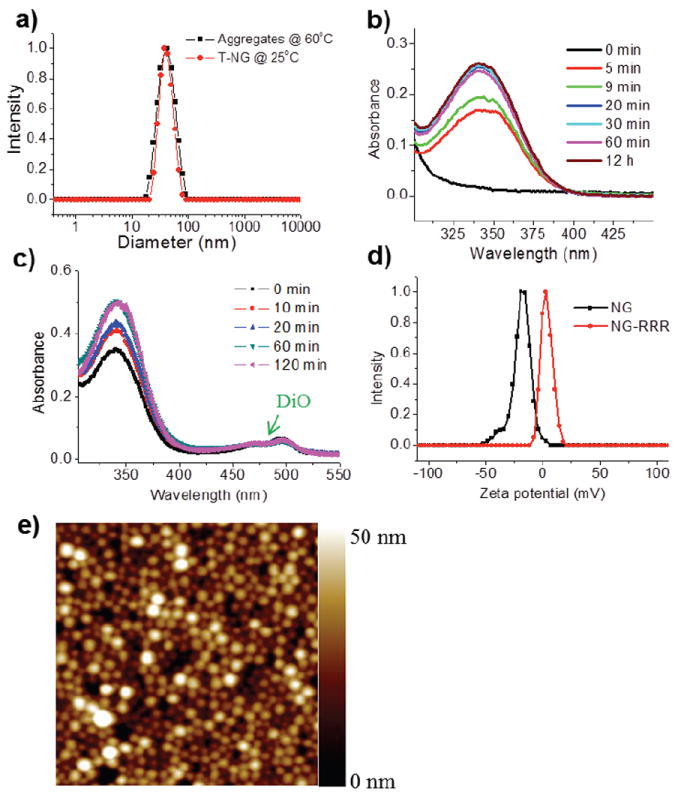
a) Dynamic light scattering of polymer aggregates (10 mg/mL) at 60 °C and T-NG (10 mg/mL) at room temperature. UV absorbance of pyridothione b) during nanogel synthesis (0.04 mg/mL) and c) ligand modification with CRRR (0.05 mg/mL). d) Zeta potential of nanogel (NG) (1 mg/mL) and NG-RRR (1 mg/mL). e) AFM image of T-NG (2 μm ×2 μm).
The cysteine-containing ligand, cysteine-triarginine (CRRR) peptide, was added to this reaction mixture to modify the surface and attachment of the ligand was evaluated by further increase in pyridothione absorption. The peak became saturated within 60 min and it was calculated that 17 mol% of total PDS is reacted with CRRR (Figure 2c). This calculation led us to estimate that there were about seven thousand ligands per T-NG (see Supporting Information). One can question whether such a thiol-containing ligand will cleave the crosslinking disulfide bonds, resulting in size change, nanogel disassembly, and leakage of hydrophobic guest molecules during ligand modification. We found that and T-NG retained their size from the polymer aggregate at high temperature (Figure 2a), and the absorption intensity of hydrophobic dye, 3,3’-dioctadecyloxacarbocyanine (DiO) which is encapsulated prior to crosslinking and surface modification was not changed, indicating that the nanogels were sufficiently stable to retain their guest molecules during this functionalization period (Figure 2c). We confirmed surface modification of the nanogels by monitoring the change of surface charge. As shown in Figure 2d, the nanogels modified with CRRR (NG-RRR) showed positive zeta potentials (+4 mV), while the nanogels showed negative zeta potentials (-20 mV) before surface modification. It is surprising that these nanogels exhibit negative zeta-potentials, because their surface is decorated with neutral poly(ethylene glycol) units. This is not well understood at this time. However, we also note that this is apparently not unusual, as similar observations have been previously reported.26-28 The resulting T-NGs showed narrow size distribution. Atomic force microscope (AFM) images reveal well-defined spherical structures with very uniform size. The size is around 50 nm in diameter, which may be ideal for the passive targeting applications.29 Additional ligands, including cysteine-modified folic acid and cysteine-containing cyclic arginine-glycine-aspartic acid (RGD) peptides, were also linked by this synthetic method to make T-NGs, NG-FA and NG-RGD, respectively (see Supporting Information).
Several features are noteworthy in the modified synthetic method, relative to our prior report17-18 on the nanogel synthesis: (i) the current method allows for obtaining these nanogels in very short amount of time; (ii) it also allows for decorating the nanogels with targeting ligands in a single pot and the precursor polymer is crosslinked into a nanogel and decorated with targeting ligands in less than two hours; (iii) the resultant nanogels exhibit narrow polydispersity; and (iv) these nanogels also exhibit enhanced encapsulation stability (vide infra).
Encapsulation stability
To investigate the noncovalent encapsulation stabilities of the T-NGs, we probed the dynamics of guest interchange in the nanocarriers using a previously reported fluorescence resonance energy transfer (FRET) experiment.30 Two lipophilic FRET pair dye molecules, 3,3’-dioctadecyloxacarbocyanine (DiO, donor) and 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI, acceptor), were independently encapsulated in the nanogels. In this experiment, mixing of the prepared solutions will either result in FRET development due to dye molecule interchange among leaky nanocarriers or lack of FRET development due to stable encapsulation of the dyes within the nanogel cores. While significant and rapid FRET evolution was observed in the case of the polymer aggregates, the crosslinked nanogels showed no significant peak shift, indicating their high encapsulation stabilities (Figure 3). Note that these nanogels stably encapsulate these molecules at relatively low crosslink densities.
Figure 3.

Fluorescence emission spectra of mixed polymer aggregates (a) and NG (b) separately encapsulated DiI/DiO.
Cellular internalization of polymer micelle and NG
Next, we were interested in investigating the differences in cellular internalization of the polymer aggregates, nanogels and T-NGs loaded with hydrophobic dye molecules. We hypothesized that any leakiness of the nanocarriers would result in the transport of their lipophilic cargo to surrounding cellular membranes due to the hydrophobic interactions. This would permit the cellular internalization of free dye prior to that of the dye molecule’s nanocarrier. To test this, we added the polymer aggregates and nanogels, containing DiO as a hydrophobic dye, into three different cell cultures (293T, MCF-7, and HeLa cell lines). The uptake was then monitored by tracing the dye’s fluorescence using confocal microscopy (Figure 4a-b). In the case of the polymer aggregates, intense green emission inside the cells was observed within 6h across all cell lines. In contrast, the nanogels showed no emission, indicating that the nanogels are not internalized in this short time period. This result suggests that the dyes from the leaky amphiphilic polymeric aggregates might be easily moved from the carrier to cellular membrane. To confirm this, we performed in situ FRET experiments with HeLa cells using the amphiphilic aggregates in which the FRET pair DiO (green emission) and DiI (red emission) was co-encapsulated. In this case, if the polymeric aggregates were internalized through the membrane, then FRET (red emission, 585-615 nm spectral filter) would be continually observed in the cytosolic interior within a short time period. After 3h incubation, we found that green (no FRET, 505-520 nm spectral filter) fluorescence is dominant inside the cell by confocal microscopy (ex = 488 nm), indicating that the two dyes are not close together within the assembly core, but that they have released from the micelles (Figure 4c). This indicates that liphophilic guests loaded inside micellar aggregates can be transferred into any cells, resulting in non-specific delivery. Thus, high encapsulation stability is essential to prevent premature, non-triggered release and achieve selective delivery of the drug molecules to target cells.
Figure 4.
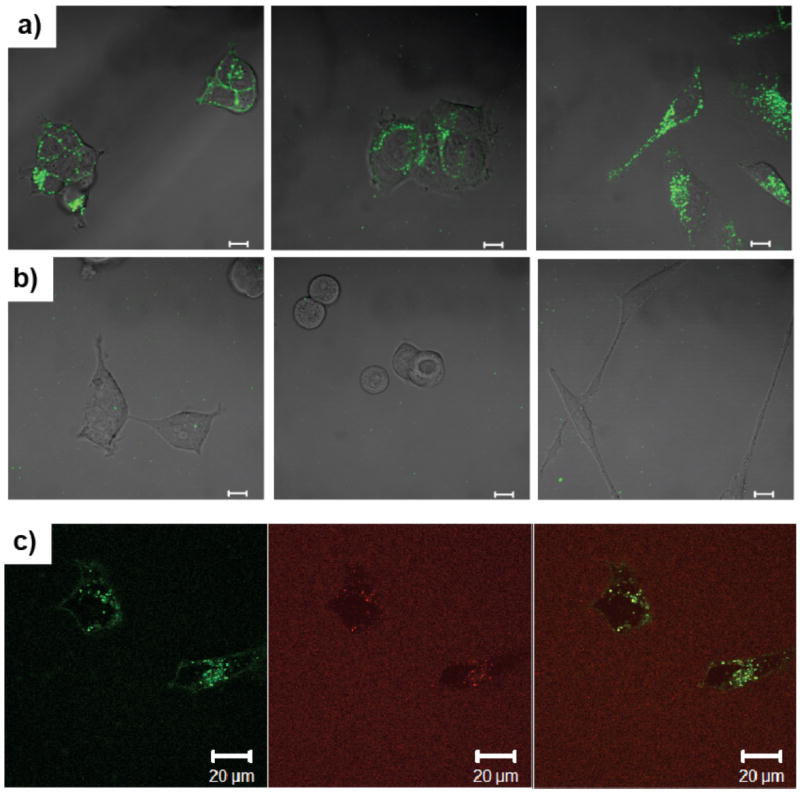
Cell internalization of DiO which is encapsulated in polymer aggregates (a) and NG (b) at different cell lines (left: 293T, middle: MCF7, right: HeLa, scale bar = 10 μm). (c) In situ cell internalization of hydrophobic dye by FRET (left: DiO channel, middle: DiI channel, right: overlap of both channel). No FRET inside cell indicates that the dyes only were transferred into the cells without polymer aggregates. Each cell line was cultured in DMEM/F12 with 10% FBS supplement.
Selective cellular internalization of T-NG
The crosslinked nanogels show highly stable encapsulation, avoiding undesirable drug leakage at off-target sites. However, the bare nanogel also suffers from very slow internalization. Such slow internalization seems to be a hurdle for drug delivery application at first sight. However, it may be that the delayed internalization of these nanogels poses an advantage in achieving selective uptake through active targeting. In an attempt to promote selective and rapid internalization into target cells, we decorated the surfaces of the slow-internalizing nanogels with specific ligands. To probe the versatility of ligand functionalization, we used three different ligands: CRRR, CRGD, and cysteine modified-folic acid. RGD is the ligand of the integrin αvβ3 which is involved in tumor angiogenesis and metastasis and is overexpressed in many solid tumor types such as breast, prostate, and ovarian cancers.31 Folic acid is one of the most widely used ligands in targeted delivery research, because folate receptors (FR) are notably overexpressed on the surfaces of many cancer cells such as ovarian, lung, and uterine tumors.32 RRR can be considered a positive control in this experiment, as this is a model cell penetrating peptide and is thus rapidly and non-selectively taken up by most cell types. These arginine-rich peptides are considered to be mimics of the well-known TAT peptides, which are known for translocating molecules and nanoscale objects across the cellular membrane.33 The nanogels which were modified with CRRR, CRGD, and cysteine-modified-folic acid are referred to as NG-RRR, NG-RGD, and NG-FA, respectively. To investigate target selectivity, we explored cellular uptake in varied cell lines: 293T kidney cells are normal cells used as a control, MCF7 is a slightly FR-positive tumor cell line and is negative to RGD, while SKOV3 and HeLa cells are positive to both ligands.
To see selective cellular internalization by receptor-mediated endocytosis, ligand-decorated nanogels encapsulating DiO as a model hydrophobic drug molecule were incubated with various cell lines for 6h and then qualitatively examined through confocal microscopy (Figure 5). In the case of NG-RRR, high green emission was observed inside cells in all cell lines within just 2h, suggesting that the nanogels were internalized rapidly because of the highly positive cell penetrating peptide, RRR. In the case of NG-FA, the nanogels exhibit fast cellular uptake with SKOV3, HeLa and MCF7 cells in which FR is overexpressed. However, the FR-negative 293T cells show negligible cellular uptake of the folic acid decorated nanogel. Similarly, with NG-RGD, HeLa and SKOV3 exhibit high internalization, as these cells are considered to overexpress integrin αvβ3. However, MCF7 and 293T cells, which are negative to integrin, show little internalization. Taken together, these results strongly support the selective internalization of the various T-NGs through receptor mediated endocytotic pathways. Thus this system has great potential as a targeted drug delivery carrier.
Figure 5.
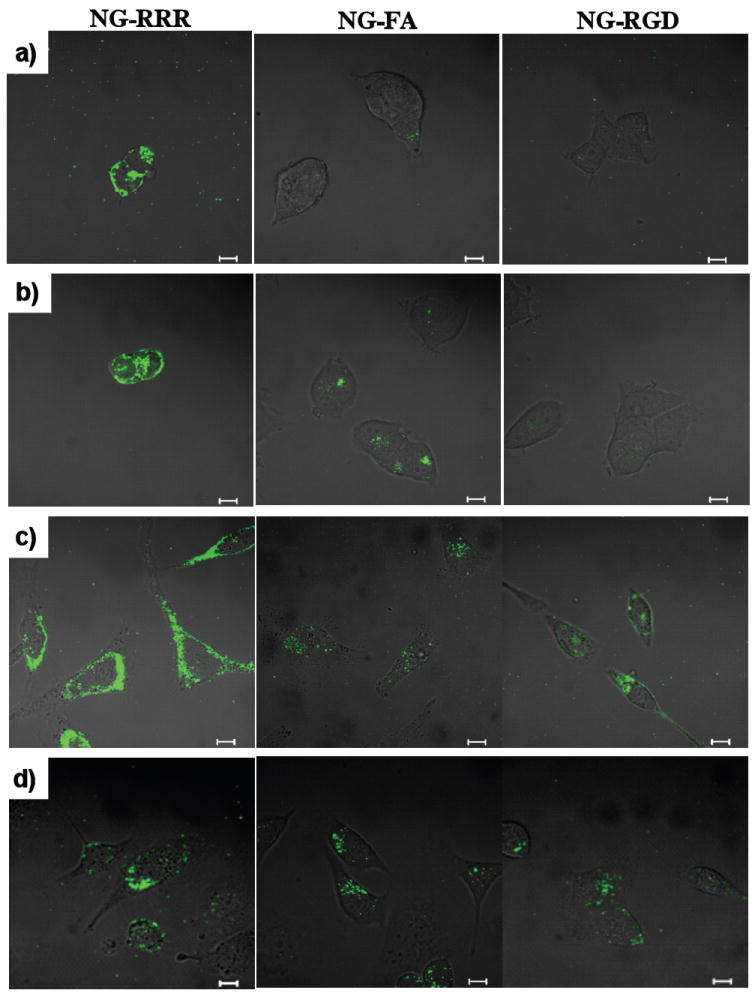
Confocal microscopy images of T-NGs containing DiO at different cell lines, a) 293T, b) MCF7, c) HeLa, and d) SKOV3. (left: NG-RRR, middle: NG-FA, right: NG-RGD, scale bar = 10 μm). Each cell line was cultured in DMEM/F12 with 10% FBS supplement.
Selective delivery of chemotherapeutic drug
Finally, we were interested in investigating whether the observed target selectivity would translate to differential cell kill efficiency of these ligand-coated nanogels encapsulating a chemotherapeutic drug molecule, paclitaxel (PTX). The PTX-loaded polymeric aggregates (polymer-PTX), NG (NG-PTX) and NG-RGD (NG-RGD-PTX) were added to MCF7 cells and HeLa cells and the extent of cell death was investigated after 72 hours. As shown in Figure 6, free PTX and polymer-PTX were highly toxic in both cell lines. This supports our hypothesis that the polymeric micellar aggregates have a leaky nature, which leads to free drug being translocated through the cellular membrane. In contrast, NG-PTX showed less toxicity, compared to both free-PTX and polymer-PTX. This is consistent with the observed slow internalization of nanogels, which should result in a reduced amount of chemotherapeutic inside the cells. Remarkably, NG-RGD-PTX showed different cell killing efficiency in two different cell lines. While MCF7 showed similar toxicity with NG-PTX, HeLa showed high toxicity that is similar to free-PTX, indicative of the selective delivery of PTX to this cell line. Together with the cellular internalization experiments, this result demonstrates that NG-RGD is highly internalized into the integrin receptor overexpressing HeLa cells, thus enhancing the efficiency of chemotherapeutics by promoting selective drug delivery.
Figure 6.
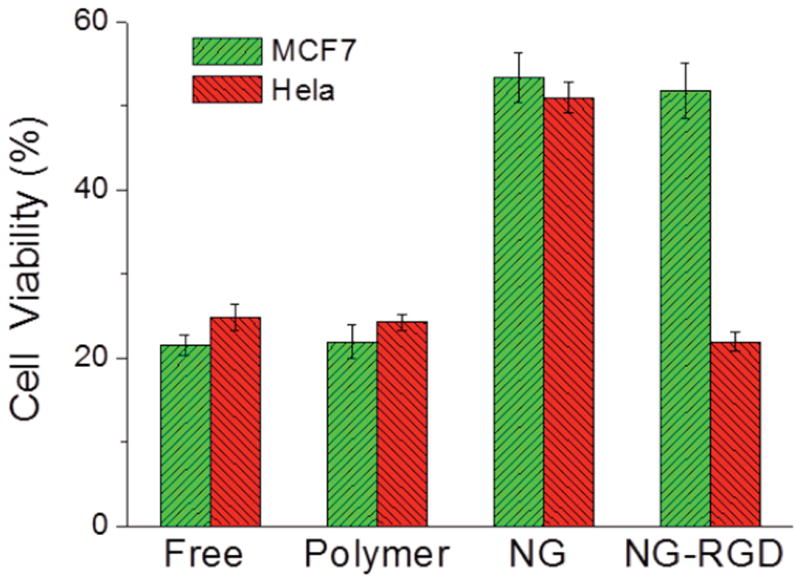
In vitro toxicity of PTX-loaded NG-RGD with MCF7 and HeLa cells after 72h incubation. The loading amount of PTX is 5 wt% for polymer and NG. 10 μM of PTX is used for all experiments.
CONCLUSIONS
In summary, we describe here a facile synthetic method for the preparation of nanogels. We show that: (i) these nanogels can be prepared in under two hours from their polymeric precursor; the reaction time includes the ligand decoration step; (ii) these nanogels exhibit high encapsulation stability of lipophilic guest molecules; (iii) the facile ligand functionalization possibility can be utilized to decorate these nanogels with cell targeting ligands; (iv) while the unfunctionalized nanogels are taken up very poorly by various cells, the ligand-decorated nanogels exhibit facilitated receptor-dependent cellular uptake, as demonstrated by selective uptake of RGD- and folic acid- decorated nanogels by cells overexpressing integrin and folate receptors; (v) functionalization of the nanogels with cell penetrating peptides caused rapid non-specific uptake by the cells, independent of the receptor; (vi) the selective internalization capability can be translated to delivering a chemotherapeutic drug molecule specifically to a specific receptor-rich cell. Overall, the reported versatile one-pot synthetic method for synthesizing the ligand-decorated nanogels, combined with the intrinsic encapsulation stability and targeting capabilities of the formed T-NGs, should open up new avenues in targeted drug delivery for crosslinked polymer nanogels.
Supplementary Material
Acknowledgments
This work was partially supported by DARPA and the NSF Center for Hierarchical Manufacturing. We are also supported by the NSF-MRSEC facility at the University of Massachusetts for the infrastructure used in characterizations of the materials.
Footnotes
Supporting Information
Spectral characterization data, turbidity experiment and plot of the absorption maximum of pyridothione, plot of FRET ratio vs. time, UV-vis absorbance of T-NG, and in vitro toxicity of polymer, empty nanogels, RGD-coated nanogels. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kabanov AV, Vinogradov SV. Angew Chem Int Ed. 2009;48:5418–5429. doi: 10.1002/anie.200900441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan R. Nat Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 3.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 4.Brannon-Peppas L, Blanchette JO. Adv Drug Delivery Rev. 2004;56:1649–1659. doi: 10.1016/j.addr.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 5.Davis ME, Chen ZG, Shin DM. Nat Rev Drug Discovery. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 6.Baban DF, Seymour LW. Adv Drug Delivery Rev. 1998;34:109–119. doi: 10.1016/s0169-409x(98)00003-9. [DOI] [PubMed] [Google Scholar]
- 7.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. J Controlled Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 8.Allen TM. J Controlled Release. 2002;2:750–763. [Google Scholar]
- 9.Byrne JD, Betancourt T, Brannon-Peppas NL. Adv Drug Delivery Rev. 2008;60:1615–1626. doi: 10.1016/j.addr.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 10.Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA, Gao J. Angew Chem Int Ed. 2004;43:6323–6327. doi: 10.1002/anie.200460800. [DOI] [PubMed] [Google Scholar]
- 11.Lee ES, Kim D, Youn YS, Oh KT, Bae YH. Angew Chem Int Ed. 2008;47:2418–2421. doi: 10.1002/anie.200704121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aluri S, Janib SM, Mackay JA. Adv Drug Delivery Rev. 2009;61:940–952. doi: 10.1016/j.addr.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudimack J, Lee RJ. Adv Drug Delivery Rev. 2000;41:147–161. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 14.Sutton D, Nasongkla N, Blanco E, Gao J. Pharm Res. 2007;24:1029–1046. doi: 10.1007/s11095-006-9223-y. [DOI] [PubMed] [Google Scholar]
- 15.Yoo HS, Park TG. J Controlled Release. 2004;96:273–283. doi: 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Xu P, Van Kirk EA, Zhan Y, Murdoch WJ, Radosz M, Shen Y. Angew Chem Int Ed. 2007;46:4999–5002. doi: 10.1002/anie.200605254. [DOI] [PubMed] [Google Scholar]
- 17.Ryu J-H, Jiwpanich S, Chacko R, Bickerton S, Thayumanavan S. J Am Chem Soc. 2010;132:8246–8247. doi: 10.1021/ja102316a. [DOI] [PubMed] [Google Scholar]
- 18.Ryu J-H, Chacko RT, Jiwpanich S, Bickerton S, Babu RP, Thayumanavan S. J Am Chem Soc. 2010;132:17227–17235. doi: 10.1021/ja1069932. [DOI] [PubMed] [Google Scholar]
- 19.Chen H, Kim S, Li L, Wang S, Park K, Cheng J-X. Proc Natl Acad Sci U S A. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vader P, van der Leonardus J, Engbersen JFJ, Storm G, Schiffelers RM. Pharm Res. 2011;28:1013–1022. doi: 10.1007/s11095-010-0344-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin C, Engbersen JFJ. Expert Opin Drug Deliv. 2009;6:421–439. doi: 10.1517/17425240902878010. [DOI] [PubMed] [Google Scholar]
- 22.Bronich TK, Ouyang M, Kabanov VA, Eisenberg A, Szoka FC, Jr, Kabanov AV. J Am Chem Soc. 2002;124:118720–11873. doi: 10.1021/ja020509p. [DOI] [PubMed] [Google Scholar]
- 23.Kim JO, Sahay G, Kabanov AV, Bronich TK. Biomacromolecules. 2010;11:919–926. doi: 10.1021/bm9013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh S, Basu S, Thayumanavan S. Macromolecules. 2006;39:5595–5597. [Google Scholar]
- 25.Kavimandan NJ, Losi E, Wilson JJ, Brodbelt JS, Peppas NA. Bioconjugate Chem. 2006;17:1376–1384. doi: 10.1021/bc050344k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas AG, Ithakissios DS. J Controlled Release. 2002;79:123–135. doi: 10.1016/s0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 27.Li Y, Pei Y, Zhang X, Gu Z, Zhou Z, Yuan W, Zhou J, Zhu J, Gao X. J Controlled Release. 2001;71:203–211. doi: 10.1016/s0168-3659(01)00218-8. [DOI] [PubMed] [Google Scholar]
- 28.Sezgin Z, Yüksel N, Baykara T. Eur J Pharm Biopharm. 2006;64:261–268. doi: 10.1016/j.ejpb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 29.Chithrani BD, Chan WCW. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 30.Jiwpanich S, Ryu J-H, Bickerton S, Thayumanavan S. J Am Chem Soc. 2010;132:10683–10685. doi: 10.1021/ja105059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haubner R, Gratias R, Diefenbach B, Goodman SL, Jonczyk A, Kessler H. J Am Chem Soc. 1996;118:7461–7472. [Google Scholar]
- 32.Lu Y, Low PS. Adv Drug Delivery Rev. 2002;54:675–693. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 33.Rothbard JB, Jessop TC, Wender PA. Adv Drug Delivery Rev. 2005;57:495–504. doi: 10.1016/j.addr.2004.10.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.