

Abstract
Doxorubicin (Dox) is an indispensable chemotherapeutic agent for the treatment of various forms of neoplasia such as lung, breast, ovarian, and bladder cancers. Cardiotoxicity is a major concern for patients receiving Dox therapy. Previous work from our laboratory indicated that glucocorticoids (GCs) alleviate Dox-induced apoptosis in cardiomyocytes. Here we have found Glucocorticoid-Induced Leucine Zipper (GILZ) to be a mediator of GC-induced cytoprotection. GILZ was found to be induced in cardiomyocytes by GC treatment. Knocking down of GILZ using siRNA resulted in cancelation of GC-induced cytoprotection against apoptosis by Dox treatment. Overexpressing GILZ by transfection was able to protect cells from apoptosis induced by Dox as measured by caspase activation, Annexin V binding and morphologic changes. Western blot analyses indicate that GILZ overexpression prevented cytochrome c release from mitochondria and cleavage of caspase-3. When bcl-2 family proteins were examined, we found that GILZ overexpression causes induction of the pro-survival protein Bcl-xL. Since siRNA against Bcl-xL reverses GC induced cytoprotection, Bcl-xL induction represents an important event in GILZ-induced cytoprotection. Our data suggest that GILZ functions as a cytoprotective gene in cardiomyocytes.
Keywords: Steroids, nuclear receptor, cell death, Bcl-xL, caspase-3, chemotherapy
Introduction
Doxorubicin (Dox) is an anthracyclin often used for cancer chemotherapy. Treatment of breast cancer, solid tumors, tissue sarcoma and aggressive lymphoma requires Dox (Keizer et al., 1990; Gewirtz, 1999; Chatterjee et al., 2010). A major side effect of Dox treatment is cardiac toxicity. Acute cardiac toxicity is presented as arrhythmias. The regiment and administering protocols for Dox have been improved to reduce acute cardiac toxicity. However, certain patients develop cardiomyopathy months or years after Dox treatment (Lipshultz et al., 1991; Krischer et al., 1997; Swain et al., 2003; Lipshultz et al., 2005). On average, about 48% of adult patients develop congestive heart failure at a cumulative dose of 700 mg/m2 and above (Swain et al., 2003; Chatterjee et al., 2010). Children and elderly patients are at a higher risk for Dox-induced cardiomyopathy (Lipshultz et al., 1991; Krischer et al., 1997; Lipshultz et al., 2005). Early studies indicate that the plasma concentration of Dox ranges from 5 to 0.1 μM following 15-min infusion of 75 mg/m2 dose in patients during chemotherapy (Greene et al., 1983). A recent study reassured that the concentration of Dox in the blood reaches over 1 μM in breast cancer patient during Dox therapy (Barpe et al., 2010). Clinically relevant doses of Dox (0.5–5 μM) have been shown to induce apoptosis in primary cultured cardiomyocytes (Tobin and Abbott, 1980; Sawyer et al., 1999). Continued loss of myocytes due to apoptosis contributes to the development of cardiomyopathies and heart failure as a result of Dox treatment. Reducing cardiotoxicity of Dox is important for increasing the lifespan of patients.
Glucocorticoids (GCs) have been used as an anti-emetic agent for patients receiving chemotherapy and are also a key component of combination therapy for lymphoma, leukemia and myeloma. The chemotherapeutic effect of GCs is related to the fact that GCs are a potent inducer of apoptosis in lymphoid-derived cells. Additional uses of GCs during cancer therapies involve the anti-inflammatory effect of GCs for cranial metastasis, anti-hypercalcemic effect and the ability to suppress tumor or chemotherapy related fevers (Herr and Pfitzenmaier, 2006; Sommer and Ray, 2008; Chen and Sun, 2009).
We have found that GCs elicit dose-dependent protection against Dox-induced apoptosis in primary cultured cardiomyocytes (Chen et al., 2005). Affymetrix microarray analyses of GC-treated cardiomyocytes revealed that 140 genes were up-regulated and 108 genes were down regulated by GC treatment (Chen et al., 2005). Among the genes upregulated by GCs are Bcl-xL and Glucocorticoid Induced Leucine Zipper (GILZ). GC-induced cytoprotection was attributed in part to the induction of the pro-survival Bcl2 family member Bcl-xL, since siRNA against Bcl-xL was able to cancel the cytoprotective effect of GCs (Chen et al., 2005).
GILZ was first identified from a thymus cDNA library during investigation of glucocorticoid induced genes (D’Adamio et al., 1997). The protein does not contain a nuclear localization signal and has been found to localize only in the cytosol (D’Adamio et al., 1997). Although GILZ may not function as a transcription factor by itself, the protein has been shown to regulate transcription through interactions with transcription factors and signaling molecules, including c-Fos, NF-kB, Ras and Raf (Riccardi et al., 1999; Ayroldi and Riccardi, 2009). GILZ was described as playing a role in mediating T-cell receptor (TCR) induced cell death in lymphocytes, T-lymphocyte activation, T-helper cell differentiation and dendritic cell function (Ayroldi et al., 2001; Delfino et al., 2004; Cohen et al., 2006; Ayroldi et al., 2007; Hamdi et al., 2007). While it has been demonstrated that GCs induce GILZ protein and elicit cytoprotection in cardiomyocytes, the functional role of GILZ in cardiomyocytes has not been determined. Here we investigate whether GILZ serves as a mediator of GC-induced cytoprotection.
Materials and Methods
Cell Culture and Treatment of Drugs
H9c2 cells were obtained from American Type Culture Collection and were kept in culture by weekly subculture in DMEM containing 10% fetal bovine serum (FBS) and 2 mM glutamine. Cells were seeded into 6-well plates at 3×105 cells per well and grown to 80% confluency before Dox treatment for 24 hr. The cells were treated with 1 μM corticosterone (CT) for 8 to 24 hr before Dox treatment.
siRNA Transfection
GILZ siRNA was generated via in vitro transcription according to Donze and Picard (Donze and Picard, 2002) with a sequence of 5′-(GAGGGUAUUCUCACGCUCCAGCUGCGA)-3′. The same method was used to generate Bcl-xL siRNA with a sequence of 5′-(GGCUGGGCGAUGAGUUUGAATT)-3′. The RNA (1 μg) was mixed with 4 μl of X-tremeGENE siRNA transfection reagent (Roche) and incubated for 1 hr in DMEM without FBS or supplements before being added to cells in 6-well plates at 70% confluency for overnight incubation.
Transient or Stable Transfection of GILZ
For transient transfection, cells seeded in 6-well plates were transfected at 30–50% confluency with 100 μL of serum-free medium containing 1 μg of DNA (pcDNA3 vector) with 4 μL Fugene 6 transfection reagent (Roche). Cells were incubated for 24 hr at which point the medium was changed to fresh DMEM containing 10% FBS and 1% penicillin/streptomycin. GILZ measurements or experiments were carried out at 48 hr after the transfection. H9c2 cells stably expressing GILZ or empty pcDNA3 vector were generated by transfection using Lonza system (Basel, Switzerland) and nucleofection “L” kit with 2 μg of Bam HI (Fermentas) linearized pcDNA3 containing full length mouse GILZ or without insert following the manufacturer’s protocol. Transfected cells in 6-well plates were grown to 70% confluency before transferring to a 10 cm dish containing DMEM supplemented with 500 μg/mL Geneticin (G418, GIBCO), 10%FBS and 1% penicillin/streptomycin. GILZ expression was verified after 4 subcultures over 4 weeks with culture medium containing 500 μg/mL G418.
Caspase Activity Assay
In 6-well plates, detached cells were collected by centrifugation at 3000 ×g for 5 min and subsequently combined with adherent cells scraped off from the same well. The cells were dissolved in 200 μl of lysis buffer (0.5% Nonidet P-40, 0.5 mM EDTA, 150 mM NaCl, 50 mM Tris pH 7.5) for measurements of caspase activity using 40 μM N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methyl coumarin (N-acetyl-DEVD-AMC; Alexis Biochemical’s, San Diego, CA) as the substrate. The released AMC was quantified as relative fluorescence units using a 96-well fluorescence plate reader (Biotek Synergy 2) with an excitation wavelength of 365 nm and an emission wavelength of 450 nm.
Annexin V Binding
H9c2 cells were seeded onto coverglass slides in 24-well plates. Detached cells in the supernatant were collected by 5 min centrifugation at 1000 r.p.m (500 ×g). After washing the detached cells and adherent cells with PBS, detached cells were combined with their corresponding group of cells remained adherent to the coverglass after addition of the labeling solution (10 mM HEPES/NaOH, pH.7.4, 140 mM NaCl, 5 mM CaCl2). Annexin V-FLOUS (Roche Applied Science, Indianapolis, IN) was added to the sample at 25 μl per well. Annexin V-positive cells were counted under an Olympus fluorescent microscope.
Western Blot
Cells in 100-mm dishes or 6-well plates were lysed by scraping in a lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM N3VO4, 1 μg/mL leupeptin; Cell Signaling, Boston MA). Protein concentration was measured by the Bradford method (Bio-Rad, Hercules, CA) for SDS-polyacrylamide gel electrophoresis (12% gel). After transferring the proteins to a PVDF membrane at 60V for 3 hr at 4°C, the membrane was blocked in 10% milk/TBS-tween solution for 2 hr. The membrane was then incubated with antibodies against GILZ, p21, or Bax (Santa Cruz Biotechnology) at a 1:200 dilution, Bcl-xL (Cell Signaling) at a 1:1000 dilution, or Vinculin (Sigma-Aldrich) at a 1:1000 dilution. The bound antibodies were detected using an Enhanced Chemiluminescence Reaction after blotting the membrane with the secondary antibodies conjugated with horseradish peroxidase. To separate cytosolic versus mitochondrial fractions, cells were collected in fractionation buffer (250 mM sucrose, 20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.2% Triton X-100, pH 7.4) and lysed by passing through a 25 gauge needle 10 times. The nuclear fraction and intact cells were removed by centrifugation at 720 ×g for 10 min at 4°C. The supernatant was collected and centrifuged at 10,000 ×g for 10 min at 4°C. The supernatant was collected as the cytosolic fraction, whereas the pellet was resuspended in fractionation buffer by passing through a 25 gauge needle 10 times. After another round of centrifugation at 10,000 ×g for 10 min at 4°C, the pellet was collected as the mitochondrial fraction.
MTT Assay for Cell Viability
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) (Sigma-Aldrich) was added to cells in 6-well plates to a final concentration of 0.5 mg/ml in culture medium. After 20-min incubation in a 37°C tissue culture incubator, culture medium was removed, and the resulting formazan crystals were dissolved in isopropanol. The solubilized formazan was quantified by a spectrophotometer for absorbance at a wavelength of 570 nm.
Real-Time RT-PCR
Total RNA extracted using TRIzol was used as templates for RT-PCR. cDNA synthesis was performed using a commercial cDNA synthesis kit (Fermentas) with random hexamers. PCR primers were purchased from Integrated DNA Technologies (San Diego, CA) with the sequences of 5′-AGCTGAACAACATAATGCGCCAGG-3′ (forward) and 5′-ATCTTGTTGTCTAGGGCCACCACA-3′ (reverse) for GILZ (108.7% primer efficiency, R2=0.985); 5′-CCCCAGAAACTGAACCA-3′ (forward) and 5′-AGTTTACCCCATCCCGAAAG-3′ (reverse) for Bcl-xL (102.4% primer efficiency, R2=0.997); 5′-AGCCATGTACGTAGCCATCC-3′ (forward) and 5′-CTCTGAGCTGTGGTGGTGAA-3′ (reverse) for β-actin (99.7% primer efficiency, R2=1.00); 5′-TCAACTTTCGATGGTAGTCGCCGT-3′ (forward) and 5′-TCCTTGGATGTGGTAGCCGTTTCT-3′ (reverse) for 18S rRNA (92.9% primer efficiency, R2=1.00); and 5′-CCTCTCTCTTGCTCTCAGTAT-3′ (forward) and 5′-GTATCCGTTGTGGATCTGACA-3′ (reverse) for GAPDH (102.5% primer efficiency, R2=1.00). Primer efficiency for each primer set was determined from 6 dilutions covering 4 log values of cDNA from control H9c2 samples. C(t) values (Y-axis) were plotted against cDNA concentrations (X-axis). All primer sets were found to fit a linear curve as shown in R2 values. Efficiency (E) is calculated by 10(−1/slope) and %E is calculated by (E − 1) × 100%.
Quantitative real time PCR was performed with the CFX96 Thermal Cycler (Bio-Rad) and Cyber Green dye (Fermentas), with initial denaturation at 95ºC for 10 min and 40 cycles of 95ºC for 15 sec for denaturation, 60ºC for 30 sec for annealing and 72ºC for 30 sec for extension. Melting curve analysis was performed at the end of PCR to verify the specificity of the product. Bio-Rad CFX Manager software was used for data analyses. The expression values were calculated by Relative Quantity (RQ) gene = Egene[CT(control)−CT(sample)] and Normalized Expression sample = RQsample/(RQreference 1 × RQreference 2 × RQreference 3)(1/3), where E = efficiency of primer set calculated by (% Efficiency × 0.01) + 1, with 100% efficiency = 2. Normfinder software was used to assess the stability of the reference genes. The stability values (M) for GAPDH (M=0.128), 18S rRNA (M=0.068), and β-actin (M=0.070) were within the acceptable stability range (M<0.150). Three technical replicates of each biological sample were performed and averaged in order to calculate relative quanitity and normalized expression for each sample.
Statistical Analysis
Results were compared using one-way analysis of variance (ANOVA) with Bonferroni post hoc test for comparison of multiple groups. For caspase assay, apoptotic cell counting, or MTT assay, the raw data from each experiment were used for ANOVA analysis. For RT-PCR and Western blot band intensity, the raw values were normalized to corresponding loading control before ANOVA. A p-value of <0.05 was considered significant.
Results
GILZ Is Essential for Corticosteroid-Induced Cytoprotection
A recent study from our laboratory indicates that corticosteroids induce GILZ in the mouse myocardium and H9c2 cells resemble primary cultured rat cardiomyocytes in regards to GILZ expression by GCs, thereby making H9c2 cells a serviceable in vitro model for studying the function of GILZ in cardiomyocytes (Aguilar et al., 2013). We investigated the role of GILZ in GC-induced cytoprotection by knocking down GILZ expression with siRNA. GILZ siRNA was able to prevent CT from inducing GILZ protein (Fig 1A). Following siRNA mediated knockdown, H9c2 cells were treated with 1 μM CT 24 hr prior to Dox treatment. The negative control, i.e. scrambled siRNA, had no effect on CT ability to protect against Dox-induced apoptosis as measured quantitatively by caspase activity, while H9c2 cells transfected with GILZ siRNA had a slightly higher basal caspase activity compared to control cells (p<0.05, Fig 1B). Furthermore, siRNA knockdown of GILZ abolished CT potential to attenuate Dox-induced caspase activation (Fig 1B), implying that GILZ is important for CT-mediated cytoprotection. We have previously shown that Bcl-xL is necessary for GC-induced cytoprotection and siRNA against Bcl-xL was able to reduce the level of Bcl-xL protein (Chen et al. 2005). With siRNA against Bcl-xL as a positive control, we found GILZ siRNA was as effective as Bcl-xL siRNA for reversing the protective effect of CT (Fig 1B). Therefore our data suggest that GILZ mediates CT induced cytoprotection.
Figure 1. GILZ Mediates CT Induced Cytoprotection.
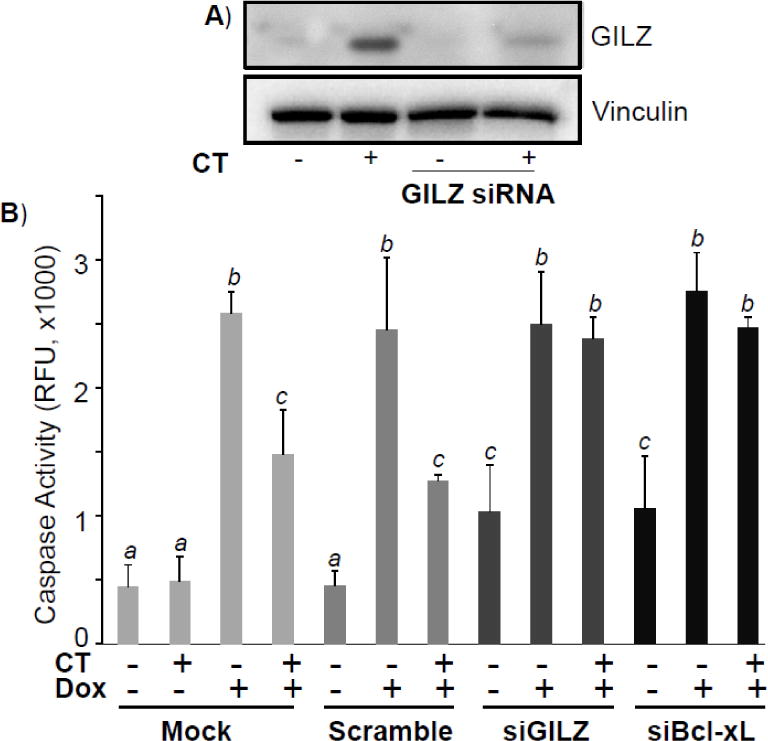
H9c2 cells were transfected with siRNA against GILZ, Bcl-xL, or scrambled control for 24 hr before corticosterone (CT, 1 μM) treatment for 8 hr. Cells were harvested for measurements of GILZ protein by Western blot with vinculin as a loading control (A). Alternatively, cells were treated with doxorubicin (Dox, 0.75 μM) for 24 hr before harvesting for measurements of caspase 3 activity (B). Results are from one experiment representative of three independent experiments (A) or means ± standard deviations from three independent experiments with triplicated samples for each experiment (B). The raw data from three independent experiments were used for statistic analyses by ANOVA. Different letters indicate significant differences between treatment groups (p<0.05). Therefore, the means labeled with “a” are significantly different from that labeled with “b” or “c”, while “b” indicates significant differences from the means labeled with “a” or “c”.
GILZ Protects Against Doxorubicin Induced Apoptosis
To address whether GILZ expression alone can alleviate Dox-induced apoptosis, a stable transfectant of H9c2 cells was established with constitutively elevated expression, approximately 3.5 fold increase of GILZ. Without GILZ transfection, H9c2 cells undergoing apoptosis show morphological changes including rounding up and detaching (Fig 2A). When apoptotic cells were quantified under a microscope, GILZ overexpression significantly reduced the number of apoptotic cells by Dox treatment (p<0.05, Fig 2B). Annexin V binding assay has been used to quantify the fraction of apoptotic cells. Compared to mock transfected controls, cells stably transfected with GILZ exhibited a decrease in Annexin V fluorescence in response to Dox treatment. Cells quantified by fluorescent microscopy indicated a significant reduction in Annexin V staining in H9c2 cells stably expressing GILZ (p<0.05, Fig 2C). Caspase 3 activity was measured to further demonstrate the protective effect of GILZ expression. As shown in Fig 3A, mock or empty vector transfected cells demonstrated a dose dependent increase of caspase activity following Dox treatment. Maximal caspase activity in empty vector and mock transfected cells was observed at 0.5 μM and 0.75 μM concentrations of Dox. Higher doses of Dox caused less caspase activation than preceding doses due to overwhelming toxicity leading to the induction of more necrosis and less apoptosis. Cells stably transfected with GILZ were resistant to Dox-induced caspase activation, exhibiting a reduction in caspase activity compared to empty vector and mock transfected cells at Dox concentrations up to 0.75 μM (p<0.05, Fig 3A). The time course of caspase activation by Dox treatment over 48 hr indicates that GILZ expression did not delay caspase activation, but indeed reduced Dox-induced caspase activation throughout the time course (Fig 3B). Maximum caspase activation occurred at 24 hr after Dox treatment in mock and empty vector cells, when the caspase activity was significantly reduced by GILZ transfection compared to mock and empty vector transfected cells (p<0.05, Fig 3B). Significant inhibition of caspase activation was also observed at 16 and 36 hr (p<0.05, Fig 3B).
Figure 2. GILZ Protects Against Dox-Induced Cell Death.
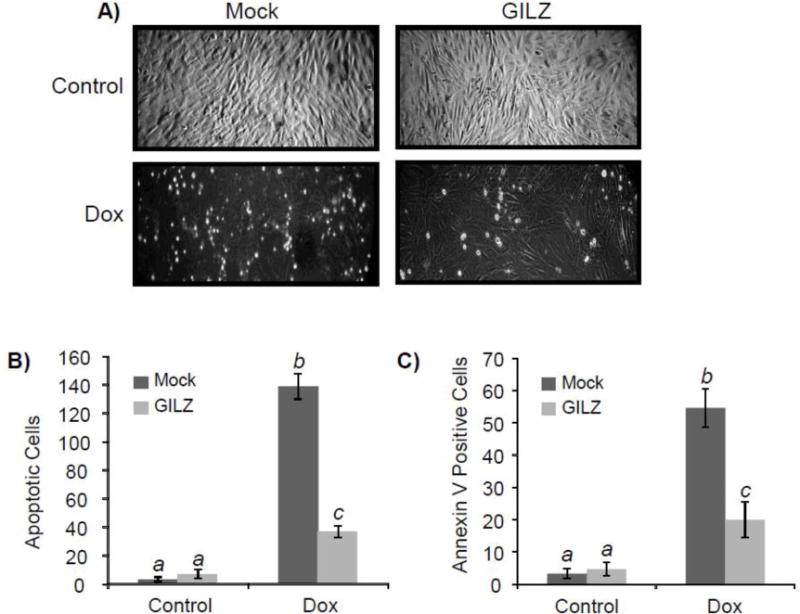
H9c2 cells stably transfected with or without pcDNA-GILZ were treated with Dox (0.75 μM) for 24 hr. The image of the cultured cells was recorded under a phase contrast microscopy (A). The number of apoptotic cells was quantitated by counting in three distinct fields under the microscope (B). Annexin V-Fluo was added to cells for fluorescence microscopy. The number of Annexin V positive cells was quantified by counting in three distinct fields under a fluorescence microscope (C). Results are from one experiment representative of three independent experiments (A) or are expressed as means ± standard deviations from three independent experiments with triplicated samples for each experiment (B, C). The raw data from three independent experiments were used for statistic analyses by ANOVA. Different letters indicate significant differences between treatment groups (p< 0.05). Therefore, the data groups labeled with “a” are significantly different from that labeled with “b” or “c”, while “b” indicates significant differences from the data groups labeled with “a” or “c”.
Figure 3. GILZ Protects Against Dox-Induced Caspase Activation.
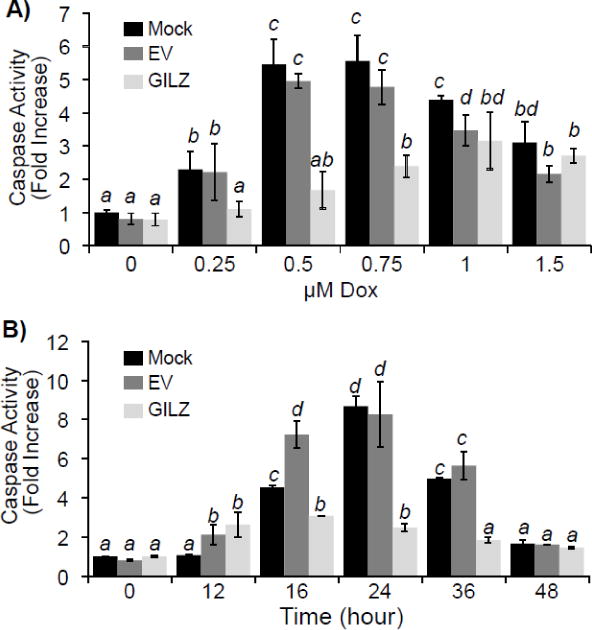
H9c2 cells stably transfected with empty vector (EV), GILZ, or without vector (Mock) were treated with Dox at the indicated doses for 24 hr (A) or with 0.75 μM Dox for the indicated times (B). Cells were collected for measurements of Caspase 3 activity. Results are expressed as means ± standard deviations from four independent experiments with triplicated samples for each experiment. The raw data from four independent experiments were used for statistic analyses by ANOVA. Different letters indicate significant differences between treatment groups (p<0.05). Therefore, the means labeled with “a” are significantly different from that labeled with “b”, “c” or “d”, while “b” indicates significant differences from the means labeled with “a”, “c” or “d”. The means labeled with two letters such as “ab” are not significant different from those labeled with either letter “a” or “b”.
We further verified an inhibition of Dox-induced apoptosis by measuring cleaved Caspase-3 and changes in cytosolic cytochrome c via Western blot analysis. The cells overexpressing GILZ exhibited a reduced Caspase-3 cleavage (p<0.05) in response to Dox treatment (Fig 4), consistent with the results of caspase activity assay. Measurements of cytosolic cytochrome c due to release from mitochondria as a result of Dox treatment also demonstrated that cells expressing GILZ released cytochrome c into the cytosol to a much lesser degree than mock or empty vector transfected cells treated with Dox (p<0.05, Fig 4).
Figure 4. GILZ Inhibits Dox-Induced Caspase Cleavage and Mitochondrial Release of Cytochrome c.
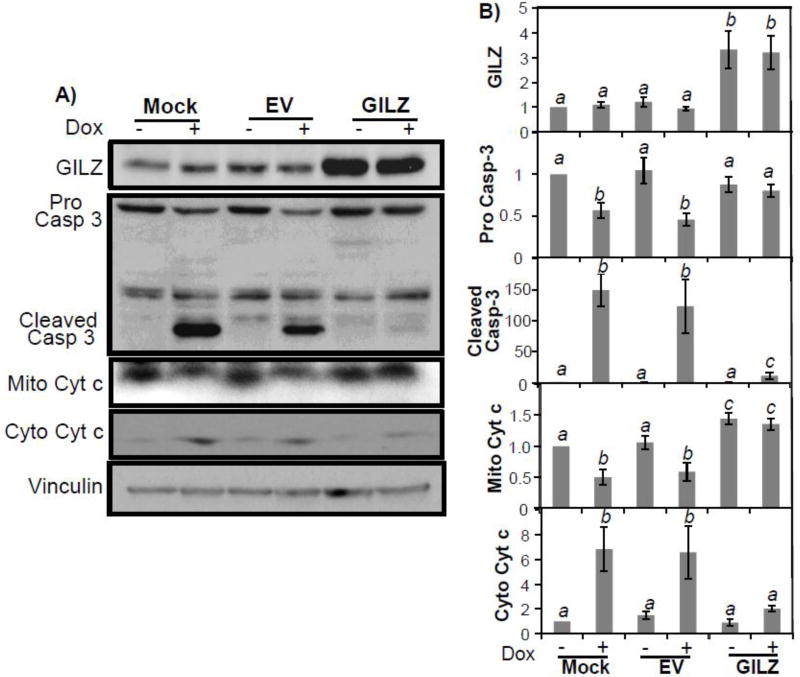
H9c2 cells stably transfected with empty vector (EV), GILZ, or no vector (Mock) were challenged with Dox (0.75 μM) for 24 hr. Cells were collected for measurements of cleaved Caspase 3 (Casp-3) and cytochrome c (Cyt c) release from mitochondria by Western blot. The data are from one experiment representative of three (A). Bar graphs represent means ± standard deviations of band intensity normalized to vinculin from 3 independent experiments with mock control being set at value “1” (B). The normalized data from three independent experiments were used for statistical analyses by ANOVA. Different letters indicate significant differences between treatment groups (p<0.05). Therefore, the means labeled with “a” are significantly different from that labeled with “b” or “c”, while “b” indicates significant differences from the data groups labeled with “a” or “c”.
The cytotoxic effects of Dox include inhibition of metabolism and induction of apoptosis or necrosis. Reduction of mitochondrial succinate dehydrogenase activity is a general measurement of inhibition of metabolism, which usually occurs in parallel or prior to apoptotic or necrotic cell death. To address whether GILZ protects cardiomyocytes against cytotoxicity of Dox in general, we performed MTT assay. Both Dox dose response and time course studies demonstrate the protective effect of GILZ expression against Dox induced loss of mitochondrial activity (Fig 5). Dox treatment caused a dose-dependent decrease in MTT formazin conversion (p<0.05) in mock and empty vector transfected cells beginning at 0.25 μM Dox (Fig 5A), This reduction in MTT formazin conversion was evident by 12 hr after Dox treatment (p<0.05) and continued for at least 48 hr (Fig 5B). Compared to mock or empty vector transfected cells, GILZ transfected cells were resistant to Dox-induced reduction of MTT formazin conversion at all doses and time points tested (p<0.05, Fig 5A&B). These data suggest that the observed ability of GILZ to reduce Dox-induced caspase activation is a result of cytoprotection and not due to a shift toward non-apoptotic cell death pathways.
Figure 5. GILZ Preserves Cell Viability as Measured by MTT Assay.
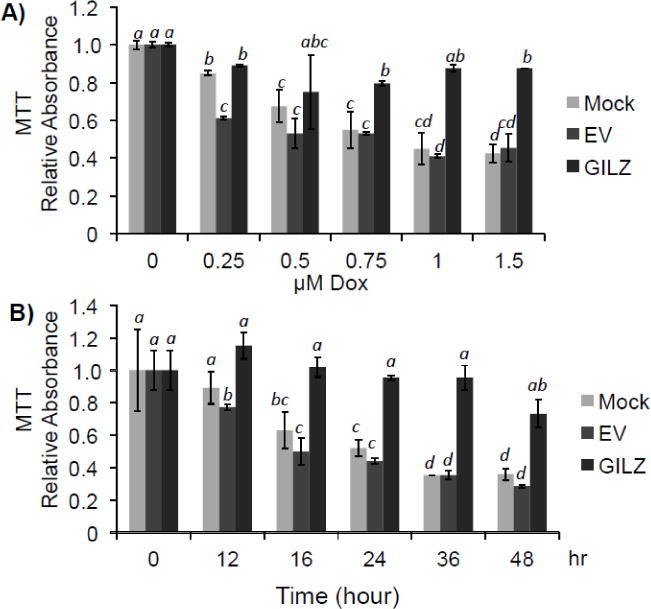
H9c2 cells stably transfected with empty vector (EV), GILZ, or no vector (Mock) were challenged with Dox at the indicated doses for 24 hr (A) or with 0.75 μM Dox for the indicated times (B). Cell viability was determined by quantifying the ability to convert 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide into formazan. Results are expressed as means ± standard deviations from four independent experiments with triplicated samples for each experiment. The raw data from four independent experiments were used for statistical analyses by ANOVA. Different letters indicate significant differences between treatment groups (p<0.05). Therefore, the data groups labeled with “a” are significantly different from that labeled with “b”, “c” or “d”, while “b” indicates significant differences from the means labeled with “a”, “c” or “d”. The means labeled with two letters such as “ab” are not significantly different from those labeled with either letter “a” or “b”. The mean labeled with three letters “abc” is not significantly different from those labeled with letter “a”, “b” or “c”.
GILZ Expression Causes an Increase of Bcl-xL Protein
Bcl-2 family members play an important role in determining cell death by apoptosis. We measured the level of bcl-2 family members in H9c2 cells transfected with empty vector or GILZ. Cells transfected with GILZ exhibited elevated levels of the anti-apoptotic protein Bcl-xL compared to mock or empty vector transfected cells (p<0.05, Fig 6). Bcl-2 protein levels were decreased in GILZ transfected cells (p<0.05), whereas Bax and Bak protein levels remained unchanged (Fig 6). To demonstrate that GILZ transfection did not produce cellular stress and therefore a possible preconditioning effect as a means of cytoprotection, we measured p21 protein levels. p21 is a cyclin dependent kinase inhibitor that is elevated in response to stress and has been shown to reduce apoptosis in some cells (Chen et al., 2000). We found that GILZ expression did not appear to increase p21 protein levels (Fig 6). These data suggest that GILZ specifically induces Bcl-xL, providing a plausible mechanism of protection against Dox-induced apoptosis.
Figure 6. Expression of Bcl-2 Family Members in GILZ Transfected Cells.
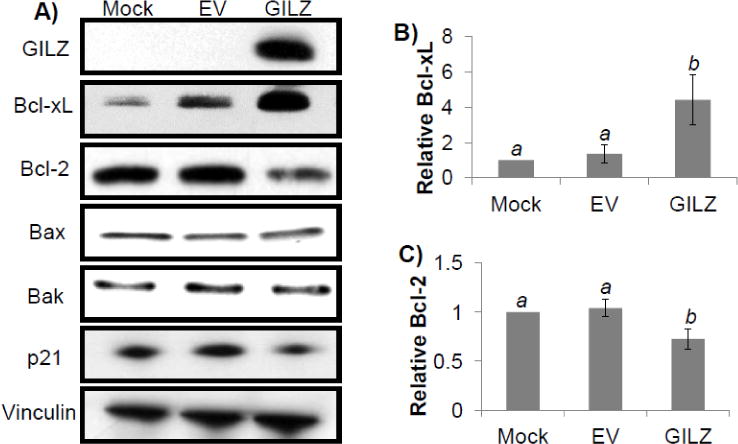
H9c2 cells from transiently transfected empty vector (EV) or GILZ vector were collected at 48 hr after transfection for Western blot analyses to determine the levels of Bcl-2 family members and p21, with vinculin as a loading control. The data are from one experiment representative of three (A). Bar graphs represent means ± standard deviations of band intensity normalized to vinculin from 3 independent experiments with the data from mock transfected cells being set at value “1” (B, C). The normalized data from three independent experiments were used for statistical analyses by ANOVA. Different letters indicate significant differences between treatment groups (p<0.05). Therefore, the means labeled with “a” are significantly different from that labeled with “b”.
We explored whether GILZ induces Bcl-xL through transcriptional or posttranscriptional means by measuring the level of Bcl-xL mRNA. Real time PCR was performed using three reference genes, GAPDH, 18S rRNA and β-actin. Normfinder analysis indicates that all three genes are stable enough for use as reference genes (Andersen et al., 2004). Whereas CT treatment causes elevated expression of Bcl-xL gene as reported (Chen et al., 2005), serving as a positive control, GILZ transfected cells did not exhibit elevated levels of Bcl-xL mRNA compared to cells transfected with empty vector (Fig 7). Therefore elevation of Bcl-xL protein due to GILZ expression is regulated at posttranscriptional levels.
Figure 7. GILZ Does Not Induce Bcl-xL mRNA.
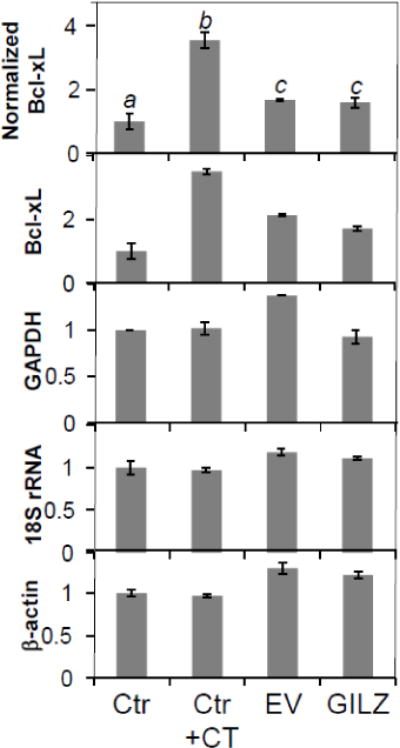
qRT-PCR analysis was used to determine Bcl-xL mRNA levels in H9c2 cells treated with corticosterone (CT, 1 μM) or stably transfected with empty vector (EV) or GILZ. Bcl-xL mRNA levels were normalized to three reference genes (GAPDH, 18S rRNA, and β-actin) as described in the Methods (A). The relative quantity of each gene is shown (B, C, D, E). Results are expressed as means ± standard deviations from three independent experiments with triplicated samples for each experiment. Different letters indicate significant differences between treatment groups (p<0.05) as determined by ANOVA. Therefore, the data groups labeled with “a” are significantly different from that labeled with “b” or “c”.
Discussion
The data presented here suggest that GILZ acts as a GC-inducible inhibitor of apoptosis in cardiomyocytes. siRNA knockdown of GILZ reversed the cytoprotective effect of CT, while overexpression of GILZ protected H9c2 cells against Dox-induced apoptosis, as measured by cell detachment, Annexin V staining, caspase activation, mitochondrial release of cytochrome c and MTT assay. The results from caspase activity assays were confirmed by Western blot analyses measuring Caspase-3 cleavage. Our data indicate that overexpression of GILZ is sufficient for eliciting cytoprotection. The observation that GILZ induces Bcl-xL protein elevation provides an explanation for the underlying mechanism of GILZ induced cytoprotection.
Bcl-xL is a well-known pro-survival gene of the bcl-2 family and normally resides on the outer membrane of the mitochondria. Bcl-xL helps to maintain mitochondrial membrane integrity, thereby preventing the release of cytochrome c from the mitochondria into the cytoplasm, where cytochrome c binds to Apaf-1 to trigger the formation of the apoptosomes. We have previously reported that Bcl-xL plays a critical role in GC-induced cytoprotection in cardiomyocytes (Chen et al., 2005). Our data here demonstrating elevated levels of Bcl-xL protein as a result of GILZ overexpression is consistent with the role of Bcl-xL in cell survival. The protective effects of Bcl-xL in cardiomyocytes have been established, as overexpression of Bcl-xL or perfusion of the heart with a peptide containing BH4 sequence of Bcl-xL reduces ischemia-reperfusion injury in rat hearts (Chen et al., 2002; Huang et al., 2003). Interestingly, GILZ overexpression caused a reduction in Bcl-2 protein levels in H9c2 cells (Fig 6). Although Bcl-2 is also an anti-apoptotic protein, Bcl-xL has an overlapping function with Bcl-2. It has been demonstrated that Bcl-xL is 10-times more potent than Bcl-2 in inhibiting Dox-induced apoptosis (Fiebig et al., 2006). This may explain the protective effect of GILZ overexpression despite the decrease in Bcl-2 protein.
The cytoprotective effect of GILZ is likely cell type dependent. In lymphocytes, GILZ induced a decrease in Bcl-xL (Delfino et al., 2004). Lack of Bcl-xL induction in HeLa and HEK293 cells (Fig 8) supports a cell type dependent effect of GILZ. GILZ has been previously reported to play dual roles in apoptosis of the immune system: mediating GC induced apoptosis in a sub-set of thymocytes, yet rescuing thymocytes from TCR induced cell death (Riccardi et al., 1999; Ayroldi and Riccardi, 2009; Beaulieu and Morand, 2011). Transgenic mice overexpressing GILZ in the thymocytes exhibit a phenotype resembling GC treatment, including apoptosis of T-lineage cells (Delfino et al., 2004). In contrast, TCR-induced cell death is inhibited in these transgenic animals (Delfino et al., 2006). This cell-type specific effect of GILZ mirrors the actions of GCs, which can act as an inducer of cell survival or cell death depending on the cell type.
Figure 8. GILZ Does Not Induce Bcl-xL Protein in HEK293 or HeLa Cells.
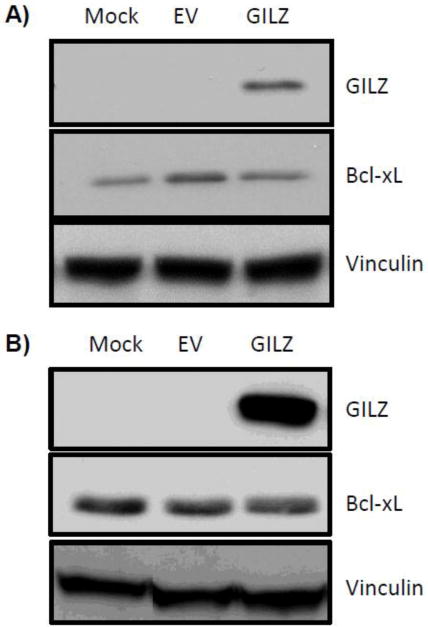
Empty vector (EV) or GILZ expression vector was introduced into HEK293 (A) or HeLa (B) cells by transient transfection. Cells were harvested 48 hr after transfection for Western blot to measure levels of GILZ, Bcl-xL or the loading control vinculin. The results are representative from two (A) or three (B) independent experiments.
Interestingly, GCs cause an elevation of Bcl-xL mRNA and Bcl-xL protein in cardiomyocytes (Chen et al., 2005), whereas GILZ overexpression induces Bcl-xL protein in the absence of increased Bcl-xL mRNA. This suggests that GCs initiate two independent mechanisms that converge to induce an elevation of Bcl-xL protein levels: 1) transcriptional activation of Bcl-xL gene and 2) induction of GILZ, which in turn causes elevation of Bcl-xL protein via a posttranscriptional mechanism. Since GCs elevated Bcl-xL mRNA and protein levels, but GILZ overexpression was only able to affect Bcl-xL protein levels, it would suggest that GILZ may play only a partial role in GC-induced protection against Dox. However, GC induces prolonged elevation of GILZ (Aguilar et al., 2013) and knockdown of GILZ with siRNA completely abolished GC-induced inhibition of caspase activation comparable to knockdown of Bcl-xL. This argues that maintenance of Bcl-xL protein elevation by GILZ is just as important as transcriptional activation of Bcl-xL for cell survival.
How GILZ causes an increase of Bcl-xL protein remains to be elucidated. GILZ protein may interact with Bcl-xL protein physically, since GILZ has been reported to interact with several proteins, including c-Fos, NF-kB, Raf and Ras. GILZ protein contains a central Leucine Zipper domain, an N-terminal domain with a Tuberous Sclerosis Complex (TSC) box; and a C-terminal domain with a proline and glutamic acid rich (PER) region. The central Leucine Zipper domain can promote homodimerization or heterodimerization with other leucine zipper proteins such as c-Fos (Mittelstadt and Ashwell, 2001). The C-terminal PER domain of GILZ can bind and inhibit NF-κB activity (Riccardi et al., 2001; Di Marco et al., 2007). The N-terminal domain of GILZ can bind to Raf, whereas the TSC box allows GILZ to interact with Ras (Ayroldi et al., 2002; Ayroldi et al., 2007). An examination of the physical interaction between Bcl-xL and GILZ may offer an insight as to how GILZ regulates Bcl-xL protein elevation. Bcl-xL has been previously reported to interact with proteins containing PER domain (Hammond et al., 2001). Sequence alignment of GILZ C-terminal PER domain with reported PER domain containing proteins supports a potential interaction of GILZ with Bcl-xL protein (Fig 9).
Figure 9. Alignment of GILZ PER domain with Bcl-xL Interacting Protein PER Splicing Factor.

GILZ protein sequence was searched against Proline Glutamic Acid Rich domains using CLUSTAL2.1 software. Three proteins were found to exhibit homology with PER domain of GILZ as shown.
Our finding of GILZ as a cytoprotective gene provides a new avenue for further investigation of methods to protect the heart from Dox cardiotoxicity. While GC-induced cytoprotection in cardiomyocytes has been demonstrated, their potential use as a pharmacological agent for cardiac protection remains controversial. A recent report supports that short term GC treatment is safe and is associated with improvement in congestion, neurohormonal status and renal function in heart failure patients (Massari et al., 2012). However, prolonged GC administration has been reported to cause cardiac hypertrophy, myocardial fibrosis, hypoxia, and ventricular dysfunction (Roy et al., 2009). Furthermore, disruption of calcium kinetics has been reported to contribute to the deleterious effect of prolonged GC treatment (De et al., 2011). Additionally, GC administration is not recommended for heart failure patients due to increased sodium and fluid retention. Clearly it remains to be addressed whether or not GILZ is cardiac protective gene. Such work is important since GILZ may provide a lead for designing new pharmacological therapies of cardiac protection to circumvent undesirable issues with GCs.
Highlights.
Corticosteroids act as a cytoprotective agent in cardiomyocytes
Corticosteroids induce GILZ expression in cardiomyocytes
Elevated GILZ results in resistance against apoptosis induced by doxorubicin
GILZ induces Bcl-xL protein without inducing Bcl-xL mRNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar DC, Strom J, Xu B, Kappeler K, Chen QM. Expression of Glucocorticoid Induced Leucine Zipper (GILZ) in Cardiomyocytes. Cardiovasc Tox. 2013;13:91–99. doi: 10.1007/s12012-012-9188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andeersen CL, Jensen JL, Orntoft TF. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, D’Adamio F, Riccardi C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001;98:743–753. doi: 10.1182/blood.v98.3.743. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009;23:3649–3658. doi: 10.1096/fj.09-134684. [DOI] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest. 2007;117:1605–1615. doi: 10.1172/JCI30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002;22:7929–7941. doi: 10.1128/MCB.22.22.7929-7941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barpe DR, Rosa DD, Froehlich PE. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur J Pharm Sci. 2010;41:458–463. doi: 10.1016/j.ejps.2010.07.015. [DOI] [PubMed] [Google Scholar]
- Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheumatol. 2011;7:340–348. doi: 10.1038/nrrheum.2011.59. [DOI] [PubMed] [Google Scholar]
- Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–162. doi: 10.1159/000265166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- Chen Q, Alexander D, Sun H, Xie L, Lin Y, Terrand J, Morrissy S, Purdom S. Corticosteroids Inhibit Cell Death Induced by Doxorubicin in Cardiomyocytes: Induction of Anti-apoptosis, Antioxidant and Detoxification Genes. Mol Pharm. 2005;67:1861–1873. doi: 10.1124/mol.104.003814. [DOI] [PubMed] [Google Scholar]
- Chen Q, Liu J, Merrett J. Apoptosis or Senescence-Like Growth Arrest: Influence of Cell Cycle Position, p53, p21 and bax in H2O2Response of Normal Human Fibroblasts. Biochem J. 2000;347:543–551. doi: 10.1042/0264-6021:3470543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun H. Glucocortidoid Pharmacotoxicological Interactions and Modulation of Toxicity. In: Harvey P, Everett D, Springall C, editors. Adrenal Toxicology. Informa; New York: 2009. pp. 205–230. [Google Scholar]
- Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, Capel F, Balian A, Naveau S, Galanaud P, Lemoine FM, Emilie D. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. 2006;107:2037–2044. doi: 10.1182/blood-2005-07-2760. [DOI] [PubMed] [Google Scholar]
- D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997;7:803–812. doi: 10.1016/s1074-7613(00)80398-2. [DOI] [PubMed] [Google Scholar]
- De P, Roy SG, Kar D, Bandyopadhyay A. Excess of glucocorticoid induces myocardial remodeling and alteration of calcium signaling in cardiomyocytes. J Endocrinol. 2011;209:105–114. doi: 10.1530/JOE-10-0431. [DOI] [PubMed] [Google Scholar]
- Delfino DV, Agostini M, Spinicelli S, Vacca C, Riccardi C. Inhibited cell death, NF-kappaB activity and increased IL-10 in TCR-triggered thymocytes of transgenic mice overexpressing the glucocorticoid-induced protein GILZ. Int Immunopharmacol. 2006;6:1126–1134. doi: 10.1016/j.intimp.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood. 2004;104:4134–4141. doi: 10.1182/blood-2004-03-0920. [DOI] [PubMed] [Google Scholar]
- Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, Donato V, Migliorati G, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007;35:517–528. doi: 10.1093/nar/gkl1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donze O, Picard D. RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase. Nucleic Acids Res. 2002;30:e46. doi: 10.1093/nar/30.10.e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig AA, Zhu W, Hollerbach C, Leber B, Andrews DW. Bcl-XL is qualitatively different from and ten times more effective than Bcl-2 when expressed in a breast cancer cell line. BMC Cancer. 2006;6:213. doi: 10.1186/1471-2407-6-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharm. 1999;57:727–741. doi: 10.1016/s0006-2952(98)00307-4. [DOI] [PubMed] [Google Scholar]
- Greene RF, Collins JM, Jenkins JF, Speyer JL, Myers CE. Plasma pharmacokinetics of adriamycin and adriamycinol: implications for the design of in vitro experiments and treatment protocols. Cancer Res. 1983;43:3417–3421. [PubMed] [Google Scholar]
- Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, Lemoine FM, Zou W, Emilie D. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood. 2007;110:211–219. doi: 10.1182/blood-2007-08-107896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond PW, Alpin J, Rise CE, Wright M, Kreider BL. In vitro selection and characterization of Bcl-X(L)-binding proteins from a mix of tissue-specific mRNA display libraries. J Biol Chem. 2001;276:20898–20906. doi: 10.1074/jbc.M011641200. [DOI] [PubMed] [Google Scholar]
- Herr I, Pfitzenmaier J. Glucocorticoid use in prostate cancer and other solid tumours: implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncol. 2006;7:425–430. doi: 10.1016/S1470-2045(06)70694-5. [DOI] [PubMed] [Google Scholar]
- Huang J, Ito Y, Morikawa M, Uchida H, Kobune M, Sasaki K, Abe T, Hamada H. Bcl-xL gene transfer protects the heart against ischemia/reperfusion injury. Biochem Biophys Res Comm. 2003;311:64–70. doi: 10.1016/j.bbrc.2003.09.160. [DOI] [PubMed] [Google Scholar]
- Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Therapeutics. 1990;47:219–231. doi: 10.1016/0163-7258(90)90088-j. [DOI] [PubMed] [Google Scholar]
- Krischer JP, Epstein S, Cuthbertson DD, Goorin AM, Epstein ML, Lipshultz SE. Clinical cardiotoxicity following anthracycline treatment for childhood cancer: the Pediatric Oncology Group experience. J Clin Oncol. 1997;15:1544–1552. doi: 10.1200/JCO.1997.15.4.1544. [DOI] [PubMed] [Google Scholar]
- Lipshultz SE, Colan SD, Gelber RD, Perez-Atayde AR, Sallan SE, Sanders SP. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. New Engl J Med. 1991;324:808–815. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, Colan SD. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. 2005;23:2629–2636. doi: 10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- Massari F, Mastropasqua F, Iacoviello M, Nuzzolese V, Torres D, Parrinello G. The glucocorticoid in acute decompensated heart failure: Dr Jekyll or Mr Hyde? Am J Emerg Med. 2012;30:517 e515–510. doi: 10.1016/j.ajem.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. 2001;276:29603–29610. doi: 10.1074/jbc.M101522200. [DOI] [PubMed] [Google Scholar]
- Riccardi C, Bruscoli S, Ayroldi E, Agostini M, Migliorati G. GILZ, a glucocorticoid hormone induced gene, modulates T lymphocytes activation and death through interaction with NF-κB. Advances Exp Med Biol. 2001;495:31–39. doi: 10.1007/978-1-4615-0685-0_5. [DOI] [PubMed] [Google Scholar]
- Riccardi C, Cifone MG, Migliorati G. Glucocorticoid hormone-induced modulation of gene expression and regulation of T-cell death: role of GITR and GILZ, two dexamethasone-induced genes. Cell Death Diff. 1999;6:1182–1189. doi: 10.1038/sj.cdd.4400609. [DOI] [PubMed] [Google Scholar]
- Roy SG, De P, Mukherjee D, Chander V, Konar A, Bandyopadhyay D, Bandyopadhyay A. Excess of glucocorticoid induces cardiac dysfunction via activating angiotensin II pathway. Cell Physiol Biochem. 2009;24:1–10. doi: 10.1159/000227803. [DOI] [PubMed] [Google Scholar]
- Sawyer DB, Fukazawa R, Arstall MA, Kelly RA. Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ Res. 1999;84:257–265. doi: 10.1161/01.res.84.3.257. [DOI] [PubMed] [Google Scholar]
- Sommer P, Ray DW. Novel therapeutic agents targeting the glucocorticoid receptor for inflammation and cancer. Curr Opin Investig Drugs. 2008;9:1070–1077. [PubMed] [Google Scholar]
- Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer. 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- Tobin TP, Abbott BC. A stereological analysis of the effect of adriamycin on the ultrastructure of rat myocardial cells in culture. J Mol Cell Cardiol. 1980;12:1207–1225. doi: 10.1016/0022-2828(80)90066-8. [DOI] [PubMed] [Google Scholar]
- Xu B, Strom J, Chen QM. Dexamethasone induces transcriptional activation of Bcl-xL gene and inhibits cardiac injury by myocardial ischemia. Eur J Pharmacol. 2011;668:194–200. doi: 10.1016/j.ejphar.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]