

Abstract
Introduction
The human immunodeficiency virus (HIV) can integrate into T cells, macrophages and dendritic cells resulting in a latent infection. Reports have also demonstrated that various microbial and host cell factors can trigger HIV reactivation leading to HIV recrudescence, potentially undermining highly active antiretroviral therapies.
Methods
This study evaluated the capacity of oral bacteria associated with chronic periodontal infections to stimulate HIV promoter activation in various cell models of HIV latency.
Results
T cells (1G5) challenged with oral bacteria demonstrated a dose–response of HIV promoter activation with a subset of the bacteria, as well as kinetics that were generally similar irrespective of the stimuli. Direct bacterial challenge of the T cells resulted in increased activation of approximately 1.5- to 7-fold over controls. Challenge of macrophages (BF24) indicated different kinetics for individual bacteria and resulted in consistent increases in promoter activation of five fold to six fold over basal levels for all bacteria except Streptococcus mutans. Dendritic cells showed increases in HIV reactivation of 7- to 34-fold specific for individual species of bacteria.
Conclusion
These results suggested that oral bacteria have the capability to reactivate HIV from latently infected cells, showing a relationship of mature dendritic cells > immature dendritic cells > macrophages ≥ T cells. Expression of various pattern recognition receptors on these various cell types may provide insight into the primary receptors/signaling pathways used for reactivation by the bacteria.
Keywords: dendritic cells, human immunodeficiency virus latency, human immunodeficiency virus type 1 promoter, macrophages, oral bacteria, T cells
Infection with the human immunodeficiency virus (HIV) results in a compromised immune system, and increased incidence of opportunistic infections with fatal consequences (28). The HIV targets and destroys CD4+ T cells, as well as infecting antigen-presenting cells such as macrophages and dendritic cells (DC) resulting in a continuous viremia and exacerbated acquired immunodeficiency syndrome (AIDS) outcomes. The advent of highly active antiretroviral therapies (HAART) has successfully transformed HIV infection from a progressive loss of immune function with associated morbidity into a chronic infection that generally manages the viremia and restores substantial T-cell immune function (27, 40). The persistence of HIV-1 latently infecting select host cells, e.g. CD4+ T cells, macrophages and DC, remains a significant obstacle in the elimination of HIV-1 infections (1).
As a critical part of the HIV-1 life cycle, reactivation of the provirus results in the production of new virions. The reactivation phase of HIV transitions from a state with no replicating virus to the production of new virions. Latency is considered a dynamic process and is likely reflected by viral activity through these processes (9). Various exogenous stimuli have been shown to reactivate HIV-1 infections (18, 22, 31). Microbial coinfections have been associated with transient bursts of HIV-1 viremia in patients. This can result from microbial activation of proinflammatory cytokines with subsequent induction of HIV-1 production by infected T cells, macrophages and/or DC (36, 47).
In addition to being a substantial viral reservoir, infected macrophages may also transfer HIV-1 to uninfected T cells (48). Although CD4+ T cells appear to be the main targets of HIV, other immune and non-immune cells with or without CD4 molecules on their surfaces can also be infected. The monocyte/macrophage has been identified to be a source of virus in infected tissues, a basis for viral transmission to target cells, and able to produce a range of cytokines/chemokines that affect both cell function and virus replication (15). It has been suggested that DC are potential first target cells in HIV-1 infections (38). These cells are also considered to be central in activating naïve T cells, which can become permissive for HIV-1 infection (38, 46). DC are innate immune cells that constitute the link between innate and adaptive immunity by recognizing pathogen-associated molecular patterns (PAMPs) on bacteria to enable uptake, processing and presentation of antigens to T cells (30). The immune function of these DC depends upon their stage of maturation, with immature dendritic cells (iDC) considered as part of the innate phase of the immune response. Mature dendritic cells (mDC) provide the linkage to the adaptive phase of the immune response, whereby they present the captured and processed antigens to prime naïve T cells for clonal expansion (2). Although it is clear that DC can engage a diverse array of microorganisms, there are minimal data concerning how HIV-latency in DC can be reactivated, and if oral bacteria or bacterial complexes have the capacity to perform this function. Lipopolysaccharide challenge of HIV-1-infected DC results in increased levels of tumor necrosis factor-α and interleukin-1β (IL-1β), as well as various β-chemokines (34).
Periodontitis is a polymicrobial disease with pathological changes that result from a complex inflammatory and immune host response generated by resident gingival cells and inflammatory cells that migrate into the infected tissues (25, 44). It has been suggested that bacterial infections and mucosal-associated bacterial translocation in HIV-1-infected individuals may reactivate HIV-1 in bystander CD4+ T cells, macrophages and DC (8, 31). This report describes studies that delineate the characteristics of HIV promoter activation in T cells, macrophages and DC in response to challenge of these cells with oral bacteria or bacterial products. We hypothesize that these cell types will demonstrate a differential response to the microbial stimuli, and that the characteristics of the oral bacteria will dictate the patterns of HIV reactivation with the different host cell reservoirs.
Materials and methods
Cellular models of HIV reactivation
A T-cell line (1G5; NIH AIDS Research and Reference Reagent Program, German-town, MD, http://www.aidsreagent.org) that had been transfected with the HIV long terminal repeat (LTR) promoter driving the luciferase gene as a reporter for promoter activation was cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and maintained at 37°C in 5% CO2. The cells were routinely split 1: 4 approximately every 5 days. Cells used in the reactivation studies were cultured for 3 days before challenge with the microbial stimuli.
The BF24 cell line (http://www.aidsre-agent.org) is a subclone of the human myelomonocytic cell line, THP-1. The BF24 cells have been transfected with the HIV LTR promoter driving a chloramphenicol acetyltransferase (CAT) gene. The cells were grown in RPMI-1640 medium (Gibco, Gaithersburg, MD) with L-glutamine and 10% FBS in 5% CO2 at 37°C. Trypan blue dye was used to reveal cell viability.
The human monocytic cell line, THP-1, which has been stably transfected with HIV-1 promoter driving the expression of luciferase gene, was a gift from Dr Seymour Klebanoff (University of Washington) (26). This cell line was cultivated in RPMI-1640 supplemented with 10% FBS and was used to generate DC. Specifically, THP-1 cells were cultured for 6 days at 105 cells per/ml in medium supplemented with 50 ng/ml recombinant granulocyte–macrophage colony-stimulating factor (rGM-CSF; R&D Systems, Minneapolis, MN) and 1000 U/ml recombinant IL-4 (rIL-4; Promega, Madison, WI). After 6 days in culture, iDC were obtained, as described previously (22). The mDC were obtained by treating iDC with tumor necrosis factor-α. The characteristics of mDC were confirmed as follows. Briefly, the DC were washed in ice-cold phosphate-buffered saline (PBS) and preincubated in blocking reagent at 4°C for 20 min. Cells were then incubated on ice for 60 min with fluorescein isothiocyanate-conjugated CD86, human leukocyte antigen (HLA) -DR, CD83, and CD80 monoclonal antibodies (BD Biosciences, San Jose, CA). Stained cells were washed twice with PBS/bovine serum albumin and analysed using an EPICS XL flow cytometer and EXPO 32 software (Beckman Coulter, Fullerton, CA). Compared with the THP-1 cells, the iDC and mDC have differential expression of CD80, CD83, CD86 and HLA-DR, with iDC being CD80hi, CD83lo, CD86lo, HLA-DRlo, and mDCs phenotypically described by CD80hi, CD83hi, CD86hi, HLA-DRhi compared with the original THP-1 cells (4, 22, 32).
Challenge of host cells
Bacteria used in these studies included: Porphyromonas gingivalis FDC W50, W83 and A7A1-28; Prevotella intermedia ATCC 25611; Fusobacterium nucleatum ATCC 25586 and ATCC 49526; Actinomyces viscosus T14V; Aggregatibacter actinomycetemcomitans JP2; Streptococcus mutans ATCC 33535; Campylobacter rectus ATCC 33238 and Tannerella forsythia ATCC 43037. All were grown as previously described (22). Bacterial sonicates were prepared from midlog-phase bacterial cultures in 500 ml of the appropriate broth. The bacterial cultures were washed three times with sterile PBS at 10,000 g for 20 min at 4°C. The pellet was resuspended in 15 ml PBS with complete ethylenediaminetetraacetic acid-free protease inhibitor cocktail (Roche, Mannheim, Germany), and finally bacteria were sonicated using an ultrasonic disrupter (Branson Sonifier model 450; Branson, Danbury, CT). The crude extract after sonication was centrifuged at 13,000 g for 10 min at 4°C and protein concentration of supernatants was determined by bicinchoninic assay (BCA; Pierce, Rockford, IL).
Lipopolysaccharides (LPS) were prepared by us from a battery of oral bacteria including P. intermedia FDC681, C. rectus strain 57, A. actinomycetemcomitans strains H66, Y4 and 7164, P. gingivalis W50, W83 and A7A1-28 and strain 21-7 (7, 16, 17, 24, 37, 39). Escherichia coli O128:B12 (L2755) and O111:B4 (L2630, J5) and Salmonella minnesota Re595 (L9764), Salmonella typhimurium (L6511) and phorbol 12-myristate 13-acetate (PMA) were obtained commercially (Sigma Chemicals Corp., St Louis, MO). The commercial LPS preparations were obtained by phenol extraction or phenol/chloroform/petroleum extraction and are reported to contain <1% protein and <2% RNA. The oral bacterial preparations contained <1% protein and <1% nucleic acid contaminants.
HIV promoter activation
Cells were cultured under various conditions of challenge with oral bacteria to assess variations in HIV promoter activation related to species, dose and time. Expression of luciferase activity driven by the HIV-1 LTR promoter in 1G5 T cells and DC was measured to quantify promoter activation. Briefly, 1 × 105 1G5 T cells or the DC were stimulated in duplicate with oral bacteria (10 μl of 109 cells/ml). Cells were harvested in 3 ml PBS, lysed using 100 μl lysis buffer and stored frozen at − 80°C before luciferase assays. One hundred microliters of the cell lysates were added in duplicate to a 96-well microtiter plate and combined with 50 μl luciferase substrate (Luciferase system, Promega). The photon/light intensities were measured with a photometer. Expression of luciferase was measured by photon/light intensities using a lumino-meter reader (MicroLumat LB96V; Molecular Devices, Inc., Union City, CA).
The production of CAT in response to the HIV promoter activation was determined by enzyme-linked immunosorbent assay (ELISA) using a commercial kit (Roche) and the extent of the reaction was determined according to the manufacturer’s instructions, using a Spectramax M2 spectrophotometer (Molecular Devices, Inc.). Briefly, the 1 × 105 BF24 cells were stimulated in duplicate with oral bacteria (10 μl 109 cells/ml). Cells were harvested in 3 ml PBS and centrifuged, the cell pellets were lysed using 100 μl lysis buffer provided by the CAT ELISA kit (Roche), and the lysates were stored frozen at − 80°C. The measurement of CAT was performed following the manufacturer’s manual. The absorbance was measured using an MRX 4000 plate reader (Dynatech Laboratories, Chantilly, VA) at 405 nm.
Statistical analyses
Analysis of the interval data for expression of luciferase or CAT was accomplished using a non-parametric Mann–Whitney Rank Sum test in comparing treatment to control cultures. Evaluation of changes in levels over time was determined using a Friedman Repeated Measures analysis of variance on ranks with post hoc Holm–Sidak testing (SigmaStat 3.0; SYSTAT Software Inc., Chicago, IL).
Results
Oral microbial activation of HIV promoter in T cells
PMA (a general T-cell activator) caused a dose–response of luminescence (Fig. 1) in the T cells with maximum responses at 100 ng/ml. The HIV promoter activation peaked at 4–6 h. Figure 1 also shows the kinetics of HIV promoter activation by E. coli LPS (100 μg/ml) with peak responses at 8–12 h. The magnitude of activation by this LPS was approximately 5% of the maximum responses noted with the PMA. These results suggest that because the HIV-1 LTR activity was increased in 1G5 T cells on stimulation with LPS from enteric bacteria, a direct stimulation of the 1G5 cells with oral bacteria may have the capacity to stimulate HIV in this model of T-cell latent infection.
Fig. 1.

Characteristics of human immunodeficiency virus (HIV) promoter activation in 1G5 T cells treated with (A) phorbol 12-myristate 13-acetate (PMA) at varying concentrations; (B) time–course of PMA stimulation; and (C) time–course of lipopolysaccharide (LPS) stimulation. HIV promoter activation determined by luciferase units. The control treatment represented the 1G5 cells alone without PMA or LPS. The points denote mean levels from triplicatate determinations in at least three independent experiments. The vertical brackets enclose 2 SD. All treatment concentrations and times with PMA were statistically different from the control and compared to baseline (e.g. 0 concentration of stimulant, 0 time) at least at P < 0.01. All time-points beginning at 4 h after treatment with LPS (100 μg/ml) were statistically different from the control at least at P < 0.01. Time-points from 4 to 16 h with LPS were significantly different from baseline at least at P < 0.01. Time-points from 24 to 72 h with LPS were significantly different from baseline at least at P < 0.05.
Stimulation of the 1G5 with bacterial sonicates demonstrated significant differences in the ability of the oral microbial species to trigger the HIV promoter (Fig. 2). Of the species tested, both gram-negative and gram-positive oral bacteria that accumulate as components of the pathogenic biofilm at sites of disease demonstrated significant stimulatory activity. Only the strain of P. intermedia that was used appeared unable to trigger HIV promoter activation under these conditions.
Fig. 2.
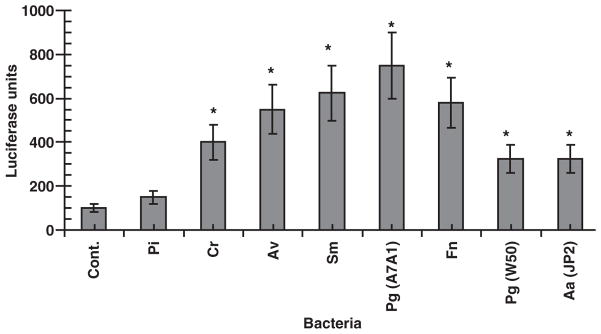
Bacterial sonicate stimulation of human immunodeficiency virus (HIV) promoter activation in 1G5 T cells. The cells were stimulated with 10 μg/ml of sonicate material from each microorganism. The control treatment represented the 1G5 cells alone without bacterial sonicate. The bars denoted mean of triplicate determinates for duplicate experiments and the vertical brackets enclose 2 SD. The asterisk denotes significantly greater than control levels at least at P < 0.01. Abbreviations used for species: Pi, Prevotella intermedia; Cr, Campylobacter rectus; Av, Actinomyces viscosus; Sm, Salmonella minnesota; Pg, Porphyromonas gingivalis; Fn, Fusobacterium nucleatum; Aa, Aggregatibacter actinomycetemcomitans.
Challenge of the 1G5 line with LPS from oral and non-oral bacteria also showed significant differences in LPS effects (Fig. 3). Three observations were notable from these data. First, LPS from some oral bacteria were as effective, and in some cases more effective, than stimulation with LPS from enteric bacteria (such as E. coli and S. typhimurium). Second, LPS preparations from different strains of the same species, whether oral or non-oral, were not uniformly stimulatory. Finally, the magnitudes of responses were generally lower with the LPS compared to bacterial sonicates from the oral species, and the relative stimulatory capabilities of the LPS and sonicates were not directly comparable across the oral bacterial species.
Fig. 3.
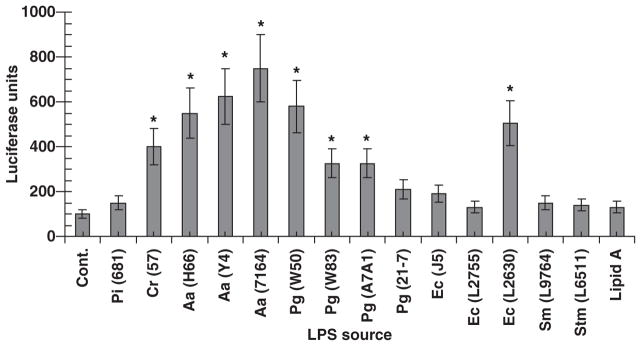
Activation of human immunodeficiency virus (HIV) promoter in 1G5 T cells following challenge with lipopolysaccharide (LPS) preparations from oral and non-oral bacteria. Cells were stimulated with 100 μg/ml of each LPS preparation and harvested for luciferase assays at 10 h. The control treatment represented the 1G5 cells alone without any LPS. The bars denote means from triplicate determinations from duplicate experiments and the asterisk denotes significantly greater than the control at least at P < 0.05. Abbreviations used for species: Pi, Prevotella intermedia; Cr, Campylobacter rectus; Aa, Aggregatibacter actinomycetemcomitans; Pg, Porphyromonas gingivalis; Ec, Escherichia coli; Sm, Salmonella minnesota; Stm, Salmonella typhimurium.
Oral microbial activation of HIV promoter in macrophages
Figure 4 presents a comparison of doses of sonicates prepared from oral bacteria on HIV promoter activation in the BF24 macrophages. The results demonstrated that F. nucleatum caused promoter activation with significant increases at a lower challenge dose. P. gingivalis sonicates also elicited a greater HIV promoter activation, at both 4 and 24 h, than Treponema denticola, with both requiring a higher dose than F. nucleatum. In contrast, S. mutans stimulated the macrophages minimally, irrespective of the concentration of sonicate used.
Fig. 4.
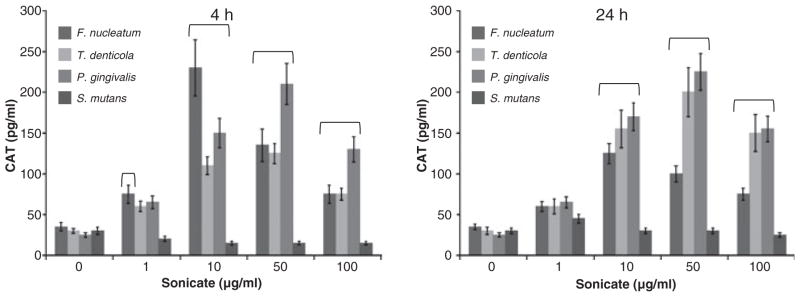
Kinetics (4 and 24 h) and dose–responses of human immunodeficiency virus (HIV) promoter activation in BF24 macrophages with sonicates from different oral bacteria. The bars denote the mean of triplicate determinations from duplicate experiments and the vertical brackets enclose 2 SD. The horizontal bracket (
 ) denotes CAT levels significantly different from control (0 μg/ml) at least at P < 0.05.
) denotes CAT levels significantly different from control (0 μg/ml) at least at P < 0.05.
Macrophages can engage live bacteria, dead bacteria and various bacterial components for subsequent antigen presentation. Figure 5 presents a comparison of the HIV promoter activation in the BF24 cells challenged with live bacteria or formalin-killed bacteria from these species. The results showed a variation in the intact bacteria triggering the HIV promoter, with differences between strains within a species similar to the findings with T-cell responses and bacterial sonicates. Additionally, the live bacteria appeared to stimulate greater HIV promoter activation, except for the P. gingivalis strains where the killed bacteria were more effective in activating the HIV promoter compared with live bacteria.
Fig. 5.
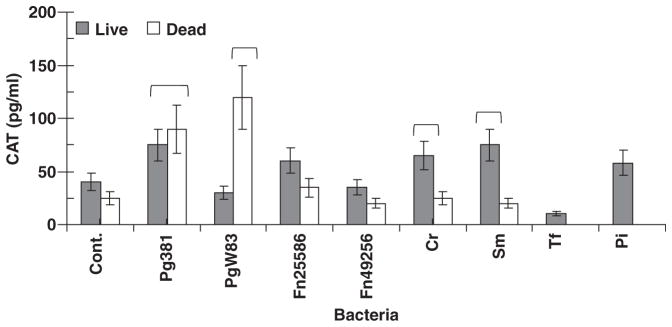
Human immunodeficiency virus (HIV) promoter activation in BF24 cells stimulated with live or formalin-killed bacteria: 1 × 105 BF24 cells were challenged with a multiplicity of infection of 100: 1 of each microbial species and chloramphenicol acetyltransferase (CAT) levels were determined at 24 h. The control treatment represented the BF24 cells alone without bacteria. The bars denote mean levels of duplicate determinations derived from two experiments. The horizontal bracket (
) denotes CAT levels significantly different than control (Cont.) at least at P < 0.05. Abbreviations used for species: Cr, Campylobacter rectus; Sm, Salmonella minnesota; Tf, Tannerella forsythia; Pi, Prevotella intermedia, Pg, Porphyromonas gingivalis; Fn, Fusobacterium nucleatum.
Oral microbial activation of HIV promoter in DC
Oral bacteria stimulated HIV promoter activation in DC with significant differences in the stimulation by the different bacteria (Fig. 6). Although all of the bacteria increased promoter activation over basal levels, P. intermedia appeared the most effective for mature DC activation, while C. rectus maximally stimulated the immature DCs.
Fig. 6.

Human immunodeficiency virus (HIV) promoter activation in T cells, macrophages and immature and mature dendritic cells (iDC and mDC) challenged with sonicates (10 μg/ml) of various oral bacteria [Porphyromona gingivalis strain W50, (Pg W50); Fusobacterium nucleatum strain 25586, (Fn 25586)]. The results are presented as fold-increase compared with untreated cultures. The bars denote triplicate determinations from at least two experiments and the vertical brackets enclose 2 SD. The solid horizontal lines connecting the bars demonstrate statistically different values at least at P < 0.01. Abbreviations used for species: Pi, Prevotella intermedia; Pg, Porphyromonas gingivalis; Fn, Fusobacterium nucleatum; Sm, Salmonella minnesota; Cr, Campylobacter rectus.
Discussion
This study demonstrated that oral bacteria have the capacity to activate HIV LTR transcription in latently infected T cells, macrophages and DC using cell culture model systems. There appeared some specificity to the magnitude of this activation dependent upon the stimulus and the cell target. As the design of these studies emphasized engagement of the bacteria related to innate immune mechanisms, the importance of various PAMP receptors, including Toll-like receptors (TLR), would be expected to be involved in these responses (3, 35). Various studies have demonstrated a significant difference in distribution of PAMPs on T cells when compared with antigen-presenting cells such as macrophages and DC (29). As a consequence, it was not unexpected that the macrophages and DC demonstrated significantly greater responses, with regards to HIV promoter activation related to microbial challenge than T cells. However, it was clear that the T cells could be triggered to substantial HIV reactivation, e.g. the response to PMA (Fig. 1), and increases in promoter activation were observed with certain bacterial stimuli. The T-cell line was not antigen-specific, which suggested that these CD4+ T cells have the capacity to engage microbial products, leading to HIV promoter activation in this latently infected cell model. Recent studies have demonstrated the presence of multiple TLRs on T cells that can engage microbial products from both gram-negative and gram-positive bacteria (21, 29), albeit the 1G5 cell line expresses only low levels of TLR2 and TLR4 (C.B. Huang, unpublished observation).
Both macrophages and DC demonstrate extensive surface expression of TLRs (5, 10). Whereas the relative abundance of these crucial innate immune receptors varies and is dependent upon cell type, stage of maturation and cell activation, a broad repertoire of these PAMPs are represented on these innate immune/antigen-presenting cells. Of particular relevance to the microbial stimuli would be the expression of surface TLR1, −2, −4 and −6. Hence, documenting the ability of a broad range of bacteria or LPS from a range of oral microorganisms to trigger HIV promoter activation in both macrophages and DC was anticipated. However, it was also clear that across the breadth of the bacteria examined, this stimulatory capacity was not uniform which suggested the capacity of the individual different microbial stimuli to interact with these cells differentially to trigger intracellular pathways leading to HIV promoter activation. Beyond the TLRs as molecular detectors of microbial challenge there are a number of other surface receptors that can engage microbial ligands and transduce signals via pathways different from the TLRs (19).
Beyond these more general observations in comparing the microbial and host cell differences in HIV reactivation potential, we noted three more specific outcomes of these in vitro studies. First, LPS from oral bacteria was in some cases significantly better at stimulating HIV promoter activation than LPS from E. coli, representing a prototype LPS. It is well described that LPS from some oral bacteria, particularly P. gingivalis, signals mainly through TLR2 differing from the TLR4 signaling induced by E. coli LPS (49). In addition, the LPS from any of the oral bacteria are not classically ‘smooth-type’ LPS with extended repeating oligosaccharide chains (23), but are more similar to what has been identified as ‘rough LPS’ so while we do not have complete evidence of the TLR affinity of LPS from other oral bacteria, it may be that the enhanced reactivation may represent a slightly different TLR engagement repertoire.
While it is clear that LPS stimulates host cells preferentially via TLR2 and/or TLR4, the 1G5 cells have very low expression of these receptors (unpublished data). However, the literature provides ample evidence that other receptors on host cells can bind LPS and trigger various intracellular signals, including scavenger receptor A (13), scavenger receptor-B1 (6), triggering receptor expressed by myeloid cells 3 and 1 (12, 33), CD55/decay accelerating factor (20), β2-integrins (14, 42), heatshock proteins 70 and 90, CXCR4 and growth differentiation factor 5 (GDF5) (45). Many of these are expressed on T cells and could contribute to the findings in this report. Although the TLRs are crucial components of the innate immune system, there is a wide array of redundancies in the system to afford protection to the host. Second, sonicates of the bacteria were superior to LPS for inducing HIV-1 LTR activation. This suggests that while the LPS from the oral bacteria may have some role in this process, as has been identified with LPS from selected enteric bacteria (41), other gram-positive and gram-negative bacterial components in the sonicates must have a role in HIV-1 LTR activation. In this regard, other bacterial components from non-oral bacteria also have capacity to reactivate HIV-1 LTR, such as lipoteichoic acid (11), and bacterial DNA (43). The characteristics of these components could be determined in future studies as potential targets for interfering with this process. Finally, it was interesting that sonicates from S. mutans significantly increased promoter activation in T cells, but a similar change was not observed in BF24 monocytes/macrophages (Figs 2 and 4). However, when live and dead S. mutans were compared with the BF24 macrophages, the live bacteria significantly stimulated HIV reactivation. These results indicated that the characteristics of the microbial challenge will impact the types of cell reservoirs that could be engaged leading to viral recrudescence in vivo.
The results suggest that chronic oral infections may provide a risk to the maintenance of treatment success in HIV-infected patients. The presence of a chronic oral infection that undermines the integrity of oral epithelial barriers can trigger responses in local T cells, macrophages and DC in gingival tissues (44). In addition, there are clear data demonstrating the capacity of oral bacteria to translocate into the systemic circulation, eliciting systemic antibody responses, creating transient bacteremias, lodging in distant tissues and inducing systemic inflammatory responses (25). Consequently, these chronic oral infections in HIV-infected subjects being treated with HAART, could contribute to the prevalence of HAART failure in the population. Future studies will be required to clarify the molecular mechanisms of these interactions related to variation in the stimuli and cell types being activated.
Acknowledgments
This study was supported by USPHS grant P20 RR020145 from the National Center for Research Resources of the National Institutes of Health (Bethesda, MD).
References
- 1.Alexaki A, Liu Y, Wigdahl B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res. 2008;6:388–400. doi: 10.2174/157016208785861195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity. 2008;29:795–806. doi: 10.1016/j.immuni.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkinson TJ. Toll-like receptors, transduction–effector pathways, and disease diversity: evidence of an immunobiological paradigm explaining all human illness? Int Rev Immunol. 2008;27:255–281. doi: 10.1080/08830180801959072. [DOI] [PubMed] [Google Scholar]
- 4.Balanescu A, Radu E, Nat R, et al. Co-stimulatory and adhesion molecules of dendritic cells in rheumatoid arthritis. J Cell Mol Med. 2002;6:415–425. doi: 10.1111/j.1582-4934.2002.tb00520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blander JM. Phagocytosis and antigen presentation: a partnership initiated by Toll-like receptors. Ann Rheum Dis. 2008;67(suppl 3):iii44–iii49. doi: 10.1136/ard.2008.097964. [DOI] [PubMed] [Google Scholar]
- 6.Bocharov AV, Baranova IN, Vishnyakova TG, et al. Targeting of scavenger receptor class B type I by synthetic amphipathic alpha-helical-containing peptides blocks lipopolysaccharide (LPS) uptake and LPS-induced pro-inflammatory cytokine responses in THP-1 monocyte cells. J Biol Chem. 2004;279:36072–36082. doi: 10.1074/jbc.M314264200. [DOI] [PubMed] [Google Scholar]
- 7.Bramanti TE, Wong GG, Weintraub ST, Holt SC. Chemical characterization and biologic properties of lipopolysaccharide from Bacteroides gingivalis strains W50, W83, and ATCC 33277. Oral Microbiol Immunol. 1989;4:183–192. doi: 10.1111/j.1399-302x.1989.tb00250.x. [DOI] [PubMed] [Google Scholar]
- 8.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 9.Brown A, Zhang H, Lopez P, Pardo CA, Gartner S. In vitro modeling of the HIV-macrophage reservoir. J Leukoc Biol. 2006;80:1127–1135. doi: 10.1189/jlb.0206126. [DOI] [PubMed] [Google Scholar]
- 10.Bullens DM, Ceuppens JL. Influence of Toll-like-receptor ligands on the dendritic cell–T cell interactions: therapeutic options for allergic diseases? Mini-review. Inflamm Allergy Drug Targets. 2008;7:211–216. doi: 10.2174/187152808786848397. [DOI] [PubMed] [Google Scholar]
- 11.Carpenter S, O’Neill LA. How important are Toll-like receptors for antimicrobial responses? Cell Microbiol. 2007;9:1891–1901. doi: 10.1111/j.1462-5822.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 12.Chung DH, Seaman WE, Daws MR. Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur J Immunol. 2002;32:59–66. doi: 10.1002/1521-4141(200201)32:1<59::AID-IMMU59>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 13.Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leukoc Biol. 1998;64:25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 14.Flaherty SF, Golenbock DT, Milham FH, Ingalls RR. CD11/CD18 leukocyte integrins: new signaling receptors for bacterial endotoxin. J Surg Res. 1997;73:85–89. doi: 10.1006/jsre.1997.5195. [DOI] [PubMed] [Google Scholar]
- 15.Freed EO, Mouland AJ. The cell biology of HIV-1 and other retroviruses. Retrovirology. 2006;3:77. doi: 10.1186/1742-4690-3-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garrison SW, Holt SC, Nichols FC. Lipopolysaccharide-stimulated PGE2 release from human monocytes. Comparison of lipopolysaccharides prepared from suspected periodontal pathogens. J Periodontol. 1988;59:684–687. doi: 10.1902/jop.1988.59.10.684. [DOI] [PubMed] [Google Scholar]
- 17.Gillespie J, Weintraub ST, Wong GG, Holt SC. Chemical and biological characterization of the lipopolysaccharide of the oral pathogen Wolinella recta ATCC 33238. Infect Immun. 1988;56:2028–2035. doi: 10.1128/iai.56.8.2028-2035.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gollapudi S, Gupta S, Thadepalli H. Salmonella typhimurium-induced reactivation of latent HIV-1 in promonocytic U1 cells is inhibited by trovafloxacin. Int J Mol Med. 2000;5:615–618. doi: 10.3892/ijmm.5.6.615. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez OA, Ebersole JL, Huang CB. Oral infectious diseases: a potential risk factor for HIV recrudescence? Oral Dis. 2009;15:313–327. doi: 10.1111/j.1601-0825.2009.01533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heine H, El-Samalouti VT, Notzel C, et al. CD55/decay accelerating factor is part of the lipopolysaccharide-induced receptor complex. Eur J Immunol. 2003;33:1399–1408. doi: 10.1002/eji.200323381. [DOI] [PubMed] [Google Scholar]
- 21.Himmel ME, Hardenberg G, Piccirillo CA, Steiner TS, Levings MK. The role of T-regulatory cells and Toll-like receptors in the pathogenesis of human inflammatory bowel disease. Immunology. 2008;125:145–153. doi: 10.1111/j.1365-2567.2008.02939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang CB, Altimova Y, Ebersole JL. HIV-1 promoter activation in dendritic cells by oral microorganisms and LPS. J Dental Res. 2009 (in press) [Google Scholar]
- 23.Kaplan JB, Perry MB, MacLean LL, Furgang D, Wilson ME, Fine DH. Structural and genetic analyses of O polysaccharide from Actinobacillus actinomycetemcomitans serotype f. Infect Immun. 2001;69:5375–5384. doi: 10.1128/IAI.69.9.5375-5384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kiley P, Holt SC. Characterization of the lipopolysaccharide from Actinobacillus actinomycetemcomitans Y4 and N27. Infect Immun. 1980;30:862–873. doi: 10.1128/iai.30.3.862-873.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinane DF, Bartold PM. Clinical relevance of the host responses of periodontitis. Periodontol 2000. 2007;43:278–293. doi: 10.1111/j.1600-0757.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- 26.Klebanoff SJ, Kazazi F, Van Voorhis WC, Schlechte KG. Activation of the human immunodeficiency virus long terminal repeat in THP-1 cells by a staphylococcal extracellular product. Proc Natl Acad Sci USA. 1994;91:10615–10619. doi: 10.1073/pnas.91.22.10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lefebvre JC, Durant J, Koechlin A, et al. HIV-1 and persistent clinical latency, TAR element analysis and improved method for in vitro viral reactivation. J Clin Virol. 2007;38:348–352. doi: 10.1016/j.jcv.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Lewin-Smith MR, Klassen MK, Frankel SS, Nelson AM. Pathology of human immunodeficiency virus infection: infectious conditions. Ann Diagn Pathol. 1998;2:181–194. doi: 10.1016/s1092-9134(98)80006-3. [DOI] [PubMed] [Google Scholar]
- 29.van Maren WW, Jacobs JF, de Vries IJ, Nierkens S, Adema GJ. Toll-like receptor signalling on Tregs: to suppress or not to suppress? Immunology. 2008;124:445–452. doi: 10.1111/j.1365-2567.2008.02871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna K, Beignon AS, Bhardwaj N. Plasmacytoid dendritic cells: linking innate and adaptive immunity. J Virol. 2005;79:17–27. doi: 10.1128/JVI.79.1.17-27.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriuchi H, Moriuchi M, Mizell SB, Ehler LA, Fauci AS. In vitro reactivation of human immunodeficiency virus 1 from latently infected, resting CD4+ T cells after bacterial stimulation. J Infect Dis. 2000;181:2041–2044. doi: 10.1086/315496. [DOI] [PubMed] [Google Scholar]
- 32.Nat R, Radu E, Regalia T, Popescu LM. Apoptosis in the immune system: 1. Fas-induced apoptosis in monocytes-derived human dendritic cells. J Cell Mol Med. 2002;6:223–234. doi: 10.1111/j.1582-4934.2002.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathan C, Ding A. TREM-1: a new regulator of innate immunity in sepsis syndrome. Nat Med. 2001;7:530–532. doi: 10.1038/87846. [DOI] [PubMed] [Google Scholar]
- 34.O’Mahony L, O’Callaghan L, McCarthy J, et al. Differential cytokine response from dendritic cells to commensal and pathogenic bacteria in different lymphoid compartments in humans. Am J Physiol Gastrointest Liver Physiol. 2006;290:G839–G845. doi: 10.1152/ajpgi.00112.2005. [DOI] [PubMed] [Google Scholar]
- 35.O’Neill LA. When signaling pathways collide: positive and negative regulation of toll-like receptor signal transduction. Immunity. 2008;29:12–20. doi: 10.1016/j.immuni.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 36.Oguariri RM, Brann TW, Imamichi T. Hydroxyurea and interleukin-6 synergistically reactivate HIV-1 replication in a latently infected promonocytic cell line via SP1/SP3 transcription factors. J Biol Chem. 2007;282:3594–3604. doi: 10.1074/jbc.M608150200. [DOI] [PubMed] [Google Scholar]
- 37.Poirier TP, Mishell R, Trummel CL, Holt SC. Biological and chemical comparison of butanol- and phenol-water extracted lipopolysaccharide from Capnocytophaga sputigena. J Periodontal Res. 1983;18:541–557. doi: 10.1111/j.1600-0765.1983.tb00391.x. [DOI] [PubMed] [Google Scholar]
- 38.Pope M. Mucosal dendritic cells and immunodeficiency viruses. J Infect Dis. 1999;179 (suppl 3):S427–S430. doi: 10.1086/314798. [DOI] [PubMed] [Google Scholar]
- 39.Progulske A, Holt SC. Isolation and characterization of the outer membrane and lipopolysaccharide from Eikenella corrodens. Infect Immun. 1984;43:166–177. doi: 10.1128/iai.43.1.166-177.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinn TC. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS. 2008;22 (suppl 3):S7–S12. doi: 10.1097/01.aids.0000327510.68503.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rittig MG, Kaufmann A, Robins A, et al. Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intra-cellular trafficking and cytokine/chemokine release in human monocytes. J Leukoc Biol. 2003;74:1045–1055. doi: 10.1189/jlb.0103015. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt A, Caron E, Hall A. Lipopolysaccharide-induced activation of beta2-integrin function in macrophages requires Irak kinase activity, p38 mitogen-activated protein kinase, and the Rap1 GTPase. Mol Cell Biol. 2001;21:438–448. doi: 10.1128/MCB.21.2.438-448.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takeshita A, Imai K, Hanazawa S. CpG motifs in Porphyromonas gingivalis DNA stimulate interleukin-6 expression in human gingival fibroblasts. Infect Immun. 1999;67:4340–4345. doi: 10.1128/iai.67.9.4340-4345.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tatakis DN, Kumar PS. Etiology and pathogenesis of periodontal diseases. Dent Clin North Am. 2005;49:491–516. v. doi: 10.1016/j.cden.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001;2:338–345. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- 46.Turville SG, Santos JJ, Frank I, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. 2004;103:2170–2179. doi: 10.1182/blood-2003-09-3129. [DOI] [PubMed] [Google Scholar]
- 47.Van Lint C, Quivy V, Demonte D, et al. Molecular mechanisms involved in HIV-1 transcriptional latency and reactivation: implications for the development of therapeutic strategies. Bull Mem Acad R Med Belg. 2004;159:176–189. [PubMed] [Google Scholar]
- 48.Wahl SM, Greenwell-Wild T, Peng G, Ma G, Orenstein JM, Vazquez N. Viral and host cofactors facilitate HIV-1 replication in macrophages. J Leukoc Biol. 2003;74:726–735. doi: 10.1189/jlb.0503220. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Q, Desta T, Fenton M, Graves DT, Amar S. Cytokine profiling of macrophages exposed to Porphyromonas gingivalis, its lipopolysaccharide, or its FimA protein. Infect Immun. 2005;73:935–943. doi: 10.1128/IAI.73.2.935-943.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]