

Abstract
Pancreatic cancer is relatively radioresistant, however, radiotherapy has been shown to provide efficacy in the treatment of local disease. To increase the effectiveness of radiotherapy in pancreatic cancer, radiosensitizing drugs are under development. In this study, we investigated the radiosensitizing activity of the anti-diabetic drug metformin on pancreatic cancer cells in vitro. We demonstrated that metformin radiosensitized MiaPaCa-2 and Panc1 cells with radiation enhancement ratios (ER) ranging from 1.33–1.45 with metformin concentrations of 30–100 μM, and in addition, we showed that metformin sensitized cells to gemcitabine alone or in combination with radiation treatment. In addition, we found that pancreatic cancer stem cell-like cells showed enhanced radiosensitization in a tumorsphere assay with a REF of 1.66. At these radiosensitizing doses, metformin alone had low toxicity (as shown by >75% clonogenic survival) and did not affect cell cycle. The combination of metformin and radiation yielded greater numbers of γ-H2AX foci after 1 h compared to radiation alone, suggesting increased DNA damage signaling. Examination of the AMPK pathway showed that pharmacological inhibition of AMPK signaling or RNAi of AMPKα1 reversed metformin-mediated radiosensitization. These studies show that metformin radiosensitization of pancreatic cancer cells at micromolar concentration acts through AMPK and may affect DNA damage signaling. The data indicate that metformin may increase the efficacy of radiation therapy for pancreatic cancer.
INTRODUCTION
Pancreatic cancer is the fourth most common cause of cancer-related death and it is estimated that there will be about 44,000 new cases of pancreatic cancer in the U.S. in the year 2011, with 37,600 estimated deaths (1). Pancreatic cancer often has a poor prognosis in all stages combined. For local disease, the 5-year survival is 23.3% while the median survival for locally advanced metastatic disease is 8.9% (2). The current treatment regimen for pancreatic cancer involves surgery, conventional radiotherapy and chemotherapy. However, the effectiveness of these treatments is limited by inherent or acquired drug resistance (3). Thus, new therapies are required to enhance the effectiveness of conventional treatments.
Therapy that sensitizes tumors to radiation is an ongoing research effort in the radiation oncology field. Metformin (N, N′-dimethylbiguanide) is a widely prescribed drug used for the management of hyperglycemia. Recent evidence now suggests that metformin has antioxidant and tumor growth inhibition activities (4–7). Patients who take metformin have lower incidence of pancreatic cancer compared to patients who do not take metformin (8, 9). Furthermore, metformin has been shown to decrease mammary adenocarcinomas in HER2/neu mice (4) and prevent growth of HCT116 p53 (negative) tumors, but not p53 (wild-type) tumors (10). In a study using a hamster model, metformin showed significant protective effect against developing pancreatic tumors induced by chemical carcinogens and a high-fat diet (11). Additionally, a retrospective study of patients taking metformin during radiotherapy for esophageal adenocarcinoma showed an association with increased response, suggesting radiosensitization (12).
In studies involving chemotherapy agents, metformin has been shown to sensitize cancer cells to paclitaxel, carboplatin or doxorubicin (13, 14). In those studies, metformin, in combination with doxorubicin, was shown to target cancer stem cells (13) and that its use could also potentially lead to a decrease in the dose of chemotherapeutic agents needed to achieve the same effect (14). Although metformin has been shown to radiosensitize breast cancer cells, it’s ability to radiosenstize pancreatic cancer cells has not been reported (15). The high radioresistance of pancreatic cancer cells and current utility of radiosensitization by gemcitabine suggest that further development of pancreatic cancer radiosensitizers may be beneficial.
One intracellular target of metformin is adenosine 5′-monophosphate-activated kinase (AMPK) (16, 17). Activation of AMPK involves phosphorylation at Thr172 of the α1/α2 subunit and liver kinase beta-1 (LKB1) has been identified as an upstream kinase of AMPK (18).
We demonstrate here that metformin decreases the survival of, and strongly radiosensitizes, pancreatic cancer cells in vitro, including cancer stem cell (CSC)-like cells. Importantly, metformin concentrations required for this effect are physiologically achievable. Metformin combined with radiation therapy resulted in increased DNA damage, compared to radiation therapy alone. Our data suggest that AMPK signaling is mechanistically involved in the radiosensitizing activity of metformin.
MATERIALS AND METHODS
Cell Lines and Reagents
MiaPaCa-2 and Panc1 were obtained from the American Type Culture Collection (ATCC). Cells were grown in high glucose Dulbecco’s modified Eagle medium (Thermo Scientific Hyclone, Logan, UT) with 10% FBS, 1% glutamine and no antibiotics. Metformin was purchased from Sigma Chemical Company (St. Louis, MO). Cells were irradiated with a Pantak 320 kV X-ray unit at 0.86 Gy/min.
Clonogenic Survival Experiments
To assess clonogenic survival, 1.0 × 105 cells were plated in T-25 flasks and cultured overnight. The next day cells were incubated with 0, 10, 30 and 100 μM metformin 1 h before irradiation with 0, 2, 4, 6 or 8 Gy. For cultures treated with gemcitabine and metformin, gemcitabine was added 1 h after metformin. For those cultures that were subsequently irradiated, gemcitabine was added 1 h before radiation exposure. After 24 h, cells were washed, trypsinized and seeded in triplicate in 60 mm plates. Colonies were incubated for 8 days before staining with crystal violet. Colonies >50 cells were counted. Clonogenic survival was calculated by dividing the plating efficiency for treated cells by the plating efficiency for untreated cells. The plating efficiency was determined by dividing the number of colonies formed by the number of cells seeded in the control dish. Survival graphs were obtained by plotting clonogenic survival versus radiation dose and the curve fit to a linear-quadratic model.
Tumorsphere Assay
Adherent cells were treated with metformin or radiation therapy before plating for tumorspheres. A tumorsphere formation assay was performed by seeding cells in 6-well plates containing methyl-cellulose (StemCell Technologies, Vancouver, Canada) containing 10% FBS, 20 ng/ml EGF (R&D Systems), 20 ng/ml bFGF (R&D Systems), 1:50 ratio of B27 (Invitrogen) and 4 μg/ml of heparin (StemCell Technologies). Tumorspheres were incubated for two weeks and those containing >50 cells were counted using a light microscope.
Gamma-H2AX Assay
To assess changes in DNA damage signaling due to metformin combined with radiation therapy, 1 × 105 cells were plated in 6-well dishes containing coverslips and incubated overnight at 37°C in growth media. The next day, cells were treated with 0 and 30 μM of metformin 1 h before irradiation with 6 Gy. Cells were collected at 1 and 24 h after irradiation. Media was removed and cells were rinsed with PBS and fixed with formalin solution containing 0.5% Triton X-100 in PBS, pH 8.2 for 15 min. After washing with PBS containing 0.5% BSA and 0.2% Tween-20, cells were blocked for 1 h using blocking buffer, then incubated with primary γ-H2AX-antibody (clone JBW101, Millipore) for 1 h at 37°C. After incubation, cells were washed with PBS and probed with secondary AlexaFluor-488 anti-mouse IgG (Invitrogen) for 45 min at room temperature. Finally, cells were washed, the coverslips taken out and mounted with ProLong® Gold antifade regent with DAPI (Invitrogen). Cells were viewed under a confocal microscope (Leica TCS SP5). The settings used to acquire confocal images with an oil immersion pinhole lens at a magnification of 63× were: a 488 and 528 excitation laser; UV 15%; smart offset 1.3%; and a gain 1,250. The images were acquired using Z-stack and each Z-step size was 1.01 μm.
Cell Cycle Studies
Cell cycle was determined by propidium iodide (PI) staining and flow cytometry (19). Five hundred thousand cells were treated with 0 or 30 μM metformin, irradiated with 6 Gy or a combination of metformin and radiation treatment. After 24–72 h, cells were trypsinized and washed with 0.5% BSA in PBS, cells were then fixed with 0.9% NaCl and 70% ethanol and stored at −20°C. On the day of analysis, cells were washed with 0.5% BSA, 0.5% Triton X-100 in PBS and were then centrifuged, after which the supernatant was removed and the pellet was resuspended with RNase A in 1× PBS at room temperature for 30 min. Finally, an aliquot of the cells was added to tubes containing PI and incubated for 15 min on ice before analysis by flow cytometry. Cell cycle quantitation was performed using ModFit (Verity Software House, Topsham, ME).
Western Blotting
One million cells were plated in 100 mm dishes one day before the treatment. The next day cells were treated with metformin concentrations ranging from 10 μM–20 mM with/without radiation treatment and collected at 1 and 24 h time points. Cells were washed with ice cold PBS, scraped and lysed with sodium dodecylsulfate (SDS) sample buffer [Halt Protease and Phosphatase Inhibitor (Sigma), Phosphatase Inhibitor Cocktail 2 and 3 (Sigma), 200 mM Na3VO4, 1 mM NaF and 2.3 mM Na2PO7]. Protein concentrations were determined using a BCA kit (Pierce Biochemical), and an equal amount of protein was separated on a 10% SDS-polyacrylamide gel (Bio-Rad, Hercules, CA). Proteins were transferred to PVDF membranes (Bio-Rad) and blocked with 5% nonfat dry milk in Tris-buffered saline [10 mM Tris-Base (pH 7.5), 150 mM NaCl with 0.1% Tween-20; TBS-T] for 30–60 min at 4°C. After blocking, the membrane was incubated with primary antibody in blocking buffer for 24 h at 4°C. The next day the membrane was washed 3 times for 10 min in TBS-T and incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. Finally after washing with TBS-T, proteins were detected using Super Signal West Pico chemiluminescent substrate (Pierce Biochemical) and exposure to film. AMPKα1, P-raptor (S792), P-AMPK (T172), P-LKB-1 (T189), P-p70S6K, AMPK, LKB-1, p70S6K, GAPDH and anti-rabbit IgG were purchased from Cell Signaling Technology (Danvers, MA).
Compound C and AMPK RNAi
Clonogenic assay was performed in the presence of compound C (6-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-3-pyridin-4-yl-pyrazolo[1,5-a] pyrimidine). MiaPaCa-2 cells were treated with 1 μM compound C 1 h prior to adding 30 μM metformin and then treated with radiation. A clonogenic assay was then performed.
RNAi of AMPKα1 (PRKAA1) in MiaPaCa-2 cells was performed using Silencer Select siRNA from Life Technologies (Grand Island, NY). The manufacturer’s instructions were followed with the exception that transfection was done on attached cells and transfection media was replaced with complete media 8 h after transfection. SiRNA sequences were sense 5′-GGA UCC AUC AUA UAU AGU UCA tt-3′ and antisense 5′-UGA ACU AUA UGA UGG AUC Ctc-3′. After 48 h of transfection, cells were treated with 0 or 30 μM metformin, irradiated with 6 Gy or 30 μM metformin and irradiated with 6 Gy, and seeded for clonogenic assay the following day. Colonies were enumerated after 10 days. To verify RNAi of AMPKα1, a Western blot was performed 48 and 72 h post-transfection.
Statistics
All experiments are the mean of at least 3 independent experiments unless otherwise noted. Error bars represent SEM. Graphs were plotted and P values were calculated, using Graphpad Prism (Graphpad Software, La Jolla, CA).
RESULTS
Metformin Radiosensitization of Pancreatic Cancer Cell Lines and Cancer Stem Cell-like Cells
Clonogenic studies were done on MiaPaCa-2 and Panc1 pancreatic cancer cells to determine whether metformin had radiosensitizing activity. When we exposed pancreatic cancer cell lines to 10–100 μM metformin 1 h before 0–8 Gy irradiation, these cells displayed dose-dependent radiosensitization (Fig. 1A–B). The KRas mutant cell lines MiaPaCa-2 and Panc1 were strongly radiosensitized by metformin with maximal radiation enhancement ratios of 1.65 and 1.42, respectively.
FIG. 1.
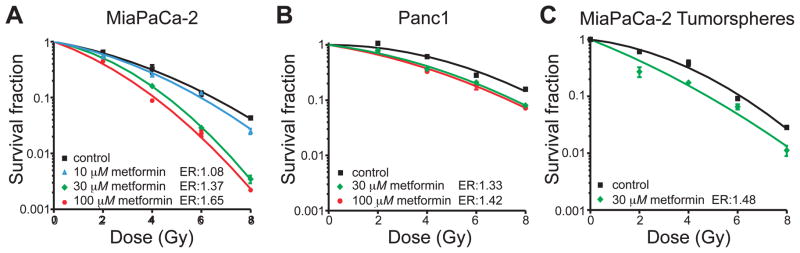
Radiosensitization of pancreatic cancer cells and cancer stem cell-like cells. Panel A: Clonogenic assay of MiaPaCa-2 cells treated with 10, 30 or 100 μM metformin 1 h before irradiation. P < 0.05 versus radiation treatment alone at all radiation doses except 10 μM metformin with 2 and 6 Gy irradiation. Panel B: Clonogenic assay of Panc1 cells treated with 30 or 100 μM metformin before radiation treatment. P < 0.05 versus radiation treatment alone at all radiation doses except 30 μM metformin plus 2 Gy irradiation. Panel C: MiaPaCa-2 cells were treated with metformin and radiation as adherent cells and then plated for tumorsphere formation. Metformin radiosensitized MiaPaCa-2 cancer stem cell-like cells with a radiation enhancement ratio (ER) of 1.48. P < 0.05 at all radiation treatment doses. Some error bars are smaller than the symbols.
Radiosensitization showed dose dependence that plateaued at 30–100 μM metformin. The radiation enhancement ratios of MiaPaCa-2 and Panc1 were reported to be 1.65 and 1.42 at a metformin concentration of 100 μM, and at a concentration of 30 μM radiation enhancement ratios were 1.37 and 1.33, respectively.
Tumorsphere-forming cells have been shown to be present in adherent cultures and are thought to be a cell type with cancer stem cell-like properties (13, 14). To measure the tumorsphere-forming cell (CSC-like cell) content in a culture, cells are incubated under nonadherent conditions in the presence of specific growth factors. We used the tumorsphere assay as a measure of cancer stem cell-like activity in MiaPaCa-2 cells treated with 30 μM metformin and 0–8 Gy ionizing radiation. Fewer tumorspheres formed in irradiated metformin-treated cultures than in cultures receiving radiation alone (Fig. 1C). Metformin treatment resulted in enhancement ratios of 1.48, which was equal to or greater than that which was observed when analyzing colony-forming cells (enhancement ratios of 1.37, Fig. 1A). This indicates metformin was at least as effective for cancer stem cell-like cells as it was for colony-forming cells.
Metformin has been shown to inhibit the growth of prostate cancer cells when used at millimolar quantities (20). To determine whether metformin had a similar effect on the growth of MiaPaCa-2 cells (exhibiting the most radiosensitization) at radiosensitizing concentrations, we performed a colony assay after incubating MiaPaCa-2 cells for 24 h with 0–10,000 μM metformin (Fig. 3). We observed a dose-dependent decrease in clonogenic survival. At 10 mM metformin treatment, clonogenic survival was 36.1 ± 5.5%. However, within the range of concentrations showing radiosensitization (≤100 μM), clonogenic survival was 74.7–86.0%. This shows that radiosensitization occurred at metformin doses that alone were only modestly cytotoxic and suggests the mechanism of radiosensitization and high-dose cytotoxicity may differ.
FIG. 3.

Effect of metformin alone on clonogenic survival. MiaPaCa-2 cells were treated with increasing doses of metformin alone and clonogenic survival was determined. There was a dose-dependent decrease in clonogenic survival up to 10 mM metformin. However, at radiosensitizing doses, the effect of metformin on clonogenic survival was minimal.
Chemosensitization of Pancreatic Cancer Cells
Gemcitabine is a standard chemotherapy given to pancreatic cancer patients and has been shown to exhibit radiosensitizing properties (21). We investigated whether metformin chemosensitizes pancreatic cancer cells to gemcitabine alone or in combination with irradiation. In clonogenic survival assays, MiaPaCa-2 cells treated with 8 Gy alone showed 4.2% clonogenic survival (Fig. 2A). The addition of 30 μM metformin or 0.2 μM gemcitabine lowered clonogenic survival to 2.5% and 2.0%, respectively (P < 0.05). When cells were treated with a combination of metformin, gemcitabine and 8 Gy of irradiation, clonogenic survival was further decreased to 1.1% (P < 0.05). This suggests that metformin increases the sensitivity of MiaPaCa-2 cells to the combination of gemcitabine with radiation treatment.
FIG. 2.
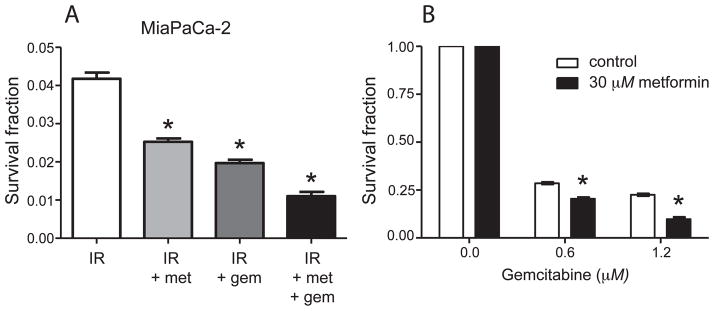
Chemosensitization of pancreatic cancer cells by metformin (met). Panel A: MiaPaCa-2 cells were treated with 8 Gy irradiation alone or in combination with 30 μM metformin and/or 0.2 μM gemcitabine and a clonogenic assay was performed. Combination metformin plus gemcitabine led to decreased clonogenic survival after irradiation, compared to treatment with either compound alone. *P < 0.05. Panel B: MiaPaCa-2 cells were treated with metformin alone or in combination with gemcitabine and a clonogenic assay performed. Metformin chemosensitized cells to gemcitabine. *P < 0.05. IR = Radiation treatment.
We also analyzed the effect of metformin on MiaPaCa-2 cells treated with gemcitabine alone to see whether sensitization would occur in the absence of radiation. As shown in Fig. 2B, the normalized survival fraction of cells treated with 0.6 μM gemcitabine alone was 0.28, while the addition of 30 μM metformin decreased the normalized survival fraction to 0.21 (P < 0.05). At 1.2 μM gemcitabine, the survival fraction was 0.22, and the addition of metformin reduced the survival fraction to 0.10 (P < 0.05). This suggests that metformin chemosensitizes pancreatic cancer cells to gemcitabine.
Effect of Metformin on Cell Cycle
Metformin has been shown in prostate and breast cancer cells to induce a cell cycle arrest (20, 22). We considered that the observed radiosensitization could be due to an effect on cell cycle. Thus, we studied cell cycle changes induced by metformin combined with radiation in MiaPaCa-2 cells because they produced the greatest radiosensitization. MiaPaCa-2 cells were analyzed for cell cycle arrest 24, 48 and 72 h after treatment with IR and 30 μM metformin (Fig. 4A–B). Radiation treatment with or without metformin induced a G2/M arrest beginning 48 h postirradiation, which was increased at 72 h postirradiation with an associated decrease in G0/G1-phase cells. However, there was no difference in cell cycle distribution between conditions of treatment with radiation alone or treatment with radiation plus metformin. Treatment with radiation alone resulted in 36.5% G2 cells while treatment with radiation plus metformin resulted in 36.1% G2/M cells when analyzed at 72 h (Fig. 4B). In contrast, untreated or metformin alone treated cells showed an equal percentage of G2/M-phase cells (18.1%). These data suggest that cell cycle does not play a role in metformin-mediated radiosensitization of pancreatic cancer cells.
FIG. 4.

Cell cycle analysis of MiaPaCa-2 treated with metformin (met) and radiation treatment (IR). Panel A: Cells were treated with 30 μM metformin 1 h prior to radiation treatment and processed at 24, 48 and 72 h for flow cytometry to analyze changes in G0/G1, S and G2/M phases. Representative histograms with ModFit analysis are shown for cells 72 h after treatment. Panel B: Time course of cell cycle changes after metformin or radiation treatment shows that metformin had no effect on cell cycle either alone or in combination with radiation treatment.
The Impact of Metformin on DNA Damage and Repair Signaling
The DNA damage signaling response includes phosphorylation of H2AX at Ser-139 and formation of γ-H2AX foci in the cell nucleus in correlation with sites of DNA strand breaks. As DNA is repaired, the number of nuclear foci decreases. To determine whether there is increased DNA damage signaling after treatment with radiation in metformin-treated cells or whether the repair of DNA is hindered by metformin, we quantified γ-H2AX foci in cells 1 and 24 h after treatment with 30 μM metformin and 6 Gy irradiation (Fig. 5A). One hour after irradiation, the number of foci per nucleus in the metformin-treated cells was higher with 4.6 ± 0.3 per nucleus, compared to cells receiving treatment with radiation alone with 3.3 ± 0.1 foci per nucleus (Fig. 5B; P < 0.05). γ-H2AX foci dissipated to similar levels 24 h after treatment with radiation plus metformin or treatment with radiation alone (0.83 vs. 0.74, respectively; P > 0.05), suggesting repair of DNA damage was similar. Also, metformin alone did not induce a significant increase in γ-HAX foci 1 h after treatment, compared to untreated cells (P > 0.05; Fig. 5C).
FIG. 5.

Analysis of DNA damage signaling. MiaPaCa-2 cells were treated with 30 μM metformin (met) and 6 Gy irradiation. DNA damage signaling was measured by γ-H2AX foci immunofluorescence. Panel A: Representative confocal images of MiaPaCa-2 at 1 and 24 h postirradiation. Green = γ-H2AX, blue = DAPI (nuclei). Panel B: γ-H2AX foci were quantified and plotted as the number of foci per nucleus. More foci were present 1 h after metformin and radiation treatment than after radiation treatment alone (*P < 0.05, t test). Panel C: γ-H2AX foci were quantified in MiaPaCa-2 cells treated with 30 μM metformin alone. No significant increase was noted (P > 0.05).
These data show that metformin combined with radiation treatment increases DNA damage signaling 1 h postirradiation by a mechanism that does not involve activation of γ-H2AX signaling by metformin itself.
AMPK and Radiosensitization
AMPK is a central protein involved in the response to perturbations in cellular energy balance. It has previously been shown that metformin treatment activates the AMPK pathway (23). It has also been shown in breast cancer cells that AMPK activation is associated with radiosensitization by metformin (15). We probed phospho-raptor, -AMPK, -LKB-1 and -p70S6K to determine the effect of radiation and metformin on pathway signaling in MiaPaCa-2 pancreatic cancer cells. Neither, metformin alone, treatment with radiation alone nor treatment with radiation and metformin had a discernible effect on phosphorylation of raptor (S792) or p70S6K (T389) (Fig. 6). Phospho-LKB-1 (T189) was decreased by metformin, treatment with radiation or treatment with radiation and metformin, but was associated with a decrease in total LKB-1 levels. Under our experimental conditions, metformin modestly increased P-AMPK (T172), but treatment with radiation alone or in combination with metformin resulted in reduced AMPK (T172) phosphorylation. Interestingly, AMPK was phosphorylated at T172 under normal culture conditions of high glucose. Overall, these data suggest complex or noncanonical signaling in MiaPaCa-2 cells exposed to metformin or radiation.
FIG. 6.

Analysis of AMPK pathway. MiaPaCa-2 cells were treated with metformin (met) 1 h prior to 6 Gy irradiation (IR) and processed for Western blot after 24 h.
Since metformin alone modestly increased P-AMPK (T172) and has been shown by others to be involved in radiosensitization of other cancer cell types, we investigated whether AMPK signaling was necessary for metformin-mediated radiosensitization of pancreatic cancer cells using an inhibitor of AMPK kinase activity (compound C) and by RNAi of AMPKα1, the catalytic subunit of AMPK. MiaPaCa-2 cells were treated with compound C with or without 30 μM metformin and 0–8 Gy irradiation for a clonogenic assay. While the radiation enhancement ratios of metformin-treated cells in the absence of compound C was 1.36, incubation of cells with compound C abrogated metformin-mediated radiosensitization with a resulting radiation enhancement ratio of 1.00 (Fig. 7A). The protein raptor is a direct phosphorylation target of AMPK (24). To confirm that compound C treatment inhibited AMPK kinase activity, we analyzed raptor phosphorylation on S792. Phospho-raptor (S792) was detected in untreated cells, as well as cells treated with metformin, radiation or metformin and radiation (Fig. 7B). Importantly, in cells treated with a combination of metformin, radiation and compound C, P-raptor (S792) was nearly undetectable. This confirms that in compound C-treated cells where metformin-mediated radiosensitization was abrogated, AMPK kinase activity was inhibited. Taken together, this suggests AMPK kinase activity is necessary for metformin-mediated radiosensitization.
FIG. 7.

Analysis of the role of AMPK in radiosensitization. Panel A: A clonogenic assay was performed on MiaPaCa-2 cells undergoing AMPK inhibition. Cells were treated with compound C 2 h prior and metformin (met) 1 h prior to irradiation (IR). Metformin radiosensitization was abrogated by AMPK inhibition with compound C (P < 0.05). Panel B: Inhibition of AMPK activity by compound C was confirmed by Western blot of Raptor, a phosphorylation target of AMPK. In the presence of compound C, phosphorylation of Raptor is greatly reduced. All lanes shown are from the same blot. Panel C: RNAi of AMPKα1 was confirmed by Western blot and shows that AMPKα1 expression is reduced 48–72 h post transfection. Ctrl = scramble siRNA transfection. All lanes shown are from the same blot. Panel D: A clonogenic assay was performed following AMPKα1 RNAi and treatment with 30 μM metformin and 6 Gy irradiation. Radiosensitization occurred in control cells but was abrogated in cells undergoing AMPKα1 RNAi. *P = 0.02, repeated measures ANOVA.
Kinase inhibitors are known to show off-target inhibition of other kinases that may lead to erroneous conclusions. To further strengthen our conclusion that radiosensitization requires AMPK signaling, we used RNAi to knockdown AMPKα1, the catalytic subunit of AMPK, in MiaPaCa-2 cells. MiaPaCa-2 cells do not express detectable levels of the α2 subunit (not shown). Transient transfection of AMPKα1 siRNA led to undetectable levels of AMPKα1 expression 2 days after transfection and protein levels began to return after 3 days (Fig. 7C). We performed clonogenic assays on cells irradiated with 6 Gy, or cells irradiated with 6 Gy and 30 μM metformin under conditions of no transfection, transfection with control siRNA or transfection with AMPKα1 siRNA. Control MiaPaCa-2 transfections of no siRNA and control siRNA showed 9.6% and 10% clonogenic survival after irradiation and metformin, compared to 15% and 14% survival without metformin, respectively (Fig. 7D). This demonstrates metformin-mediated radiosensitization under control conditions. In contrast, no radiosensitization was observed in cells undergoing RNAi of AMPKα1 with 12% clonogenic survival of cells treated with radiation and metformin, compared to 14% in cells treated with radiation alone. Clonogenic survival of AMPKα1-knockdown cells was significantly greater than control siRNA-transfected cells (P = 0.02, repeated measures ANOVA, n = 3). These results are consistent with the compound C experimental results and confirm that AMPK is necessary for metformin-mediated radiosensitization of pancreatic cancer cells.
DISCUSSION
The comorbidities of type II diabetes and pancreatic cancer are increasingly drawing attention from the medical community. Many studies have shown that diabetes is a risk factor for pancreatic cancer (25–27). The rate of pancreatic cancer in people with type II diabetes is elevated by a factor of six compared to the general population (27). However, it has been noted that type II diabetic patients treated with metformin had a 62% lower risk of developing pancreatic cancer (28).
The biguanide metformin is the most widely prescribed drug for the treatment of type II diabetes. Metformin reduces blood glucose levels by decreasing hepatic glycogenesis and increasing glucose uptake in skeletal muscle and adipose tissue (29). At therapeutic concentrations, metformin is known to stimulate the activation of AMPK (30), a conserved regulator of the cellular response to low energy that is activated when ATP concentrations decrease and 5′-AMP concentrations increase in response to nutrient deprivation, hypoxia and metformin administration (31). Metformin induces activation of AMPK, which in turn inhibits mTOR function by TSC2 and raptor phosphorylation (24, 32–34).
We hypothesized that metformin would radiosensitize pancreatic cancer cells based on its perturbation of metabolic regulation and signaling. We showed that metformin was able to radiosensitize K-Ras mutant pancreatic cancer cells (MiaPaCa-2 and Panc-1) in vitro (Fig. 1). Importantly, radiosensitization occurs at clinically relevant concentrations. It has been shown that the plasma concentration of metformin in treated type II diabetic patients is in the range of 6–30 μM (35), thus administering therapeutic levels of metformin may significantly benefit cancer patients undergoing radiotherapy.
Changes in AMPK activity are thought to play an important role in the radiosensitizing effect of metformin in vitro. In addition to inducing clonogenic death, the exposure of metformin reduced the size of the colonies formed and decreased the cell density in the colonies (data not shown) suggesting that metformin not only kills cancer cells but also suppresses the proliferation of surviving cancer cells, which is in agreement with the reports of other investigators (15, 16).
Our results indicate that metformin caused a decrease in tumorsphere formation when combined with radiation treatment. The radiation enhancement factors indicated similar radiosensitization in tumorsphere-forming cells as in colony forming cells (1.48 vs. 1.37). This suggests that metformin also radiosensitizes cancer stem cells. Similar results have been obtained in breast cancer cell lines, where the population of cancer stem cells quantified by using CD44+/CD24− markers showed marked decrease in cancer stem cells when 1 mM metformin was combined with radiation treatment (15).
We investigated DNA damage signaling and cell cycle as potential mechanisms for radiosensitization. A γ-H2AX foci formation assay revealed that when metformin is combined with radiation treatment, greater numbers of γ-H2AX foci persist 1 h postirradiation, suggesting altered DNA damage signaling. However, dissipation of foci 24 h after irradiation occurred similarly in metformin and untreated cells, suggesting that DNA repair occurred efficiently in the presence of metformin. A similar increase in γ-H2AX has been seen in prostate cancer cells treated with AICAR, with or without radiation treatment (36). This was attributed to an effect on pyrimidine nucleotide pools. In Fig. 5D, we show a slight increase in γ-H2AX foci from metformin-alone treatment, but this was not statistically significant (P =0.4). It has been shown that 5–10 mM metformin causes a cell cycle arrest in the G1 phase (20, 22). We also analyzed the cell cycle distribution as a possible mechanism of radiosensitization. At a radiosensitizing dose (30 μM), metformin alone or in combination with radiation treatment had no measureable impact on cell cycle. From our studies on DNA damage signaling and cell cycle, we conclude that at micromolar concentrations, metformin potentiates early DNA damage signaling and this may lead to other downstream effects on cell survival. Our findings do not support a hypothesis of a defect in DNA repair.
Other investigators have shown that the AMPK agonist metformin sensitizes cells to further damage by activating ataxia telangiectasia mutated (ATM)-mediated DNA damage response in A431 epithelial cancer (37, 38). Dian et al. (39) also reported that treatment with pharmacological agents increasing the AMP/ATP ratio (i.e., AICAR and metformin) on human diploid fibroblast induces activation of ATM at Ser-1981, increases the overall levels of ATM protein and activates AMPK. However, Halicka et al. (40) reported that treatment of normal mitogen-stimulated lymphocytes or tumor cell lines treated with metformin had attenuated ATM activation and constitutive γ-H2AX phosphorylation. These differences in results may be due to experimental variability and conditions, thus further analysis should be done to clarify metformin’s role in activating ATM.
AMPK has been shown to be stimulated by metformin and others have suggested it can play a role in radiosensitization of cancer cells (16). Metformin or radiation led to unexpected phosphorylation changes to AMPK (T172), raptor (S792), LKB-1 (T189) and p70S6K (T389) phosphorylation. Crosstalk with ATM as described above or with AKT may complicate linear signaling in this pathway, or other phosphorylation sites on these proteins may play a greater role in reacting to metformin or radiation (37, 38). Also, under our experimental conditions of high glucose media, AMPK (T172) was constitutively phosphorylated. This may be specific to pancreatic cancer cells, since similar results in other pancreatic cancer cell lines have been found by another group (41). Therefore, we chose to investigate the role of AMPK in mediating radiosensitization by metformin using two independent methods. Pharmacological inhibition of AMPK activity or RNAi of AMPKα1 negated any radiosensitizing effect of metformin in a clonogenic assay. This shows that radiosensitization, in part, requires AMPK kinase activity. Our results are consistent with other investigators’ findings that showed reversal of the effect of metformin-induced radiosensitization when AMPK activity was inhibited in lung cancer cell lines (16).
In conclusion, we showed that metformin radiosensitizes pancreatic cancer cells in vitro at clinically relevant concentrations and that cancer stem cell-like cells exhibited radiosensitization. Metformin induced greater γ-H2AX foci in radiation-treated cells 1 h after exposure, suggesting an influence on DNA damage signaling. Cell cycle was unaffected by metformin at radiosensitizing doses. Radiosensitization by metformin was found to require AMPK activity. Metformin radiosensitization may therefore be mediated through AMPK and involve altered DNA damage signaling, resulting in decreased clonogenic survival. These data suggest metformin may be useful when combined with radiotherapy for pancreatic cancer.
Acknowledgments
This work was supported by an Angelika Burger Award from the Karmanos Cancer Institute (SZ, MH). The Microscopy, Imaging and Cytometry Resources Core is supported, in part, by NIH Center grant P30CA22453 to The Karmanos Cancer Institute, Wayne State University and the Perinatology Research Branch of the National Institutes of Child Health and Development, Wayne State University.
References
- 1.Hariharan D, Saied A, Kocher HM. Analysis of mortality rates for pancreatic cancer across the world. HPB (Oxford) 2008;10:58–62. doi: 10.1080/13651820701883148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Altekruse SF, et al., editors. SEER Cancer Statistics Review, 1975–2009. Bethesda: National Cancer Institute; 2012. [Google Scholar]
- 3.Bhardwaj V, Tadinada MS, Lai CK, Bhushan A. Failure of pancreatic cancer chemotherapy: consequences of drug resistance mechanisms. In: Srivastava SK, editor. Pancreatic cancer. molecular mechanism and targets. Rijeka, Croatia: InTech; 2012. –144.pp. 60 [Google Scholar]
- 4.Anisimov VN, Semenchenko AV, Yashin AI. Insulin and longevity: antidiabetic biguanides as geroprotectors. Biogerontology. 2003;4:297–307. doi: 10.1023/a:1026299318315. [DOI] [PubMed] [Google Scholar]
- 5.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 6.Bonnefont-Rousselot D, Raji B, Walrand S, Gardes-Albert M, Jore D, Legrand A, et al. An intracellular modulation of free radical production could contribute to the beneficial effects of metformin towards oxidative stress. Metabolism. 2003;52:586–9. doi: 10.1053/meta.2003.50093. [DOI] [PubMed] [Google Scholar]
- 7.Dilman VM, Berstein LM, Zabezhinski MA, Alexandrov VA, Bobrov JF, Pliss GB. Inhibition of DMBA-induced carcinogenesis by phenformin in the mammary gland of rats. Arch Geschwulstforsch. 1978;48:1–8. [PubMed] [Google Scholar]
- 8.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care. 2006;29:1990–1. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- 9.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–52. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 11.Schneider MB, Matsuzaki H, Haorah J, Ulrich A, Standop J, Ding XZ, et al. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology. 2001;120:1263–70. doi: 10.1053/gast.2001.23258. [DOI] [PubMed] [Google Scholar]
- 12.Skinner HD, McCurdy MR, Echeverria AE, Lin SH, Welsh JW, O’Reilly MS, et al. Metformin use and improved response to therapy in esophageal adenocarcinoma. Acta Oncol. 2013;52:1002–9. doi: 10.3109/0284186X.2012.718096. [DOI] [PubMed] [Google Scholar]
- 13.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011;71:3196–201. doi: 10.1158/0008-5472.CAN-10-3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, et al. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep. 2012;2:362. doi: 10.1038/srep00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2012;78:221–9. doi: 10.1016/j.ijrobp.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–48. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zielske SP, Gerson SL. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther. 2003;7:325–33. doi: 10.1016/s1525-0016(03)00005-4. [DOI] [PubMed] [Google Scholar]
- 20.Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–72. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence TS, Chang EY, Hahn TM, Hertel LW, Shewach DS. Radiosensitization of pancreatic cancer cells by 2′,2′-difluoro-2′-deoxycytidine. Int J Radiat Oncol Biol Phys. 1996;34:867–72. doi: 10.1016/0360-3016(95)02134-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51:2420–5. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 24.Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;3:214–26. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chow WH, Gridley G, Nyren O, Linet MS, Ekbom A, Fraumeni JF, Jr, et al. Risk of pancreatic cancer following diabetes mellitus: a nationwide cohort study in Sweden. J Natl Cancer Inst. 1995;87:930–1. doi: 10.1093/jnci/87.12.930. [DOI] [PubMed] [Google Scholar]
- 26.Everhart J, Wright D. Diabetes mellitus as a risk factor for pancreatic cancer. A meta-analysis. J Am Med Assoc. 1995;273:1605–9. [PubMed] [Google Scholar]
- 27.Gullo L, Pezzilli R, Morselli-Labate AM. Diabetes and the risk of pancreatic cancer. N Engl J Med. 1994;331:81–4. doi: 10.1056/NEJM199407143310203. [DOI] [PubMed] [Google Scholar]
- 28.Li D, Yeung SC, Hassan MM, Konopleva M, Abbruzzese JL. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology. 2009;137:482–8. doi: 10.1053/j.gastro.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardie DG. AMP–activated protein kinase as a drug target. Annu Rev Pharmacol Toxicol. 2007;47:185–210. doi: 10.1146/annurev.pharmtox.47.120505.105304. [DOI] [PubMed] [Google Scholar]
- 31.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 32.Chan SO, Wong SS, Yeung DC. Expression of c-Ki-ras in developing rat liver. Comp Biochem Physiol B. 1992;102:111–7. doi: 10.1016/0305-0491(92)90281-u. [DOI] [PubMed] [Google Scholar]
- 33.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–68. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 34.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 35.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, et al. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–40. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 36.Isebaert SF, Swinnen JV, McBride WH, Begg AC, Haustermans KM. 5-aminoimidazole-4-carboxamide riboside enhances effect of ionizing radiation in PC3 prostate cancer cells. Int J Radiat Oncol Biol Phys. 2011;81:1515–23. doi: 10.1016/j.ijrobp.2011.06.1964. [DOI] [PubMed] [Google Scholar]
- 37.Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Martin-Castillo B, Menendez JA. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011;10:1499–501. doi: 10.4161/cc.10.9.15423. [DOI] [PubMed] [Google Scholar]
- 38.Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108:2021–32. doi: 10.1038/bjc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan X, Ponomareva L, Veeranki S, Choubey D. IFI16 induction by glucose restriction in human fibroblasts contributes to autophagy through activation of the ATM/AMPK/p53 pathway. PloS One. 2011;6:e19532. doi: 10.1371/journal.pone.0019532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halicka HD, Zhao H, Li J, Traganos F, Zhang S, Lee M, et al. Genome protective effect of metformin as revealed by reduced level of constitutive DNA damage signaling. Aging. 2011;3:1028–38. doi: 10.18632/aging.100397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low concentrations of metformin selectively inhibit CD133(+) cell proliferation in pancreatic cancer and have anticancer action. PLoS One. 2013;8:e63969. doi: 10.1371/journal.pone.0063969. [DOI] [PMC free article] [PubMed] [Google Scholar]