

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease involving motoneuron (MN) axonal withdrawal and cell death. Previously, we established that facial MN (FMN) survival levels in the SOD1G93A transgenic mouse model of ALS are reduced and nerve regeneration is delayed, similar to immunodeficient RAG2-/- mice, after facial nerve axotomy. The objective of this study was to examine the functionality of SOD1G93A splenic microenvironment, focusing on CD4+ T cells, with regard to defects in immune-mediated neuroprotection of injured MN. We utilized the RAG2-/- and SOD1G93A mouse models, along with the facial nerve axotomy paradigm and a variety of cellular adoptive transfers, to assess immune-mediated neuroprotection of FMN survival levels. We determined that adoptively transferred SOD1G93A unfractionated splenocytes into RAG2-/- mice were unable to support FMN survival after axotomy, but that adoptive transfer of isolated SOD1G93A CD4+ T cells could. Although WT unfractionated splenocytes adoptively transferred into SOD1G93A mice were able to maintain FMN survival levels, WT CD4+ T cells alone could not. Importantly, these results suggest that SOD1G93A CD4+ T cells retain neuroprotective functionality when removed from a dysfunctional SOD1G93A peripheral splenic microenvironment. These results also indicate that the SOD1G93A central nervous system microenvironment is able to re-activate CD4+ T cells for immune-mediated neuroprotection when a permissive peripheral microenvironment exists. We hypothesize that dysfunctional SOD1G93A peripheral splenic microenvironment may compromise neuroprotective CD4+ T cell activation and/or differentiation, which, in turn, results in impaired immune-mediated neuroprotection for MN survival after peripheral axotomy in SOD1G93A mice.
Keywords: motoneuron, T cell, APC, axotomy, SOD1, ALS, immune
Introduction
In previous studies, we have established an immune-mediated model of endogenous neuroprotection following facial nerve axotomy in wild-type (WT) and immunodeficient recombinase activating gene-2 knock-out (RAG2-/-) mice lacking functionally mature B and T cells, but intrinsically maintaining antigen presentation by MHC class II-expressing peripheral antigen-presenting cells (APC; Serpe et al., 1999; Serpe et al., 2003). Key to immune-mediated neuroprotection after axotomy is the generation of neuroprotective CD4+ T cells that are antigen-speifc and require: 1) initial activation peripherally, via interaction with MHC class II-expressing APC, and 2) re-activation centrally, via interaction with MHC class II-expressing microglia (Byram et al., 2004).
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease resulting in motoneuron degeneration and accompanied by neuroinflammation involving reactive microglia and astrocytes centrally and immune activation peripherally (Appel et al., 2010; Philips and Robberecht, 2011). The most widely used transgenic mouse model of ALS, involving the overexpression of human mutant superoxide dismutase-1 (SOD1G93A), develops disease pathology similar to that in familial and sporadic ALS patients (Rosen et al., 1993; Gurney, 1994; Gurney et al., 1994). An axonal die-back response precedes MN cell death in SOD1 mice (Kennel et al., 1996; Fischer et al., 2004; Hegedus et al., 2007), resulting in a cascade of events similar to that observed in WT mice after peripheral nerve injury. Specifically, axonal degeneration, denervated neuromuscular junctions, afferent presynaptic stripping surrounding MN cell bodies in CNS, immune cell activation peripherally, and glial activation centrally are responses that occur both as a result of axonal die-back in ALS and peripheral nerve injury (Moran and Graeber, 2004; Jones et al., 2005; Zang et al., 2005; Chiu et al., 2009; Jinno and Yamada, 2011).
SOD1G93A mice demonstrate significantly increased FMN cell death following either a facial nerve transection or crush axotomy, relative to WT (Mesnard et al., 2011; Mesnard et al., 2013). Interestingly, while axotomized SOD1G93A FMN respond with a pro-regenerative response similar to WT, a dysregulated response to axotomy exists in the microenvironment surrounding the SOD1G93A FMN cell bodies (Mesnard et al., 2011). Importantly, target disconnection via disease or facial nerve axotomy in SOD1G93A mice results in comparable motoneuron- and glial-specific molecular changes within the facial nucleus (Haulcomb et al., 2014). Furthermore, SOD1G93A FMN exhibit a delayed functional recovery response to facial nerve crush axotomy, relative to WT mice (Mesnard et al., 2013), that resembles the delayed functional recovery response of FMN in immunodeficient mice following facial nerve crush (Serpe et al., 2002). Therefore, both peripheral and central immune cell irregularities appear to impact SOD1G93A FMN survival and functionality after facial nerve axotomy.
The main objective of the current study was to begin to define whether an immune defect in SOD1G93A CD4+ T cell development, activation, or re-activation is associated with the increased susceptibility of SOD1 FMN to axotomy-induced cell death or the defect lies within the previously identified central glial response (Mesnard et al., 2011). Through a variety of adoptive transfer experiments utilizing SOD1G93A and RAG2-/- mice, our results suggest that a defective SOD1G93A peripheral microenvironment and/or response, rather than a defect in the CD4+ T cell itself, may underscore the impaired immune-mediated neuroprotection required for motoneuron survival and regeneration.
Materials & Methods
Animals
Female, C57Bl/6 wild-type (WT) and transgenic SOD1 (SOD1G93A) were obtained from Jackson, and recombination activating-2 gene knock-out (RAG2-/-) from Taconic, at 6 weeks of age and permitted 1 week to acclimate prior to experimental manipulation. The mice were provided autoclaved pellets and water ad libitum, and housed under a 12 h light/dark cycle in microisolater cages contained within a laminar flow system to maintain a pathogen-free environment.
Cellular adoptive transfers
Cellular adoptive transfers were completed at 7 weeks of age and 1 week prior to undergoing facial nerve axotomy. Spleens were removed from WT or SOD1G93A mice, and the splenocytes were isolated as previously described by our laboratory (Serpe et al., 1999; Byram et al., 2003; Serpe et al., 2003). Specifically, WT or SOD1G93A splenocytes were collected for adoptive transfer at a concentration of 50 × 106 splenocytes/100 μL PBS via tail vein injection per animal. Naïve WT or SOD1G93A CD4+ T cells were isolated from the splenocyte samples via autoMACS magnetic cell sorting for adoptive transfer at a concentration of 5 × 106 CD4+ T cells/100 μL PBS per animal. Axotomy-activated CD4+ T cells (Byram et al., 2004) were isolated from WT or SOD1G93A spleens 3 days after facial nerve axotomy via autoMACS. Ten specific groups (N = 3-6/group) were utilized for FMN survival analyses, including WT, SOD1G93A RAG2-/-, SOD1G93A + WT splenocytes, RAG2-/- + SOD1G93A splenocytes, RAG2-/- + WT CD4+ T cells, RAG2-/- + SOD1G93A CD4+ T cells, SOD1G93A + WT CD4+ T cells, SOD1G93A + WT CD4-depleted splenocytes, and SOD1G93A + Axot WT CD4+ T cells. All SOD1G93A mice were utilized by 12 weeks of age, and pre-symptomatic.
Facial nerve axotomy
A right facial nerve transection axotomy was performed in mice at 8 weeks of age. All surgical procedures were completed in accordance with National Institutes of Health guidelines on the care and use of laboratory animals for research purposes. Using aseptic techniques, mice were anesthetized with 3% isoflurane and maintained at 1.5%, and the right facial nerve was exposed and completely transected at its exit from the stylomastoid foramen (Jones and LaVelle, 1985; Serpe et al., 1999). The proximal and distal facial nerve stumps were manually pushed away from each other in order to prevent reconnection. The left facial nerve remained intact, leaving the left facial nucleus to serve as an internal control for comparison purposes.
Facial motoneuron (FMN) counts
For each experiment, FMN survival levels were assessed at four weeks following facial nerve transection axotomy, where surviving FMN were counted in the ipsilateral facial nucleus and compared to the contralateral uninjured (control) facial nucleus. At 4 weeks post-axotomy, 25 μm cryosections were collected throughout the rostrocaudal extent of the facial motor nucleus (Mesnard et al., 2011). FMN were counted in thionin-stained tissue sections, and after application of the Abercrombie correction factor, results were presented as the average percent of FMN survival ± the standard error of the mean (SEM), as described in detail (Serpe et al., 1999; Mesnard et al., 2011). Representative photomicrographs of facial motor nuclei were obtained with an Olympus microscope and Image-Pro software. Statistical analysis was accomplished using a one-way ANOVA, followed by the Student-Newman-Keuls post-hoc multiple comparison test, with significance at p < 0.05.
Results
Significant FMN cell loss in SOD1G93A and RAG2-/- mice after axotomy
Compared to the uninjured facial nucleus in WT mice (92 neurons ± 2; Figure 1A), baseline FMN counts were not altered in transgenic mice containing overexpression of the human mutant SOD1G93A gene (90 neurons ± 2; Figure 1B) or deletion of the RAG2 gene (90 neurons ± 3; Figure 1C) at 12 weeks of age (quantitative data not shown). However, at 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of WT mice (84% ± 2.0; Figures 1D and 2) were significantly higher compared to both SOD1G93A (68% ± 1.0; Figures 1E and 2) and RAG2-/- mice in (57% ± 2.5; Figures 1F and 2), relative to the respective uninjured facial nuclei.
Figure 1. Representative photomicrographs of coronal control or axotomized facial motor nuclei sections from WT, and SOD1G93A, and RAG2-/- mice ± cellular adoptive transfers collected 4 weeks post-axotomy.
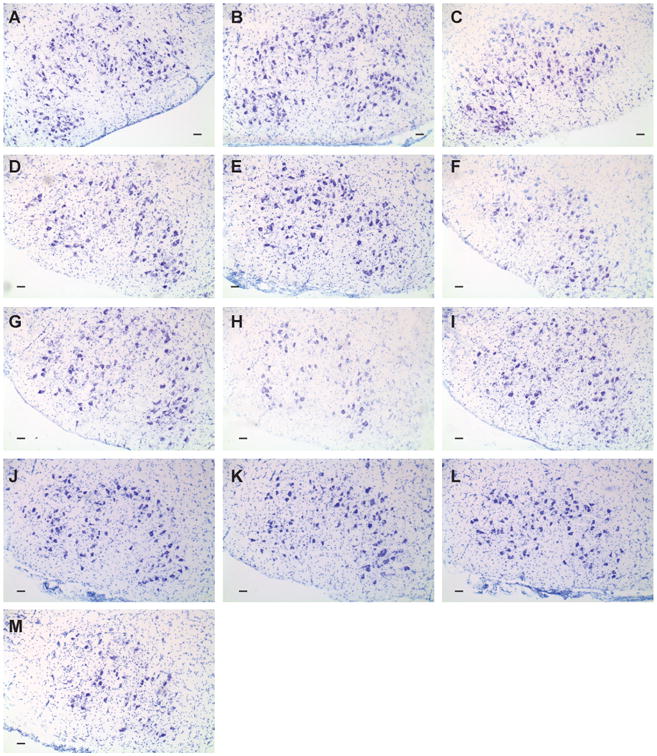
Representative photomicrographs are shown of control facial nuclei from (A) WT, (B) SOD1G93A, (C) RAG2-/-, and of axotomized facial nuclei from (D) WT, (E) SOD1G93A, (F) RAG2-/-, (G) SOD1G93A + WT splenocytes, (H) RAG2-/- + SODG93A splenocytes, (I) RAG2-/- + WT CD4+ T cells, (J) RAG2-/- + SOD1G93A CD4+ T cells, (K) SOD1G93A + WT CD4+ T cells, (L) SOD1G93A + WT CD4-depleted splenocytes, (M) SOD1G93A + prior axotomy-activated WT CD4+ T cells. Scale bar indicates 120 μm.
Figure 2. FMN survival in WT, and SOD1G93A and RAG2-/- mice ± cellular adoptive transfers 4 weeks post-axotomy.

Average percent survival ± SEM of FMN from axotomized facial nucleus of WT, SOD1G93A, RAG2-/-, RAG2-/- + SOD1G93A splenocytes, RAG2-/- + WT CD4+ T cells, and RAG2-/- + SOD1G93A CD4+ T cells, relative to the uninjured nucleus. Significance denoted by *compared to WT, **compared to SOD1G93A, and ***compared to RAG2-/- at p < 0.05.
WT splenocytes adoptively transferred into SOD1G93A mice maintain axotomized FMN survival levels
To determine whether the increased axotomy-induced cell death in SOD1G93A mice could be prevented with the addition of naïve peripheral immune cells, WT unfractionated splenocytes were adoptively transferred via tail vein into SOD1G93A mice prior to axotomy. At 4 weeks post-axotomy, adoptive transfer of WT splenocytes into SOD1G93A mice (SOD1G93A + WT splenocytes) prior to axotomy resulted in maintenance of FMN survival levels at 76% ± 1.8 in the axotomized facial nucleus, relative to the uninjured facial nucleus (Figures 1G and 2). The FMN survival levels in the SOD1G93A + WT splenocytes group were similar to the levels observed in WT, and significantly elevated compared to SOD1G93A mice without adoptive transfer. These results suggest that a potential defect exists within the intrinsic peripheral immune cells of SOD1G93A mice, and that the introduction of extrinsic WT peripheral immune cells into SOD1G93A mice prior to axotomy prevents the axotomy-induced cell death.
SOD1G93A splenocytes adoptively transferred into RAG2-/- mice unable to sustain WT levels of axotomized FMN
To determine whether SOD1G93A splenocytes were capable of restoring WT FMN survival levels in RAG2-/- mice, as observed with WT unfractionated splenocytes (Serpe et al., 2000), unfractionated splenocytes from SOD1G93A mice were adoptively transferred into RAG2-/- mice prior to axotomy. At 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of RAG2-/- mice that received an adoptive transfer of SOD1G93A unfractionated splenocytes (RAG2-/- + SOD1G93A splenocytes) were 60% ± 3.4, relative to the uninjured facial nucleus (Figures 1H and 2), comparable to FMN survival levels in the RAG2-/- group and significantly reduced relative to WT group. Thus, splenocytes from SOD1G93A mice were not capable of the immune-mediated neuroprotection required to maintain RAG2-/- FMN survival levels.
SOD1G93A CD4+ T cells adoptively transferred into RAG2-/- mice as effective as WT CD4+ T cells in restoring FMN survival levels after axotomy
In previous studies, B, CD4+ T, CD8+ T, CD4+CD25+ Treg, NKT, and NK cellular populations were separately isolated from WT splenocytes and adoptively transferred into RAG2-/- mice prior to axotomy (Serpe et al., 2000; Byram et al., 2003; Serpe et al., 2003; DeBoy et al., 2006a; Deboy et al., 2006b). Only CD4+ T cells were found to provide the necessary immune-mediated support required to maintain RAG2-/- FMN survival levels to those of WT after axotomy (Serpe et al., 2003). The immune-mediated neuroprotective ability of SOD1G93A CD4+ T cells was compared to WT CD4+ T cells via adoptive transfer of the respective CD4+ T cells into RAG2-/- mice prior to axotomy. At 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of RAG2-/-mice that received an adoptive transfer of WT CD4+ T cells (RAG2-/- + WT CD4+ T cells) were 82% ± 2.1 (Figures 1I and 2) and in the axotomized facial nucleus of RAG2-/- mice that received an adoptive transfer of SOD1G93A CD4+ T cells (RAG2-/- + SOD1G93A CD4+ T cells) were 78% ± 1.9 (Figures 1J and 2), relative to the uninjured facial nucleus. FMN survival levels in the RAG2-/- + WT CD4+ T cells and the RAG2-/- + SOD1G93A CD4+ T cells groups were significantly increased compared to the RAG2-/- group, and comparable to the WT group. These results indicate that SOD1G93A CD4+ T cells are as effective as WT cells in mediating neuroprotection of axotomized FMN, when placed in a non-SOD1G93A peripheral microenvironment (i.e., RAG2-/-), and support the concept that other cells in the SOD1G93A central and/or peripheral immune compartment(s) may prevent neuroprotective SOD1G93A CD4+ T cell development after axotomy.
Increased naïve CD4+ T cells, from WT mice, adoptively transferred to SOD1G93A mice unable to support WT levels of axotomized FMN
To investigate whether the addition of isolate WT CD4+ T cells could prevent the increased susceptibility of SOD1G93A FMN to axotomy-induced cell death, WT CD4+ T cells were adoptively transferred into SOD1G93A mice prior to axotomy. Thus, the number of CD4+ T cells in the intrinsic peripheral immune compartment of SOD1G93A mice was increased in an attempt to determine if this would prevent the susceptibility of SOD1G93A FMN to axotomy-induced cell death. At 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of SOD1G93A mice that received an adoptive transfer of WT CD4+ T cells (SOD1G93A + WT CD4+ T cells) were 72% ± 2.8, relative to the uninjured facial nucleus (Figures 1K and 2), which was significantly reduced compared to WT FMN survival levels and no different from SOD1G93A FMN survival levels. These results indicate that the addition of WT CD4+ T cells alone could not restore WT FMN survival levels after axotomy.
Increased non-CD4+ naïve peripheral splenocytes, from WT mice, adoptively transferred to SOD1G93A mice unable to support WT levels of axotomized FMN
In an effort to alter the intrinsic peripheral immune compartment of pre-symptomatic SOD1G93A mice and provide a peripheral microenvironment that could potentially activate intrinsic SOD1G93A CD4+ T cells after axotomy, WT splenocytes depleted of CD4+ T cells were adoptively transferred into SOD1G93A mice prior to axotomy. At 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of SOD1G93A mice that received an adoptive transfer of WT splenocytes depleted of CD4+ T cells (SOD1G93A + CD4-depleted WT splenocytes) were 72% ± 4.5, relative to the uninjured facial nucleus (Figures 1L and 2). The FMN survival levels in the SOD1G93A + CD4-depleted WT splenocytes group were comparable to SOD1G93A levels and significantly reduced compared to WT. These results indicate that the addition of WT CD4-depleted splenocytes could not support WT FMN survival levels after axotomy, as observed with the addition of WT unfractionated splenocytes, or prime SOD1G93A CD4+ T cells to mediate neuroprotection.
Bypassing the SOD1G93A peripheral microenvironment with adoptive transfer of prior axotomy-activated WT CD4+ T cells did not result in WT levels of axotomized FMN
To evaluate whether the addition of prior activated CD4+ T cells from axotomized WT mice could bypass the initial activation requirement after axotomy in the SOD1G93A peripheral immune compartment, prior axotomy-activated (i.e., facial nerve antigen-primed; Byram et al., 2004) CD4+ T cells were isolated from the cervical lymph nodes and spleens of WT mice after axotomy and adoptively transferred into SOD1G93A mice prior to axotomy. At 4 weeks post-axotomy, FMN survival levels in the axotomized facial nucleus of SOD1G93A mice that received an adoptive transfer of prior axotomy-activated WT CD4+ T cells (SOD1G93A + Axot WT CD4+ T cells) were 59% ± 5.1, relative to the uninjured facial nucleus (Figures 1M and 2). The SOD1G93A + Axot WT CD4+ T cells group exhibited FMN survival levels comparable to SOD1G93A levels and significantly reduced compared to WT levels. Prior activation of WT CD4+ T cells was unable to restore SOD1G93A FMN survival levels via bypassing the initial activation in the SOD1G93A peripheral immune compartment.
Discussion
Molecular abnormalities in the microenvironment surrounding FMN of SOD1G93A mice are evident prior to and following facial nerve transection (Mesnard et al., 2011). The significant delay in SOD1G93A peripheral nerve regeneration (Mesnard et al., 2013) and reduced FMN survival levels (Mesnard et al., 2011) mirror the response of immunodeficient mice to facial nerve crush or cut axotomy (Serpe et al., 1999; Serpe et al., 2000; Serpe et al., 2002), and implicate a potential defect in immune-mediated neuroprotection. In immunodeficient mice, we have established a crucial role for the CD4+ T cell, initially activated in the peripheral immune compartment, and then reactivated in the CNS by MHCII-expressing glial cells, once FMN have been experimentally disconnected from their target musculature (Byram et al., 2004). Thus, the functionality of CD4+ T cells, peripheral APC, and central glia, is critical to the mechanism underlying immune-mediated neuroprotection.
The objective of this study was to utilize the facial nerve axotomy model to begin to elucidate underlying immune-related defects that contribute to disease in the SOD1G93A mouse model of ALS WT levels of FMN survival after axotomy require functional APC in both peripheral (i.e., B cells, macrophages, or dendritic cells) and central (i.e., microglia) compartments (Byram et al., 2004). If SOD1G93A FMN survival levels after axotomy indeed resemble RAG2-/- mice due to a defect in immune-mediated neuroprotection, this would suggest a defect in either the central glial or peripheral immune response.
Interestingly, our data indicate that WT unfractionated splenocytes adoptively transferred into SOD1G93A mice are able to maintain WT levels of FMN survival after axotomy. These results eliminate the possibility of a defect located centrally in the SOD1G93A mice because the re-activation of the peripherally-activated CD4+ T cell, which is essential for FMN survival (Byram et al., 2004), was able to occur and maintain FMN survival levels to that of WT after axotomy in the SOD1G93A mice when WT mouse splenocytes were adoptively transferred. However, SOD1G93A unfractionated splenocytes adoptively transferred into RAG2-/- mice fail to support WT levels of FMN survival. Yet, isolated SOD1G93A CD4+ T cells are equally as effective in maintaining FMN survival levels as WT CD4+ T cells when adoptively transferred into RAG2-/- mice. Therefore, separation of the SOD1G93A CD4+ T cell from its peripheral microenvironment before adoptive transfer is necessary for the SOD1G93A CD4+ T cell to functionally mediate neuroprotection. However, increasing the number of naïve CD4+ T cells, increasing the number of naïve non-CD4+ splenocytes, or attempting to bypass the intrinsic SOD1G93A peripheral immune compartment with prior activated WT CD4+ T cells all failed to maintain FMN survival levels in SOD1G93A mice when adoptively transferred prior to axotomy. Collectively, these data indicate that a peripheral microenvironment defect exists in the SOD1G93A mouse that may involve the prevention and/or reversal of the appropriate neuroprotective CD4+ T cell development or activation through an active inhibitory mechanism.
Interestingly, polyclonally-activated WT CD4+ T cells, as opposed to naïve CD4+ T cells, are effective in altering disease progression when adoptively transferred to SOD1G93A mice (Banerjee et al., 2008), further suggesting that a defective SOD1G93A peripheral microenvironment prevents naïve T cell activation. Immunodeficient SOD1G93A mice, crossed onto the RAG2-/- background, possess a shortened life-span (Beers et al., 2008), which can be restored with the addition of WT or SOD1G93A bone marrow, or adoptive transfer of WT or SOD1G93A CD4+ T cells (Beers et al., 2008; Beers et al., 2011). Two alternative interpretations of these results are that either the SOD1G93A B and CD4+ T cells contribute a degree of neuroprotection in the SOD1G93A mouse model of ALS or the remaining peripheral microenvironment is neurodestructive without functional interactions with CD4+ T cells. Both interpretations are supported by the results obtained in the current study. Moreover, the adoptive transfer of anti-CD3-activated WT CD4+ T cells into SOD1G93A mice crossed with RAG2-/- mice delays disease progression (Banerjee et al., 2008), as do WT CD4+ (Chiu et al., 2008) or CD4+Foxp3+ (Beers et al., 2008) T cells. These reports further suggest that exogenous CD4+ T cells are capable of mediating neuroprotection in SOD1G93A mice to regulate the ALS disease process, but do not address the functionality of the SOD1G93A CD4+ T cells specifically.
In the current study, our novel finding was that SOD1G93A CD4+ T cells indeed possess the capability of mediating neuroprotection and supporting MN survival, suggesting that their development is not interrupted but, rather, that their activation is inhibited or disrupted. Therefore, a peripheral splenic microenvironment-mediated defect may underlie the heightened susceptibility of SOD1G93A FMN to axotomy-induced cell death, and a combination of WT CD4+ T cells and WT APC is required to overcome the suppressive or dysfunctional SOD1G93A peripheral microenvironment. Collectively, this study demonstrates that the SOD1G93A CD4+ T cell is able to respond normally to an antigen-specific response initiated by facial nerve axotomy, but is prevented from functioning in a neuroprotective manner due to a complex immune regulatory mechanism that exists in SOD1G93A mice and modulated only with WT unfractionated splenocytes.
In summary, an overall disrupted homeostatic balance in the peripheral microenvironment (i.e., macrophages, dendritic cells, B cells, T cell subsets, etc.) and central microenvironment (i.e., microglia, astrocytes, infiltrating immune cells, etc.) appears to play a role in ALS motor neuron degeneration, disease onset and progression (Troost et al., 1992; O'Reilly et al., 1995; Graves et al., 2004; Henkel et al., 2004; Turner et al., 2004; Rafalowska et al., 2010; Sanagi et al., 2010; Mesnard et al., 2011). Our results point toward a problem in ALS CD4+ T cell activation, peripheral antigen presentation, and/or antigen-presenting cell development in the periphery that, in turn, contributes to MN death when disconnection, by disease or experimentally by axotomy, from target musculature occurs.
Acknowledgments
This study was supported by NIH NS40433 (KJJ and VMS) and the Muscular Dystrophy Association MDA202906 (JX).
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Cited Literature
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- Appel SH, Beers DR, Henkel JS. T cell-microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends in immunology. 2010;31:7–17. doi: 10.1016/j.it.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Mosley RL, Reynolds AD, Dhar A, Jackson-Lewis V, Gordon PH, Przedborski S, Gendelman HE. Adaptive immune neuroprotection in G93A-SOD1 amyotrophic lateral sclerosis mice. PloS one. 2008;3:e2740. doi: 10.1371/journal.pone.0002740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15558–15563. doi: 10.1073/pnas.0807419105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, Appel SH, Henkel JS. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain, behavior, and immunity. 2011;25:1025–1035. doi: 10.1016/j.bbi.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram SC, Carson MJ, DeBoy CA, Serpe CJ, Sanders VM, Jones KJ. CD4-positive T cell-mediated neuroprotection requires dual compartment antigen presentation. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:4333–4339. doi: 10.1523/JNEUROSCI.5276-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byram SC, Serpe CJ, Pruett SB, Sanders VM, Jones KJ. Natural killer cells do not mediate facial motoneuron survival after facial nerve transection. Brain, behavior, and immunity. 2003;17:417–425. doi: 10.1016/s0889-1591(03)00089-8. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, Brown RH, Jr, Carroll MC. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17913–17918. doi: 10.1073/pnas.0804610105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Phatnani H, Kuligowski M, Tapia JC, Carrasco MA, Zhang M, Maniatis T, Carroll MC. Activation of innate and humoral immunity in the peripheral nervous system of ALS transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20960–20965. doi: 10.1073/pnas.0911405106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoy CA, Byram SC, Serpe CJ, Wisuri D, Sanders VM, Jones KJ. CD4+CD25+ regulatory T cells and CD1-restricted NKT cells do not mediate facial motoneuron survival after axotomy. Journal of neuroimmunology. 2006a;176:34–38. doi: 10.1016/j.jneuroim.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Deboy CA, Xin J, Byram SC, Serpe CJ, Sanders VM, Jones KJ. Immune-mediated neuroprotection of axotomized mouse facial motoneurons is dependent on the IL-4/STAT6 signaling pathway in CD4(+) T cells. Experimental neurology. 2006b;201:212–224. doi: 10.1016/j.expneurol.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Experimental neurology. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F, van Kooten C, Vinters HV. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2004;5:213–219. doi: 10.1080/14660820410020286. [DOI] [PubMed] [Google Scholar]
- Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. The New England journal of medicine. 1994;331:1721–1722. doi: 10.1056/NEJM199412223312516. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Haulcomb MM, Mesnard NA, Batka RJ, Alexander TD, Sanders VM, Jones KJ. Axotomy-induced target disconnection promotes an additional death mechanism involved in motoneuron degeneration in ALS transgenic mice. The Journal of comparative neurology. 2014 doi: 10.1002/cne.23538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiology of disease. 2007;28:154–164. doi: 10.1016/j.nbd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Annals of neurology. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Holmoy T. T cells in amyotrophic lateral sclerosis. European journal of neurology : the official journal of the European Federation of Neurological Societies. 2008;15:360–366. doi: 10.1111/j.1468-1331.2008.02065.x. [DOI] [PubMed] [Google Scholar]
- Jinno S, Yamada J. Using comparative anatomy in the axotomy model to identify distinct roles for microglia and astrocytes in synaptic stripping. Neuron glia biology. 2011;7:55–66. doi: 10.1017/S1740925X11000135. [DOI] [PubMed] [Google Scholar]
- Jones KJ, LaVelle A. Changes in nuclear envelope invaginations in axotomized immature and mature hamster facial motoneurons. Brain Res. 1985;353:241–249. doi: 10.1016/0165-3806(85)90212-3. [DOI] [PubMed] [Google Scholar]
- Jones KJ, Serpe CJ, Byram SC, Deboy CA, Sanders VM. Role of the immune system in the maintenance of mouse facial motoneuron viability after nerve injury. Brain, behavior, and immunity. 2005;19:12–19. doi: 10.1016/j.bbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. The American journal of pathology. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- Kennel PF, Finiels F, Revah F, Mallet J. Neuromuscular function impairment is not caused by motor neurone loss in FALS mice: an electromyographic study. Neuroreport. 1996;7:1427–1431. doi: 10.1097/00001756-199605310-00021. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle & nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- Mesnard NA, Hauclomb MM, Tanzer L, Sanders VM, Jones KJ. Delayed functional recovery in presymptomatic mSOD1-G93A mice following facial nerve crush axotomy. J Neurodegen Regen. 2013;4:21–25. [PMC free article] [PubMed] [Google Scholar]
- Mesnard NA, Sanders VM, Jones KJ. Differential gene expression in the axotomized facial motor nucleus of presymptomatic SOD1 mice. The Journal of comparative neurology. 2011;519:3488–3506. doi: 10.1002/cne.22718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LB, Graeber MB. The facial nerve axotomy model. Brain research Brain research reviews. 2004;44:154–178. doi: 10.1016/j.brainresrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- O'Reilly SA, Roedica J, Nagy D, Hallewell RA, Alderson K, Marklund SL, Kuby J, Kushner PD. Motor neuron-astrocyte interactions and levels of Cu,Zn superoxide dismutase in sporadic amyotrophic lateral sclerosis. Experimental neurology. 1995;131:203–210. doi: 10.1016/0014-4886(95)90042-x. [DOI] [PubMed] [Google Scholar]
- Philips T, Robberecht W. Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. Lancet neurology. 2011;10:253–263. doi: 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- Rafalowska J, Dziewulska D, Gadamski R, Chrzanowska H, Modrzewska-Lewczuk M, Grieb P. Is the spinal cord motoneuron exclusively a target in ALS? Comparison between astroglial reactivity in a rat model of familial ALS and in human sporadic ALS cases. Neurological research. 2010;32:867–872. doi: 10.1179/174313209X414542. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sanagi T, Yuasa S, Nakamura Y, Suzuki E, Aoki M, Warita H, Itoyama Y, Uchino S, Kohsaka S, Ohsawa K. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. Journal of neuroscience research. 2010;88:2736–2746. doi: 10.1002/jnr.22424. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Coers S, Sanders VM, Jones KJ. CD4+ T, but not CD8+ or B, lymphocytes mediate facial motoneuron survival after facial nerve transection. Brain, behavior, and immunity. 2003;17:393–402. doi: 10.1016/s0889-1591(03)00028-x. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunodeficient (scid) mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1999;19:RC7. doi: 10.1523/JNEUROSCI.19-11-j0004.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpe CJ, Sanders VM, Jones KJ. Kinetics of facial motoneuron loss following facial nerve transection in severe combined immunodeficient mice. Journal of neuroscience research. 2000;62:273–278. doi: 10.1002/1097-4547(20001015)62:2<273::AID-JNR11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Serpe CJ, Tetzlaff JE, Coers S, Sanders VM, Jones KJ. Functional recovery after facial nerve crush is delayed in severe combined immunodeficient mice. Brain, behavior, and immunity. 2002;16:808–812. doi: 10.1016/s0889-1591(02)00017-x. [DOI] [PubMed] [Google Scholar]
- Troost D, Das PK, van den Oord JJ, Louwerse ES. Immunohistological alterations in muscle of patients with amyotrophic lateral sclerosis: mononuclear cell phenotypes and expression of MHC products. Clinical neuropathology. 1992;11:115–120. [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiology of disease. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Zang DW, Lopes EC, Cheema SS. Loss of synaptophysin-positive boutons on lumbar motor neurons innervating the medial gastrocnemius muscle of the SOD1G93A G1H transgenic mouse model of ALS. Journal of neuroscience research. 2005;79:694–699. doi: 10.1002/jnr.20379. [DOI] [PubMed] [Google Scholar]
- Zhao W, Beers DR, Liao B, Henkel JS, Appel SH. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiology of disease. 2012;48:418–428. doi: 10.1016/j.nbd.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]