

Abstract
A critical step for the development of biosensors is the immobilization of the biorecognition element to the surface of a substrate. Among other materials that can be used as substrates, block copolymers have the untapped potential to provide significant advantages for the immobilization of proteins. To explore such possibility, this manuscript describes the fabrication and characterization of thin-films of polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP). These films were then used to investigate the immobilization of glucose oxidase, a model enzyme for the development of biosensors. According to the results presented, the nanoporous films can provide significant increases in surface area of the substrate and the immobilization of larger amounts of active enzyme. The characterization of the substrate-enzyme interface discussed in the manuscript aims to provide critical information about relationship between the surface (material, geometry, and density of pores), the protein structure, and the immobilization conditions (pH, ionic strength, and protein concentration) required to improve the catalytic activity and stability of the enzymes. A maximum normalized activity of 3300 ± 700 U m−2 was achieved for the nanoporous film of PS-b-P2VP.
Keywords: polystyrene-block-poly(2-vinylpyridine), glucose oxidase, ellipsometry, atomic force microscopy, nanoporous materials, adsorption, entrapment
1. Introduction
A critical consideration in the development of biosensors is the method selected for the immobilization of the biorecognition element on the surface of a substrate. Besides cross-linking, entrapment, and microencapsulation [1], physical adsorption is one of the simplest and most benign immobilization methods because it is fast and avoids harsh reaction conditions or additional components (such as entrapping polymers). Moreover, since adsorption is the first interaction step, it affects most other immobilization routes [2] and can play a key role in the rational development of biocatalytic surfaces. In general, the adsorption of biorecognition elements (and more so, proteins) to solid surfaces is driven by a combination of hydrophobic and electrostatic interactions as well as other processes such as redistribution of charged residues, arrangement of ions and solvent molecules, and structural modifications [3]. These processes ultimately determine the conformation and functionality of the adsorbed molecules. As recently described [4], the change in a protein’s native conformation upon interaction with a surface depends on a balance between favorable entropic and unfavorable enthalpic changes. In the case of weakly attractive conditions, favorable entropic changes dominate the process yielding to the stabilization of the structure of the protein while maintaining (most of) its footprint on the surface. With enhanced surface-protein interactions, enthalpic effects become dominant, the native structure is destabilized, and the proteins tend to experience structural rearrangements during the adsorption process. As a consequence, a significant number of proteins adsorbed to solid surfaces can be affected by surface-induced structural changes, leading to spreading (unfolding and refolding) which can be detrimental to the functionality of the adsorbed proteins. In addition, chemical modifications of the protein, use of hydrophilic substrates [5], and prudent selection of specific experimental conditions (that maximize protein immobilization rate) [6] can limit the spreading and help to preserve the activity of enzymes [7, 8].
Interestingly, only a handful of papers have described the use of nanostructured cavities for biosensing applications [8] and just a few papers have described protein-related applications of nanopores. Among them, Ma and Young [9] (considering proteins as hard entities that would not adsorb to the substrate) reported that the entrapment process is mostly dependent on the membrane pore diameter. Aiming to measure translocation (not adsorption) of single protein molecules, platforms based on voltage-biased silicon nitride films [10–12] or protein-based nanopores have also been used [10–13]. Although these reports take advantage of a very clever experimental design and highlight the scientific relevance of investigating the interaction of biomolecules with restricted domains [14], protein-pore interactions are often neglected, the experiments are performed at high ionic strength (> 2M NaCl), and a potential difference across the pore is applied. In addition, protein-based nanopores are rather fragile, expensive, and require specific resources for fabrication and assembly [15, 16].
To overcome these limitations, increased attention has been directed to the use of block copolymers (BCPs) [17, 18]. These materials are composed of two or more chemically distinct, and frequently immiscible, polymer blocks covalently linked [19]. BCPs have the capacity to self-assemble into nanostructures with various conformations, requiring short processing times and low-cost facilities for fabrication. Moreover, thin films of polystyrene-block-poly(methyl methacrylate) (PS-b-PMMA) have enabled the selective deposition of proteins [20, 21] and nanoparticles [22] on the surface of one of the components. In such cases, proteins have shown a preference for the hydrophobic polystyrene (PS) microdomain regions over the PMMA. An interesting system composed of polystyrene-block-poly(2-vinylpyridine) (PS-b-P2VP) can self-assemble into a rich variety of nano/microphase-separated morphologies including spheres (micelles), cylinders, gyroids, and lamellas creating a nanoporous surface [23]. More importantly, the nanostructured surface of these block copolymers could provide significant advantages for the immobilization of proteins by increasing surface area and increasing surface curvature to limit protein spreading [24]. Therefore, and considering the potential that nanoporous films of PS-b-P2VP have for the development of nanocomposites, this manuscript describes the fabrication and characterization of the substrates, the development of an optical model to describe their optical properties, the immobilization of glucose oxidase (GOx), and the measurement of the catalytic activity of the immobilized enzyme. GOx was selected because it can be used for the removal of glucose or oxygen from food products, production of gluconic acid, as an antibiotic, and for the development of glucose biosensors [25, 26]. GOx is a dimeric globular enzyme with only one non-covalently bound flavin adenine dinucleotide per monomer (active-site), which under denaturing conditions, can be released from the protein [27]. In all these cases, improving the catalytic activity of the surfaces modified with GOx is essential.
2. Materials and Methods
2.1. Reagents and Solutions
All chemicals were analytical-reagent grade and used as received. Hydrogen peroxide (35%) was purchased from Columbus Chemical (Columbus, WI). Sodium acetate was purchased from Mallinckrodt Baker, Inc. (acquired by Avantor Performance Materials, Center Valley, PA). Hydrochloric acid and sodium hydroxide were purchased from EMD Millipore (Billerica, MA). Block copolymers of polystyrene and poly-(2-vinylpyridine) (MWPS = 101,000 Da, MWP2VP = 29,000 Da, Mw/Mn = 1.6) were purchased from Polymer Source Inc. (Dorvel, Quebec). Citric acid, ethanol, GOx from Aspergillus niger (17.3 U·mg−1), horseradish peroxidase (HRP, 199 purpurogallin units mg−1), N,N-dimethylformamide (DMF), o-xylene, PS (MW = 95,800 Da), and α-D-Glucose were purchased from Sigma Aldrich (St. Louis, MO). O-dianisidine dihydrochloride was purchased from TCI America (Portland, OR) and sulfuric acid was purchased from VWR (Radnor, PA). The pH of the aqueous solutions was adjusted using either 1 M NaOH or 1 M HCl and measured using a glass electrode and a digital pH meter (Orion 420A+, Thermo; Waltham, MA). All aqueous solutions were prepared using 18 MΩcm water (NANOpure Diamond, Barnstead; Dubuque, IA). Unless otherwise stated, experiments were conducted at room temperature (22 ± 2 °C).
2.2. Preparation of the nanostructured films
Standard <111> silicon wafers (Sumco; Phoenix, AZ) were initially scored using a computer-controlled engraver (Gravograph IS400, Gravotech; Duluth, GA). The process defined substrates of 1 cm in width and 4–6 cm in height that were then manually cut and cleaned in piranha solution (30% hydrogen peroxide and 70% sulfuric acid) at 90°C for 30 min. After thorough rinsing with water, the substrates were dried under a stream of nitrogen and stored in clean vials until coated. In order to obtain the nanostructured films, solutions of PS and PS-b-P2VP (0.5% w/v in o-xylene and 0.5% w/v in DMF) were prepared and then filtered through a 0.2 μm poly(tetrafluoroethylene) (PTFE) membrane at least three times to remove any aggregates. The solution containing the selected block copolymer was spin-coated onto silicon substrates (wafers) using a Laurell Technologies WS-400A-6NPP/LITE spin coater (North Wales, PA), depositing a monolayer of arranged micelles [28]. The wafer is then dried at room temperature and immersed in a glass petri dish containing ethanol to induce micelle opening while stirring at 60 rpm on an Innova 2000 Platform Shaker (Enfield, CT). After the selected time, the substrates were removed from the vial and the ethanol was allowed to evaporate under a gentle stream of nitrogen.
2.3. Spectroscopic ellipsometry
Determination of the thickness of the PS-b-P2VP films as well as the immobilization kinetics were performed by spectroscopic ellipsometry (WVASE, J.A. Woollam Co., Lincoln, NE) following a procedure described elsewhere [6, 29–31]. As described by Equation 1,
| Equation 1 |
the ellipsometric angles (Ψ, amplitude and Δ, phase difference) can be related to changes in amplitude and phase difference between the parallel (RP) and perpendicular (RS) components of a polarized light beam upon reflection from a surface [32]. Under these conditions, the technique has proven suitable to calculate thickness of the immobilized film and therefore to investigate the kinetics of the immobilization of proteins. The sensitivity of the technique [33] was also considered appropriate for the purpose of the present study. Collected data (ellipsometric angles as function of time, angle, and/or wavelength) were modeled using the WVASE software package (J. A. Woollam Co., Lincoln, NE) using the mean square error (MSE) to quantify the difference between the experimental and model-generated data. The ellipsometry experiments herein discussed were performed in either air or an ad-hoc cell [34] by performing spectroscopic scans in the 300 to 800 nm range (with 10 nm steps) using air or the corresponding aqueous buffer as the ambient medium.
2.4. Atomic Force Microscopy
In order to gain insight about the topography as well as to verify the thickness of the block-di-polymer film, atomic force microscopy (AFM) was used. AFM was performed in the tapping mode with a Nanoscope V (multimode, VEECO Instruments, USA) and using Aspire conical AFM tips (Nanoscience Instruments, Phoenix, AZ). The AFM tip has a conical shape with a height of 15 μm and a radius of curvature of 8 nm. Topography analysis to determine surface features was determined by using various functions in the NanoScope Analysis v1.40 and Gwyddion v2.31 software. It is also worth mentioning AFM measurements underestimate actual surface roughness due to the tip’s finite size [35–37] and limited measured area.
2.5. Enzymatic Activity
The activity of the GOx immobilized to the SiO2, PS, and PS-b-P2VP thin films was measured using a modified version of a previously reported procedure [38], based on the reaction of β-D-glucose (Sigma-Aldrich, Saint Louis, MO). In the presence of GOx, the oxidation reaction produced β-D-glucono-∂-lactone and H2O2. The latter was then used to oxidize o-dianisidine in the presence of HRP, developing a color change that was monitored spectrophotometrically at 500 nm. In order to determine the effect of the proposed films on the activity of immobilized GOx, the PS-b-P2VP-coated substrates (2 cm2 geometric area) were first immersed in a solution containing 0.5 mg mL−1 of GOx (in 10 mM citrate buffer, pH = 4.2) for 30 min. These conditions were selected because the solution provides high buffer capacity around the isoelectric point of the protein, therefore maximizing the initial adsorption rate [5]. Then, the PS-b-P2VP/GOx substrates were gently rinsed with buffer (to remove any GOx loosely bound to the substrate), scanned using ellipsometry to determine the effective thickness, and placed in a quartz cuvette previously filled with a mixture of glucose, o-dianisidine, and HRP. To homogenize the solution, a magnetic bar was placed inside the quartz cuvette and stirred at a constant rate using a magnetic stirrer (Spinette, Sterna; Atascadero, CA, USA). The enzymatic activity was calculated by following the change in absorbance at 500 nm, monitored for at least 15 min using a spectrophotometer (Genesys 10 UV, Thermo Scientific, USA).
3. Results and Discussion
3.1. Characterization of the Nanostructured Films
In order to investigate the topography of the substrates, AFM was used. Figure 1 shows representative images obtained with the selected polymers. In the first case (Figure 1A, control), the Si/SiO2 wafer was coated with a solution of PS (disolved in o-xylene) and rendered a rather flat and featureless surface. Figure 1B shows the surface of the Si/SiO2 wafer upon spin-coating a solution containing the selected PS-b-P2VP block-copolymer (also prepared in o-xylene). As it can be observed, a higher density of nanostructures was obtained on the surface, forming a micellar array consisting of a hydrophobic PS matrix and hydrophilic P2VP clusters [39]. The contact angle, measured using a 10 μL drop of DI water and a standard digital camera, was approximately 86°. As demonstrated in Figure 1C, the P2VP clusters can be opened upon the immersion of the substrate in ethanol, yielding an array of nanopores. A contact angle of approximately 75° was measured thus further confirming the micellar formation and bursting of hydrophilic chains of the block copolymer proposed by Chen et al. [39] Although similar films of PS-b-P2VP can be prepared in DMF (spin-coating onto wafers and immersed in ethanol for 1.5 h), the resulting AFM images showed large features of various shapes and sizes, which were deemed as unsuitable for this study (data not shown). In order to provide comparative information related to the immobilization process, Figure 1D shows the resulting topography of the substrate after the interaction with GOx.
Figure 1.

3D AFM images corresponding to Si/SiO2 wafers modified with polystyrene (A), as-coated PS-b-P2VP (B), the nanoporous block copolymer formed upon the immersion in ethanol for 2 h (C), and GOx immobilized to the nanoporous block polymer (D). Conditions: polymer concentration: 5 mg mL−1 in o-xylene, spin-coated at 3000 rpm.
Although it is known that the dimensions (chain length and molecular weight) [19] of the selected components of the block copolymer can affect the topographical features of the film, our study showed that spin rate of the spin coater and ethanol soaking time can also affect the size and density of the nanoporous array (Figure 2). Experiments were conducted to study the pore count and pore diameter as a function of spin rate as well as roughness and surface area as a function of ethanol soaking (calculated using the Nanoscope software functions). According to the results shown in Figure 2A, pores with an average diameter ranging from 33 ± 10 nm to 42 ± 6 nm were formed when the substrates were spin-coated at speeds between 2000 and 4000 rpm. The density of nanopores in the array, also depended on the spin rate and varied from 660 ± 30 pores μm−2 to 816 ± 4 pores μm−2. It was also observed that spin rates of 2000 rpm do not provide uniform deposits of PS-b-P2VP onto the wafer, leaving the edges uncoated and resulting in pore diameters with a higher standard deviation. Based on these results, films produced at 2000 rpm were considered inappropriate for the goals of this project.
Figure 2.
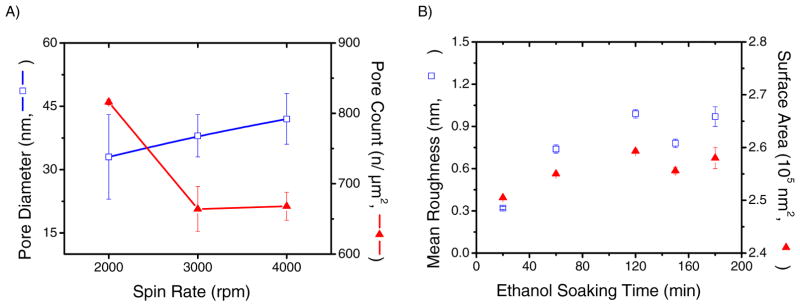
The pore diameters and count were evaluated as a function of spin rate (A). The mean roughness and surface area of the block copolymer spin-coated at 3000 rpm was evaluated as a function of ethanol soaking time (B). The projected surface area of the AFM image size is 250,000 nm2.
According to the results shown in Figure 2B, both roughness and surface area of the nanoporous polymer increase with increasing ethanol soaking time. A minimum of 120 min of ethanol soaking time is required to ensure roughness and surface area are maximized (fluctuations within the maximized range were observed for ethanol times greater than 120 min). Due to the reproducibility and a balance between experimental efficiency and optimum conditions, a spin rate of 3000 rpm and ethanol soaking time of 120 min were considered the most appropriate for the proposed experiments. In order to demonstrate the stability of the nanoporous substrates, the substrates were stored over the course of 28 days and sequential AFM images were obtained every seven days to show that the “pores” remained intact after the selected amount of time (results shown in the Supplementary Information).
Developing a model that can accurately describe the optical properties of the PS-b-P2VP substrates in terms of refractive index (n), extinction coefficient (k), and thickness (d) is critical to interpret the data collected by spectroscopic ellipsometry. Besides considering that the measurement can be affected by the microstructure of the film, an additional problem associated with thin layers is that the three parameters (n, k, and d) can be coupled leading, in such case, to incorrect interpretations. In order to avoid this problem, a series of substrates coated with a layer of PS-b-P2VP (before and after immersion in ethanol) were used to develop an optical model that adequately represents the optical properties of the material. As a result, the substrate was described using a model comprising layers of silicon (bulk; d = 1 mm), SiO2 (d = 1.57 ± 0.02 nm), and a transparent layer (representing the nanoporous PS-b-P2VP) described using a Bruggeman Effective Media Approximation (EMA) layer composed of the polymer substrate (described by a Cauchy function) and void space (Figure 3A). After a first approximation to estimate the thickness (that was later confirmed by AFM using a scratch test shown in the Supplementary Information), the computer-calculated factors of the model (n(λ) = A + Bλ−2 + Cλ−4) were also allowed to fit to further improve the optical model. The resulting fitted parameters of the Cauchy function for as-coated (A = 1.1565, B = 0.0107 and C = 0) and nanoporous block copolymer (A = 1.5506, B = 0.0102 and C = 0) yielded a very good agreement between their respective sets of data (experimental and model-generated) and allowed calculating the average thickness of the as-coated films of 20.2 ± 0.4 nm and nanoporous films of 20.5 ± 0.7 nm (Figure 3B). The roughness of the surface, incorporated using the % void in the EMA layer, was always < 5%.
Figure 3.
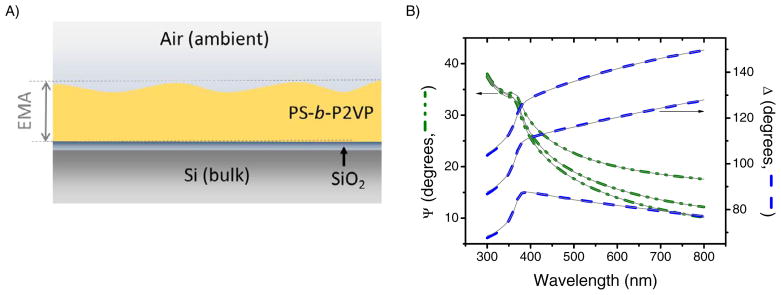
The proposed optical model (A) and the spectroscopic scan (B) obtained for three angles (65, 70, and 75°) of the nanoporous substrate in air (MSE = 3.814). The gray lines indicate the results generated by the proposed optical model.
Considering that the block copolymer is composed by a section of PS (101 kDa) and a section of P2VP (29 kDa), it is reasonable to assume that optical parameters of the two sections of the block copolymer would not be significantly different. Upon verifying the thickness of the PS-b-P2VP layer by AFM, the presented approach allowed investigating the immobilization of the enzyme to the selected substrates.
3.2 Immobilization of GOx
In order to calculate the amount of GOx immobilized on the PS-b-P2VP film, the optical properties of the substrates were measured by spectroscopic ellipsometry. For these experiments, the substrate coated with the selected film was aligned in the cell and a spectroscopic scan was performed to determine the initial thickness. Next, a solution containing GOx was introduced into the cell and the ellipsometric angles followed using 500 nm as the incident wavelength. Once the signal was stabilized, an additional spectroscopic scan was performed to determine the final thickness and the microstructure of the sample. In order to maximize the initial immobilization rate and minimize the possibility of inducing conformational changes, experiments were conducted at the isoelectric point of the protein (pI = 4.2) and at a protein concentration of 0.5 mg mL−1. As it can be observed in Figure 4A, significant changes were observed in the ellipsometric angles after the substrate was exposed to the solution containing GOx, indicating that the protein was immobilized to the nanoporous polymer.
Figure 4.
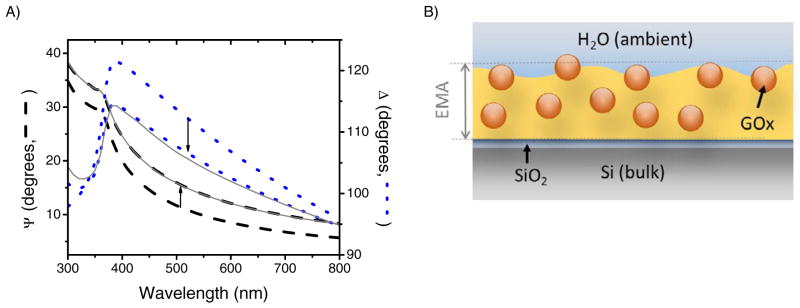
Spectroscopic scan (A) before and after 3 h of 0.5 mg mL−1 GOx immobilization in 0.010 M citrate buffer, pH = 4.2. Directions of arrows indicate the change in ellipsometric angles from before to after GOx immobilization. The gray lines indicate the results generated by the proposed optical model, after the immobilization of GOx (MSE = 3.302). The proposed optical model (B) with immobilized GOx.
As it was expected to observe proteins penetrating into the pores (because the pores are larger than the protein molecules), the proposed optical model was initially allowed to consider the adsorption of proteins on the surface of the film [6], replacing void space (defined by the nanopores) with protein. However, when multiple experiments were analyzed, significant increases in the MSE value were obtained, indicating that the proposed model was not able to accurately describe the optical properties of the sample and therefore may not represent the physical meaning of the measurement. Therefore, and in order to consider the possibility of GOx penetrating into the structure of the polymer, the EMA layer was modified to include the protein (also described with a Cauchy function, n(λ) = 1.45 + 0.01·λ−2) [33, 40, 41] as an additional component. The resulting model, schematically shown in Figure 4B, allowed calculating changes in the total thickness of the substrates as well as the relative contribution of the protein to the film.
As shown in Figure 4A, a very good agreement (MSE = 3.302) was obtained between the experimental and the model-generated data for the spectroscopic scan, indicating that the proposed model was able to describe the immobilization of GOx to the nanoporous polymer. In this case, the resulting EMA layer was measured to be approximately 41.0 nm and contained 33.9 ± 0.4% of GOx. Based on these results, the amount of GOx immobilized corresponds to an effective thickness of 13.9 ± 0.2 nm. Considering that GOx is a globular (long ovoid) protein with approximate dimensions of 7 nm × 5.5 nm × 8 nm [42] that most often adsorbs in single layers [43], our results are in agreement with the hypothesis that protein molecules are not only able to adsorb on the surface of the film but also penetrate the polymer matrix, yielding to a rather uniform nanocomposite. It is important to note that although this optical model allowed the description of the resulting system from the spectroscopic scans, it did not allow calculating the distribution of the enzyme within the film, probably due to the similarity of the optical properties of the polymer and protein.
In order to obtain information about the stability of the immobilized protein layer, desorption experiments were performed by switching from the GOx-containing solution to the background electrolyte. In all cases (data not shown), a small fraction (< 7%) of the immobilized protein was released from the surface after a 80-min rinsing step with buffer. This observation suggests that the affinity of GOx for the polymer is relatively high and is compatible with the semi-soft nature of GOx, also associated with potential structural changes in the adsorbed state [27].
Control experiments performed with a nanoporous polymer exposed to a solution (citrate buffer, pH = 4.2) with no GOx, showed only slight changes in Psi; suggesting a swelling rate for the bare polymer of 0.005 nm min−1.
3.3 Activity of Immobilized GOx
As previously stated, the goal of this project was to mitigate post-immobilization conformational changes and maximize the catalytic activity of the resulting nanocomposite. Such information is critical for the development of catalytic surfaces and biosensors because the conditions selected for the immobilization can yield GOx films with a wide range of activity [5, 44–47]. Thus, the normalized catalytic activity of GOx immobilized to the selected films was evaluated spectrophotometrically. As shown in Figure 5, the lowest activity was obtained for the bare SiO2 substrate followed by the substrate coated with PS, the as-coated PS-b-P2VP, and the nanoporous PS-b-P2VP. These results clearly evidence the advantages of using block copolymers for immobilization of enzymes.
Figure 5.

Normalized enzymatic activity of GOx immobilized to Si/SiO2 wafers modified with PS, as-coated PS-b-P2VP (*), and the nanoporous PS-b-P2VP (formed upon the immersion in ethanol). Conditions for the immobilization: 0.010 M Buffer citrate, pH = 4.2, enzymatic activity measured as described in the experimental section.
4. Conclusions
In this report, the results related to the use of nanoporous block copolymers of PS-b-P2VP as substrates for the development of nanocomposites are described. Thin-films (thickness in the order of 20 nm) were produced, yielding a relatively ordered structure with cavities of ranging from approximately 23 to 48 nm in diameter and about 3 to 4 nm in depth. Such films were then used as substrates to investigate the immobilization of GOx, an enzyme that has been extensively used as a model for biosensing applications. To interpret the results collected by spectroscopic ellipsometry, an optical model was developed. According to the results presented, the films provided increases in surface area and curvature and favored the immobilization of the enzyme with a normalized maximum activity of 3300 ± 700 U m−2 for the nanoporous film of PS-b-P2VP. The immobilization of GOx demonstrated that (at least some) proteins should not be considered proteins as hard spheres and that their interaction with the inside of the cavity may not be the most probable route but rather a combination of both entrapment and adsorption to the polymer matrix and surface is more likely.
Supplementary Material
Highlights.
AFM and ellipsometry used to characterize thin-films of (PS-b-P2VP)
Model describing the optical properties of the substrates
GOx immobilized on the block copolymer by means of adsorption and entrapment
Highest enzymatic activity obtained with the nanoporous PS-b-P2VP substrate
Acknowledgments
The authors gratefully acknowledge the financial support provided by the University of Texas at San Antonio and the National Institutes of Health through the National Institute of General Medical Sciences (1SC3GM081085, 2SC3GM081085), the Research Centers at Minority Institutions (G12MD007591), and the Partnership for Research and Education in Materials at the University of Texas at San Antonio (NSF-DMR-0934218). Authors would also like to thank Karin Chumbimuni-Torres for performing the preliminary experiments related to this project.
Abbreviations
- PS-b-P2VP
polystyrene-block-poly(2-vinylpyridine)
- GOx
glucose oxidase
- BCPs
block copolymers
- PS-b-PMMA
polystyrene-block-poly(methyl methacrylate)
- PS
polystyrene
- P2VP
poly-(2-vinylpyridine)
- MW
molecular weight
- Mw/Mn
distribution ratio
- HRP
horseradish peroxidase
- DMF
N,N-dimethylformamide
- PTFE
poly(tetrafluoroethylene)
- n
refractive index
- k
extinction coefficient
- d
thickness
- Ψ
amplitude
- Δ
phase difference
- RP
parallel component
- RS
perpendicular component
- VASE
variable angle spectroscopic ellipsometry
- MSE
mean square error
- AFM
atomic force microscopy
- EMA
Effective Media Approximation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mikkelsen SR, Cortón E. Bioanalytical chemistry. John Wiley & Sons; Hoboken, N.J: 2004. [Google Scholar]
- 2.Gao Y, Kyratzis I. Bioconjugate Chem. 2008;19:1945. doi: 10.1021/bc800051c. [DOI] [PubMed] [Google Scholar]
- 3.Karajanagi SS, Vertegel AA, Kane RS, Dordick JS. Langmuir. 2004;20:11594. doi: 10.1021/la047994h. [DOI] [PubMed] [Google Scholar]
- 4.Vaitheeswaran S, Garcia AE. J Chem Phys. 2011;134:125101. doi: 10.1063/1.3558776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nejadnik MR, Francis L, Garcia CD. Electroanal. 2011;23:1462. doi: 10.1002/elan.201000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felhofer JL, Caranto JD, Garcia CD. Langmuir. 2010;26:17178. doi: 10.1021/la103035n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Norde W. In: Biopolymers at Interfaces (Surfactant Sci) Malmsten M, editor. Marcel Dekker; New York: 2003. [Google Scholar]
- 8.Norde W. Colloids Surf B. 2008;61:1. doi: 10.1016/j.colsurfb.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 9.Ma C, Yeung ES. Anal Chem. 2010;82:478. doi: 10.1021/ac902487c. [DOI] [PubMed] [Google Scholar]
- 10.Talaga DS, Li J. J Am Chem Soc. 2009;131:9287. doi: 10.1021/ja901088b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han A, Creus M, Schürmann G, Linder V, Ward TR, de Rooij NF, Staufer U. Anal Chem. 2008;80:4651. doi: 10.1021/ac7025207. [DOI] [PubMed] [Google Scholar]
- 12.Fologea D, Ledden B, McNabb DS, Li J. Appl Phys Lett. 2007;91 doi: 10.1063/1.2767206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han A, Schurmann G, Mondin G, Bitterli RA, Hegelbach NG, Rooij NFd, Staufer U. Appl Phys Lett. 2006;88:093901. [Google Scholar]
- 14.Pedone D, Langecker M, Abstreiter G, Rant U. Nano Letters. 2011;11:1561. doi: 10.1021/nl104359c. [DOI] [PubMed] [Google Scholar]
- 15.Shim JW, Gu LQ. J Phys Chem A. 2008;112:8354. doi: 10.1021/jp0775911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawano R, Osaki T, Sasaki H, Takinoue M, Yoshizawa S, Takeuchi S. J Am Chem Soc. 2011;133:8474. doi: 10.1021/ja2026085. [DOI] [PubMed] [Google Scholar]
- 17.Xue L, Zhang J, Han Y. Prog Polym Sci. 2012;37:564. [Google Scholar]
- 18.Escalé P, Rubatat L, Billon L, Save M. Eur Polym J. 2012;48:1001. [Google Scholar]
- 19.Darling SB. Prog Polym Sci. 2007;32:1152. [Google Scholar]
- 20.Kumar N, Hahm J-i. Langmuir. 2005;21:6652. doi: 10.1021/la050331v. [DOI] [PubMed] [Google Scholar]
- 21.Li, Hitchcock AP, Robar N, Cornelius R, Brash JL, Scholl A, Doran A. J Phys Chem A. 2006;110:16763. doi: 10.1021/jp062442o. [DOI] [PubMed] [Google Scholar]
- 22.Jung JM, Kwon KY, Ha TH, Chung BH, Jung HT. Small. 2006;2:1010. doi: 10.1002/smll.200600005. [DOI] [PubMed] [Google Scholar]
- 23.Lee DH, Cho H, Yoo S, Park S. Journal of Colloid and Interface Science. 2012;383:118. doi: 10.1016/j.jcis.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 24.Vertegel AA, Siegel RW, Dordick JS. Langmuir. 2004;20:6800. doi: 10.1021/la0497200. [DOI] [PubMed] [Google Scholar]
- 25.Ahmad A, Akhtar MS, Bhakuni V. Biochemistry. 2001;40:1945. doi: 10.1021/bi001933a. [DOI] [PubMed] [Google Scholar]
- 26.Akhtar MS, Ahmad A, Bhakuni V. Biochemistry. 2002;41:3819. doi: 10.1021/bi0116700. [DOI] [PubMed] [Google Scholar]
- 27.Georganopoulou DG, Williams DE, Pereira CM, Silva F, Su TJ, Lu JR. Langmuir. 2003;19:4977. [Google Scholar]
- 28.Segalman RA. Materials Science and Engineering: R: Reports. 2005;48:191. [Google Scholar]
- 29.Mora MF, Giacomelli CE, Garcia CD. Anal Chem. 2009;81:1016. doi: 10.1021/ac802068n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soetedjo H, Mora MF, Garcia CD. Thin Solid Films. 2010;518:3954. doi: 10.1016/j.tsf.2010.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wehmeyer J, Bizios R, Garcia CD. Mat Sci Eng C. 2010;30:277. doi: 10.1016/j.msec.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujiwara H. Principles and applications. J. Wiley & Sons; West Sussex, England: 2007. Spectroscopic ellipsometry. [Google Scholar]
- 33.Nejadnik MR, Garcia CD. Colloids Surf B. 2011;82:253. doi: 10.1016/j.colsurfb.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mora MF, Reza Nejadnik M, Baylon-Cardiel JL, Giacomelli CE, Garcia CD. J Colloid Interface Sci. 2010;346:208. doi: 10.1016/j.jcis.2010.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Westra K, Thomson D. Journal of Vacuum Science & Technology B. 1995;13:344. [Google Scholar]
- 36.Odin C, Aime J, El Kaakour Z, Bouhacina T. Surface Science. 1994;317:321. [Google Scholar]
- 37.Tranchida D, Piccarolo S, Deblieck RAC. Meas Sci Technol. 2006;17:2630. [Google Scholar]
- 38.Bergmeyer HU. Methods of enzymatic analysis, Verlag Chemie. Academic Press; Weinheim New York: 1974. [Google Scholar]
- 39.Chen Z, He C, Li F, Tong L, Liao X, Wang Y. Langmuir. 2010;26:8869. doi: 10.1021/la904623e. [DOI] [PubMed] [Google Scholar]
- 40.Felhofer JL, Caranto J, Garcia CD. Langmuir. 2010;26:17178. doi: 10.1021/la103035n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chumbimuni-Torres KY, Coronado RE, Mfuh AM, Castro-Guerrero C, Silva MF, Negrete GR, Bizios R, Garcia CD. RSC Advances. 2011:706. doi: 10.1039/C1RA00198A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hecht HJ, Schomburg D, Kalisz H, Schmid RD. Biosensors and Bioelectronics. 1993;8:197. doi: 10.1016/0956-5663(93)85033-k. [DOI] [PubMed] [Google Scholar]
- 43.Benavidez TE, Garcia CD. Langmuir. 2013;29:14154. doi: 10.1021/la4029657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Georganopoulou DG, Williams DE, Pereira CM, Silva F, Su TJ, Lu JR. Langmuir. 2003;19:4977. [Google Scholar]
- 45.Cullen SP, Mandel IC, Gopalan P. Langmuir. 2008;24:13701. doi: 10.1021/la8024952. [DOI] [PubMed] [Google Scholar]
- 46.Sun S, Ho-Si PH, Harrison DJ. Langmuir. 1991;7:727. [Google Scholar]
- 47.Svensson O, Arnebrant T. J Colloid Interface Sci. 2012;368:533. doi: 10.1016/j.jcis.2011.11.034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.