

Summary
Mycobacterial Clp-family proteases function via collaboration of the heteromeric ClpP1P2 peptidase with a AAA+ partner, ClpX or ClpC1. These enzymes are essential for M. tuberculosis viability and are validated antibacterial drug targets, but the requirements for assembly and regulation of functional proteolytic complexes are poorly understood. Here, we report the reconstitution of protein degradation by mycobacterial Clp proteases in vitro and describe novel features of these enzymes that distinguish them from orthologs in other bacteria. Both ClpX and ClpC1 catalyze ATP-dependent unfolding and degradation of native protein substrates in conjunction with ClpP1P2, but neither mediates protein degradation with just ClpP1 or ClpP2. ClpP1P2 alone has negligible peptidase activity, but is strongly stimulated by translocation of protein substrates into ClpP1P2 by either AAA+ partner. Interestingly, our results support a model in which both binding of a AAA+ partner and protein-substrate delivery are required to stabilize active ClpP1P2. Our model has implications for therapeutically targeting ClpP1P2 in dormant M. tuberculosis, and our reconstituted systems should facilitate identification of novel Clp protease inhibitors and activators.
Introduction
The emergence of bacterial pathogens resistant to conventional antibiotics has led to an urgent need for novel therapeutics (Neu, 1992, Levy & Marshall, 2004, Howard et al., 2013). This need is especially pressing for Mycobacterium tuberculosis, a globally significant pathogen that continues to acquire resistance to the limited armamentarium of antibiotics (Jassal & Bishai, 2009, Gandhi et al., 2010). The Clp proteases have the potential to address this clinical need as novel and orthogonal antibiotic targets (Brotz-Oesterhelt et al., 2005, Raju et al., 2012, Brötz-Oesterhelt & Sass, 2013). These enzymes are essential for viability of M. tuberculosis, which is also sensitive to unregulated activation of Clp activity, making these proteases especially attractive antibiotic targets (Sassetti et al., 2003, Griffin et al., 2011, Ollinger et al., 2012, Raju et al., 2012, Personne et al., 2013). The Clp proteases consist of a self-compartmentalized peptidase and an ATP-dependent AAA+ unfoldase (ATPases associated with diverse cellular activities). These components collaborate to carry out regulated protein degradation in a variety of physiological processes, including homeostatic protein quality control, responses to environmental stress, and virulence in pathogenic bacteria (Gaillot et al., 2000, Frees et al., 2003, Kwon et al., 2004, Sauer et al., 2004, Fernandez et al., 2012, Frees et al., 2014).
To degrade protein substrates, the ClpP peptidase functions with a partner AAA+ unfoldase (ClpX, ClpC, or ClpA). ClpP forms heptameric rings, which stack face-to-face to form a tetradecameric barrel-shaped enzyme (Flanagan et al., 1995, Wang et al., 1997). The ClpP catalytic sites face an interior solvent-filled degradation chamber, which is separated from bulk solvent by a narrow axial pore in each heptameric ring. The AAA+ partner enzymes form ring hexamers, which interface coaxially with one heptameric face the ClpP barrel (Grimaud et al., 1998, Maurizi et al., 1998, Kim & Kim, 2003). Binding of a AAA+ partner to ClpP induces widening of the axial pore (Lee et al., 2010a, Lee et al., 2010b, Li et al., 2010). Protein substrates are recognized by the AAA+ partner, which uses cycles of ATP binding, hydrolysis, and product release to mechanically unfold and translocate the polypeptide through the axial pore of ClpP and into its proteolytic chamber for degradation.
Most proteobacteria and firmicutes encode a single clpP ortholog (Yu & Houry, 2007), which self-assembles into a catalytically active tetradecamer even in the absence of a AAA+ partner (Böttcher & Sieber, 2008, Lee et al., 2010a, Lee et al., 2010b, Li et al., 2010). By contrast, other bacterial phyla, including all actinobacteria, encode multiple clpP paralogs. Although several parologous ClpP systems have been characterized in vitro (Stanne et al., 2007, Zeiler et al., 2011, Akopian et al., 2012, Tryggvesson et al., 2012), the assembly, regulation and function of multi-ClpP enzymes in protein degradation may differ across bacterial phyla. Indeed, phylogenetic comparisons suggest that clpP paralogs evolved independently in different bacterial lineages and that actinobacterial ClpP paralogs, in particular, are conserved and distinct from the ClpP enzymes of other phyla (Fig. S1).
M. tuberculosis, an actinobacterium, encodes two ClpP paralogs, ClpP1 and ClpP2, which are expressed from a bicistronic operon (Raju et al., 2012). Each paralog independently assembles into inactive homo-heptameric rings (ClpP1 or ClpP2), which then combine to form the double-ring ClpP1P2 tetradecamer (Benaroudj et al., 2011, Akopian et al., 2012). Curiously, ClpP1P2 tetradecamer formation and cleavage of small peptide substrates in vitro requires the presence of N-blocked hydrophobic peptides or peptide aldehydes, which act as agonists that are not themselves cleaved (Akopian et al., 2012, Compton et al., 2013). Agonists act by a mechanism distinct from AAA+ partners and acyldepsipeptide (ADEP) antibiotics, which are not needed for cleavage of small peptides by E. coli ClpP and related homomeric enzymes but activate cleavage of larger peptides and polypeptides by enlarging the axial ClpP pore (Lee et al., 2010b, Li et al., 2010). Why agonists are required for mycobacterial Clp activity is not clear, nor has it been established whether agonists are necessary for degradation of folded proteins by ClpP1P2 in conjunction with ClpX or ClpC1, the endogenous mycobacterial AAA+ partners. To understand how Clp proteases in M. tuberculosis and other actinobacteria function and are regulated, we sought to reconstruct mycobacterial Clp proteases capable of degrading folded protein substrates.
Results
Enzymatic characterization of ClpX and ClpC1
We were able to clone and purify full-length M. tuberculosis ClpX bearing an N-terminal SUMO tag that improved stability and solubility (MtbsClpX) but failed to clone M. tuberculosis ClpC1. We were, however, able to clone and purify M. smegmatis ClpC1 (MsmClpC1), which is 94% identical to M. tuberculosis ClpC1. Moreover, the regions of MsmClpC1 and MtbClpC1 responsible for nucleotide binding and hydrolysis and for interactions with ClpP are identical in both proteins. Both MtbsClpX and MsmClpC1 hydrolyzed ATP in the absence of ClpP (Fig. 1). MsmClpC1 had a relatively high KM for ATP hydrolysis (~3.6 mM) and an ATPase rate that depended on enzyme concentration, with half-maximal activity at a hexamer concentration of ~60 nM (Fig. 1A,B). The latter result suggests that MsmClpC1 hexamers dissociate to inactive species at low concentrations. MtbsClpX had a lower KM for ATP hydrolysis (~0.4 mM), but formed a less stable hexamer, with half-maximal ATPase activity at ~500 nM hexamer (Fig. 1C,D). Removing the SUMO tag with Ulp1 peptidase did not alter the concentration dependence of ClpX ATPase activity (Fig. 1D), indicating that hexamer instability is an intrinsic property and not a consequence of the tag.
Figure 1. ATPase activity and hexamer stability of mycobacterial ClpX and ClpC1.

A MsmClpC1 (0.5 μM) ATPase activity was measured as a function of ATP concentration and fit to a Hill equation (apparent KM = 3.6 ± 0.6 mM; Hill n = 1.3 ± 0.2).
B Dependence of ATPase activity on MsmClpC1 concentration. The line is a fit to a Hill equation (K0.5 = 58 ± 1.9 nM; n = 3.4 ± 0.4).
C MtbsClpX (0.5 μM) ATPase activity was measured as a function of ATP concentration and fit to a Hill equation (apparent KM = 0.4 ± 0.006 mM; n = 2.7 ± 0.09).
D Dependence of ATPase activity on MtbsClpX concentration. The line is a fit to a Hill equation (K0.5 = 0.54 ± 0.09 μM; n = 1.4 ± 0.3 nM). Removal of the N-terminal SUMO domain from MtbsClpX by cleavage with the Ulp1 protease (confirmed by SDS-PAGE) had little effect on hexamer stability (K0.5 = 0.66 ± 0.23 μM; n = 1.8 ± 0.8).
Values in all panels are averages (N = 3) ± 1 standard deviation (SD).
AAA+ partners and agonist stimulate ClpP1P2 activity synergistically
We tested if ClpP1, ClpP2, or ClpP1P2 catalyzed cleavage of a fluorogenic decapeptide, and whether this activity was affected by binding of MtbsClpX, MsmClpC1, and/or Z-Ile-Leu agonist (Fig. 2A, S2A). In the absence of AAA+ partners, ClpP1, ClpP2, and ClpP1P2 exhibited negligible decapeptide cleavage with or without agonist. Similarly, little cleavage activity was observed in the absence of agonist, even when a AAA+ partner was present. Strikingly, however, the combination of agonist and either AAA+ partner strongly stimulated ClpP1P2 cleavage of the decapeptide. Robust stimulation occurred only in the presence of both ClpP1 and ClpP2, and much lower activities were observed with ClpP1 or ClpP2 alone (Fig. S2A). ClpP1P2 activation by MtbsClpX occurred in the presence ATP or ATPγS, but not with ADP or in the absence of nucleotide (Fig. S2D), consistent with other bacterial Clp systems where only ATP- or ATPγS-bound ATPases interact with ClpP (Seol et al., 1995, Joshi et al., 2004). Curiously, under the conditions of this assay, MsmClpC1 strongly activated ClpP1P2 only in the presence of ATPγS (Fig. S2E), similar to the behavior the distantly related AAA+ enzyme PAN, which interacts with the 20S peptidase more tightly in the presence of ATPγS than ATP (Barthelme & Sauer, 2012). MsmClpC1 also supported a low level of ClpP1P2 peptidase activity in the presence of ATP and an ATP regeneration system (Fig. S3E).
Figure 2. Robust peptidase activity requires ClpP1P2, agonist, and ClpC1 or ClpX.

A, B Cleavage of a fluorogenic decapeptide (15 μM, A) or Z-GGL-AMC tripeptide (50 μM, B) was assayed for different combinations of ClpP1P2 (1 μM), Z-Ile-Leu agonist (1 mM), and MsmClpC1 (1 μM; 1 mM ATPγS) or MtbsClpX (1 μM; 1 mM ATPγS). Robust cleavage of either peptide was only observed if ClpP1 and ClpP2 were both present (Fig. S2A,B) and if ATPγS and a AAA+ partner were present (Fig. S2D,E). A contaminant in MtbsClpX preparation catalyzed slow ClpP1P2-independent peptide cleavage (Fig. S2C). Values are averages (N ≥ 3) ± 1 SD.
In principle, AAA+ partners and agonist might stimulate decapeptide cleavage by M. tuberculosis ClpP1P2 by stabilizing a catalytically active form of the peptidase and/or by enhancing peptide diffusion into the degradation chamber by opening the axial pore (Lee et al., 2010a, Lee et al., 2010b, Li et al., 2010). To test if pore opening alone is responsible for activity enhancement, we assayed the rate of cleavage of a tripeptide, which should freely diffuse into the ClpP chamber, with different combinations of ClpP1P2, AAA+ partners, and agonist (Fig. 2B, Fig. S2B). As observed with decapeptide cleavage, robust activity was observed only in the presence ClpP1P2, agonist, and a AAA+ partner. Thus, agonist and AAA+ partners appear to synergistically stabilize an active conformation of ClpP1P2. Because the combination of ClpP1 and ClpP2 alone is inactive in peptide degradation, these experiments do not resolve whether pore opening is also required for decapeptide cleavage.
ClpX and ClpC1 collaborate with ClpP1P2 to degrade folded substrates
We next tested if mycobacterial ClpP-family AAA+ proteases are capable of degrading natively folded proteins in vitro, as previous studies only demonstrated degradation of unstructured substrates (Barik et al., 2010, Akopian et al., 2012). Michaelis-Menten analysis showed that MtbsClpX•ClpP1P2 degrades native green-fluorescent protein bearing a M. tuberculosis ssrA tag (GFP-ssrA) with a Vmax of ~0.1 min−1 enz−1 and a KM of ~3 μM in the presence of Z-Ile-Leu (Fig. 3A). Because ClpP1P2 alone is inactive, this degradation activity is evidence of a direct interaction between MtbsClpX and ClpP1P2. An apparent affinity of 380 μM for ClpP1P2 interaction with MtbsClpX was measured using GFP-ssrA degradation as a proxy for binding (Fig. 3B). By contrast, no substantial degradation of GFP-ssrA was observed with agonist and MtbsClpX and ClpP1, MtbsClpX and ClpP2, or MtbsClpX and E. coli ClpP (EcoClpP; Fig. 3C). The inability of MtbsClpX to collaborate with mycobacterial ClpP1 or ClpP2 in protein degradation was consistent with the decapeptide-cleavage experiments described above. However, its inability to function with EcoClpP was surprising, given that a variant of E. coli ClpX lacking the N-domain (EcoClpXΔN; 43% identical to the corresponding region of M. tuberculosis ClpX) forms a functional protease in conjunction with MtbClpP1P2 (Fig. 3D; see Discussion).
Figure 3. Native protein degradation by ClpX•ClpP1P2 and ClpC1•ClpP1P2.

A MtbsClpX (0.5 μM) and ClpP1P2 (1 μM) degrade GFP bearing a mycobacterial ssrA tag. The line is a fit to the Michaelis-Menten equation (KM = 2.9 ± 0.2 μM; Vmax = 0.097 GFP•min−1•ClpX−1). Degradation of different concentrations of GFP-ssrA was assayed by loss of native fluorescence as described (Kim et al., 2000).
B Degradation of GFP-ssrA (10 μM) was measured as a function of ClpP1P2 concentration in the presence of MtbsClpX (0.5 μM; gray circles) or MsmClpC1 (0.5 μM; white circles). Fitting to a Hill equation gave apparent affinities of 0.38 ± 0.03 μM for MtbsClpX and 1.7 ± 0.2 μM for MsmClpC1, with Hill constants of 1.3 ± 0.1 and 1.2 ± 0.1, respectively).
C MtbsClpX (0.5 μM) functioned with ClpP1P2 (0.75 μM) but not with ClpP1 (0.75 μM), ClpP2 (0.75 μM), or EcoClpP (0.75 μM) to degrade GFP-ssrA (10 μM).
D EcoClpXΔN (0.2 μM) degraded GFP-ssrA at similar rates in combination with ClpP1P2 (0.75 μM) or EcoClpP (0.75 μM), but not with ClpP1 (0.75 μM) or ClpP2 (0.75 μM).
E MsmClpC1 (1 μM) functioned with ClpP1P2 (2 μM) but not ClpP1 (2 μM) or ClpP2 (2 μM) to degrade GFP-ssrA (10 μM).
F H6-TEV-MsmClpC1 was degraded in an ATP-dependent reaction in the presence of ClpP1P2 (0.5 μM). Untagged MsmClpC1 was not degraded when ClpP1P2 and ATP were present.
Values in panels A, B, C, D, and E are averages (N = 3) ± 1 SD, and Z-Ile-Leu was present in all reactions at a concentration of 0.5 mM.
At saturating GFP-ssrA, the rate of degradation by MtbsClpX•ClpP1P2 was more than 10-fold slower than degradation by EcoClpXΔN•ClpP1P2 or EcoClpXΔN•EcoClpP (Fig. 3C,D). Thus, the overall rate of degradation of GFP-ssrA by MtbsClpX•ClpP1P2 appears to be limited by the rate of MtbsClpX unfolding rather than by the rate of ClpP1P2 proteolysis. Slower unfolding by MtbsClpX may be a consequence of its relatively low rate of ATP hydrolysis, ~15% that of EcoClpX (Joshi et al., 2004), as GFP-ssrA is unfolded and degraded by EcoClpXP much more efficiently at high ATPase rates (Martin et al., 2008).
MsmClpC1•ClpP1P2 also degraded GFP-ssrA at a modest maximal rate (0.12 min−1 enz−1; Fig. 3E) in the presence of agonist, suggesting that low degradation activity may be an intrinsic characteristic of the mycobacterial AAA+ ClpP-family proteases or that a component missing from our biochemical assays promotes faster degradation in vivo. Again, GFP-ssrA degradation was observed with MsmClpC1 and ClpP1P2 but not with ClpP1 or ClpP2 alone (Fig. 3E). By assaying GFP-ssrA degradation in the presence of fixed MsmClpC1 and varying ClpP1P2 concentrations, we determined a Kapp of 1.7 μM for the interaction of MsmClpC1 with ClpP1P2 (Fig. 3B). Interestingly, we also observed ATP-dependent and ClpP1P2-dependent auto-proteolysis of ClpC1 bearing an N-terminal H6-TEV tag (Fig. 3F). Removal of the tag prevented auto-proteolysis, suggesting recognition of the unstructured tag near the ClpC1 N-terminus.
Using GFP-ssrA degradation as an assay, we tested the effects of inactivating mutations in the catalytic serines of ClpP1 (S98A), ClpP2 (S110A), or both ClpP1 and ClpP2 (Fig. 4A, B, C). In conjunction with any of the AAA+ partners, proteases with one active and one inactive ClpP ring were able to degrade protein substrate, indicating that both ClpP1 and ClpP2 are active in substrate degradation. Curiously, MsmClpC1•ClpP1P2 had higher activity when one ClpP ring was inactive (Fig. 4C), and these mutant ClpP rings bound more tightly to MsmClpC1 (Fig. 4D). Thus, the S98A and S110A active-site mutations appear to stabilize a ClpP1P2 conformation that binds MsmClpC1 more tightly either directly or indirectly, for example by strengthening binding of Z-Ile-Leu. As anticipated, complexes with active-site mutations in both ClpP1 and ClpP2 were unable to degrade protein substrate (Fig. 4A, B, C).
Figure 4. The active sites of both the P1 and P2 rings in ClpP1P2 contribute to substrate degradation.

A Degradation of GFP-ssrA (10 μM) by EcoClpXΔN (0.2 μM) and ClpP1P2 (2 μM) bearing catalytic SA mutations in neither ring, one or the other ring, or both rings.
B Degradation assayed as in A but with MtbsClpX (0.5 μM).
C Degradation assayed as in A but with MsmClpC1 (0.5 μM).
D Degradation of GFP-ssrA (10 μM) was assayed in the presence of MsmClpC1 (0.5 μM) and varying concentrations of ClpP1SAP2 (black circles) or ClpP1P2SA (gray circles). Fitting to a Hill equation gave an apparent affinity of 0.68 ± 0.08 μM and a Hill constant of 2.0 ± 0.4 for ClpP1SAP2 (black line), and an apparent affinity of 0.73 ± 0.07 μM and a Hill constant of 1.9 ± 0.3 for ClpP1P2SA (gray line). The fit for degradation in the presence of wild-type ClpP1P2 (dashed line, from Fig. 3B) is shown for comparison.
All experiments contained ATP (2.5 mM and a regeneration system) and Z-Ile-Leu (0.5 mM). Values are averages (N = 3) ± 1 SD.
Protein substrates delivered by ClpX stimulate ClpP1P2 peptidase activity
We found that GFP-ssrA degradation by EcoClpXΔN•ClpP1P2, MtbsClpX•ClpP1P2, or MsmClpC1•ClpP1P2 occurred in the absence of the Z-Ile-Leu agonist, but at 30–80% of the rate determined with agonist (Fig. 5A). This slower protease activity correlated with weaker apparent affinities between the AAA+ ATPases and ClpP1P2 in the absence of agonist (Fig. S3). By contrast, when agonist was omitted from decapeptide or tripeptide cleavage reactions, cleavage activities were only 7% or less of the stimulated reaction (Fig. 2). We hypothesized that protein substrates translocated into the ClpP1P2 degradation chamber might substitute for agonist in stimulating cleavage activity. In the absence of agonist, this model predicts that active degradation of a folded substrate would stimulate peptide degradation in trans. Indeed, we found that degradation of a non-fluorescent model substrate, V15P-titinI27-ssrA, by MtbsClpX•ClpP1P2 or EcoClpXΔN•ClpP1P2 stimulated degradation of the Z-GGL-AMC tripeptide in the absence of agonist (Fig. 5B). Stimulation by MtbsClpX•ClpP1P2 proteolysis was supported by ATP but not ATPγS, indicating that active unfolding and translocation of substrate into the ClpP degradation chamber is necessary for stimulation of peptide cleavage. Several observations support a model in which faster protein degradation results in a higher level of stimulation of peptidase activity. First, proteolysis of GFP-ssrA by EcoClpXΔN•ClpP1P2 enhanced peptide cleavage ~3-fold more than proteolysis by MtbsClpX•ClpP1P2, correlating with faster protein degradation by the former protease (Fig. 3C,D). Second, less stable titinI27-ssrA proteins that are degraded more rapidly by EcoClpXP (Kenniston et al., 2003) stimulated higher levels of peptide cleavage by MtbsClpX•ClpP1P2 (Fig. S2F). Third, GFP-ssrA also weakly stimulated peptide cleavage by EcoClpXΔN•ClpP1P2 in the presence of ATPγS, in accord with the ability of EcoClpXΔN to hydrolyze ATPγS slowly (Burton et al., 2003).
Figure 5. Protein substrates stimulate ClpP1P2 peptide-cleavage activity.

A MtbsClpX (0.5 μM), EcoClpXΔN (0.2 μM), or MsmClpC1 (1 μM) supported ClpP1P2 (2 μM) degradation of GFP-ssrA (10 μM) in the absence of agonist peptide at ~30–80% of the rate in the presence of Z-Ile-Leu agonist (0.5 mM).
B Active degradation of native V15P-titinI27-ssrA (10 μM) by ClpP1P2 (0.3 μM) and MtbsClpX (0.5 μM) or EcoClpXΔN (0.5 μM) stimulated ClpP1P2 cleavage of Z-GGL-AMC (75 μM) in the absence and presence of agonist (0.5 mM).
C In the upper panel, EcoClpXΔN•ClpP1P2 (0.5 μM) degradation of GFP-ssrA (4 μM; black circles; left axis) and cleavage of Z-GGL-AMC (50 μM; gray triangles; right axis) were monitored simultaneously. Peptide cleavage ceased at essentially the same time (arrow) that protein degradation stopped because GFP-ssrA was depleted (the residual GFP fluorescence is from protein lacking a functionalssrA tag). In the lower panel, GFP-ssrA was replaced with agonist (0.5 mM) and peptide cleavage by EcoClpXΔN•ClpP1P2 was continuous.
D In the upper, panel EcoClpXΔN•ClpP1P2 (0.5 μM) degradation of GFP-ssrA (10 μM) and cleavage of Z-GGL-AMC (50 μM) were monitored in the presence of limiting ATP (1 mM) with no ATP-regeneration system. When the ATP concentration fell below a level required to support GFP-ssrA degradation (arrow), peptide cleaved ceased with similar kinetics. In the lower panel, GFP-ssrA was replaced with agonist (0.5 mM) and peptide cleavage by EcoClpXΔN•ClpP1P2 was continuous over 35 min. Over a longer time scale, peptide cleavage also effectively ceased (Fig. S4) because ATP is required to support EcoClpXΔN activation of ClpP1P2 (Fig. S3).
Values in panels A and B are averages (N = 3) ± 1 SD.
To test further the model that polypeptide translocation into the proteolytic chamber of ClpP1P2 stabilizes a functional conformation of the ClpP1P2 active sites, we simultaneously monitored GFP-ssrA degradation and cleavage of a fluorogenic peptide by EcoClpXΔN•ClpP1P2, in the absence of agonist, under conditions where the protein substrate (Fig. 5C, upper panel) or ATP (Fig. 5D, upper panel) was depleted within ~10 min. In both experiments, protein degradation and peptide cleavage ceased with the same kinetics. By contrast, in parallel reactions where agonist was present in place of GFP-ssrA, peptide cleavage was continuous over the same time course (Fig. 5C, D, lower panels). The ability of protein substrates to stimulate peptide-cleavage activity of the ClpX•ClpP1P2 complex supports a model in which the peptidase activity of ClpP1P2 is reversibly regulated by substrate delivery by a AAA+ partner (Fig. 6).
Figure 6. A model for mycobacterial Clp protease regulation.
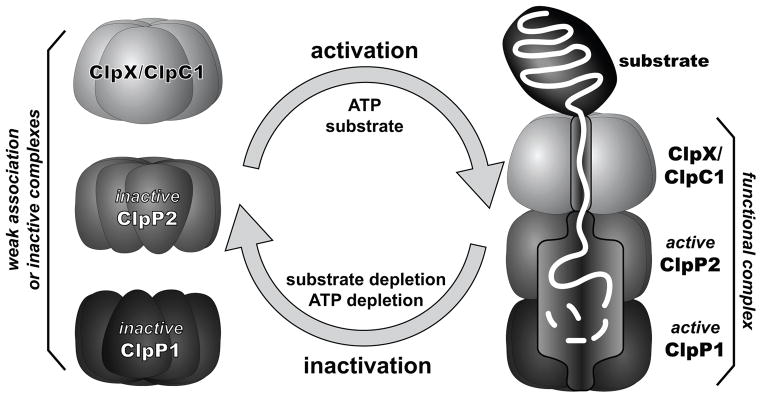
In the absence of protein substrate, the ClpP1 ring, the ClpP2 ring, and a ClpX/ClpC1 ring interact weakly or form proteolytically inactive complexes. In the presence of ATP and protein substrate, AAA+ partner binding and ATP-fueled translocation of the substrate polypeptide into the degradation chamber of ClpP1P2 stabilize the functional conformation of the peptidase active sites, allowing degradation. Low concentrations of protein substrate and/or ATP favor inactivation of ClpP1 and ClpP2. A ring of ClpX or ClpC1 is shown bound to the ClpP2 ring but could bind to ClpP1 or to both peptidase rings.
Discussion
We find that the mycobacterial Clp proteases are tightly regulated by ATP-dependent binding of AAA+ partners and by protein-substrate availability. This behavior contrasts markedly with orthologous Clp systems in which ClpP forms catalytically active tetradecamers in the absence of interaction partners, and unregulated degradation of substrates is restricted solely by the narrow axial pores of ClpP. In those systems, large peptides and unfolded polypeptides can slowly diffuse through the ClpP pores (Lee et al., 2010b), resulting in a low level of unregulated proteolysis. By contrast, mycobacterial ClpP1P2 adopts a catalytically inactive conformation under conditions where its AAA+ partners fail to bind or actively unfold and translocate protein substrates (e.g., low ATP). These restrictions in vitro likely mimic conditions inside the cell during periods of low metabolic activity. Under such conditions, the inactivity of ClpP1P2 could reduce wasteful degradation of nascent polypeptides or transiently disordered proteins and permit cells to carry out low levels of protein synthesis and chaperone-mediated protein folding without interference. These characteristics may confer a fitness advantage to slow-growing organisms adapted to nutrient-poor conditions and may contribute to the long-term viability of M. tuberculosis during latent infection. These regulatory characteristics are likely to be conserved across actinobacteria, given the conservation of the ClpP1 and ClpP2 paralogs. Substrate-binding may analogously regulate assembly and activity of more distantly related orthologs, such as human mitochondrial ClpP, which is similar to the mycobacterial system in that it forms inactive heptamers by itself but forms active tetradecamers in conjunction with ClpX (Kang et al., 2005).
Our findings are consistent with a dual allosteric model in which both AAA+ binding to ClpP1P2 and protein-substrate binding to the ClpP1P2 active sites regulate protease activity (Fig. 6). The enzyme components that form the functional AAA+ proteases are in dynamic equilibrium and appear to be predominantly inactive in the absence of protein substrates or at low ATP concentrations. Under these conditions, for example, most ClpP1 and ClpP2 would form inactive heptameric rings or an inactive ClpP1P2 tetradecamer with malformed substrate-binding pockets and/or catalytic triads. Similarly, ClpX and ClpC1 would largely be inactive hexamers or smaller oligomers at low ATP concentrations. As in any dynamic equilibrium, a small population of functional ClpX•ClpP1P2 or ClpC1•ClpP1P2 would exist. In the presence of saturating ATP and abundant protein substrate, AAA+ mediated recognition, unfolding, and translocation by these active complexes could have several effects. First, the translocating substrate could stabilize the complex between the AAA+ hexamer and ClpP1P2 by making contacts with both components. Second, the translocating substrate could stabilize the active conformation of ClpP1P2, allowing degradation. Third, both of these reactions would result in a shift of more components in the population into assembled and active proteolytic complexes. These active complexes are likely to be transient, and would disassemble or revert to inactive forms once ATP or protein substrates were depleted.
The transition between inactive and active forms of ClpP is likely to involve global conformational changes in ClpP1 and ClpP2 that simultaneously affect AAA+ partner binding, peptide-cleavage activity, and heptamer association. This model is supported by structural evidence from ClpP orthologs that show a dynamic interface between the two heptameric rings and demonstrate a correlation between the interface conformation and properly formed catalytic triads and substrate-binding pockets (Gribun et al., 2005, Kimber et al., 2010, Lee et al., 2011, Religa et al., 2011, Gersch et al., 2012). Crystal structures in which the interface has an extended conformation also show functional active-site conformations, whereas structures with a disordered or compressed interface, including that of M. tuberculosis ClpP1, reveal blocked substrate pockets and active-site residues positioned improperly for catalysis (Wang et al., 1997, Ingvarsson et al., 2007, Geiger et al., 2011, Lee et al., 2011).
Our model provides an explanation for the apparent requirement of non-physiological agonist peptides for peptide hydrolysis by ClpP1P2 (Akopian et al., 2012). Specifically, agonist peptides serendipitously mimic protein substrates, such that sub-stoichiometric occupancy of active sites by agonist provides binding energy to stabilize the active ClpP1P2 conformation. This interpretation is supported by the prior observation that high concentrations of agonist partially block covalent modification of the ClpP1P2 active-site serines (Akopian et al., 2012).
In addition to the substrate-mediated regulation described above, we note the potential for regulation through the partner AAA+ ATPases. The relatively weak KM of ClpC1 for ATP hydrolysis, and its apparent inhibition by ADP, may serve to sense the growth state of mycobacteria. Measurements of intracellular nucleotide concentration suggest that M. tuberculosis ATP levels are six-fold higher in actively dividing than in dormant cells (Rao et al., 2008), which could serve as a switch for ClpC1 activity. By contrast, ClpX binds ATP more tightly, but exhibits relatively weak hexamer stability. Thus, ClpX mediated proteolysis may require high expression levels of this enzyme, or its activity may require stabilizing interactions with ClpP, adaptors, or substrates.
The observation that both ClpX•ClpP1P2 and ClpC1•ClpP1P2 degrade ssrA-tagged substrates suggests that these enzymes have overlapping substrate specificities and that both could contribute to the turnover of ssrA-tagged proteins in the cell. E. coli ClpA, a double-ring AAA+ ATPase related to ClpC1, also supports ClpP degradation of ssrA-tagged proteins (Gottesman et al., 1998), although B. subtilis ClpC apparently does not (Wiegert & Schumann, 2001). We did not detect ClpC1 mediated degradation of constructs incorporating the B. subtilis ClpC adaptor and substrate, MecA (Turgay et al., 1998, Wang et al., 2011), homologs of which are absent in mycobacteria. Thus, we infer significant functional divergence between mycobacterial ClpC1 and B. subtilis ClpC.
Divergence is also apparent in the interactions between ClpX and ClpP, given the observation that EcoClpXΔN productively collaborates with both M. tuberculosis and E. coli ClpP enzymes, whereas MtbsClpX only forms a functional protease with M. tuberculosis ClpP1P2. The structural determinants of the interaction between ClpX and ClpP are not understood in detail, making the reason for this specificity difference unclear. We note that the E. coli ClpX IGF loops and pore-2 loops, which are important determinants of ClpP binding (Kim et al., 2001, Joshi et al., 2004, Martin et al., 2007), are similar in M. tuberculosis ClpX, suggesting that additional regions of this enzyme may be critical for the interaction with ClpP1P2.
Our work indicates that ClpP1P2 is the functional form of mycobacterial ClpP, supporting a model proposed by Akopian and colleagues (Akopian et al., 2012, Raju et al., 2012). However there are notable differences between our results and this prior study. In our assays Z-Ile-Leu alone did not support ClpP1P2 activity, whereas Akopian and colleagues found that a ten-fold higher concentration of a similar peptide, Z-Leu-Leu, activated ClpP1P2 in the absence of ATPase partner (Akopian et al., 2012). This result suggests that tighter-binding agonists or higher agonist concentrations provide sufficient binding energy to stabilize a substantial equilibrium population of active ClpP1P2. Akopian and coworkers also found that agonist was required for degradation of FITC-casein by M. tuberculosis ClpC1 and ClpP1P2, whereas we observed degradation of GFP-ssrA by MsmClpC1•ClpP1P2 even in the absence of Z-Ile-Leu. This discrepancy may be the result of the different substrates, slightly different buffer conditions, the relatively low concentration of ClpP1P2 used in the prior study, or differences between ClpC1 orthologs of M. tuberculosis and M. smegmatis.
Importantly, our model for ClpP1P2 function has implications for attempts to kill M. tuberculosis and other bacterial pathogens with active-site inhibitors of ClpP1P2, an area of active research (Zeiler et al., 2011, Compton et al., 2013, Gersch et al., 2013a, Gersch et al., 2013b). Dormant M. tuberculosis almost certainly carries out less Clp-mediated protein degradation, with ClpP1 and ClpP2 existing predominantly in inactive conformations. The inactive forms of these enzymes would be resistant to inhibitors that require an active conformation of the catalytic triad and substrate-binding pockets. However, our model predicts that simultaneous treatment with dysregulatory activators such as ADEPs could stabilize active ClpP1P2, rendering dormant cells more susceptible to active-site inhibitors of ClpP. Indeed, preliminary studies in vitro show that agonist peptides cause a substantial increase in ADEP activation of ClpP1P2 and that a combination of agonist and ADEP also enhances chemical modification of the active sites of ClpP1P2 by inhibitors (K. Schmitz, D. Carney, J. Sello, and R. Sauer, in preparation).
Our studies reveal that mycobacterial Clp-family AAA+ proteases utilize novel mechanisms of regulation, linking protein substrate delivery with ClpP peptidase activity. We anticipate that this mechanistic characterization along with the ability to functionally reconstruct these mycobacterial systems in vitro will aid in identifying and characterizing potential antibiotics against M. tuberculosis.
Experimental Procedures
Protein expression and purification
M. tuberculosis ClpP1 and ClpP2, bearing C-terminal His6-tag fusions, were cloned, expressed, and purified as described (Compton et al., 2013). Variants of titinI27 bearing C-terminal E. coli ssrA tags (AANDENYALAA) were cloned, expressed, and purified as described (Kenniston et al., 2005). Full-length M. tuberculosis ClpX was amplified by PCR from M. tuberculosis H37Rv genomic DNA (ATCC). An N-terminal fusion encoding a His7 tag and the yeast SUMO homolog Smt3p was added by PCR. The resulting construct (MtbsClpX) was cloned into pET-21b (Novagen). Full-length M. smegmatis ClpC1 (MsmClpC1) was amplified by PCR from M. smegmatis MC2155 genomic DNA (ATCC) and cloned into pET-21b (Novagen) in frame with an N-terminal His6-TEV tag. Plasmids encoding MtbsClpX or MsmClpC1 were transformed into ER2566 E. coli (NEB). GFP-ssrA ((Martin et al., 2005) was modified to incorporate a C-terminal mycobacterial ssrA tag (ADSNQRDYALAA) and cloned into pET-21b frame with an N-terminal His6-TEV tag. The plasmid encoding GFP-ssrA was transformed into the clpP− E. coli strain JK10 (Kenniston et al., 2005). Proteins were overexpressed in a 1:1 mixture of LB and 2xYT broth at RT, following induction with 0.5 mM isopropyl β-D-I-thiogalactopyranoside (IPTG; Teknova); 1 mM ZnCl2 was included for overexpression of MtbsClpX. Lysates were clarified by centrifugation, and proteins were purified by metal affinity (HisPur Ni-NTA agarose, Thermo), anion exchange (MonoQ, GE Healthcare), and size-exclusion chromatography (Superdex 200, GE Healthcare). The His6-TEV tag was removed from MsmClpC1 prior to anion exchange chromatography by overnight incubation with 1 μg tobacco etch virus protease per 10 μg protein in 25 mM HEPES, 50 mM NaCl, 5% glycerol, 1 mM DTT, 1 mM EDTA, pH 8.0. Purified MtbsClpX and MsmClpC1 were concentrated to ≥ 20 μM hexamer, and GFP-ssrA to 600 μM, in storage buffer (25 mM HEPES, 150 mM NaCl, 10% glycerol, pH 7.5).
Enzymatic assays
Assays were performed at 30°C in PD buffer (25 mM HEPES, 100 mM KCl, 10% glycerol, 5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, pH 7.5) on a SpectraMax M5 microplate reader (Molecular Devices). ATPase assays included 2.5 mM ATP (unless otherwise indicated; Sigma) and a NADH-coupled regeneration system (Nørby, 1988). Protease assays included 2.5 mM ATP and a regeneration system comprising 16 mM creatine phosphate (MP Biomedicals) and 0.32 mg/mL creatine phosphokinase (Sigma). ATP hydrolysis was monitored via decrease in 340 nm NADH absorbance. Peptidase assays followed hydrolysis of a fluorogenic decapeptide, Abz-KASPVSLGYNO2D (Lee et al., 2010b), by increase in 420 nm fluorescence upon 320 nm excitation, or of a fluorogenic tripeptide (Z-GGL-AMC; Enzo Life Sciences) by increase in 460 nm fluorescence upon 380 nm excitation. GFP-ssrA degradation was monitored by loss of 511 nm emission following excitation at 450 nm. Z-Ile-Leu and ADP were obtained from Sigma. ATPγS was obtained from EMD.
Supplementary Material
Figure S1. ClpP paralogs in actinobacteria are phylogentically distinct from other multi-ClpP systems.
A phylogram of representative bacterial ClpP enzymes suggests that multi-ClpP systems arose independently in different bacterial lineages. Actinobacteria, including M. tuberculosis (Mtb) encode conserved ClpP1 and ClpP2 paralogs. Cyanobacteria and chlamidiae each possess distinct multi-ClpP systems that are conserved within these phyla. The multiple ClpP enzymes in Bacillus thuringiensis (Bth) are likely the result of a recent gene duplication event, whereas those of Pseudomonas aeruginosa (Pae) and Listeria monocytogenes (Lmo) are likely to have arisen from horizontal gene transfer. Ordinal suffixes have been added (asterisks) to multi-ClpP systems on the basis of phylogeny in cases where no suffix was included in the protein annotation.
Species abbreviations: Apl: Arthrospira platensis NIES-39; Bsu: Bacillus subtilis; Bth: Bacillus thuringiensis; Blo: Bifidobacterium longum subsp. longum BBMN68; Cfe: Chlamydophila felis Fe/C-56; Cpn: Chlamydophila pneumoniae TW-183; Eco: Escherichia coli str. K12 substr. W3110; Fal: Frankia alni ACN14a; Gva: Gardnerella vaginalis ATCC 14019; Gsu: Geobacter sulfurreducens KN400; Lmo: Listeria monocytogenes; Mtb: Mycobacterium tuberculosis H37Rv; Neu: Nitrosomonas eutropha C91; Nfa: Nocardia farcinica IFM 10152; Npu: Nostoc punctiforme PCC 73102; Pae: Pseudomonas aeruginosa PAO1; Req: Rhodococcus equi 103S; Rri: Rickettsia rickettsii; Sde: Saccharophagus degradans; Sen: Salmonella enterica subsp. enterica serovar Agona str. 62.H.72; Sne: Simkania negevensis; Sco: Streptomyces coelicolor A3(2); Syn: Synechocystis sp. PCC 6803; Tfu: Thermobifida fusca YX; Twh: Tropheryma whipplei TW08/27.
Figure S2. Robust peptide-cleavage activity requires ClpP1, ClpP2, agonist, ClpX or ClpC1, and nucleotide.
A High levels of cleavage of a fluorogenic decapeptide substrate (15 μM) required ClpP1 (0.25 μM), ClpP2 (0.25 μM), Z-Ile-Leu agonist (1 mM), and MsmClpC1 (0.75 μM; 1 mM ATPγS) or MtbsClpX (0.75 μM; 1 mM ATPγS).
B Robust cleavage of Z-GGL-AMC (75 μM) also required ClpP1 (0.25 μM), ClpP2 (0.25 μM), Z-Ile-Leu agonist (1 mM), and MsmClpC1 (0.75 μM; 1 mM ATPγS) or MtbsClpX (0.75 μM; 1 mM ATPγS).
C Our preparation of MtbsClpX (0.5 μM) catalyzed a low level of cleavage of Z-GGL-AMC (75 μM), presumably because of a contaminating peptidase activity, which was inhibited by Z-Ile-Leu.
D Stimulation of robust ClpP1P2 (0.5 μM) cleavage of Z-GGL-AMC (75 μM) by MtbsClpX (0.5 μM) was supported by ATPγS (1 mM). A lower rate of cleavage was supported by ATP (2.5 mM), but not by ADP (2.5 mM) or no nucleotide.
E Simulation of ClpP1P2 (0.5 μM) cleavage of Z-GGL-AMC (75 μM) by MsmClpC1 (0.5 μM) was robustly supported by ATPγS (1 mM) and weakly supported by ATP (2.5 mM) in the presence of an ATP regeneration system, but not by ATP alone (2.5 mM) or ADP (2.5 mM), suggesting that ADP (resulting from ATP hydrolysis or as a contaminant in the ATP) destabilizes the active complex.
F Active degradation of titinI27-ssrA constructs (10 μM) by ClpP1P2 (0.3 μM) and MtbsClpX (0.5 μM) stimulated ClpP1P2 cleavage of Z-GGL-AMC (75 μM) in the absence of agonist. The magnitude of peptidase stimulation correlates with the rate at which E. coli ClpXP degrades these substrates (Kenniston et al., 2003).
Values are averages (N = 3) ± 1 SD.
Figure S3. Binding of AAA+ proteins to ClpP1P2 in the absence of Z-Ile-Leu.
Degradation of GFP-ssrA (10 μM) was assayed in the absence of Z-Ile-Leu agonist and the presence of MtbsClpX (0.5 μM; gray circles) or MsmClpC1 (0.5 μM; white circles) and varying concentrations of ClpP1P2. Fitting to a Hill equation gave an apparent affinity of 4.5 ± 3.1 μM and a Hill constant of 1.0 ± 0.2 for MtbsClpX (solid line), and an apparent affinity of 3.5 ± 2.4 μM and a Hill constant of 0.9 ± 0.2 for MsmClpC1 (dashed line).
Figure S4. GFP-ssrA degradation and activation of ClpP1P2 by ClpX are ATP-dependent.
A Degradation of 50 μM Z-GGL-AMC by 0.5 μM EcoClpXΔN•ClpP1P2 was assayed in the presence of 0.5 mM Z-Ile-Leu agonist and either 1 mM ATP (black circles) or 10 mM ATP (gray triangles). Depletion of 1 mM ATP resulted in cessation of protease activity, likely as a result of dissociation of ADP-bound EcoClpXΔN from ClpP.
B Degradation of 10 μM GFP-ssrA by 0.5 μM EcoClpXΔN•ClpP1P2 was assayed in the presence of 0.5 mM Z-Ile-Leu agonist and either 1 mM ATP (black circles) or 10 mM ATP (gray triangles). At the lower ATP concentration, GFP-ssrA degradation was slower and ceased before most substrate was degraded.
Acknowledgments
We thank D. Carney, B. Hall, J. Sello, B. Stinson, and O. Yosefson for advice. Supported by NIH grant GM-101988.
References
- Akopian T, Kandror O, Raju RM, Unnikrishnan M, Rubin EJ, Goldberg AL. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012 doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik S, Sureka K, Mukherjee P, Basu J, Kundu M. RseA, the SigE specific anti-sigma factor of Mycobacterium tuberculosis, is inactivated by phosphorylation-dependent ClpC1P2 proteolysis. Molecular Microbiology. 2010;75:592–606. doi: 10.1111/j.1365-2958.2009.07008.x. [DOI] [PubMed] [Google Scholar]
- Barthelme D, Sauer R. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science (New York, NY ) 2012;337:843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaroudj N, Raynal B, Miot M, Ortiz-Lombardia M. Assembly and proteolytic processing of mycobacterial ClpP1 and ClpP2. BMC Biochem. 2011;12:61. doi: 10.1186/1471-2091-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher T, Sieber SA. β-Lactones as Specific Inhibitors of ClpP Attenuate the Production of Extracellular Virulence Factors of Staphylococcus aureus. Journal of the American Chemical Society. 2008;130:14400–14401. doi: 10.1021/ja8051365. [DOI] [PubMed] [Google Scholar]
- Brotz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Sass P. Bacterial Cell Stress Protein ClpP: A Novel Antibiotic Target. In: Henderson B, editor. Moonlighting Cell Stress Proteins in Microbial Infections. Springer; Netherlands: 2013. pp. 375–385. [Google Scholar]
- Burton RE, Baker TA, Sauer RT. Energy-dependent degradation: Linkage between ClpX-catalyzed nucleotide hydrolysis and protein-substrate processing. Protein Sci. 2003;12:893–902. doi: 10.1110/ps.0237603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton C, Schmitz K, Sauer R, Sello J. Antibacterial Activity of and Resistance to Small Molecule Inhibitors of the ClpP Peptidase. ACS chemical biology. 2013;8:2669–2677. doi: 10.1021/cb400577b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez L, Breidenstein EB, Song D, Hancock RE. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2012;56:1128–1132. doi: 10.1128/AAC.05336-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JM, Wall JS, Capel MS, Schneider DK, Shanklin J. Scanning transmission electron microscopy and small-angle scattering provide evidence that native Escherichia coli ClpP is a tetradecamer with an axial pore. Biochemistry. 1995;34:10910–10917. doi: 10.1021/bi00034a025. [DOI] [PubMed] [Google Scholar]
- Frees D, Gerth U, Ingmer H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. International Journal of Medical Microbiology. 2014;304:142–149. doi: 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Frees D, Qazi SNA, Hill PJ, Ingmer H. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Molecular Microbiology. 2003;48:1565–1578. doi: 10.1046/j.1365-2958.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Molecular Microbiology. 2000;35:1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x. [DOI] [PubMed] [Google Scholar]
- Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, et al. Multidrug-resistant and extensively drug-resistant tuberculosis: a threat to global control of tuberculosis. The Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- Geiger SR, Bottcher T, Sieber SA, Cramer P. A Conformational Switch Underlies ClpP Protease Function. Angew Chem Int Ed Engl. 2011;50:5749–5752. doi: 10.1002/anie.201100666. [DOI] [PubMed] [Google Scholar]
- Gersch M, Gut F, Korotkov V, Lehmann J, Böttcher T, Rusch M, et al. The Mechanism of Caseinolytic Protease (ClpP) Inhibition. Angewandte Chemie (International ed in English) 2013a;52:3009–3014. doi: 10.1002/anie.201204690. [DOI] [PubMed] [Google Scholar]
- Gersch M, Kolb R, Alte F, Groll M, Sieber S. Disruption of Oligomerization and Dehydroalanine Formation as Mechanisms for ClpP Protease Inhibition. Journal of the American Chemical Society. 2013b doi: 10.1021/ja4082793. [DOI] [PubMed] [Google Scholar]
- Gersch M, List A, Groll M, Sieber SA. Insights into the structural network responsible for oligomerization and activity Of the bacterial virulence regulator caseinolytic protease P (ClpP) J Biol Chem. 2012 doi: 10.1074/jbc.M111.336222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S, Roche E, Zhou Y, Sauer RT. The ClpXP and ClpAP proteases degrade proteins with carboxy-terminal peptide tails added by the SsrA-tagging system. Genes Dev. 1998;12:1338–1347. doi: 10.1101/gad.12.9.1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic acids research. 2010;38:9. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribun A, Kimber MS, Ching R, Sprangers R, Fiebig KM, Houry WA. The ClpP double ring tetradecameric protease exhibits plastic ring-ring interactions, and the N termini of its subunits form flexible loops that are essential for ClpXP and ClpAP complex formation. J Biol Chem. 2005;280:16185–16196. doi: 10.1074/jbc.M414124200. [DOI] [PubMed] [Google Scholar]
- Griffin J, Gawronski J, Dejesus M, Ioerger T, Akerley B, Sassetti C. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS pathogens. 2011;7 doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaud R, Kessel M, Beuron F, Steven A, Maurizi M. Enzymatic and structural similarities between the Escherichia coli ATP-dependent proteases, ClpXP and ClpAP. The Journal of biological chemistry. 1998;273:12476–12481. doi: 10.1074/jbc.273.20.12476. [DOI] [PubMed] [Google Scholar]
- Howard SJ, Catchpole M, Watson J, Davies SC. Antibiotic resistance: global response needed. The Lancet Infectious Diseases. 2013;13:1001–1003. doi: 10.1016/S1473-3099(13)70195-6. [DOI] [PubMed] [Google Scholar]
- Ingvarsson H, Mate MJ, Hogbom M, Portnoi D, Benaroudj N, Alzari PM, et al. Insights into the inter-ring plasticity of caseinolytic proteases from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr D Biol Crystallogr. 2007;63:249–259. doi: 10.1107/S0907444906050530. [DOI] [PubMed] [Google Scholar]
- Jassal M, Bishai WR. Extensively drug-resistant tuberculosis. Lancet Infect Dis. 2009;9:19–30. doi: 10.1016/S1473-3099(08)70260-3. [DOI] [PubMed] [Google Scholar]
- Joshi SA, Hersch GL, Baker TA, Sauer RT. Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol. 2004;11:404–411. doi: 10.1038/nsmb752. [DOI] [PubMed] [Google Scholar]
- Kang S, Dimitrova M, Ortega J, Ginsburg A, Maurizi M. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. The Journal of biological chemistry. 2005;280:35424–35432. doi: 10.1074/jbc.M507240200. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Fernandez JM, Sauer RT. Linkage between ATP consumption and mechanical unfolding during the protein processing reactions of an AAA+ degradation machine. Cell. 2003;114:511–520. doi: 10.1016/s0092-8674(03)00612-3. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT. Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci U S A. 2005;102:1390–1395. doi: 10.1073/pnas.0409634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DY, Kim KK. Crystal structure of ClpX molecular chaperone from Helicobacter pylori. J Biol Chem. 2003;278:50664–50670. doi: 10.1074/jbc.M305882200. [DOI] [PubMed] [Google Scholar]
- Kim Y, Burton R, Burton B, Sauer R, Baker T. Dynamics of substrate denaturation and translocation by the ClpXP degradation machine. Molecular cell. 2000;5:639–648. doi: 10.1016/s1097-2765(00)80243-9. [DOI] [PubMed] [Google Scholar]
- Kim YI, Levchenko I, Fraczkowska K, Woodruff RV, Sauer RT, Baker TA. Molecular determinants of complex formation between Clp/Hsp100 ATPases and the ClpP peptidase. Nat Struct Biol. 2001;8:230–233. doi: 10.1038/84967. [DOI] [PubMed] [Google Scholar]
- Kimber MS, Yu AY, Borg M, Leung E, Chan HS, Houry WA. Structural and theoretical studies indicate that the cylindrical protease ClpP samples extended and compact conformations. Structure. 2010;18:798–808. doi: 10.1016/j.str.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Kwon HY, Ogunniyi AD, Choi MH, Pyo SN, Rhee DK, Paton JC. The ClpP protease of Streptococcus pneumoniae modulates virulence gene expression and protects against fatal pneumococcal challenge. Infect Immun. 2004;72:5646–5653. doi: 10.1128/IAI.72.10.5646-5653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin M, Blackshields G, Brown N, Chenna R, McGettigan P, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics (Oxford, England) 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lee BG, Kim MK, Song HK. Structural insights into the conformational diversity of ClpP from Bacillus subtilis. Mol Cells. 2011;32:589–595. doi: 10.1007/s10059-011-0197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, et al. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol. 2010a;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]
- Lee ME, Baker TA, Sauer RT. Control of substrate gating and translocation into ClpP by channel residues and ClpX binding. J Mol Biol. 2010b;399:707–718. doi: 10.1016/j.jmb.2010.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SB, Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med. 2004 doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, et al. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature. 2005;437:1115–1120. doi: 10.1038/nature04031. [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. Distinct static and dynamic interactions control ATPase-peptidase communication in a AAA+ protease. Mol Cell. 2007;27:41–52. doi: 10.1016/j.molcel.2007.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT. Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat Struct Mol Biol. 2008;15:139–145. doi: 10.1038/nsmb.1380. [DOI] [PubMed] [Google Scholar]
- Maurizi MR, Singh SK, Thompson MW, Kessel M, Ginsburg A. Molecular properties of ClpAP protease of Escherichia coli: ATP-dependent association of ClpA and clpP. Biochemistry. 1998;37:7778–7786. doi: 10.1021/bi973093e. [DOI] [PubMed] [Google Scholar]
- Neu HC. The Crisis in Antibiotic Resistance. Science. 1992;257:1064–1073. doi: 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- Nørby J. Coupled assay of Na+,K+-ATPase activity. Methods in enzymology. 1988;156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- Ollinger J, O'Malley T, Kesicki EA, Odingo J, Parish T. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J Bacteriol. 2012;194:663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personne Y, Brown A, Schuessler De, Parish T. Mycobacterium tuberculosis ClpP proteases are co-transcribed but exhibit different substrate specificities. PloS one. 2013;8 doi: 10.1371/journal.pone.0060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, et al. Mycobacterium tuberculosis ClpP1 and ClpP2 Function Together in Protein Degradation and Are Required for Viability in vitro and During Infection. PLoS Pathog. 2012;8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. FigTree. Institute of Evolutionary Biology, University of Edinburgh; 2012. [Google Scholar]
- Rao SPS, Alonso S, Rand L, Dick T, Pethe K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences. 2008;105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Religa TL, Ruschak AM, Rosenzweig R, Kay LE. Site-Directed Methyl Group Labeling as an NMR Probe of Structure and Dynamics in Supramolecular Protein Systems: Applications to the Proteasome and to the ClpP Protease. Journal of the American Chemical Society. 2011;133:9063–9068. doi: 10.1021/ja202259a. [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell. 2004;119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol J, Woo K, Kang M, Ha D, Chung C. Requirement of ATP hydrolysis for assembly of ClpA/ClpP complex, the ATP-dependent protease Ti in Escherichia coli. Biochemical and biophysical research communications. 1995;217:41–51. doi: 10.1006/bbrc.1995.2743. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson T, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular systems biology. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanne TM, Pojidaeva E, Andersson FI, Clarke AK. Distinctive types of ATP-dependent Clp proteases in cyanobacteria. J Biol Chem. 2007;282:14394–14402. doi: 10.1074/jbc.M700275200. [DOI] [PubMed] [Google Scholar]
- Tryggvesson A, Stahlberg FM, Mogk A, Zeth K, Clarke AK. Interaction specificity between the chaperone and proteolytic components of the cyanobacterial Clp protease. Biochem J. 2012;446:311–320. doi: 10.1042/BJ20120649. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature. 2011;471:331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- Wang J, Hartling JA, Flanagan JM. The structure of ClpP at 2.3 A resolution suggests a model for ATP-dependent proteolysis. Cell. 1997;91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- Wiegert T, Schumann W. SsrA-mediated tagging in Bacillus subtilis. J Bacteriol. 2001;183:3885–3889. doi: 10.1128/JB.183.13.3885-3889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AYH, Houry WA. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. FEBS Letters. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- Zeiler E, Braun N, Böttcher T, Kastenmüller A, Weinkauf S, Sieber S. Vibralactone as a tool to study the activity and structure of the ClpP1P2 complex from Listeria monocytogenes. Angewandte Chemie (International ed in English) 2011;50:11001–11004. doi: 10.1002/anie.201104391. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. ClpP paralogs in actinobacteria are phylogentically distinct from other multi-ClpP systems.
A phylogram of representative bacterial ClpP enzymes suggests that multi-ClpP systems arose independently in different bacterial lineages. Actinobacteria, including M. tuberculosis (Mtb) encode conserved ClpP1 and ClpP2 paralogs. Cyanobacteria and chlamidiae each possess distinct multi-ClpP systems that are conserved within these phyla. The multiple ClpP enzymes in Bacillus thuringiensis (Bth) are likely the result of a recent gene duplication event, whereas those of Pseudomonas aeruginosa (Pae) and Listeria monocytogenes (Lmo) are likely to have arisen from horizontal gene transfer. Ordinal suffixes have been added (asterisks) to multi-ClpP systems on the basis of phylogeny in cases where no suffix was included in the protein annotation.
Species abbreviations: Apl: Arthrospira platensis NIES-39; Bsu: Bacillus subtilis; Bth: Bacillus thuringiensis; Blo: Bifidobacterium longum subsp. longum BBMN68; Cfe: Chlamydophila felis Fe/C-56; Cpn: Chlamydophila pneumoniae TW-183; Eco: Escherichia coli str. K12 substr. W3110; Fal: Frankia alni ACN14a; Gva: Gardnerella vaginalis ATCC 14019; Gsu: Geobacter sulfurreducens KN400; Lmo: Listeria monocytogenes; Mtb: Mycobacterium tuberculosis H37Rv; Neu: Nitrosomonas eutropha C91; Nfa: Nocardia farcinica IFM 10152; Npu: Nostoc punctiforme PCC 73102; Pae: Pseudomonas aeruginosa PAO1; Req: Rhodococcus equi 103S; Rri: Rickettsia rickettsii; Sde: Saccharophagus degradans; Sen: Salmonella enterica subsp. enterica serovar Agona str. 62.H.72; Sne: Simkania negevensis; Sco: Streptomyces coelicolor A3(2); Syn: Synechocystis sp. PCC 6803; Tfu: Thermobifida fusca YX; Twh: Tropheryma whipplei TW08/27.
Figure S2. Robust peptide-cleavage activity requires ClpP1, ClpP2, agonist, ClpX or ClpC1, and nucleotide.
A High levels of cleavage of a fluorogenic decapeptide substrate (15 μM) required ClpP1 (0.25 μM), ClpP2 (0.25 μM), Z-Ile-Leu agonist (1 mM), and MsmClpC1 (0.75 μM; 1 mM ATPγS) or MtbsClpX (0.75 μM; 1 mM ATPγS).
B Robust cleavage of Z-GGL-AMC (75 μM) also required ClpP1 (0.25 μM), ClpP2 (0.25 μM), Z-Ile-Leu agonist (1 mM), and MsmClpC1 (0.75 μM; 1 mM ATPγS) or MtbsClpX (0.75 μM; 1 mM ATPγS).
C Our preparation of MtbsClpX (0.5 μM) catalyzed a low level of cleavage of Z-GGL-AMC (75 μM), presumably because of a contaminating peptidase activity, which was inhibited by Z-Ile-Leu.
D Stimulation of robust ClpP1P2 (0.5 μM) cleavage of Z-GGL-AMC (75 μM) by MtbsClpX (0.5 μM) was supported by ATPγS (1 mM). A lower rate of cleavage was supported by ATP (2.5 mM), but not by ADP (2.5 mM) or no nucleotide.
E Simulation of ClpP1P2 (0.5 μM) cleavage of Z-GGL-AMC (75 μM) by MsmClpC1 (0.5 μM) was robustly supported by ATPγS (1 mM) and weakly supported by ATP (2.5 mM) in the presence of an ATP regeneration system, but not by ATP alone (2.5 mM) or ADP (2.5 mM), suggesting that ADP (resulting from ATP hydrolysis or as a contaminant in the ATP) destabilizes the active complex.
F Active degradation of titinI27-ssrA constructs (10 μM) by ClpP1P2 (0.3 μM) and MtbsClpX (0.5 μM) stimulated ClpP1P2 cleavage of Z-GGL-AMC (75 μM) in the absence of agonist. The magnitude of peptidase stimulation correlates with the rate at which E. coli ClpXP degrades these substrates (Kenniston et al., 2003).
Values are averages (N = 3) ± 1 SD.
Figure S3. Binding of AAA+ proteins to ClpP1P2 in the absence of Z-Ile-Leu.
Degradation of GFP-ssrA (10 μM) was assayed in the absence of Z-Ile-Leu agonist and the presence of MtbsClpX (0.5 μM; gray circles) or MsmClpC1 (0.5 μM; white circles) and varying concentrations of ClpP1P2. Fitting to a Hill equation gave an apparent affinity of 4.5 ± 3.1 μM and a Hill constant of 1.0 ± 0.2 for MtbsClpX (solid line), and an apparent affinity of 3.5 ± 2.4 μM and a Hill constant of 0.9 ± 0.2 for MsmClpC1 (dashed line).
Figure S4. GFP-ssrA degradation and activation of ClpP1P2 by ClpX are ATP-dependent.
A Degradation of 50 μM Z-GGL-AMC by 0.5 μM EcoClpXΔN•ClpP1P2 was assayed in the presence of 0.5 mM Z-Ile-Leu agonist and either 1 mM ATP (black circles) or 10 mM ATP (gray triangles). Depletion of 1 mM ATP resulted in cessation of protease activity, likely as a result of dissociation of ADP-bound EcoClpXΔN from ClpP.
B Degradation of 10 μM GFP-ssrA by 0.5 μM EcoClpXΔN•ClpP1P2 was assayed in the presence of 0.5 mM Z-Ile-Leu agonist and either 1 mM ATP (black circles) or 10 mM ATP (gray triangles). At the lower ATP concentration, GFP-ssrA degradation was slower and ceased before most substrate was degraded.