

Abstract
Pre-eclampsia is the leading cause of fetal and maternal morbidity and mortality. Early onset pre-eclampsia (EOPE) is a disorder that has severe maternal and fetal outcomes, whilst its etiology is poorly understood. We hypothesize that epigenetics plays an important role to mediate the development of EOPE and conducted a case–control study to compare the genome-wide methylome difference between chorioamniotic membranes from 30 EOPE and 17 full-term pregnancies using the Infinium Human Methylation 450 BeadChip arrays. Bioinformatics analysis tested differential methylation (DM) at CpG site level, gene level, and pathway and network level. A striking genome-wide hypermethylation pattern coupled with hypomethylation in promoters was observed. Out of 385 184 CpG sites, 9995 showed DM (2.6%). Of those DM sites, 91.9% showed hypermethylation (9186 of 9995). Over 900 genes had DM associated with promoters. Promoter-based DM analysis revealed that genes in canonical cancer-related pathways such as Rac, Ras, PI3K/Akt, NFκB and ErBB4 were enriched, and represented biological functional alterations that involve cell cycle, apoptosis, cancer signaling and inflammation. A group of genes previously found to be up-regulated in pre-eclampsia, including GRB2, ATF3, NFKB2, as well as genes in proteasome subunits (PSMA1, PMSE1, PSMD1 and PMSD8), harbored hypomethylated promoters. Contrarily, a cluster of microRNAs, including mir-519a1, mir-301a, mir-487a, mir-185, mir-329, mir-194, mir-376a1, mir-486 and mir-744 were all hypermethylated in their promoters in the EOPE samples. These findings collectively reveal new avenues of research regarding the vast epigenetic modifications in EOPE.
Keywords: pre-eclampsia, epigenetics, DNA methylation, chorioamniotic membranes, bioinformatics
Introduction
Pre-eclampsia is a pregnancy specific condition characterized by hypertension (>140/90 mmHg) and proteinuria (>0.3 g/day) which affects 5–8% of all pregnancies (Srinivas et al., 2009) and is the leading cause of fetal/neonatal and maternal morbidity and mortality (Maynard et al., 2003). In early onset pre-eclampsia (EOPE), the underlying cause of pre-eclampsia is hypothesized to be shallow implantation of the placenta into the uterine wall (Jung et al., 2012). During normal pregnancy, spiral arteries in the endometrium and decidua (the uterine lining that forms the maternal part of the placenta) are remodeled by extravillous trophoblasts into low-pressure vessels. However in EOPE, the remodeling is restricted to the decidua which leads to poor placenta development and function (Pijnenborg et al., 2006; Jung et al., 2012). Because of the shallow implantation and the lack of transformation of maternal arteries, hypoxia and ischemic conditions result which trigger re-oxygenation and inflammatory stress, apoptotic pathways and deregulated proteasome activities within the trophoblast (Allaire et al., 2000; Leung et al., 2001; Ishihara et al., 2002; Tal, 2012). These events induce production of circulating factors by the placenta and synctiotrophic apoptotic vesicles, which culminate in the clinical manifestations of pre-eclampsia (Stillman and Karumanchi, 2007). In mothers, endothelial damage underlies the clinical and pathologic manifestations which include hypertension, thrombocytopenia, hemolysis, seizures, acute atherosis, renal failure and proteinuria (Boos and Lip, 2006; Nizet and Johnson, 2009; Palazón et al., 2012). In the fetus, the result is intrauterine growth restriction and pre-term delivery with the associated consequences such as neurodevelopmental disorders (Batalle et al., 2012) and adult onset disorders.
Pre-eclampsia may develop at any time after 20 weeks of pregnancy and can be categorized by EOPE (<34 weeks) and late onset pre-eclampsia (LOPE). Although some clinical features of EOPE and LOPE overlap, they have distinct etiologies, biochemical markers and maternal and fetal outcomes (Raymond and Peterson, 2011). EOPE is considered a more severe fetal disorder and it is typically associated with inadequate placental implantation and subsequent placental dysfunction, intrauterine growth restriction, low birthweight, perinatal death, and adverse maternal and fetal outcomes. In contrast, LOPE is considered a maternal disorder often associated with a normal placenta, larger placental volume, normal fetal growth and favorable maternal and fetal outcomes. Given the increased morbidity of EOPE and differing etiologies between EOPE and LOPE, we elected to study EOPE cases.
It is now emerging that epigenetic processes, especially DNA methylation, are involved in the development and normal functioning of pregnancies (Schroeder et al., 2013). Under normal pregnancy, the human placental landscape of partially methylated domains (PMD) had little variation across all three trimesters among individuals (Schroeder et al., 2013). However, it is suspected that epigenetic alteration plays a role in mediating pregnancy-related complications (Choudhury and Friedman, 2012). A few studies have investigated the alteration of methylation and epigenetic factors in pre-eclampsia (Bourque et al., 2010; Yuen et al., 2010; Gao et al., 2011; Kulkarni et al., 2011). Several studies have measured overall levels of methylation in pre-eclampsia (Gao et al., 2011; Kulkarni et al., 2011) and others have measured site-specific methylation by targeted PCR sequencing (Chelbi et al., 2007; Wang et al., 2010; Yuen et al., 2010; Gao et al., 2011; Mousa et al., 2012) or CpG island microarrays (Jia et al., 2012). Hypomethylation in promoters of multiple non-imprinted genes in EOPE was reported, including tissue inhibitor of metalloproteinase 3 (TIMP3) and serine protease inhibitor 3 (SERPINA3), whereas hypermethylation was also observed in other promoter regions, such as that of H19 the maternally imprinted non-coding RNA (Gao et al., 2011). Using Alu and LINE-1 repeats as surrogates to measure overall DNA methylation levels, it was found that EOPE placentas had significantly higher genome-wide methylation compared with normal placentas; however, the detailed methylome change was not examined (Kulkarni et al., 2011). Most recently, a case–control study of DNA methylome and gene expression profiles from 20 EOPE placentas and 20 pre-term non-EOPE placentas reported widespread DNA hypomethylation at enhancer regions in the EOPE placentas (Blair et al., 2013). However, it is still unclear if and to what extent, this pattern persists between pre-term EOPE and full-term delivered placentas, and what is an underlying cause versus consequence of the disease process.
In this study, we used the Infinium Human Methylation 450K BeadChip to quantitatively analyze methylation on over 485 000 CpG sites in the chorioamniotic membrane tissues from EOPE (cases) and full-term uncomplicated (controls) placentas. Chorioamniotic membranes are the amnion and chorion, which surround and protect a developing fetus. They represent the innermost layer of the placenta, composed of three layers, the amnion, chorion and adherent decidua (Niknejad et al., 2008). These structures develop early in the pregnancy and are less differentiated. Thus, they reflect early methylation patterns set in the developing embryo, and are presumably less prone to methylation change as gestation advances, which is seen in the villous part of the placenta due to metabolic demands (Novakovic et al., 2011). Thus, analyzing the genome-wide methylation changes in chorioamniotic membrane tissues is informative of epigenetic alternations occurring during early implantation that influences the development of EOPE.
Methods
Patient specimens
We utilized samples from the IRB-approved RMATRIX Hawaii Biorepository (HiBR) which has its own application process and a scientific review committee. From this repository, a total of 36 cases and 36 controls were initially identified. Cases had early onset pre-eclampsia at <34 weeks of gestation and controls were full-term pregnancies without pre-eclampsia. To ensure the quality of the samples, patients with known medical or obstetrical disorders associated with pre-eclampsia were excluded, including diabetes mellitus or gestational diabetes mellitus, hypertension, renal disease, hyperthyroidism, systemic lupus erythematous and twins. Formalin fixed paraffin embedded fetal chorioamniotic membrane rolls were used for DNA extraction. The samples were processed by the University of Hawaii Cancer Center (UHCC) Pathology Shared Resource, and then delivered to the Genomics Core at UHCC for the methylation study.
DNA isolation
DNA was extracted from the archived chorioamniotic membranes tissue using the AllPrep DNA/RNA FFPE kit (Qiagen). DNA was quantified using the Quant-iT Pico Green dsDNA Assay Kit (Invitrogen). The DNA samples were stored at −80°C until used.
Genome-scale methalome profiling using Human Methylation 450 Beadchip
Methylation profiling was done using the Illumina Infinium methylation assay and the Human Methylation 450 BeadChip. First, the DNA samples were pre-screened using the quantitative PCR-based Infinium HD FFPE QC Kit to evaluate if they were usable for the Methylation 450 assay. As a result, only 30 early onset pre-eclamptic patients (cases) and 17 normal patients without pre-eclampsia (controls) were actually analyzed due to poor DNA extraction in the remaining samples. DNA samples were treated with bisulfite using EZ DNA Methylation kit (Zymo Research), and 100 ng of bisulfite-converted DNA was restored using the Infinium HD FFPE Restore Kit and then hybridized onto the Infinium Human Methylation 450 BeadChip following the IIlumina Infinium HD Methylation protocol.
Methylation data processing
Raw intensity data were extracted using the ‘minfi’ package in R. Background controls were subtracted from the data and raw data that did not pass detection P value of 0.05 were removed. For each CpG site, the methylation score was initially calculated as beta value, a fluorescence intensity ratio between 0 and 1. It was then transformed as M value, the inverse logit transformation of beta value for normalization by a SWAN method (Maksimovic et al., 2012). We elected to use M-values, instead of beta values for the normalization, because M-values allow more reliable true-positive identification (Du et al., 2010; Zhuang et al., 2012). CpG sites whose probes had known underlying SNPs were removed from analysis due to potential confounding. CpG sites associated with X and Y chromosomes were also removed to eliminate the gender effect.
Statistical analysis of differential methylation
Before the test for differential expression between case and control, a submodel selection was conducted on all clinical parameters, using a linear mixed model in R via Akaike Information Criterion (AIC). The AIC score was maximized most often by removing all variables except the pre-eclampsia variable. To verify the result, a source of variance analysis on the marginal F-stats was performed, using a linear mixed model that includes multiple clinical variables. Pre-eclampsia had the largest contribution to variance in the data (i.e. the highest F stat). Due to the non-normal distribution of M values, the non-parametric Mann–Whitney–Wilcoxon ranked tests were used to calculate statistical significance of CpG sites between cases and controls. Due to the significant difference associated with pre-eclampsia, Bonferroni correction was then used to adjust for multiple hypotheses tests (MHTs). CpG sites with corrected P < 0.05 were considered significant. Other graphic representations, such as correlation heatmaps, PCA plots, dot plots, volcano plots and pie charts were all generated in R 3.0.2.
Ingenuity pathway analysis
IPA was used to identify significant relationship networks, canonical pathways and associated genes related to pre-eclampsia. Given the importance of promoters in gene regulation, we elected to infer gene-level methylation with the summed beta values across all CpG sites in the transcript-specific promoter. To measure the overall methylation difference in the promoters of the transcripts, the summed beta values in the TSS200 region of each transcript were calculated. Mann–Whitney–Wilcoxon ranked tests were conducted to calculate statistical significance of TSS200 regions between cases. Bonferroni correction was then used to adjust for MHT in order to choose significant promoters with corrected P < 0.05. These genes were inputs for gene-level IPA analysis. The averaged beta values were taken from all CpG sites located within the TSS200 promoter regions. The fold changes were calculated as the ratios of the beta values in cases over controls. Negative fold change represents hypomethylation and positive fold change represents hypermethylation.
Pyrosequencing validation
Pyrosequencing validation was done using the PyroMark PCR kit (Qiagen) following the suggested protocol on the PyroMark Gold Q24 instrument using the PyroMark Gold Q24 Reagents. Primer design was carried out using the PyroMark Assay Design 2.0 software. One of the primers was biotinylated to enable capture by Streptavidin Sepharose. Bisulfite-treated DNA was amplified and the single-stranded biotinylated DNA was processed according to the manufacturer's protocol. The primers for the selected CpG site validation are listed in Supplementary data, Table SI.
Results
Pre-eclampsia placenta characterized by genome-wide CpG hypermethylation
The relevant demographic and clinical information is summarized in Table I, including patient age, body mass index (BMI), mother's age, baby's gender, weight and gestational period. We did not observe statistical differences in maternal age, BMI, parity and sex of babies between cases and controls. However, both gestational age and mean weight of the babies were significantly different between EOPE and full-term controls (32.17 versus 39.18 weeks, P < 0.01 and 1654 g versus 3322 g, P < 0.01, respectively).
Table I.
Relevant clinical characteristics of the placenta samples in the study.
| Clinicopathologic characteristics | Case | Control | P-value |
|---|---|---|---|
| Total number of samples | 30 | 17 | N/A |
| Age of mother (years) | 29.8 ± 5.99 | 29.1 ± 4.76 | 0.629 |
| BMI (kg/m2) | 28.3 ± 8.17 | 26.5 ± 6.24 | 0.425 |
| Gestational weeks | 32.2 ± 1.97 | 39.2 ± 0.73 | <0.01 |
| Weight of baby (g) | 1654 ± 596 | 3322 ± 440 | <0.01 |
| Parity | 1.31 ± 1.32 | 1.12 ± 1.27 | 0.644 |
| Sex of baby (no. of females/males) | 16/14 | 11/6 | 0.652 |
Age of mother, sex of baby, pre-eclampsia status, mother's BMI, gestational age and baby's weight at birth were gathered. Clinical parameters were tested for between-group differences by two-sample t-tests. Weight of the baby and gestational age were significantly different between cases and control (P < 0.01), whereas age of the mother and BMI were not (P = 0.629 and P = 0.425, respectively).
After pre-processing, 385 184 CpG sites remained as useful loci for differential methylation analysis. Principal component analysis (PCA) of the data based on methylation M-value distances showed a good distinction between EOPE cases and controls, using only the first two principal components (Fig. 1A). The samples also showed a clear distinction in genome-wide methylation levels (average M-value and average beta across all probes) between two groups (Supplementary data, Figs S7 and S8). To examine the contribution of potential confounding factors, including mother's age, parity, BMI as well as the characteristics of the babies such as sex and birthweight, we performed a source of variance analysis. The marginal F-statistics in a linear ANOVA model shows that pre-eclampsia status is the dominating factor that contributed the most variance (Fig. 1B). Pre-eclampsia status had an average F-statistic of 3.3, while other factors had much less effects with F-statistics <1.5. To choose an appropriate set of parameters, we used a step-down approach to AIC on the ANOVA model with factors shown in Fig. 1B. Pre-eclampsia was the only parameter chosen by AIC, confirming its importance and sufficiency to explain the differences in the data. We next sought differentially methylated (DM) CpG regions between pre-eclampsia and controls. Using a non-parametric Wilcoxon rank-based test followed by Bonferroni correction (P < 0.05), we obtained 9995 sites that showed differential methylation between EOPE and control samples, and of those the vast majority (91.9%) showed hypermethylation in EOPE (Fig. 1C). Among these DM sites, a majority of them (9186 sites) were hypermethylated in pre-eclampsia, whereas far fewer loci (809 sites) were hypomethylated (Fig. 2A). We selected four CpG sites (cg18236464 from PAPPA2, cg1842256 from PSMD8, cg23227945 from NFKB2 and cg24244854 from GRB2) for technical validation using pyrosequencing. Combining the overall validation results, the R2 correlation value between pyrosequencing methylation and 450K beta values is 0.848 (Fig. 1D). The individual CpG site comparisons between the two platforms are shown in Supplementary data, Fig. S9.
Figure 1.

Global CpG hypermethylation is associated with pre-eclampsia. (A) Principle component analysis (PCA) plot representing differences between pre-eclamptic versus non-pre-eclamptic placentas. The PCA was based on the distance matrix of the methylation intensity values of all samples, measured by the Illumina Human Methylation450 BeadChip. The pre-eclamptic placenta cases are labeled as red and the controls are labeled as blue. The horizontal scale is the first principal component (PCA1) and the vertical scale is the second principal component (PCA2). (B) Source of variance plot of clinical factors using the methylation M values in all chorioamniotic membrane samples. Marginal F-stats were taken from a linear regression ANOVA model comprising pre-eclampsia, age of the mother (Mother_age), sex of the baby (Sex_of_baby), BMI of the mother (BMI) and weight of the baby at birth (BabyWgt) as independent factors. The F-stats were averaged over all CpG sites. (C) Volcano plot of the probes in the Illumina Human Methylation4 50 BeadChip. The x-axis is the average M value difference between the cases and controls. The y-axis is the negative log of the P-values, using the Wilcoxon rank test. The dashed black line represents the α threshold for a significance of 0.05 before Bonferroni correction and the dashed red line is the significance of 0.05 after Bonferroni correction. (D) Pyrosequencing validation results on four selected CpG sites. Y-axis: averaged percentage of methylation per sample in pyrosequencing. X-axis: averaged beta values per sample from Illumina 450K methylation array.
Figure 2.


Comparison of CpG site methylation between pre-eclamptic and normal full-term chorioamniotic membranes. (A) The number of significant versus non-significant CpG sites using Wilcoxon ranked test after Bonferroni correction (left) and the overall distribution of hyper- versus hypomethylated sites (right). (B) CpG site distribution by location relative to genes. Distribution of all CpG sites on the Illumina Human Methylation 450 BeadChip array (top left), distribution of all DM CpG sites (top right), distribution of hypomethylated CpG sites (bottom left) and distribution of hypermethylated CpG sites (bottom right). (C) CpG site distribution by location relative to CpG islands and nearby regions. Distribution of all CpG sites on the Illumina Human Methylation 450 BeadChip array (top left), distribution of DM CpG sites (top right), distribution of hypomethylated CpG sites (bottom left) and distribution of hypermethylated CpG sites (bottom right). P-values for differences in the categorical proportions between differential CpG sites and expected background (all CpG sites) were calculated with χ2 tests, and labeled with asterisks for the significance level of P < 0.001. P-values for differences in categorical proportions between hypomethylation and hypermethylation were calculated and labeled in the same way.
Genome-wide patterns of DM CpG sites in CpG islands and surrounding areas
The probes on the Illumina 450K methylation platform have a sampling of roughly 1/3 sites in CpG islands, 1/3 in open sea and the rest of 1/3 in shores and shelves of CpG islands (Fig. 2B). Per the annotation of Illumina, CpG islands are defined based on the following criteria: GC content of 50% or greater, length >200 bp and the ratio of observed CpGs to the expected number of CpGs >0.6 (Gardiner-Garden and Frommer, 1987). CpG shores are defined as 0–2 kb upstream (north) or downstream (south) of the CpG islands and CpG shelves as 2–4 kb upstream or downstream of the CpG islands. We define the ‘open sea’ regions as regions not located in any of the above-mentioned regions relating to CpG islands. The DM loci show an increased representation in CpG shelves and open sea, but decreased representation in CpG islands and shores. The hypomethylated sites and hypermethylated sites have striking and significant complementarity in CpG categories. As shown in Fig. 2B, the majority of the hypomethylated CpG sites (603/809 sites) are in CpG islands and are nearly absent in shelves (1/809 sites), whereas the majority of the hypermethylated CpG sites (5134/9186) are located in the open seas with a very small proportion of CpG islands (445/9186 sites). The open seas are mostly associated with gene bodies and intergenic regions, whereas CpG islands are mostly associated with promoters (P < 0.001, χ2 test). To further elucidate the methylation patterns relevant to CpG islands, we used unsupervised hierarchical clustering and generated heatmaps in individual CpG island regions (Fig. 3). All clustering stratified by region produced good separation of samples by pre-eclampsia status, confirming the clustering found in the PCA (Fig. 1A). Comparatively, CpG islands show the lowest level of methylation overall (average beta 0.21), whereas open seas have the highest level of methylation (average beta 0.67).
Figure 3.


Heatmaps based on the relative position of significant CpG sites to CpG islands (north shelves, north shores, within CpG islands, south shores, south shelves and open seas). The color scale is based on methylation fraction beta values varying from 0 (red) to 1 (blue). The placenta samples are labeled by pre-eclampsia cases (blue) and controls (green). Both CpG sites (rows) and samples (columns) are ordered per unsupervised hierarchical clustering.
Global patterns of DM CpG sites in association with genes
Of the probes with CpG sites on the Illumina 450K methylation platform, roughly 40% sites are distributed in the promoter proximal regions (TSS1500, TSS200 and 5′UTR), 42% in gene body and first exons and 18% in 3′ UTR and intergenic regions (Fig. 2C), noting that some loci have multiple gene-associated memberships due to their locations in overlapping transcripts. Our DM loci show an increased representation of gene body, 3′UTR and intergenic sites, but decreased representation of promoter proximal regions including TSS1500, TS200 and 5′UTR, as well as first exons (Fig. 2C, P < 0.001, χ2 test). The hypomethylated CpG sites and hypermethylated sites have strikingly complementary gene-associated locations. As shown in Fig. 2C, a majority of the hypomethylated CpG sites (603/809 sites) are in the promoter regions including TSS200 (461/1691 sites) and TSS1500 (326/1691 sites), whereas nearly 50% of the hypermethylated CpG sites (6691/13 787) are located in the gene bodies. To further reveal the methylation patterns relevant to gene regions, we generated heatmaps in each genomic region associated with the gene (Fig. 4). All clustering based on associations to genes produced good separation between pre-eclampsia and control samples. TSS200 and first exons showed the lowest average levels of methylation (average beta 0.166 and 0.195 respectively), whereas gene body and 3′UTR regions showed the highest levels of methylation (average beta 0.60 and 0.72 respectively). Such strong hypomethylation proximal to promoters and hypermethylation of gene transcripts may suggest a globally activated gene expression program in EOPE.
Figure 4.

Continued.
Figure 4.

Heatmaps based on the relative position of significant CpG sites to gene locations (TSS1500, TSS200, 5′UTR, first exon, gene body and 3′UTR). The color scale is based on beta values varying from 0 (red) to 1 (blue). The placenta samples are labeled by pre-eclampsia case (blue) and control (green). Both axes are ordered by unsupervised hierarchical clustering.
As examples, we selected the most statistically significant 20 hypomethylated and 20 hypermethylated CpG sites with an average beta value difference >0.2 and show their dot plots in Supplementary data, Figs S1 and S2. The top three hypermethylated probes were cg12503717 (not associated with a gene), cg16645440 (associated with BAT2L2) and cg15149117 (associated with OR52H1). The top three hypomethylated probes were cg15653194 (associated with LFNG), cg16268778 (associated with PNMT) and cg11528984 (associated with C1orf69). The dot plots show consistent differential methylation of these CpG sites between pre-eclampsia cases and controls. Some sites are associated with genes, ranging from a wide variety of functional annotations, including inflammation (IRF6), cytoskeleton (MAP2 and SYNPO2), cancer-related genes (RAB4A and ST20) and transcription factors (HLX and ZNF165).
Promoters associated network and pathway analysis
Given the promoter localization of the majority of the CpG islands, as well as the importance of promoters to regulate gene expression, we decided to focus the analysis on proximal promoter regions (TSS200), similar to that of others (West et al., 2013). To measure the overall methylation difference in the promoters of the transcripts, we summed the beta values in the TSS200 region of each transcript and then conducted Wilcoxon rank-based tests. Using this approach, we detected 1161 transcripts with DM proximal promoters after Bonferroni correction (P < 0.05). These transcripts mapped to 888 genes, of which, 599 gene promoters were hypomethylated, and 290 gene promoters were hypermethylated. One gene (FZR1) had double counts as one isoform was significantly hypomethylated; however, another isoform was significantly hypermethylated due to significantly different promoter regions between them.
To identify top networks and canonical pathways that are altered in EOPE, we used ingenuity pathway analysis (IPA) to analyze the transcripts with DM promoters. The top five networks had these functions: (1) cancer, cell morphology, cell-to-cell signaling and interaction, (2) cell death and survival, inflammatory response, cellular development, (3) gene expression, cellular function and maintenance, developmental disorder and (4) cell cycle, embryonic development, cellular assembly and organization and (5) cell cycle, cancer, hematological disease (Table II and Fig. 5). The top five canonical pathways were: (1) Rac signaling, (2) PI3 K/AKT signaling, (3) actin cytoskeleton signaling, (4) Rho family GTPases signaling and (5) ErbB4 signaling (Table II and Supplementary data, Figs S3–S6). Many genes and protein complexes previously shown relevant to pre-eclampsia were DM in the top pathways and networks. Some genes of interest that had DM promoters are listed in Table III. They include some genes previously known to have altered expression in pre-eclampsia: activating transcription factor 3 (ATF3, with mean betas of 0.275 and 0.07 in controls and EOPE, respectively, and adjusted P-value = 0.003), ADP-ribosylation factor 1 (ARF1, with mean betas of 0.070 and 0.035 in controls and EOPE, respectively, and adjusted P-value = 0.01), growth factor receptor-bound protein 2 (GRB2, with mean betas of 0.164 and 0.087 in controls and EOPE, respectively, and adjusted P-value = 0.004), nuclear factor of kappa light polypeptide gene enhancer in B-cells 2 (NFκB2, with mean betas of 0.254 and 0.134 in controls and EOPE, respectively, and adjusted P-value = 0.04), death inducer-obliterator-1 (DIDO1, with mean betas of 0.222 and 0.073 in controls and EOPE, respectively, and adjusted P-value = 0.008), and proteosomal subunits (PSMA1, PSME1, PSMD1 and PSMD8).
Table II.
Summary of the top five most significant networks and pathways inferred by IPA.
| Network functions | IPA score | Number of genes found |
|---|---|---|
| Cancer, cell morphology, cell-to-cell signaling and interaction | 43 | 33 |
| Cell death and survival, inflammatory response, cellular development | 32 | 28 |
| Gene expression, cellular function and maintenance, developmental disorder | 32 | 28 |
| Cell cycle, embryonic development, cellular assembly and organization | 30 | 27 |
| Cell cycle, cancer, hematological disease | 25 | 24 |
Significant genes were determined by significant methylation differences in the promoters of genes. Summed beta values of the probes in the TSS200 region of promoters were used for the Wilcoxon ranked test at the transcript level. A gene was defined significantly methylated if P < 0.05 after Bonferroni correction. IPA scores are negative log P-values, and higher IPA scores indicate more significance.
Figure 5.

Top networks in IPA analysis. (A) Network of ‘cancer, cell morphology, cell-to-cell signaling and interaction’. (B) Network of ‘cell death and survival, inflammatory response, cellular development’. (C) Network of ‘cell cycle, embryonic development, cellular assembly and organization’. (D) Network of ‘hereditary disorder, skeletal and muscular disorders, connective tissue disorders’. Red and green colors represent hypermethylation and hypomethylation in the promoter of the gene, respectively.
Table III.
List of genes of interest involved in pre-eclampsia with DM promoters.
| Gene name | Refseq ID | Promoter (TSS200) |
Body (first exon and body) |
Long gene name | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean control | Mean EOPE | Delta beta | Fold change | Number of probes in TSS200 | Adjusted P-value | Mean control | Mean EOPE | Absolute delta beta | Relative fold change | Number of probes in body | Adjusted P-value | Location in cell | |||
| ARF1 | NM_001658 | 0.07 | 0.04 | 0.03 | −1.98 | 3 | 0.01 | 0.07 | 0.05 | 0.02 | 0.72 | 2 | 0.10 | Cytoplasm | ADP-ribosylation factor 1 |
| PAPPA2 | NM_020318 | 0.11 | 0.26 | 0.15 | 2.28 | 1 | 0.00 | 4.06 | 4.74 | 0.68 | 1.17 | 7 | 0.00 | Extracellular space | Pappalysin 2 |
| IL1RAP | NM_001167928 | 0.13 | 0.03 | 0.09 | −3.90 | 4 | 0.02 | 3.78 | 4.05 | 0.27 | 1.07 | 6 | 0.01 | Plasma membrane | Interleukin 1 receptor accessory protein |
| NRAS | NM_002524 | 0.13 | 0.08 | 0.05 | −1.67 | 1 | 0.00 | 0.02 | 0.01 | 0.01 | 0.60 | 2 | 0.04 | Plasma membrane | Neuroblastoma RAS viral (v-ras) oncogene homolog |
| GRB2 | NM_203506 | 0.16 | 0.09 | 0.08 | −1.88 | 6 | 0.00 | 9.92 | 10.29 | 0.38 | 1.04 | 13 | 0.03 | Cytoplasm | Growth factor receptor-bound protein 2 |
| PSMA1 | NM_148976 | 0.17 | 0.06 | 0.11 | −2.69 | 3 | 0.03 | 4.01 | 4.45 | 0.44 | 1.11 | 23 | 0.00 | Cytoplasm | Proteasome (prosome, macropain) subunit, alpha type, 1 |
| REL | NM_002908 | 0.18 | 0.08 | 0.10 | −2.20 | 3 | 0.03 | 0.07 | 0.03 | 0.04 | 0.48 | 1 | 0.00 | Nucleus | v-rel reticuloendotheliosis viral oncogene homolog (avian) |
| PIK3R3 | NM_001114172 | 0.20 | 0.07 | 0.13 | −2.88 | 4 | 0.04 | 2.23 | 2.52 | 0.29 | 1.13 | 7 | 0.00 | Cytoplasm | Phosphoinositide-3-kinase, regulatory subunit 3 (gamma) |
| GNGT1 | NM_021955 | 0.21 | 0.44 | 0.24 | 2.14 | 1 | 0.00 | NA | NA | NA | NA | 0 | NA | Plasma Membrane | Guanine nucleotide-binding protein (G protein), gamma transducing activity polypeptide 1 |
| PSMD1 | NM_002807 | 0.22 | 0.09 | 0.13 | −2.46 | 4 | 0.01 | 5.28 | 5.96 | 0.69 | 1.13 | 11 | 0.00 | Cytoplasm | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 1 |
| DIDO1 | NM_033081 | 0.22 | 0.07 | 0.15 | −3.04 | 3 | 0.01 | 7.45 | 8.01 | 0.56 | 1.08 | 11 | 0.00 | Nucleus | Death inducer-obliterator 1 |
| NFKBIA | NM_020529 | 0.23 | 0.10 | 0.13 | −2.31 | 4 | 0.03 | 0.58 | 0.66 | 0.08 | 1.14 | 8 | 0.37 | Cytoplasm | Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| NFKB2 | NM_001077493 | 0.25 | 0.13 | 0.12 | −1.90 | 6 | 0.04 | 0.31 | 0.28 | 0.04 | 0.89 | 6 | 0.97 | Nucleus | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 2 (p49/p100) |
| CFHR5 | NM_030787 | 0.26 | 0.49 | 0.23 | 1.91 | 1 | 0.00 | 0.69 | 0.69 | 0.00 | 1.00 | 1 | 0.94 | Extracellular Space | Complement factor H-related 5 |
| ANXA2 | NM_004039 | 0.27 | 0.15 | 0.12 | −1.76 | 5 | 0.03 | 2.60 | 2.94 | 0.34 | 1.13 | 5 | 0.01 | Plasma membrane | Annexin A2 |
| HNF4G | NM_004133 | 0.27 | 0.52 | 0.25 | 1.94 | 1 | 0.01 | NA | NA | NA | NA | 0 | NA | Nucleus | Hepatocyte nuclear factor 4, gamma |
| IL12B | NM_002187 | 0.27 | 0.55 | 0.28 | 2.03 | 1 | 0.00 | 1.17 | 1.36 | 0.18 | 1.16 | 3 | 0.05 | Extracellular Space | Interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation factor 2, p40) |
| ATF3 | NM_001040619 | 0.27 | 0.07 | 0.20 | −3.90 | 2 | 0.00 | 0.04 | 0.04 | 0.01 | 0.87 | 2 | 0.34 | Nucleus | Activating transcription factor 3 |
| GFI1B | NM_004188 | 0.28 | 0.54 | 0.26 | 1.92 | 1 | 0.02 | 3.15 | 3.51 | 0.37 | 1.12 | 4 | 0.00 | Nucleus | Growth factor independent 1B transcription repressor |
| PSME1 | NM_006263 | 0.28 | 0.12 | 0.16 | −2.33 | 5 | 0.02 | 0.27 | 0.19 | 0.08 | 0.71 | 4 | 0.05 | Cytoplasm | Proteasome (prosome, macropain) activator subunit 1 (PA28 alpha) |
| SPINK7 | NM_032566 | 0.29 | 0.56 | 0.27 | 1.95 | 1 | 0.01 | 1.27 | 1.43 | 0.16 | 1.12 | 2 | 0.28 | Extracellular Space | Serine peptidase inhibitor, Kazal type 7 (putative) |
| GABRA1 | NM_001127647 | 0.30 | 0.59 | 0.29 | 1.98 | 1 | 0.00 | NA | NA | NA | NA | 0 | NA | Plasma Membrane | Gamma-aminobutyric acid (GABA) A receptor, alpha 1 |
| RASA2 | NM_006506 | 0.32 | 0.12 | 0.19 | −2.58 | 2 | 0.00 | 3.29 | 3.93 | 0.64 | 1.19 | 10 | 0.00 | Cytoplasm | RAS p21 protein activator 2 |
| CCL20 | NM_004591 | 0.36 | 0.68 | 0.32 | 1.90 | 1 | 0.00 | 1.75 | 1.85 | 0.10 | 1.06 | 2 | 0.36 | Extracellular Space | Chemokine (C-C motif) ligand 20 |
| mir-185 | NR_029968 | 0.41 | 0.64 | 0.23 | 1.55 | 2 | 0.04 | NA | NA | NA | NA | 0 | NA | Cytoplasm | mir-185 |
| PIK3R4 | NM_014602 | 0.44 | 0.16 | 0.28 | −2.75 | 2 | 0.02 | 1.82 | 1.94 | 0.12 | 1.07 | 7 | 0.00 | Cytoplasm | Phosphoinositide-3-kinase, regulatory subunit 4 |
| PSMD8 | NM_002812 | 0.51 | 0.23 | 0.28 | −2.24 | 6 | 0.03 | 1.51 | 1.62 | 0.11 | 1.07 | 7 | 0.02 | Cytoplasm | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 8 |
| mir-519a1 | NR_030218 | 0.58 | 0.88 | 0.30 | 1.51 | 2 | 0.00 | NA | NA | NA | NA | 0 | NA | Cytoplasm | mir-519a1 |
| POPDC2 | NM_022135 | 0.64 | 1.25 | 0.61 | 1.96 | 2 | 0.00 | 2.37 | 2.59 | 0.21 | 1.09 | 3 | 0.01 | Plasma Membrane | Popeye domain containing 2 |
| mir-301a | NR_029842 | 0.78 | 0.92 | 0.14 | 1.18 | 2 | 0.02 | 0.87 | 0.91 | 0.04 | 1.04 | 1 | 0.50 | Cytoplasm | mir-301a |
| RBCK1 | NM_006462 | 0.80 | 0.48 | 0.31 | −1.64 | 6 | 0.03 | 5.12 | 5.33 | 0.22 | 1.04 | 7 | 0.01 | Cytoplasm | RanBP-type and C3HC4-type zinc finger containing 1 |
| mir-329-2 | NR_029706 | 1.30 | 1.66 | 0.36 | 1.28 | 4 | 0.02 | NA | NA | NA | NA | 0 | NA | Cytoplasm | mir-329-2 |
| mir-487a | NR_030162 | 1.50 | 1.73 | 0.24 | 1.16 | 4 | 0.04 | 0.91 | 0.93 | 0.02 | 1.02 | 1 | 0.37 | Cytoplasm | mir-487a |
| IL3 | NM_000588 | 2.23 | 2.68 | 0.44 | 1.20 | 3 | 0.04 | 0.81 | 0.94 | 0.13 | 1.17 | 1 | 0.00 | Extracellular Space | Interleukin 3 (colony-stimulating factor, multiple) |
| UTRN | NM_007124 | 2.66 | 2.82 | 0.17 | 1.06 | 3 | 0.03 | 24.26 | 25.87 | 1.61 | 1.07 | 31 | 0.00 | Plasma Membrane | Utrophin |
| SUMO4 | NM_001002255 | 4.41 | 4.80 | 0.39 | 1.09 | 5 | 0.00 | 0.88 | 0.95 | 0.07 | 1.09 | 1 | 0.00 | Cytoplasm | Small ubiquitin-like modifier 4 |
To check if the methylation trend we obtained from the promoter region is qualitatively correct in the context of the entire gene, we graphically present all the CpG sites that are within the gene boundaries (from TSS1500 to 3′UTR), and label the TSS200 promoter region by two adjacent red marks on transcripts (Fig. 6). We observed the following patterns: (1) Indeed, the TSS200 and nearby 3′ downstream regions are densely packed with significantly DM (mostly hypomethylated) CpG sites, relative to other regions of the gene; (2) the differential methylation densities among the promoters of isoforms of the same gene vary, and this may be associated with different isoform expression statuses. For example, DIDO1 has 2 TSS200 regions, but we only see differential methylation signals from the promoter of the three longest transcripts and (3) TSS 200 and gene bodies appear to have correlated but opposite methylation patterns: the majority of the listed genes have lower methylation level in the TSS200 but higher methylation level in the gene bodies, when comparing the cases and the controls (Figs 6 and 7). Moreover, the cases versus control groups are well separated in the patterns of gene body average methylation versus TSS200 average methylation (Fig. 7).
Figure 6.

CpG methylation site maps among genes and microRNAs of interest. The X axis represents transcripts of a gene that span from the TSS1500 5′ terminal to transcription end sites. Along the direction of the row for each transcript, tick marks on each transcript represent TSS1500 5′ terminal (black), TSS200 5′ end (red) and transcription start site (red), and the arrow at the end represents the transcription start site (TES). The Y-axis represents the methylation beta values varying from 0 (no methylation) to 1 (complete methylation). Each CpG site with probes located within the gene is marked by vertical lines: red color represents average beta values in cases and thick blue color averaged beta values in control samples. The CpG sites that have significant P-values < 0.001 (after Bonferroni correction) are labeled by (*).
Figure 7.

Correlation of CpG methylation between sites located in proximal promoters (TSS200) and sites in gene bodies. Y axis is the averaged beta values among all sites in the gene body and x axis is the averaged eta values among all sites in the TSS200 region. The cases are labeled in red and controls in blue. The linear regression was performed and the correlation and R-square (R2) are shown.
Differential methylation in the non-coding genomic regions
Understanding the functions of non-coding genomic regions (ncGR), such as enhancers and non-coding RNAs (ncRNA) has recently become a field of active interest. We asked the question if the methylation patterns observed in our study are much different between ncGR and RefSeq genes. Using Ensembl database information, we re-annotated the probes and categorized 15 570 sites associated with long intergenic non-coding RNA (lincRNA) and 3755 sites associated with pre-microRNAs. RefSeq gene region and enhancers account for the majority of the CpG sites in the Illumina 450K platform. Comparatively, lincRNAs have a lower frequency of overall DM sites (P < 0.001, Fisher's test) and hypermethylated sites (P < 0.001, Fisher's test), relative to the expected frequency (Fig. 8). This suggests that lincRNAs may be un-favored by or refractory to DNA methylation modification in pre-eclampsia, opposite to RefSeq genes.
Figure 8.
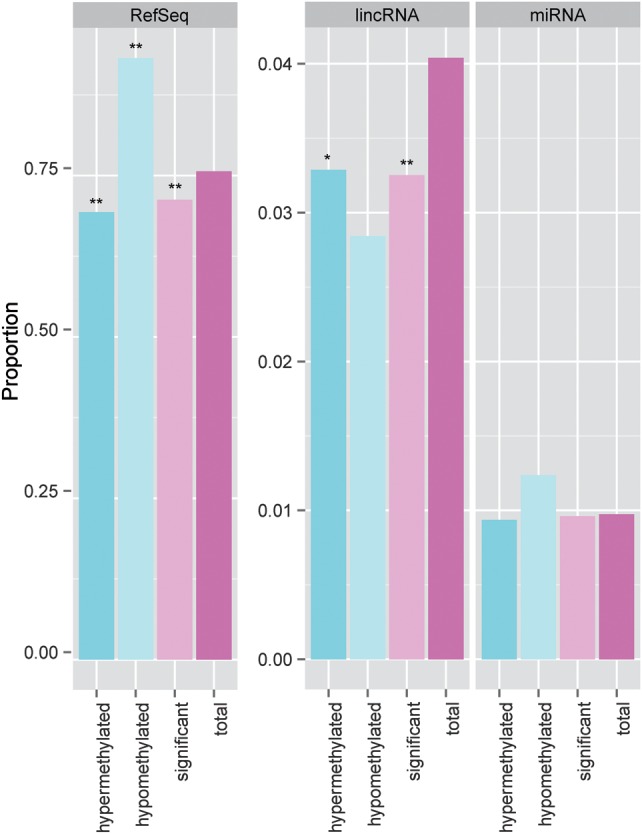
Comparison of proportions of candidate CpG sites from different genomic regions relative to the whole array. Three genomic regions are shown: RefSeq protein coding genes (RefSeq), long intergenic non-coding RNA (lincRNA) and pri-microRNA (miRNA). The proportion of candidate site (total, significant, etc) in each genomic region is calculated through the division of the number of qualified CpG sites in that genomic region by the total number of qualified CpG sites on the whole array. The columns labeled by ‘total’ represent the expected ‘background’ level in specific genomic regions. P-values of each category in comparison with the expected background are shown as asterisks. (*P < 0.001 and **P < 0.0001).
Relative to lincRNAs, microRNAs have better characterized functions. We thus included microRNAs in the network analysis. Mir-519a1 was the microRNA with the largest single CpG site level methylation difference between cases and control (P = 1.57e-5, and delta beta > 0.3) among microRNAs that have CpG probes (Fig. 6), and it also had significant hypermethylation at promoter level (Table III, P = 0.04). Mir-519a1 is involved in cell death, survival and inflammation (Fig. 6) in the IPA network analysis. It was also reported by others that mir-519a1 expression were elevated in pre-eclamptic placentas (Ishihara et al., 2002). IPA also showed that a network involved in hereditary disorder, skeletal and muscular disorders and connective tissue disorders had significant contributions from microRNAs (total 25 microRNAs), among which mir-301a, mir-487a, mir-185, mir-329, mir-194, mir-376a1, mir-486 and mir-744 were collectively hypermethylated in their promoters (Fig. 5). Among them, miR-301a was previously shown to be significantly elevated in placentas of PE and was indicated to play important roles in trophoblast proliferation during early pregnancy (Wang et al., 2012). MiR-185 and miR-329 were shown to inhibit cell proliferation, in which miR-185 targets RhoA, Cdc42, Six1 and c-Myc (Qu et al., 2013), whereas miR-329 targets transcription factor E2F1 (Xiao et al., 2013). MiR-329 also suppresses angiogenesis by targeting CD146. Additionally, miR-487a belongs to the large imprinted microRNA gene cluster at the Dlk1-Gtl2 domain that spans roughly 200 kb in the human chr14 14q32.q region (Seitz et al., 2004), and its DM indicates additional epigenetic regulation due to pre-eclampsia. Comprehensively, the methylation alteration also affects the promoters of microRNAs, which may function as the additional level of gene regulation in pre-eclampsia and lead to changes in cell proliferation and vascularization.
Discussion
Comparison with other methylation studies
In this study, we present in-depth the significant genome-wide hypermethylation coupled with promoter hypomethylation in chorioamniotic membranes of EOPE, based on Illumina Human Methylation 450 BeadChip methylation arrays. The chorioamniotic membranes develop early in gestation and do not undergo further developmental changes during gestation, thus the methylation pattern seen should reflect methylation that occurs early after the phase of genome-wide demethylation and subsequent remethylation in the early blastocyst. Our study confirms and expands the earlier methylation studies in pre-eclamptic placentas which had lower throughput and lower resolutions. Using Illumina GoldenGate Methylation panel, Yuen et al. analyzed 1505 CpG sites associated with 807 genes in placentas of EOPE, LOPE, intrauterine growth restriction (IUGR) and control subjects. They found that 34 loci in promoters were all hypomethylated (Yuen et al., 2010). In another study, Gao et al. used 5-methylcytosine immunohistochemistry and Alu and LINE-1 repeat pyrosequencing to measure the global methylation changes. They found that both the global DNA methylation level and the DNA (cytosine-5) methyltransferase 1 mRNA level were significantly higher in the EOPE placentas, compared with the normal controls (Gao et al., 2011). Similarly, Kulkarni et al. observed increased homocysteine and global DNA methylation levels in the pre-eclampsia group (term and pre-term PE) (Kulkarni et al., 2011). Interestingly, White et al. recently demonstrated that a similar genome-wide hypermethylation pattern exists in pre-eclamptic maternal leukocyte DNAs, using Illumina Human Methylation-27 K arrays (White et al., 2013), indicating that such significant epigenomic changes affect multiple organs and could be systematic. The agreement between our study and others suggest that PE have broad effects on abnormal DNA methylation during placentation, in both fetal-derived tissues as well as maternal tissues.
In contrast is the study of Blair et al. (2013) in which they reported extremely significant genome-wide hypomethylation (over 100 000 CpG loci at FDR < 0.05) comparing placentas of EOPE and pre-term normal controls (mean gestational ages were 31.8 weeks in both groups). One potential source to explain differences between Blair et al. and other studies including ours is the selection criteria for the control group. Rather than using full-term placentas with no complications as controls, such as our study and others' mentioned above, Blair et al. selected pre-term placentas. Although, there is weak association between gestational age and global DNA methylation level observed in a study with a small sample size (Chavan-Gautam et al., 2011), the source of variance analysis in our data showed that pre-eclampsia, rather than other factors such as gestational age, is primarily responsible for the observed differential methylation, supporting the suitability of using full-term placentas as controls. Moreover, full-term delivery is the expected biologically normal event, and pre-term delivery obtained placentas may have methylation differences due to the pathologic factors that lead to spontaneous pre-term birth. Other differences may be due to the small sample size of both studies, and other underlying factors such as ethnicity differences between the two studies; our data consisted of mainly Asians, while the race composition of Blair's study is mainly Caucasian (Personal communication).
We compared the methylation patterns in our chorioamniotic samples with the placental PMDs determined in the study of Schroeder et al. (2013) (Supplementary data, Fig. S7). Following the procedure in their paper, we first removed probes found in CpG islands, promoters and shores. We then calculated the beta value running average of 100 probes across each chromosome and overlayed the placental PMD regions. We found vast regions of reduced methylation in our chorioamniotic membrane samples that were consistent across samples (Supplementary data, Fig. S7). However, these regions did not share a strong overlap with the placental PMDs found in Schroeder et al.'s study. This is potentially due to tissue specificity, as chorioamniotic membranes and placental tissues are distinct, albeit closely related tissues.
Methylation changes related to hypoxia
Hypoxia is a feature of pre-eclampsia (Tal, 2012). As a result of hypoxia, the placenta launches an innate immune response leading to inflammation through the hypoxia-inducible factor complex 1 (HIF-1) (Tal, 2012), which leads to hypertension and acute arthrosclerosis (Boos and Lip, 2006; Nizet and Johnson, 2009; Palazón et al., 2012). Although HIF-1 did not appear to be DM in our study, several genes and transcription factors closely associated with HIF-1 were. One of these genes, ATF3, was significantly hypomethylated in the promoter (Table III). To our knowledge, methylation of ATF3 had never been reported in relation to pre-eclampsia prior to our current study. Corroborating our results, ATF3 was previously shown to be one of the four most consistently up-regulated genes in pre-eclampsia from the meta-analysis of gene expression array data sets, with an average fold change of 1.95 (Moslehi et al., 2013). ATF3 could up-regulate FLT1 transcription (Moslehi et al., 2013), through binding to the promoter region of soluble vascular endothelial growth factor receptor-1 (FLT1), one of the downstream effects of HIF-1 (Nevo et al., 2006; Jung et al., 2012). Consequently, FLT1 inhibits vascular development (Nevo et al., 2006; Jung et al., 2012) by binding and inhibiting vascular endothelial growth factor and placental growth factor (VEGF-A and PIGF). Additionally, ARF1, which was shown to be required for the secretion of FLT1 (Jung et al., 2012) was also significantly hypomethylated in the promoter region in our data set. Therefore, it is plausible that the up-regulation of FLT1 may be indirectly affected by promoter hypomethylation of associated factors such as ATF3 and ARF1, even though it is not DM.
Methylation changes related to inflammation
Changes of CpG methylation related to the inflammatory process are clearly evident in our data. In the second IPA network (Fig. 6) we see that a large number of DM genes contribute directly or indirectly to the nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) complex, including subunits NFκB2 and REL, its inhibitor IκB (NFκBIA) as well as activators IL1RAP and RBCK1. It is known that increased oxidative stress exists in pre-eclampsia placentas (Sikkema et al., 2001; Dechend et al., 2003) and produces increased amount of reactive oxidative species (ROS), likely due to the ischemic conditions and the lack of antioxidant/nutrients such as vitamin E (Takacs et al., 2001; Dechend et al., 2003). Thus, one mechanism of NF-κB activation in pre-eclampsia (Dechend et al., 2003) may involve promoter hypomethylation as a consequence of ROS. In short, the observed differential methylation in the promoter of genes related to the NF-κB complex may provide one mechanism for the previously observed over-expression of NF-κB in pre-eclampsia.
Methylation changes in signaling pathways
Adaptor protein GRB2 appears in our methylation data as a gene with hypomethylated promoters. GRB2 is a key protein that links several top ranked pathways found in our data. While the absolute delta beta difference between EOPE and control samples is not extreme (delta beta of 0.077), the P-value after Bonferroni correction is very significant (P = 0.004). Corresponding to the hypomethylation observed in our study, GRB2 was shown to be up-regulated by 36–65% in the placenta of pre-eclampsia patients at the gene expression level (Anteby et al., 2005). Through the SH2 domain, GRB2 binds to EGF and receptor tyrosine kinases (RTK) in the Ras pathway, and activates Ras through the activation of protein Son of Sevenless (SOS). Moreover, Ras itself was also hypomethylated in pre-eclampsia in our data, confirming the earlier hypothesis that Ras signaling might play a role in pre-eclampsia (Anteby et al., 2005). C-Raf is also hypomethylated, indicating that Ras-Raf-Mek-Erk signal transduction cascade could be involved as well. GRB2 is connected with the p85 subunit of PI3K/AKT pathway through scaffolding protein GRB2-associated binding protein (GAB), as well as actin cytoskeleton signaling through Ras.
The PI3K/AKT pathway is ranked second in canonical IPA pathways. It is involved in the transcription of multiple cellular processes and is downstream of VEGF receptors (VEGFR-1 and VEGFR-2) (Dayanir et al., 2001). Under hypoxic conditions such as in pre-eclampsia, the PI3K/AKT pathway is inactivated (Chiang et al., 2009; Zhou et al., 2013). We found that the p85 subunits of PI3K, encoded by the PIK3R3 and PIK3R4 genes, were hypomethylated in our data. p85 is an inhibitory subunit to the phosphorylation activity of the p110 kinase subunit of PI3K (Yu et al., 1998; Gavrieli and Murphy, 2006), which catalyzes a reaction cascade that eventually leads to AKT phosphorylation. Thus, the promoter hypomethylation of p85 could be a reason for the inactivation of PI3K/AKT pathway. Additionally, Pi3K-p85 has also been shown to have kinase-independent activity for controlling actin organization (Jimenez et al., 2000), and therefore could partially explain the emergence of actin cytoskeleton signaling, along with the PI3K/AKT pathway in pre-eclampsia.
Methylation changes related apoptosis and protein degradation
Apoptosis in pre-eclampsia is thought to be due to ischemic/hypoxic conditions (Leung et al., 2001; Ishihara et al., 2002). We observed evidence of differential methylation of genes involved in apoptosis. Death inducer-obliterator-1 (DIDO1) is an apoptotic factor significantly hypomethylated in the promoter from our analysis. DIDO1 was previously shown to be associated with caspase promoters, resulting in caspase up-regulation and caspase mediated apoptosis (García-Domingo et al., 1999, 2003).
Perhaps the most surprising finding in our study is the strong evidence of hypomethylation in promoters of two 20S subunits PSMA1 and PSME1, two 26S subunits PSMD1 and PSMD8, as well as ubiquitin-conjugating enzyme UBE2W. The 20S proteasome is responsible for metabolizing tissue-damaging oxidatively stressed proteins in pre-eclampsia. Intriguingly, a reduced overall 20S proteasome activity of 20% in pre-eclampsia has been previously reported, possibly related to the accumulation of oxidatively damaged proteins and thus metabolic interference in pre-eclampsia (Rajakumar et al., 2003, 2008). The mechanisms for reduced proteasomal activity in the pathological placentas need to be explored further in order to explain the apparent discrepancy between proteasome subunit hypomethylation and reduced enzymatic activity. One source of complexity could be attributed to the 20S proteasome. It is a relatively large complex of 700 kDa with many subunits and proteasome assays using 2D-SDS PAGE have limited ability to characterize adequately the abundance of proteasomal subunits, as previously suggested (Rajakumar et al., 2003, 2008). Another possibility could be that proteasomal activity may be post-transcriptionally inactivated by the overproduction of reactive oxygen species or other oxidatively modified proteins, besides the epigenetic modification that could lead to transcript level changes.
Novel findings of the study
In this study, we corroborate expression data with methylation data at locus level, gene level, as well as the network and pathway level. With the assumption that hypomethylation in CpG islands and promoters, and hypermethylation in gene bodies are associated with gene activation, the methylation pattern observed in this study suggests an activated gene expression program in the chorioamniotic membranes with early onset pre-eclampsia. Network and pathway analysis through IPA using the overall methylation levels of the promoter regions reveals striking complementarity between gene-level methylation networks and gene expression differences reported in existing literature. Recently, Liu et al. (2013) integrated multiple placenta gene expression studies with serum proteomes, and developed serum biomarker panels for both EOPE and LOPE that outperform the biomarker of serum sFlt-1/PIGF ratio. It will be interesting to test if some of the identified methylation differences in our study also exist in maternal serum DNA, with the potential of methylation biomarkers. We also report for the first time the genome-wide methylation profiles in promoters of non-coding regions such as the lincRNA and microRNAs. We find that lincRNAs are un-favored by DNA methylation modification in pre-eclampsia, compared with the RefSeq protein coding genes. Additionally, we discovered a group of microRNAs that are collectively hypomethylated in promoters and are associated with functional disorders in pre-eclampsia. In summary, the patterns of genome-wide hypermethylation coupled with hypomethylation in promoters are widespread in the chorioamniotic membranes of EOPE. These findings unfold new areas of research regarding the importance of epigenetic determinants of pre-eclampsia.
Open questions and future work
To our knowledge, this study is the first to address the genome-wide methylation changes in the chorioamniotic membranes of the EOPE patients. Chorioamniotic membranes consist of three layers, the amnion, chorion and adherent decidua. The amnion and chorion are fetal in origin, but the decidua layer is composed of maternal decidua cells and invading trophoblasts. Thus, the samples are predominantly fetal. Differences in cellular composition may affect methylation changes. The cellular composition of the chorioamniotic membranes does not change significantly in the third trimester, except for the small percentage of influx of leukocytes (less than 10%) associated with the labor process (Gomez-Lopez et al., 2010). The samples in the study were obtained from the biorepository with the DNA extracted from paraffin imbedded blocks of membranes. Thus, we could not have histologic evaluation of the samples used to determine the percentage of inflammation present. However, this influx is not expected to significantly alter the cellular composition. Likewise we also miss the information on the location of the membranes in relation to the placenta and the site of membrane rupture. In the future prospective studies with fresh samples, we plan to determine which layer of the membrane and where in the membranes the differential methylation occur, coupled with genome-wide gene expression analysis.
Supplementary data
Supplementary data are available at http://molehr.oxfordjournals.org/.
Authors’ roles
L.X.G. supervised the work. T.C. and L.X.G. wrote the manuscript. T.C. analyzed the data and designed primers of pyrosequencing. M.S. and M.T. performed the 450K assays. J.M. performed the bisulfite conversion and pyrosequencing technical validation. M.B. and D.T. initiated the project and oversaw the sample collection. All authors have read, revised and approved the manuscript.
Funding
The work was partially supported by RMATRIX award U54MD007584 from the National Institute on Minority Health and Health Disparities, National Institutes of Health (NIH) and research funding provided by the University of Hawaii Cancer Center (UHCC) to L.X. Garmire. M.B. is supported by NIH grants RO1 DK47320 and G12 MD007601. The Genomics Shared Resource (GSR) and the Pathology Shared Resource (PSR) at UHCC is supported by CCSG P30-CA071789-14.
Conflict of interest
There are no conflicts of interests with any of the authors with regard to the content of this paper.
Supplementary Material
Acknowledgements
We especially thank Dr Alika Maunakea for valuable suggestions and discussions during the preparation of the manuscript. We thank the University of Hawaii Biorepository team, including Dr Abby Collier, Dr Timothy Dye, Dr Joshua Astern, Will Chen and others for providing samples and resources, and Mr Hugh Luk from Pathology Shared Resource (GSR) at UHCC for the technical help.
References
- Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA. Placental apoptosis in preeclampsia. Obstet Gynecol. 2000;96:271–276. doi: 10.1016/s0029-7844(00)00895-4. [DOI] [PubMed] [Google Scholar]
- Anteby EY, Ayesh S, Shochina M, Hamani Y, Schneider T, Al-Shareef W, Hochberg A, Ariel I. Growth factor receptor-protein bound 2 (GRB2) upregulation in the placenta in preeclampsia implies a possible role for ras signalling. Eur J Obstet Gynecol Reprod Biol. 2005;118:174–181. doi: 10.1016/j.ejogrb.2004.04.029. [DOI] [PubMed] [Google Scholar]
- Batalle D, Eixarch E, Figueras F, Muñoz-Moreno E, Bargallo N, Illa M, Acosta-Rojas R, Amat-Roldan I, Gratacos E. Altered small-world topology of structural brain networks in infants with intrauterine growth restriction and its association with later neurodevelopmental outcome. NeuroImage. 2012;60:1352–1366. doi: 10.1016/j.neuroimage.2012.01.059. [DOI] [PubMed] [Google Scholar]
- Blair JD, Yuen RKC, Lim BK, McFadden DE, von Dadelszen P, Robinson WP. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early-onset pre-eclampsia. Mol Hum Reprod. 2013;19:697–708. doi: 10.1093/molehr/gat044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos CJ, Lip GY. Is hypertension an inflammatory process? Curr Pharm Des. 2006;12:1623–1635. doi: 10.2174/138161206776843313. [DOI] [PubMed] [Google Scholar]
- Bourque DK, Avila L, Penaherrera M, Von Dadelszen P, Robinson WP. Decreased placental methylation at the H19/IGF2 imprinting control region is associated with normotensive intrauterine growth restriction but not preeclampsia. Placenta. 2010;31:197–202. doi: 10.1016/j.placenta.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Chavan-Gautam P, Sundrani D, Pisal H, Nimbargi V, Mehendale S, Joshi S. Gestation-dependent changes in human placental global DNA methylation levels. Mol Reprod Dev. 2011;78:150. doi: 10.1002/mrd.21296. [DOI] [PubMed] [Google Scholar]
- Chelbi ST, Mondon F, Jammes H, Buffat C, Mignot T-M, Tost J, Busato F, Gut I, Rebourcet R, Laissue P. Expressional and epigenetic alterations of placental serine protease inhibitors SERPINA3 is a potential marker of preeclampsia. Hypertension. 2007;49:76–83. doi: 10.1161/01.HYP.0000250831.52876.cb. [DOI] [PubMed] [Google Scholar]
- Chiang M-H, Liang F-Y, Chen C-P, Chang C-W, Cheong M-L, Wang L-J, Liang C-Y, Lin F-Y, Chou C-C, Chen H. Mechanism of hypoxia-induced GCM1 degradation implications for the pathogenesis of preeclampsia. J Biol Chem. 2009;284:17411–17419. doi: 10.1074/jbc.M109.016170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury M, Friedman JE. Epigenetics and microRNAs in preeclampsia. Clin Exp Hypertens N Y N 1993. 2012;34:334–341. doi: 10.3109/10641963.2011.649931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayanir V, Meyer RD, Lashkari K, Rahimi N. Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol 3-kinase and cell proliferation. J Biol Chem. 2001;276:17686–17692. doi: 10.1074/jbc.M009128200. [DOI] [PubMed] [Google Scholar]
- Dechend R, Viedt C, Müller DN, Ugele B, Brandes RP, Wallukat G, Park J-K, Janke J, Barta P, Theuer J. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107:1632–1639. doi: 10.1161/01.CIR.0000058200.90059.B1. [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Li D, Xiao Z, Liao Q, Yang H, Li Y, Ji L, Wang Y. Detection of global DNA methylation and paternally imprinted H19 gene methylation in preeclamptic placentas. Hypertens Res. 2011;34:655–661. doi: 10.1038/hr.2011.9. [DOI] [PubMed] [Google Scholar]
- García-Domingo D, Leonardo E, Grandien A, Martínez P, Albar JP, Izpisúa-Belmonte JC, Martínez-A C. DIO-1 is a gene involved in onset of apoptosis in vitro, whose misexpression disrupts limb development. Proc Natl Acad Sci USA. 1999;96:7992–7997. doi: 10.1073/pnas.96.14.7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Domingo D, Ramírez D, de Buitrago GG, Martínez-A C. Death inducer-obliterator 1 triggers apoptosis after nuclear translocation and caspase upregulation. Mol Cell Biol. 2003;23:3216–3225. doi: 10.1128/MCB.23.9.3216-3225.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Gavrieli M, Murphy KM. Association of Grb-2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem Biophys Res Commun. 2006;345:1440–1445. doi: 10.1016/j.bbrc.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Gomez-Lopez N, Laresgoiti-Servitje E, Olson DM, Estrada-Gutiérrez G, Vadillo-Ortega F. The role of chemokines in term and premature rupture of the fetal membranes: a review. Biol Reprod. 2010;82:809–814. doi: 10.1095/biolreprod.109.080432. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T. Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol. 2002;186:158–166. doi: 10.1067/mob.2002.119176. [DOI] [PubMed] [Google Scholar]
- Jia R-Z, Zhang X, Hu P, Liu X-M, Hua X-D, Wang X, Ding H-J. Screening for differential methylation status in human placenta in preeclampsia using a CpG island plus promoter microarray. Int J Mol Med. 2012;30:133–141. doi: 10.3892/ijmm.2012.983. [DOI] [PubMed] [Google Scholar]
- Jimenez C, Portela RA, Mellado M, Rodriguez-Frade JM, Collard J, Serrano A, Martinez-A C, Avila J, Carrera AC. Role of the Pi3k regulatory subunit in the control of actin organization and cell migration. J Cell Biol. 2000;151:249–262. doi: 10.1083/jcb.151.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J-J, Tiwari A, Inamdar SM, Thomas CP, Goel A, Choudhury A. Secretion of soluble vascular endothelial growth factor receptor 1 (sVEGFR1/sFlt1) requires Arf1, Arf6, and Rab11 GTPases. PloS One. 2012;7:e44572. doi: 10.1371/journal.pone.0044572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A, Chavan-Gautam P, Mehendale S, Yadav H, Joshi S. Global DNA methylation patterns in placenta and its association with maternal hypertension in pre-eclampsia. DNA Cell Biol. 2011;30:79–84. doi: 10.1089/dna.2010.1084. [DOI] [PubMed] [Google Scholar]
- Leung DN, Smith SC, To KF, Sahota DS, Baker PN. Increased placental apoptosis in pregnancies complicated by preeclampsia. Am J Obstet Gynecol. 2001;184:1249–1250. doi: 10.1067/mob.2001.112906. [DOI] [PubMed] [Google Scholar]
- Liu LY, Yang T, Ji J, Wen Q, Morgan AA, Jin B, Chen G, Lyell DJ, Stevenson DK, Ling XB. Integrating multiple'omics’ analyses identifies serological protein biomarkers for preeclampsia. BMC Med. 2013;11:236. doi: 10.1186/1741-7015-11-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J, Gordon L, Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard SE, Min J-Y, Merchan J, Lim K-H, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslehi R, Mills JL, Signore C, Kumar A, Ambroggio X, Dzutsev A. Integrative transcriptome analysis reveals dysregulation of canonical cancer molecular pathways in placenta leading to preeclampsia. Sci Rep. 2013;3:2407. doi: 10.1038/srep02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousa AA, Strauss JF, Walsh SW. Reduced methylation of the thromboxane synthase gene is correlated with its increased vascular expression in preeclampsia. Hypertension. 2012;59:1249–1255. doi: 10.1161/HYPERTENSIONAHA.111.188730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo O, Soleymanlou N, Wu Y, Xu J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol-Regul Integr Comp Physiol. 2006;291:R1085–R1093. doi: 10.1152/ajpregu.00794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niknejad H, Peirovi H, Jorjani M, Ahmadiani A, Ghanavi J, Seifalian AM. Properties of the amniotic membrane for potential use in tissue engineering. Eur Cell Mater. 2008;15:88–99. doi: 10.22203/ecm.v015a07. [DOI] [PubMed] [Google Scholar]
- Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009;9:609–617. doi: 10.1038/nri2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakovic B, Yuen RK, Gordon L, Penaherrera MS, Sharkey A, Moffett A, Craig JM, Robinson WP, Saffery R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genomics. 2011;12:529. doi: 10.1186/1471-2164-12-529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazón A, Aragonés J, Morales-Kastresana A, de Landázuri MO, Melero I. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. 2012;18:1207–1213. doi: 10.1158/1078-0432.CCR-11-1591. [DOI] [PubMed] [Google Scholar]
- Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. doi: 10.1016/j.placenta.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Qu F, Cui X, Hong Y, Wang J, Li Y, Chen L, Liu Y, Gao Y, Xu D, Wang Q. MicroRNA-185 suppresses proliferation, invasion, migration, and tumorigenicity of human prostate cancer cells through targeting androgen receptor. Mol Cell Biochem. 2013;377:121–130. doi: 10.1007/s11010-013-1576-z. [DOI] [PubMed] [Google Scholar]
- Rajakumar A, Doty K, Daftary A, Harger G, Conrad KP. Impaired oxygen-dependent reduction of HIF-1α and-2α proteins in pre-eclamptic placentae. Placenta. 2003;24:199–208. doi: 10.1053/plac.2002.0893. [DOI] [PubMed] [Google Scholar]
- Rajakumar A, Michael HM, Daftary A, Jeyabalan A, Gilmour C, Conrad KP. Proteasomal activity in placentas from women with preeclampsia and intrauterine growth restriction: Implications for expression of HIF-α proteins. Placenta. 2008;29:290–299. doi: 10.1016/j.placenta.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Raymond D, Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv. 2011;66:497–506. doi: 10.1097/OGX.0b013e3182331028. [DOI] [PubMed] [Google Scholar]
- Schroeder DI, Blair JD, Lott P, Yu HOK, Hong D, Crary F, Ashwood P, Walker C, Korf I, Robinson WP, et al. The human placenta methylome. Proc Natl Acad Sci USA. 2013;110:6037–6042. doi: 10.1073/pnas.1215145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz H, Royo H, Bortolin M-L, Lin S-P, Ferguson-Smith AC, Cavaillé J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res. 2004;14:1741–1748. doi: 10.1101/gr.2743304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkema JM, Van Rijn BB, Franx A, Bruinse HW, De Roos R, Stroes ESG, Van Faassen EE. Placental superoxide is increased in pre-eclampsia. Placenta. 2001;22:304–308. doi: 10.1053/plac.2001.0629. [DOI] [PubMed] [Google Scholar]
- Srinivas SK, Edlow AG, Neff PM, Sammel MD, Andrela CM, Elovitz MA. Rethinking IUGR in preeclampsia: dependent or independent of maternal hypertension? J Perinatol. 2009;29:680–684. doi: 10.1038/jp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stillman IE, Karumanchi SA. The glomerular injury of preeclampsia. J Am Soc Nephrol. 2007;18:2281–2284. doi: 10.1681/ASN.2007020255. [DOI] [PubMed] [Google Scholar]
- Takacs P, Kauma SW, Sholley MM, Walsh SW, Dinsmoor MJ, Green K. Increased circulating lipid peroxides in severe preeclampsia activate NF-κB and upregulate ICAM-1 in vascular endothelial cells. FASEB J. 2001;15:279–281. doi: 10.1096/fj.00-0549fje. [DOI] [PubMed] [Google Scholar]
- Tal R. The role of hypoxia and hypoxia-inducible factor-1alpha in preeclampsia pathogenesis. Biol Reprod. 2012;87:134. doi: 10.1095/biolreprod.112.102723. [DOI] [PubMed] [Google Scholar]
- Wang Z, Lu S, Liu C, Zhao B, Pei K, Tian L, Ma X. Expressional and epigenetic alterations of placental matrix metalloproteinase 9 in preeclampsia. Gynecol Endocrinol. 2010;26:96–102. doi: 10.3109/09513590903184100. [DOI] [PubMed] [Google Scholar]
- Wang D, Song W, Na Q. The emerging roles of placenta-specific microRNAs in regulating trophoblast proliferation during the first trimester. Aust N Z J Obstet Gynaecol. 2012;52:565–570. doi: 10.1111/j.1479-828X.2012.01481.x. [DOI] [PubMed] [Google Scholar]
- West J, Beck S, Wang X, Teschendorff AE. An integrative network algorithm identifies age-associated differential methylation interactome hotspots targeting stem-cell differentiation pathways. Sci Rep. 2013;3:1630. doi: 10.1038/srep01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White WM, Brost B, Sun Z, Rose C, Craici I, Wagner SJ, Turner ST, Garovic VD. Genome-wide methylation profiling demonstrates hypermethylation in maternal leukocyte DNA in preeclamptic compared to normotensive pregnancies. Hypertens Pregnancy Off J Int Soc Study Hypertens Pregnancy. 2013;32:257–269. doi: 10.3109/10641955.2013.796970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Tan L, He B, Liu Z, Xu R. MiRNA-329 targeting E2F1 inhibits cell proliferation in glioma cells. J Transl Med. 2013;11:172. doi: 10.1186/1479-5876-11-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Zhang Y, McIlroy J, Rordorf-Nikolic T, Orr GA, Backer JM. Regulation of the p85/p110 phosphatidylinositol 3?-Kinase: stabilization and Inhibition of the p110? Catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. doi: 10.1128/mcb.18.3.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Penaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18:1006–1012. doi: 10.1038/ejhg.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Gormley MJ, Hunkapiller NM, Kapidzic M, Stolyarov Y, Feng V, Nishida M, Drake PM, Bianco K, Wang F. Reversal of gene dysregulation in cultured cytotrophoblasts reveals possible causes of preeclampsia. J Clin Invest. 2013;123:0–0. doi: 10.1172/JCI66966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Widschwendter M, Teschendorff AE. A comparison of feature selection and classification methods in DNA methylation studies using the Illumina Infinium platform. BMC Bioinformatics. 2012;13:59. doi: 10.1186/1471-2105-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.