

Abstract
Megazol (7) is a 5-nitroimidazole that is highly active against Trypanosoma cruzi and Trypanosoma brucei, as well as drug-resistant forms of trypanosomiasis. Compound 7 is not used clinically due to its mutagenic and genotoxic properties, but has been largely used as a lead compound. Here, we compared the activity of 7 with its 4H-1,2,4-triazole bioisostere (8) in bloodstream forms of T. brucei and T. cruzi and evaluated their activation by T. brucei type I nitroreductase (TbNTR) enzyme. We also analysed the cytotoxic and genotoxic effects of these compounds in whole human blood using Comet and fluorescein diacetate/ethidium bromide assays. Although the only difference between 7 and 8 is the substitution of sulphur (in the thiadiazole in 7) for nitrogen (in the triazole in 8), the results indicated that 8 had poorer antiparasitic activity than 7 and was not genotoxic, whereas 7 presented this effect. The determination of Vmax indicated that although 8 was metabolised more rapidly than 7, it bounds to the TbNTR with better affinity, resulting in equivalent kcat/KM values. Docking assays of 7 and 8 performed within the active site of a homology model of the TbNTR indicating that 8 had greater affinity than 7.
Keywords: megazol, Trypanosoma cruzi, Trypanosoma brucei, T. brucei, nitroreductase, genotoxicity
Protozoan parasites belonging to the genus Trypanosoma are responsible for two major infections in humans; namely, Chagas disease (or American trypanosomiasis) and human African trypanosomiasis (HAT), which is the sleeping sickness. Estimates indicate that more than 10 million people worldwide are infected by the causative agents, Trypanosoma cruzi and Trypanosoma brucei, resulting in considerable morbidity and mortality, especially in developing countries (Barrett et al. 2003, WHO 2010, Wilkinson et al. 2011). Current therapies targeting these pathogens have proven to be unsatisfactory because of many of these therapeutic agents have limited efficacy, some serious side effects and resistance in the clinical context has emerged (Castro et al. 2006, Wilkinson & Kelly 2009, Rassi-Jr et al. 2012). In addition, in HAT intravenous administration of drugs, the time required to complete the course of treatment and the cost associated with them remain highly limiting (Barrett et al. 2003, Nwaka & Hudson 2006). Chagas disease treatment is focused on the use of two nitroheterocyclic prodrugs: nifurtimox (1), a 5-nitrofuran and benznidazole (2), a 2-nitroimidazole (Fig. 1). Neither of these agents is ideal due to undesirable secondary side effects. Variable results have been observed with these agents depending on the phase of the disease (they are only effective in the acute and recent chronic phases of the infection), the dose and duration of treatment, the patient’s age and the endemic region (Castro et al. 2006, Soeiro & de Castro 2011). Five approved treatments for HAT exist, including four monotherapies, pentamidine (3), suramin (4), melarsoprol (5) and eflornithine (6) (Fig. 1) and a nifurtimox-eflornithine combination therapy. Benefits and disadvantages have been associated with each medication (Wilkinson & Kelly 2009, Jacobs et al. 2011). The arsenical compound melarsoprol (5) is of particular note, as it is highly toxic, causing reactive encephalopathy in 5-10% of treated patients, of which, 50% will die as a result of this adverse reaction (El-Sayed et al. 2005, Bolognesi et al. 2008). Despite these drawbacks, melarsoprol (5) is still in use and represents the only drug that can target the neurological phase of all cases of African sleeping sickness.
Fig. 1. : chemical structures of nifurtimox (1) and benznidazole (2), the drugs currently used to treat Chagas disease, and pentamidine (3), suramin (4), melarsoprol (5) and eflornithine (6), the agents currently used to treat African sleeping sickness.

Megazol (7) is a 5-nitroimidazole that contains a 1,3,4-thiadiazole substituent group at position 2 (Fig. 2) and displays significant activity against a wide range of microbes. When screened on trypanosomes, it was shown to be more effective against T. cruzi than compounds 1 or 2, displaying activity towards strains that were refractory to these two nitroheterocycles (Filardi & Brener 1987). It also displayed considerable growth inhibitory activity against T. brucei, in which it was demonstrated to promote parasite clearance in animals after the administration of a single dose (Bouteille et al. 1995, Enanga et al. 1999, 2000).
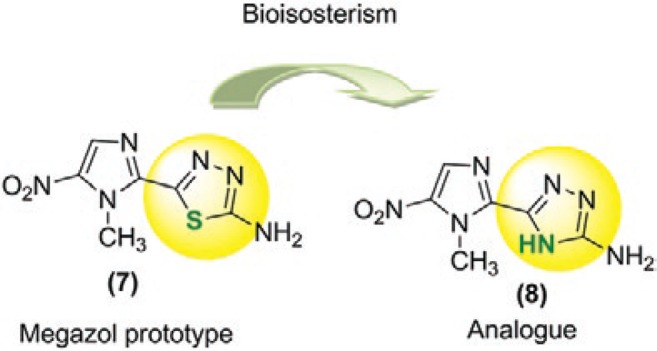
Fig. 2. : chemical structures of megazol (7) and its triazole analog [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8)].
These initial results have stimulated research aimed at revealing the mechanism by which compound 7 mediates its antiparasitic activities. Several studies have clearly indicated that compound 7 enters the parasite by passive diffusion (Barrett et al. 2003) and once inside the cell, it undergoes activation by nitroreductase enzymes (NTR). These enzymes catalyse one or two electron reduction of the conserved nitro group found on the imidazole ring (Viodé et al. 1999, Wilkinson & Kelly 2009). The subsequent downstream mechanisms accounting for the mode of action of compound 7 are unclear. Based on studies using T. cruzi, it has been proposed that compound 7 may interfere with oxygen metabolism (Viodé et al. 1999), possibly by affecting intracellular thiol levels, especially trypanothione (Maya et al. 2003) and/or via the inhibition of NADH fumarate reductase activity (Turrens et al. 1996). Alternatively, compound 7 has clearly been shown to mediate DNA damage in T. brucei mutants lacking RAD51, a key component of the parasite’s homologous DNA repair pathway (Enanga et al. 2003). Subsequent reports provided evidence that compound 7 was mutagenic in a bacterial Ames assay (Ferreira & Ferreira 1986) and promoted chromosomal aberrations in mammalian cells (Nesslany et al. 2004). Further investigations into using this compound as a trypanocidal agent were discontinued. Thus, compound 7 is a promising target compound that can be used to search for new analogs that lack these side effects.
Nowadays, many efforts have been done in the search of new synthetic lead compounds such as triazoles, quinolines and others (Papadopoulou et al. 2011, 2013, da Silva et al. 2012) and synthetic nitroimidazole derivatives (Mital 2009, Paula et al. 2009).
Our research group has been conducting studies with this aim in mind (Boechat et al. 2001, Carvalho et al. 2004, 2006, 2007, 2008, Salomão et al. 2010) and we have identified several promising prototypes (Boechat et al. 2001, Carvalho et al. 2004, 2006, 2007, 2008). The triazole nucleus is one of the most important heterocycles and has well-known trypanocidal activity (Boechat et al. 2012). Using a triazole nucleus as an isostere of thiadiazole, which is contained in the megazol prototype, we synthesised 5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8), as shown in Fig. 2 (Carvalho et al. 2007). Unfortunately, a dramatic loss of activity against T. cruzi was observed. However, the results of a preliminary cytotoxicity study revealed that compound 8 was not toxic, encouraging us to study the physical-chemical and biological properties of bioisostere 8. Our goal is not only to find active compounds against T. cruzi and T. brucei, but also explore the so well documented relationship of the nitro group with the biological and mutagenic activities. Thus, the purpose of the present study was to compare the in vitro activities of bioisosteres 7 and 8 in T. cruzi and T. brucei, as well as their genotoxicity and cytotoxicity. We also aimed to study the in vitro and in silico activation process of these compounds by NTR of T. brucei (TbNTR).
SUBJECTS, MATERIALS AND METHODS
Chemistry - Benznidazole (2) was provided by Hoffmann-La Roche Inc. Megazol (7) and its bioisostere 5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8) was synthesised from 2-cyano-5-nitroimidazole by condensation in trifluoroacetic acid with thiosemicarbazide to generate the thiadiazole ring or with aminoguanidine to generate the triazole ring, as previously described (Carvalho et al. 2007).
Parasites - T. cruzi (Y strain) bloodstream form (BSF) trypomastigotes were obtained from infected male Swiss Webster mice (18-20 g in body weight) at the peak of parasitaemia and isolated using differential centrifugation (de Castro et al. 1987). T. brucei brucei (MITat 427 strain; clone 221a) BSF trypomastigotes were grown at 37ºC under a 5% (v/v) CO2 atmosphere in modified Iscove’s medium, as previously described (Hirumi & Hirumi 1989).
T. cruzi BSF were suspended to a concentration of 1 x 107 parasites mL-1 in Dulbecco’s modified Eagle’s medium containing 10% (v/v) foetal calf serum. This suspension (100 μL) was added to the same volume of compound 7 or 8, which had been previously prepared at double the desired final concentrations in 96-well microplates and was incubated for 24 h at 37ºC under a 5% (v/v) CO2 atmosphere. Cell counts were then carried out using a Neubauer chamber and the compound concentration that killed 50% of the parasites (LD50) was determined. Untreated and benznidazole-treated parasites were used as controls.
T. brucei BSF were seeded at 1 x 103 parasites mL-1 in growth medium containing different concentrations of compound 7 or 8 and aliquots (200 μL) were dispensed into the wells of 96-well microplates. After incubation at 37ºC for three days, alamarBlue™ (20 µl) (Life Technologies Ltd, Paisley, UK) was added to each well and the plates were incubated for another 16 h. The fluorescence of each culture was then determined using a Gemini fluorescent plate reader (Molecular Devices Ltd, Wokingham, UK) at an excitation wavelength of 530 nm, an emission wavelength of 585 nm and a filter cut-off at 550 nm. The colour change resulting from dye reduction is proportional to the number of live cells. The concentration of each compound that inhibited parasite growth by 50% (IC50) was then established.
Enzyme activity of the TbNTR - Enzyme activity was measured spectrophotometrically by following NADH oxidation (λ = 340 nm; ε = 6,220 M-1cm-1) using recombinant TbNTR, which had been purified as previously described (Hall et al. 2010). A standard reaction mixture (1 mL) containing 50 mM Tris-Cl, pH 7.5, NADH (100 µM) and substrate (0-200 µM) was incubated at room temperature (RT) for 5 min. The background rate of NADH oxidation was determined and the reaction was initiated by the addition of trypanosomal enzyme (20 µg). The results were evaluated via a non-linear regression analysis using GraphPad Prism 5 (GraphPad Software).
Toxicity to whole blood - Heparinised human blood was obtained by venipuncture immediately prior to the assays. Whole blood was treated for 2 h at 37ºC with different concentrations of compounds 7 (380-4000 µM) or 8 (149-6400 µM) in 5% (v/v) dimethyl sulfoxide (DMSO) (solvent-control) and then used in the assays. Cell viability was determined at the end of drug treatment using the fluorescein diacetate (FDA)/ethidium bromide (EtBr) assay (Hartmann & Speit 1997). Whole blood (50 µL) was mixed with an equal volume of the freshly prepared staining solution, which consisted of 30 µg/mL of FDA plus 8 µg/mL of EtBr in phosphate buffered saline (PBS). The samples (50 µL) were spread on microscope slides and covered with coverslips. Viable cells appeared as green-fluorescent, whereas orange-stained nuclei indicated dead cells. A total of 200 cells were analysed for each treatment.
Genotoxicity in an in vitro alkaline Comet assay - DNA damage in whole blood was evaluated at the end of a 2 h treatment in duplicate with compound 7 or 8, at the same concentrations indicated above using the alkaline Comet assay (Tice et al. 2000, Speit & Hartmann 2006). Methyl methane-sulfonate (MMS) (160 µM) Milwaukee, WI, USA) was used as a positive control. Aliquots of 5 µL of whole blood were mixed with 120 µL of 0.5% (w/v) low melting-point agarose (LMPA) in PBS (Sigma-Aldrich, St. Louis, MO, USA) at 37ºC and added to microscope slides that had been previously covered with 1.5% (w/v) normal melting-point agarose (Sigma-Aldrich). Four slides per treatment were prepared (2 slides per culture). Immediately afterward, the slides were covered with a coverslip and after LMPA solidification for 3 min at 4-5ºC, the coverslips were removed. The slides were then immersed in cold lysis buffer [2.5 M NaCl, 100 µM mM Na2 ethylenediamine tetraacetic acid (EDTA), 10 mM µM Tris, 1% (w/v) N-lauroylsarcosine sodium salt, 1% (v/v) Triton X-100 and 10% (v/v) DMSO, pH 10] for a minimum of 1 h at 4ºC and protected from light. The slides were placed on a horizontal gel electrophoresis unit that had been filled with freshly made alkaline buffer at a pH > 13 (300 mM NaOH and 1 mM EDTA). The duration of alkali exposure and DNA electrophoresis (0.86 V/cm and 300 mA) was 20 min each in an ice bath. The samples were neutralised by washing them three times (5 min per wash) in Tris buffer (0.4 M Tris, pH 7.5), drained, fixed in absolute ethanol [99.8% (v/v)] for approximately 10 min and left at RT overnight to dry. EtBr (30 µL of a 20 µg/mL stock) was then added to each slide. The slide was then covered with a coverslip and analysed using a fluorescence microscope at 400X magnification. Fifty randomly selected cells per slide (200 cells per treatment) were analysed visually and divided into one of four DNA damage classes based on tail size: 0 (undamaged; i.e., no visible tail), 1 (slightly damaged), 2 (moderately damaged) and 3 (maximally damaged; i.e., the head of the comet was very small and the majority of the DNA was in the tail). The DNA damage was expressed as the percentage of cells in four classes and as arbitrary units (AU) according to the formula: AU = (0 x nº of comets in Class 0) + (1 x nº of comets in Class 1) + (2 x nº of comets in Class 2) + (3 x nº of comets in Class 3). The total DNA damage score in AU (TAU) for 200 comets can range from 0 TAU (undamaged) to 600 TAU (maximally damaged). Differences between the mean values of TAU from three and two independent experiments under the same conditions, respectively, for each concentration of compounds 7 and 8 were tested for significance (p < 0.05) in relation to the solvent-control group using Student’s one-tailed t test. In addition, the effects of megazol and compound 8 on the intercellular distribution of DNA damage were tested for statistical significance using one-way ANOVA, followed by a Dunnett’s multiple comparison test to compare each concentration of the compounds.
Molecular modelling - Theoretical calculations were carried out to investigate the stereoelectronic properties of megazol and compound 8 and were aimed to characterise the electronic features of the designed molecules that may be important for their bioactivity. The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies were calculated using the MOPAC2009 software package (OpenMOPAC.net) using the semiempirical PM3 method with the full geometry optimisation. The ejection fraction (EF) routine was used for the minimum search. Default values were taken for the other parameters. The Cartesian coordinates of megazol and compound 8 were built using the Avogadro Program (Hanwell et al. 2012).
So far, no experimental (NMR or crystallographic) structure of the TbNTR is available in the Protein Data Bank (PDB) (Bernstein et al. 1977). Therefore, the construction of a homology model was necessary for the docking studies. The amino acid sequence of the TbNTR was retrieved from the National Center for Biotechnology Information (Geer et al. 2010) under the XP_846343.1 code. This sequence was subsequently blasted against the PDB, yielding structures belonging to the nitro-flavin mononucleoside (FMN)-reductase superfamily. The crystal structure of the nitroreductase family protein from Streptococcus pneumoniae Tigr4 (PDB code 2B67) (Kim et al. 2011) and the structure of Escherichia coli nitroreductase bound to the antibiotic nitrofurazone (PDB code 1YKI) (Race et al. 2005) were used as templates. The multiple-sequence alignment between the target and the templates was carried out using the Multiple Alignment using Fast Fourier Transform server (Katoh et al. 2002).
The homology model was built using the Modeller 9.v.8 program (Eswar et al. 2006) and the resulting structure was submitted for evaluation to the Swiss-model workspace (Arnold et al. 2006).
Docking experiments of megazol and compound 8 in the active site of the modelled TbNTR were performed using AutoDock VINA software (Trott & Olson 2010). Gasteiger-Hückel charges and AutoDock (Morris et al. 1996) molecular mechanics force fields were used. One hundred solutions were generated for each compound using different pseudorandom seeds. In each run 200 binding modes were created, out of which only the top 20, ranked by the VINA scoring function, were accessible to the user. The search was conducted in a cubic volume of 25 x 25 x 25 Å3, centred on the coordinates of the crystallographic compound, with a value of 150 for exhaustiveness.
All calculations were carried out on an Intel® CoreTM i5 CPU machine and graphics were generated using the Jmol (jmol.org/) and PyMOL (pymol.org/) software packages.
Ethics - All experiments were carried out in accordance with the guidelines established by the Oswaldo Cruz Foundation Ethical Committee for the Use of Animals (LW 16-13).
RESULTS
Megazol (7) and its bioisostere 5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8) were synthesised from 2-cyano-5-nitroimidazole as previously described (Carvalho et al. 2007).
To evaluate whether the TbNTR could metabolise compounds 7 and 8 (compound 2 was also tested), recombinant protein was purified from E. coli cells and then used in NADH-based oxidation assays containing the nitroimidazole as a substrate (Hall et al. 2010). These results can be found in Table.
TABLE . Biological properties of the nitroimidazoles.
|
Tb NTR |
Mammalian cells
|
|||||
|---|---|---|---|---|---|---|
| Compound | Trypanosoma cruzi LD50 (µM) | Trypanosoma brucei IC50 (µM) | Vmax a | kcat/KM b | Genotoxicity | Cytotoxicity |
| 7 | 9.9 ± 0.8 | 0.14 ± 0.01 | 475.7 ± 21 | 4.0 x 104 | p < 0.01 | Absence |
| 8 | 256.8 ± 53.0 | > 40.0 | 768.8 ± 28 | 2.4 x 104 | Absence | Absence |
| 2 | 23.8 | 22.1 ± 0.9 | 625.4 ± 50 | 2.6 x 103 | ND | ND |
a : activity expressed as µmol NADH oxidised min-1mg-1; b : expressed as M-1s-1; compound 2: benznidazole; compound 7: megazol; compound 8: 5-(1-methyl-5-nitro-1 H -imidazol-2-yl)-4 H -1,2,4-triazol-3-amine; IC: inhibitory concentration; LD50: lethal dose; ND: not determined; Tb NTR: nitroreductase enzymes of T. brucei.
The biological activities of compounds 7 and 8 were then evaluated in parasitic and mammalian cells (Table).
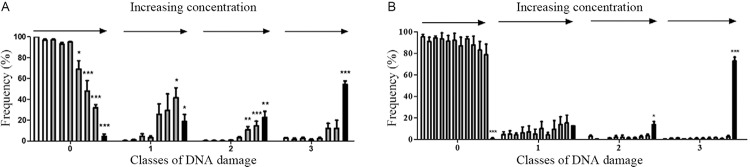
The cytotoxicity of compounds 7 and 8 and their ability to induce DNA damage in human blood cells are described in Fig. 3, Table and the degree of damage are in Fig. 4.
Fig. 3. : DNA damage induction in human whole blood as analysed using the Comet assay. A: megazol (7) (n = 3); B: triazole analog [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8)] (n = 2). The white bars correspond to the untreated control culture and the solvent-control [5% (v/v) dimethyl sulfoxide (DMSO)], the grey bars correspond to the tested compounds and the black bars correspond to the positive control (160 µM methyl methane-sulfonate). The bars represent the standard error of the means of the total arbitrary units (AU). For Student’s one tailed t test, the asterisks indicate significance at 1% (**) and 0.1% (***) levels. compound 7: megazol; compound 8: 5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine.
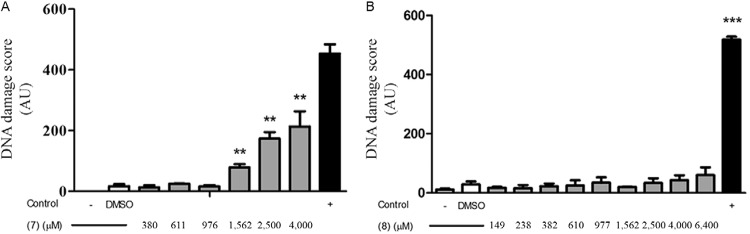
Fig. 4. : intercellular distribution of DNA damage in human whole blood, as analysed using the Comet assay. A: megazol (7) at concentrations of 380, 611, 976, 1,562, 2,500 and 4,000 µM (arrows) (n = 3); B: the triazole analog [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine (8)] at concentrations of 149, 238, 382, 610, 977, 1,562, 2,500, 4,000 and 6,400 µM (arrows) (n = 2). The white bars correspond to the untreated control culture and the solvent-control [5% (v/v) dimethyl sulfoxide], the grey bars correspond to the tested compounds and the black bars correspond to the positive control (160 µM methyl methane-sulfonate). The bars represent the standard error of the mean frequency (%) of the cells in the different classes of DNA damage. For the Dunnett’s test, the asterisks indicate significance at 5% (*), 1% (**) and 0.1% (***) levels.
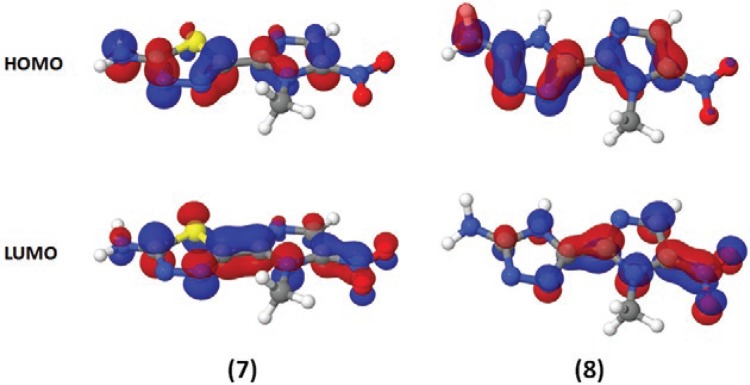
Theoretical calculations for the HOMO and the LUMO frontier orbitals revealed that the HOMO energies were -9.42 and -9.62 eV and that the LUMO energies were -1.64 and -1.72 eV for compounds 7 and 8, respectively (Fig. 5).
Fig. 5. : electronic distributions for megazol (7) and compound 8 [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine]. Red indicates an electron-rich highest occupied molecular orbital (HOMO) [or lowest unoccupied molecular orbital (LUMO)] region, while blue indicates an electron-poor region. Semi-empirical calculations were performed using the MOPAC2009 software package (OpenMOPAC.net) with the semiempirical PM3 method and full geometric optimisation. The ejection fraction routine was used for the minimum search.
DISCUSSION
Most nitroheterocyclic compounds act as prodrugs and must be activated before their cytotoxic effects can be evaluated. Initially, it was proposed that trypanocidal action occurs by inducing oxidative stress through the one-electron reduction of its nitro group and the subsequent formation of superoxide by a futile cycle. However, more recent studies have indicated that this process does not occur to an extent that would be toxic to the parasites and that a type I NTR is responsible for the trypanocidal activity of nifurtimox (1) and benznidazole (2). This enzyme has been shown to play a key role in activating many other antitrypanosomal nitroheterocyclic drugs (Wilkinson & Kelly 2009, Bot et al. 2010, Hall et al. 2010). NTR mediates a series of two-electron reduction reactions, resulting in the fragmentation of the heterocyclic ring and the production of toxic metabolites. Because the type I NTR, which is absent in most eukaryotes except trypanosomes, can catalyse the activation of nitroheterocyclic prodrugs, these compounds have been investigated for their application as antiparasitic agents (Wilkinson & Kelly 2009, Bot et al. 2010, Hall et al. 2010).
To evaluate whether the TbNTR could metabolise compounds 7 and 8 (compound 2 was also tested), recombinant protein was purified from E. coli cells and then used in NADH-based oxidation assays containing the nitroimidazole as a substrate (Hall et al. 2010). This test demonstrated that the parasite enzyme metabolised all three of the compounds evaluated. Based on the enzyme kinetic values, the activity (apparent Vmax) indicated that compound 8 was metabolised at a faster rate than compound 7, it bounds to the TbNTR with a greater affinity, resulting in these two compounds having equivalent kcat/KM values (Table). Interaction of the TbNTR with compound 2 indicated that although a reasonable Vmax value was generated, this 2-nitroimidazole displayed the lowest affinity for the parasite enzyme of the compounds screened, resulting in a kcat/KM value that was one order of magnitude lower than that of the 5-nitroimidazole-based agents.
When tested in T. brucei, compound 8 did not affect the growth of BSF cells (40 µM was the upper limit used in these experiments), whereas compound 7 significantly inhibited BSF proliferation, with an IC50 value of 0.14 µM. A similar trend was observed when the activities of compounds 7 and 8 in T. cruzi were investigated: both compounds killed the parasites, with the LD50 of compound 7 being 25-fold lower than that of compound 8. These disparities in the trypanocidal activity of the two compounds may indicate differences in their uptake by the parasites or may suggest that once in the pathogen the two compounds have distinct mechanisms of action, since those compound 8 bonds to the TbNTR with higher affinity.
The alkaline Comet assay (single cell gel electrophoresis assay) combines the simplicity of biochemical techniques that detect DNA single and double-strand breaks and alkali-labile sites with a single cell approach, which is typical of cytogenetic assays (Singh et al. 1988). The advantages of this assay include its sensitivity in detecting DNA damage, the ability to analyse data at the level of the individual cell and the ability to use extremely small cell samples (Anderson et al. 1998, Speit & Hartmann 2006, Lynch et al. 2011). Using the Comet and FDA/EtBr assays, we compared the cytotoxicity of compounds 7 and 8 and their ability to induce DNA damage in human blood cells (Fig. 3). When high concentrations (> 1,500 µM) of compound 7 were used, significant (p < 0.01) DNA damage was detected in whole blood cells compared to treatment with 5% (v/v) DMSO alone; cells that had been incubated with MMS (0.16 µM) also displayed DNA damage. The degree of damage was quantified by comparing the amount of DNA in the comet head with that present within the tail (see the Materials and Methods section) (Fig. 4). In this system, Class 0 is equivalent to no DNA damage (with the entire nuclear DNA contained within the comet head and no visible tail), while Class 3 corresponds to maximum damage (with little DNA in the comet head and the majority contained in the tail). Classes 1 and 2 represent intermediate levels of damage, such that Class 1 indicates slight damage and Class 2 indicates moderate damage. In cells treated with 2,500 or 4,000 µM concentrations of compound 7, significant (p < 0.05) Class 1 (42% for 4,000 µM) and highly significant (p < 0.01) Class 2 (11% and 14% for 2,500 and 4,000 µM, respectively) of damage were observed, with some of the cells displaying Class 3 (12%) DNA damage. In contrast, significant Class 3 damage was detected following treatment with MMS (1,600 µM), with 54% of the cells displaying this level of damage. The genotoxicity observed following treatment with high concentrations of compound 7 was not associated with cytotoxicity, as whole blood cell viability studies using 1,562 µM, 2,500 µM and 400 µM concentrations resulted in 5%, 7% and 9% of cells being non-viable, respectively; under the same conditions, treatment with 5% (v/v) DMSO alone resulted in an 11% decrease in cell viability.
When the above experiments were extended to compound 8, no significant DNA damage in human blood cells (p > 0.1) was detected in relation to the solvent-control (Fig. 3). In addition, no significant differences in the percentage of cells in the four classes of DNA damage (Fig. 4) were observed. When cell viability studies were conducted, no cytotoxicity in human blood cells was observed. Together, these results clearly indicated a genotoxic negative result for compound 8 and a genotoxic positive, concentration-dependent result that was not associated with cytotoxicity to human blood cells for compound 7 (Table).
The HOMO energies, -9.42 and -9.62 eV, and LUMO energy, -1.64 and -1.72 eV, were very similar for both compounds 7 and 8 (Fig. 5). From the electronic distribution perspective, it is difficult to explain the discrepancies in the behaviours of compounds 7 and 8.
From a structural viewpoint, a more detailed analysis of the molecular interactions between these compounds and the receptor binding site was performed based on a homology model for the TbNTR. Sequence alignment between the targets and the templates resulted in identity values of 26% and 24% for 2B67 and 1YKI, respectively. In contrast, sequence alignment between the templates revealed that despite these proteins not having very similar sequences (24% identity and 40% similarity), their structural alignment resulted in a root mean square deviation of 3.73 Å, indicating that a considerable degree of conservation exists in their secondary structures and overall shapes. According to the PROCHECK program (Laskowski et al. 1993), both chains of the TbNTR homodimer homology model displayed 90% of their residues in the allowed regions of the Ramachandran plot. The QMEAN6 (Benkert et al. 2009) scores for chains A and B of the homodimer were 0.52 and 0.57, respectively, indicating average quality values for both structures. In addition, the DFIRE (Zhou & Zhou 2002) pseudoenergy values were -250.17 (chain A) and -256.57 (chain B), respectively, revealing energy optimised structures. Finally, the coordinates of the FMN cofactor were transferred from the 1YKI template into the TbNTR homology model, based on the sequence conservation of the neighbouring residues. The whole system was energy minimised with 10,000 steps by applying steepest descent algorithm.
The interaction energies of docked ligands within the active site of the TbNTR suggested a stronger interaction with compound 8 than that of compound 7. According to the Vina score function, the minimum binding energy values were -5.8 and -7.2 kcal/mol for compounds 7 and 8, respectively. The proposed plausible binding mode of compound 7 inside the TbNTR is shown in Fig. 6. It is noteworthy that the lower energy-docked solutions for both compounds projected their nitroimidazole rings into the bulk. This observed orientation was similar to what was observed for the bactericidal nitrofurazone in the binding site of the E. coli NTR (which was used as a template) (Race et al. 2005). In addition, Van der Waals contacts were observed between compound 8 and the Leu197 and Tyr200 residues in chain B. The nitroimidazole ring was positioned over the polar ring of FMN while the aminotriazole ring was located over the central ring of the cofactor. The secondary amine hydrogen atom bound to the Phe125 carbonyl oxygen and Thr227 hydroxyl in chain A. It is noteworthy that this H-bond is equivalent to the one in the 1YKI PDB structure, where the nitrofurazone antibiotic hydrogen bonds to the Thr41 of the E. coli NTR. In addition, the nitrogen atoms (1 and 2) of the triazole ring formed hydrogen bonds with one OH group of FMN (Fig. 6A).
Fig. 6. : putative binding modes for compound 8 [5-(1-methyl-5-nitro-1H-imidazol-2-yl)-4H-1,2,4-triazol-3-amine] (A) and megazol (B) within the modelled Trypanosoma brucei type I nitroreductase binding site. The flavin mononucleoside cofactor is also depicted in cyan. Residues Thr227 (chain A - purple) and Phe114 (chain B - yellow) are shown as lines. The H-bond distances between the ligands and proteins are shown in yellow and the units are given in angstroms (Å).
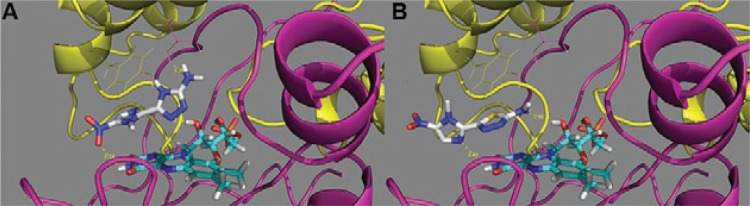
In contrast, the thiadiazole ring was located over the polar ring of FMN and the NH2 was attached (hydrogen bonded) to one of the hydroxyl groups of the cofactor and the oxygen atoms belonging to the side chain of Glu266. Similar to compound 8, compound 7 was encompassed by both chains of the TbNTR homodimer and its nitro group was more exposed to the solution than that of compound 8, projecting itself into the Tyr199 and Tyr200 residues. Finally, we observed that the replacement of the triazole ring sulphur atom by a NH group allowed compound 8 to penetrate further into the binding site, enhancing the possibility of making more specific interactions with the receptor (Fig. 6B).
In conclusion, we compared the trypanocidal activity of compound 7 with its 4H-1,2,4-triazole 8 bioisostere and demonstrated that both compounds were effective substrates for the TbNTR. A homology structural model of the TbNTR was built and used for docking assays of compounds 7 and 8. The results of the molecular docking assays confirmed an energetic preference for the binding of bioisostere 8 over compound 7 within the active site of the nitroreductase. Additionally, in agreement with the results obtained by the enzymatic assay, the results also revealed that the anti-trypanosomal activity is not likely related to the isolated action on the TbNTR enzyme as compound 8 had a superior affinity for this target than compound 7.
The disparities in the trypanocidal activity of the two compounds may indicate differences in their uptake by the parasite or may suggest that once in the pathogen, the two compounds can have distinct mechanisms of action.
We have also observed that the replacement of the sulphur atom of the triazole ring with a NH group has allowed that compound 8 can penetrate further into the binding site, enhancing the possibility of making more specific interactions with the receptor.
Although the genotoxic properties of compound 7 have been well documented (Ferreira & Ferreira 1986, Nesslany et al. 2004) and assumed to be due to the nitro group, we conclude that the absence of genotoxicity observed for the nitro derivative compound 8 demonstrates that this group is not solely responsible for this activity. The present study will help to guide future research focusing on obtaining new nitroimidazoles with high trypanocidal activity, such as compound 7, that are not genotoxic, like compound 8. Our found is not a “negative result”, but can really contribute with the better understanding of the importance of nitro group in the medicinal chemistry of nitroimidazoles.
ACKNOWLEDGEMENTS
To Dr Nelilma Romeiro (UFRJ, Macaé, Brazil), for her critical contributions to the molecular modelling studies.
Footnotes
Financial support: FAPERJ, CNPq, FIOCRUZ, Queen Mary University of London
REFERENCES
- Anderson D, Yu TW, McGregor DB. Comet assay responses as indicators of carcinogen exposure. Mutagenesis. 1998;13:539–555. doi: 10.1093/mutage/13.6.539. [DOI] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. The trypanosomiases. Lancet. 2003;362:1469–1480. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- Benkert P, Schwede T, Tosatto SCE. QMEANclust: estimation of protein model quality by combining a composite scoring function with structural density information. BMC Struct Biol. 2009;9:35–52. doi: 10.1186/1472-6807-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein FC, Koetzle TF, Williams GJ, Meyer EE, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- Boechat N, Carvalho AS, Fernandez-Ferreira E, Soares ROA, Souza AS, Gibaldi D, Bozza M, Pinto AC. Novel nitroimidazoles with trypanocidal and cell growth inhibition activities. Cytobios. 2001;105:83–90. [PubMed] [Google Scholar]
- Boechat N, Pinheiro LCS, Silva TS, Aguiar ACC, Carvalho AS, Bastos MM, Costa CCP, Pinheiro S, Pinto AC, Mendonça JC, Dutra KDDB, Valverde A, Santos OA, Filho, Ceravolo SP, Krettli AU. New trifluoromethyl triazolopyrimidines as anti-Plasmodium falciparum agents. Molecules. 2012;17:8285–8302. doi: 10.3390/molecules17078285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi ML, Lizzi F, Perozzo R, Brun R, Cavalli A. Synthesis of a small library of 2-phenoxy-1,4-naphthoquinone and 2-phenoxy-1,4-anthraquinone derivatives bearing anti-trypanosomal and anti-leishmanial activity. Bioorg Med Chem Lett. 2008;18:2272–2276. doi: 10.1016/j.bmcl.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Bot C, Hall BS, Bashir N, Taylor MC, Helsby NA, Wilkinson SR. Trypanocidal activity of aziridinyl nitrobenzamide prodrugs. Antimicrob Agents Chemother. 2010;54:4246–4252. doi: 10.1128/AAC.00800-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouteille B, Marie-Daragon A, Chauvière G, de Albuquerque C, Enanga B, Dardé ML, Vallat JM, Périé J, Dumas M. Effect of megazol on Trypanosoma brucei brucei acute and subacute infections in Swiss mice. Acta Trop. 1995;60:73–80. doi: 10.1016/0001-706x(95)00109-r. [DOI] [PubMed] [Google Scholar]
- Carvalho AS, Gibaldi D, Pinto AC, Bozza M, Boechat N. Synthesis and trypanocidal evaluation of news 5-[N-(3-(5- substituted)-1,3,4-thiadiazolyl)]amino-1-methyl-4- nitroimida zoles. Lett Drug Des Discov. 2006;3:98–101. [Google Scholar]
- Carvalho AS, Menna-Barreto RFS, Romeiro NC, de Castro SL, Boechat N. Design, synthesis and activity against Trypanosoma cruzi of azaheterocyclic analogs of megazol. Med Chem. 2007;3:460–465. doi: 10.2174/157340607781745519. [DOI] [PubMed] [Google Scholar]
- Carvalho SA, Lopes FAS, Salomão K, Romeiro NC, Wardell SMVS, de Castro SL. Studies toward the structural optimization of new brazilizone-related trypanocidal 1,3,4-thiadiazole-2-arylhydrazone derivatives. Bioorg Med Chem. 2008;16:413–421. doi: 10.1016/j.bmc.2007.09.027. [DOI] [PubMed] [Google Scholar]
- Carvalho SA, Silva EF, Santa-Rita RM, de Castro SL, Fraga CAM. Synthesis and antitrypanosomal profile of new functionalyzed 1,3,4-thiadiazole-2-arylhydrazone derivatives, designed as non-mutagenic megazol analogues. Bioorg Med Chem Lett. 2004;14:5967–5970. doi: 10.1016/j.bmcl.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Castro JA, de Mecca MM, Bartel LC. Toxic side effects of drugs used to treat Chagas disease (American trypanosomiasis) Hum Exp Toxicol. 2006;25:471–479. doi: 10.1191/0960327106het653oa. [DOI] [PubMed] [Google Scholar]
- da Silva FC, Ferreira SB, da Rocha DR, Ferreira VF. Chagas disease: challenges in developing new trypanocidal lead compounds. Rev Virtual Quim. 2012;4:46–72. [Google Scholar]
- de Castro SL, Meirelles MNL, Oliveira MM. Trypanosoma cruzi: adrenergic modulation of cAMP role in proliferation and differentiation of amastigotes in vitro. Exp Parasitol. 1987;64:368–375. doi: 10.1016/0014-4894(87)90049-x. [DOI] [PubMed] [Google Scholar]
- El-Sayed NM, Myler PJ, Blandin G, Berriman M, Crabtree J, Aggarwal G, Caler E, Renauld H, Worthey EA, Hertz-Fowler C, Ghedin E, Peacock C, Bartholomeu DC, Haas BJ, Tran AN, Wortman JR, Alsmark UC, Angiuoli S, Anupama A, Badger J, Bringaud F, Cadag E, Carlton JM, Cerqueira GC, Creasy T, Delcher AL, Djikeng A, Embley TM, Hauser C, Ivens AC, Kummerfeld SK, Pereira-Leal JB, Nilsson D, Peterson J, Salzberg SL, Shallom J, Silva JC, Sundaram J, Westenberger S, White O, Melville SE, Donelson JE, Andersson B, Stuart KD, Hall N. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- Enanga B, Ariyanayagam MR, Stewart ML, Barrett MP. Activity of megazol, a trypanocidal nitroimidazole, is associated with DNA damage. Antimicrob Agents Chemother. 2003;47:3368–3370. doi: 10.1128/AAC.47.10.3368-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enanga B, Boudra H, Chauvière G, Labat C, Bouteille B, Dumas M, Houin G. Pharmacokinetics, metabolism and excretion of megazol, a new potent trypanocidal drug in animals. Arzneimittelforschung. 1999;49:441–447. doi: 10.1055/s-0031-1300440. [DOI] [PubMed] [Google Scholar]
- Enanga B, Ndong JM, Boudra H, Debrauwer L, Dubreuil G, Bouteille B, Chauvière G, Labat C, Dumas M, Périé J, Houin G. Pharmacokinetics, metabolism and excretion of megazol in a Trypanosoma brucei gambiense primate model of human African trypanosomiasis. Preliminary study. Arzneimittelforschung. 2000;50:158–162. doi: 10.1055/s-0031-1300182. [DOI] [PubMed] [Google Scholar]
- Eswar N, Marti-Renom MA, Webb B, Madhusudhan MS, Eramian D, Shen M, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinformatics. 2006;(15):5.6.1–5.6.30. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira RCC, Ferreira LCS. CL 64,855, a potent anti-Trypanosoma cruzi drug, is also mutagenic in the Salmonella/microsome assay. Mem Inst Oswaldo Cruz. 1986;81:49–52. doi: 10.1590/s0074-02761986000100006. [DOI] [PubMed] [Google Scholar]
- Filardi LS, Brener Z. Susceptibility and natural resistance of Trypanosoma cruzi strains to drugs used clinically in Chagas disease. Trans R Soc Trop Med Hyg. 1987;81:755–759. doi: 10.1016/0035-9203(87)90020-4. [DOI] [PubMed] [Google Scholar]
- Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH. The NCBI BioSystems database. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BS, Wu X, Hu L, Wilkinson SR. Exploiting the drug-activating properties of a novel trypanosomal nitroreductase. Antimicrob Agents Chemother. 2010;54:1193–1199. doi: 10.1128/AAC.01213-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR. Avogadro: an advanced semantic chemical editor, visualization and analysis platform. J Cheminform. 2012;4:1–17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Speit G. The contribution of cytotoxicity to DNA - effects in the single cell gel test (comet assay) Toxicol Lett. 1997;90:183–188. doi: 10.1016/s0378-4274(96)03847-7. [DOI] [PubMed] [Google Scholar]
- Hirumi H, Hirumi K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol. 1989;75:985–989. [PubMed] [Google Scholar]
- Jacobs RT, Nare B, Phillips MA. State of the art in African trypanosome drug discovery. Curr Top Med Chem. 2011;11:1255–1274. doi: 10.2174/156802611795429167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acid Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Volkart L, Abdullah J, Collart F, Joachimiak A. Crystal structure of the nitroreductase family protein from Streptococcus pneumoniae TIGR4. 2011 rcsb.org/pdb/explore.do?structureId=2b67
- Laskowski RA, Macarthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Lynch AM, Sasaki JC, Elespuru R, Jacobson-Kram D, Thybaud V, Boeck M, Aardema MJ, Aubrecht J, Benz RD, Dertinger SD, Douglas GR, White PA, Escobar PA, Fornace A, Jr, Honma M, Naven RT, Rusling JF, Schiestl RH, Walmsley RM, Yamamura E, van Benthem J, Kim JH. New and emerging technologies for genetic toxicity testing. Environ Mol Mutagen. 2011;52:205–223. doi: 10.1002/em.20614. [DOI] [PubMed] [Google Scholar]
- Maya JD, Bollo S, Nuñez-Vergara LJ, Squella JA, Repetto Y, Morello A, Périé J, Chauvière G. Trypanosoma cruzi: effect and mode of action of nitroimidazole and nitrofuran derivatives. Biochem Pharmacol. 2003;6:999–1006. doi: 10.1016/s0006-2952(02)01663-5. [DOI] [PubMed] [Google Scholar]
- Mital A. Synthetic nitroimidazoles: biological activities and mutagenicity relationships. Sci Pharm. 2009;77:497–520. [Google Scholar]
- Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J Comput Aided Mol Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- Nesslany F, Brugier S, Mouries MA, Le Curieux F, Marzin D. In vitro and in vivo chromosomal aberrations induced by megazol. Mutat Res. 2004;560:147–158. doi: 10.1016/j.mrgentox.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- Papadopoulou MV, Bloomer WD, Rosenzweig HS, Kaiser M, Chatelain E, Ioset J-R. Novel 3-nitro-1H-1,2,4-triazole-based piperazines and 2-amino-1,3-benzothiazoles as antichagasicagents. Bioorg Med Chem. 2013;21:6600–6607. doi: 10.1016/j.bmc.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulou MV, Trunz BB, Bloomer WD, McKenzie C, Wilkinson SR, Prasittichai C, Brun R, Kaiser M, Torreele E. Novel 3-nitro-1H-1,2,4-triazole-based aliphatic and aromatic amines as anti-chagasicagents. J Med Chem. 2011;54:8214–8223. doi: 10.1021/jm201215n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paula FR, Serrano SHP, Tavares LC. Aspectos mecanísticos da bioatividade e toxicidade de nitrocompostos. Quim Nova. 2009;32:1013–1020. [Google Scholar]
- Race PR, Lovering AL, Green RM, Ossor A, White SA, Searle PF, Wrighton CJ, Hyde EI. Structural and mechanistic studies of Escherichia coli nitroreductase with the antibiotic nitrofurazone. Reversed binding orientations in different redox states of the enzyme. J Biol Chem. 2005;280:13256–13264. doi: 10.1074/jbc.M409652200. [DOI] [PubMed] [Google Scholar]
- Rassi A, Jr, Rassi A, de Rezende JM. American trypanosomiasis (Chagas disease) Infect Dis Clin North Am. 2012;26:275–291. doi: 10.1016/j.idc.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Salomão K, de Souza EM, Carvalho AS, Silva EF, Fraga CAM, Barbosa HS, Castro SL. In vitro and in vivo activity of 1,3,4-thiadiazole-2-arylhydrazone derivatives of megazol on Trypanosoma cruzi. Antimicrob Agent Chemother. 2010;54:2023–2031. doi: 10.1128/AAC.01241-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res. 1988;175:184–191. doi: 10.1016/0014-4827(88)90265-0. [DOI] [PubMed] [Google Scholar]
- Soeiro MNC, de Castro SL. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo studies. Open Med Chem J. 2011;5:21–30. doi: 10.2174/1874104501105010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speit G, Hartmann A. The comet assay: a sensitive genotoxicity test for the detection of DNA damage and repair. Methods Mol Biol. 2006;314:275–286. doi: 10.1385/1-59259-973-7:275. [DOI] [PubMed] [Google Scholar]
- Tice RR, Aqurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryn JC, Sasaki YF. Single cell gel comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(sici)1098-2280(2000)35:3<206::aid-em8>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. Software news and update autodock vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF, Watts BP, Jr, Zhong L, Docampo R. Inhibition of Trypanosoma cruzi and Trypanosoma brucei NADH fumarate reductase by benznidazole and anthelmintic imidazole derivatives. Mol Biochem Parasitol. 1996;82:125–129. doi: 10.1016/0166-6851(96)02722-3. [DOI] [PubMed] [Google Scholar]
- Viodé C, Bettache N, Cenas N, Krauth-Siegel RL, Chauvière G, Bakalara N, Périé J. Enzymatic reduction studies of nitroheterocycles. Biochem Pharmacol. 1999;57:549–557. doi: 10.1016/s0006-2952(98)00324-4. [DOI] [PubMed] [Google Scholar]
- WHO - World Health Organization Working to overcome the global impact of neglected tropical diseases: first WHO report on neglected tropical diseases. WHO; Geneva: 2010. pp. 1–172. [Google Scholar]
- Wilkinson SR, Bot C, Kelly JM, Hall BS. Trypanocidal activity of nitroaromatic prodrugs: current treatments and future perspectives. Curr Topic Med Chem. 2011;11:2072–2084. doi: 10.2174/156802611796575894. [DOI] [PubMed] [Google Scholar]
- Wilkinson SR, Kelly JM. Trypanocidal drugs: mechanisms, resistance and new targets. Expert Rev Mol Med. 2009;11: doi: 10.1017/S1462399409001252. [DOI] [PubMed] [Google Scholar]
- Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002;11:2714–2726. doi: 10.1110/ps.0217002. [DOI] [PMC free article] [PubMed] [Google Scholar]