

Abstract
Enterococcus mundtii QU 25, a non-dairy bacterial strain of ovine faecal origin, can ferment both cellobiose and xylose to produce l-lactic acid. The use of this strain is highly desirable for economical l-lactate production from renewable biomass substrates. Genome sequence determination is necessary for the genetic improvement of this strain. We report the complete genome sequence of strain QU 25, primarily determined using Pacific Biosciences sequencing technology. The E. mundtii QU 25 genome comprises a 3 022 186-bp single circular chromosome (GC content, 38.6%) and five circular plasmids: pQY182, pQY082, pQY039, pQY024, and pQY003. In all, 2900 protein-coding sequences, 63 tRNA genes, and 6 rRNA operons were predicted in the QU 25 chromosome. Plasmid pQY024 harbours genes for mundticin production. We found that strain QU 25 produces a bacteriocin, suggesting that mundticin-encoded genes on plasmid pQY024 were functional. For lactic acid fermentation, two gene clusters were identified—one involved in the initial metabolism of xylose and uptake of pentose and the second containing genes for the pentose phosphate pathway and uptake of related sugars. This is the first complete genome sequence of an E. mundtii strain. The data provide insights into lactate production in this bacterium and its evolution among enterococci.
Keywords: Enterococcus mundtii, xylose, l-lactic acid, mundticin
1. Introduction
Optically pure lactic acid is necessary for the production of the bioplastic polylactic acid. The use of cellulosic biomass instead of food crops should lower the cost for commercial production of this green plastic. Enterococcus mundtii QU 25 is a non-dairy bacterial strain that was originally isolated from ovine faeces.1 Unlike most lactic acid bacteria, strain QU 25 can ferment both cellobiose and xylose to produce l-lactic acid.1,2 This strain metabolizes a mixture of glucose and cellobiose simultaneously without apparent carbon catabolite repression (CCR),1 and it produces optically pure l-lactate (≥99.9%) with a yield of 1.41 mol/mol xylose consumed, without by-products such as acetic acid or ethanol.1,2 Moreover, high productivity of l-lactic acid in an open repeated batch fermentation system under non-sterile conditions was demonstrated.3 Therefore, the use of strain QU 25 is highly desirable for the economical production of l-lactate from renewable biomass substrates. Furthermore, determination of the genome sequence of this bacterium is necessary to generate optimized, recombinant strains for commercial use.
Draft genome sequences of microorganisms can be obtained rapidly and cost-effectively by using second-generation sequencing technologies. Owing to its relatively short read length of second-generation platforms (100–700 bp), obtaining a complete genome sequence requires additional costs and time-consuming finishing steps such as scaffolding and gap closing. By comparison, third-generation single molecule real time (SMRT) sequencing technology developed by Pacific Biosciences (PacBio) can produce considerably long sequences (∼23 kb). Complete genome sequencing using only PacBio sequence data was recently reported.4
To date, there have been only two draft genome sequences available for E. mundtii (strains CRL16565 and ATCC 882). Here, we report the complete genome sequence of strain QU 25, whose chromosome sequence was determined using only PacBio sequence data. This is the first complete genome sequence of an E. mundtii strain. The data reveal useful insights on lactic acid fermentation in this bacterium, as well as phylogenetic relationships with other Enterococcus species.
2. Materials and methods
2.1. Culture conditions and preparation of genomic DNA for sequencing
Enterococcus mundtii QU 25 cells were grown to mid-log phase in GM17.6 Cells from 100-ml culture were harvested, suspended in 25 ml of a solution containing 2 g of polyethylene glycol 2000, 62.5 mg of egg white lysozyme, and 5 mM Tris–hydrochloride (pH 8.0), and incubated for 60 min at 37°C. After centrifugation, the cells were resuspended in 12.5 ml of TES buffer (50 mM Tris–hydrochloride [pH 7.6], 20 mM sodium ethylenediaminetetraacetic acid, and 25% sucrose), treated with 8 µg/ml of ribonuclease A for 60 min at 37°C, and then lysed with heat at 60°C for 2 h in the presence of 40 µg/ml of proteinase K and 1.7% sodium dodecyl sulphate. Total DNA was extracted gently from the lysate with PCI mixture (phenol, chloroform, and isoamyl alcohol in a ratio of 25 : 24 : 1, respectively) three times and precipitated with ethanol.
2.2. Genome sequencing
For PacBio RS sequencing, two types of SMRTbell DNA template libraries were created with 1- and 10-kb sheared genomic DNA, and prepared using the standard PacBio RS sample preparation methods with C2 chemistry specific to each insert size. The 10-kb library was sequenced on eight SMRT cells with a 1 × 120-min collection protocol, generating 189 953 post-filtered continuous long reads (CLRs; mean length, 3702 bp; maximum length 20 405 bp; ∼234× coverage). The 1-kb library was sequenced on eight SMRT cells with a 2 × 55-min collection protocol, generating 258 068 post-filtered circular consensus sequences (CCS; mean length, 660 bp; ∼57× coverage).
For the second-generation Roche/454 sequencing, a 1.6-kb fragment library was sequenced using the GS-FLX+ system. A total of 302 056 reads (mean length, 455 bp; ∼34× coverage) was generated and assembled using Newbler (ver. 2.6). To estimate the plasmid copy number, a 500-bp paired-end library was sequenced using the Illumina Genome Analyzer IIx, generating 76-bp paired-end reads (48 724 736 reads, ∼1234× coverage). The copy number of plasmid per chromosome was estimated based on the coverage ratio between the corresponding contig and the chromosome contig using CLC Genomics workbench 6 (CLC bio, Aarhus, Denmark).
2.3. Error correction, assembly, and optical mapping
The error correction of CLRs was performed using the command pacBioToCA.7 CCS (57× length coverage) reads were used for correction. After error correction, we selected reads named PacBio-corrected Reads (PBcRs) for assembly. Assembly was performed using Celera Assembler (ver. 7.0).8 To confirm the correctness of the assembly, DNA from QU 25 cells was digested using NcoI and a whole-genome optical map (OpGen, Inc., Gaithersburg, MD, USA) was generated.9
2.4. Genome analysis
At first, the genome sequence was automatically annotated using the MiGAP, Microbial Genome Annotation Pipeline (www.migap.org) with the g-MiGAP level. In the pipeline, open reading frames (ORFs) were identified using MetaGeneAnnotator,10 and genes for tRNAs and rRNAs were identified by tRNAscan-SE and RNAmmer, respectively.11 Predicted ORFs were annotated using BLASTP searches against other Enterococcus genomes, and National Center of Biological Information (NCBI) nr12 with an E-value of 1 × 10−10. Additional annotation was performed using InterProScan13 and KEGG pathway analyses.14 Regions containing prophage were predicted by a phage search tool, PHAST (http://phast.wishartlab.com/)15 and further manual inspection. The insertion sequence (IS) was detected by ISsaga.16 CRISPR loci were detected by CRISPRfinder.17 For comparative genomes of other Enterococcus spp., a draft genome sequence of E. mundtii ATCC 882 was obtained from the Broad Institute website (https://olive.broadinstitute.org/genomes/ente_mund_atcc882.1). In addition, complete genome sequences of five Enterococcus spp. with (E. casseliflavus EC2018: NC_021023; E. faecalis V58319: NC_004668, NC_004669, NC_004670, and NC_004671; E. faecium Aus000420: NC_017022, NC_017023, NC_017024, and NC_017032; E. faecium DO21: NC_017960, NC_017961, NC_017962, and NC_017963; E. hirae ATCC 979022: NC_015845 and NC_018081) were obtained from the NCBI website for genome comparisons. GenomeMatcher23 and In Silico Molecular Cloning Genomics Edition (IMC-GE) software (In Silico Biology, Japan) were also used for intra- and inter-QU 25 genome comparisons.
2.5. Vancomycin resistance assay
Enterococcus mundtii QU 25 was propagated in MRS medium (Oxoid, Basingstoke, UK) at 30°C for 12 h as a pre-culture. One hundred microliters of pre-culture were inoculated in 10 ml of MRS medium containing various concentrations of vancomycin (2–100 µg/ml, Sigma, St. Louis, MO, USA). After incubation at 30°C for 24 h, the minimum concentration of vancomycin that resulted in the absence of growth was considered to be the minimum inhibitory concentration (MIC).
2.6. Bacteriocin activity assay
Lactobacillus sakei JCM 1157T and E. faecalis JCM 5803T were employed as indicator strains for bacteriocin activity. Taken together with E. mundtii QU 25, E. mundtii QU 2 (mundticin producer)24 and JCM 8731T (non-bacteriocin producer) were tested as positive and negative controls, respectively. The three E. mundtii strains were also used as indicator strains for cross- and self-immunity. All strains were propagated in MRS medium at 30°C for 12–18 h before use. Strains QU 25, QU 2, and JCM 8731T were cultured in MRS medium at 30°C for 12 h for bacteriocin production. Bacteriocin activity assay was performed by the spot-on-lawn method, as described previously.25 Briefly, 10 µl of each cell-free culture supernatant were spotted onto a double-layered agar plate containing 5 ml of Lactobacilli Agar AOAC (BD, Sparks, MD, USA) inoculated with an overnight culture of an indicator strain as an upper layer, and 10 ml of MRS medium supplemented with 1.2% agar as a bottom layer. After overnight incubation at appropriate temperatures for indicator strains, bacterial lawns were checked for inhibition zones.
2.7. Catalase and haemolysin activity assays
Strain QU 25 was tested for catalase activity by two methods. First, cells cultured in MRS liquid medium at 30°C for 12 h were collected by centrifugation and examined for catalase activity by the addition of 3% hydrogen peroxide solution. Secondly, colonies formed on the sheep blood-containing agar plates (Eiken Chemical, Tokyo, Japan) after incubation at 30°C for 24 h were examined by the addition of 3% hydrogen peroxide solution. Generation of oxygen bubbles was considered to be indicative of catalase activity. Colonies formed on blood agar plates were also examined for haemolysin activity. Discolouration or clearing of blood agar in the vicinity of the colonies was regarded as haemolysis.
2.8. Nucleotide sequence accession numbers
The complete genome sequence for E. mundtii QU 25 was deposited in GenBank/DDBJ/EMBL under accession numbers AP013036–AP013041.
3. Results and discussion
3.1. Genome summary
To determine the full genome sequence of E. mundtii QU 25, we combined two sequencing platforms: PacBio and Roche/454. As a result, the complete chromosome sequence of E. mundtii QU 25 was determined using only PacBio RS sequencing. This technology generates two types of sequences: CLRs and CCS reads.26 The read length of CLRs can reach up to 23 kb; however, the average base accuracy is only 82.1–84.4%.7 CCS reads are consensus sequences obtained from multiple passes on a single sequence with relatively short lengths (∼2 kb) and a low error rate.27 The PBcR algorithm can improve read accuracy of CLRs from 80% to over 99.9% by using CCS reads.7
Error correction of CLRs was done using high-accuracy sequences of CCS with the PBcR algorithm.7 After error correction, the resulting 6806 PBcRs of at least 7-kb length (mean length, 8517 bp; maximum length, 16 733 bp; ∼20× coverage) were assembled, generating five contigs. Comparison between the NcoI digest of the whole-genome optical map and in silico-generated physical maps of contigs showed that the largest contig (3 Mb) was mapped onto the chromosome, confirming the correctness of the assembly (Fig. 1A). Thus, we concluded that the remaining four contigs were plasmids. The presence of the 2.5-kb plasmid, which had been detected as an extrachromosomal element by agarose gel electrophoresis (data not shown), was not included in the five contigs generated by PacBio, due to its assembly threshold of 7-kb read length. Therefore, we adapted one contig produced using Roche/454.
Figure 1.

Analysis of the Enterococcus mundtii QU 25 complete genome. (A) Linearized NcoI-digested whole-genome optical map of QU 25 compared with the in silico-derived NcoI-digest map, demonstrating correct chromosome assembly. (B) Genome map of the QU 25 strain. In the outermost circle, three prophages of phiEmqu1, phiEmqu2, and phiEmqu3, replication origin (dnaA), and terminus (dif) are shown. In the second circle, the ORFs transcribed in a clockwise manner are shown as bars. The third circle shows ORFS transcribed in a counter-clockwise manner. The fourth to ninth circles depict the results of ortholog analyses (BLASTP E-value ≤1 × 10−10) with E. mundtii ATCC 882, E. faecium DO, E. faecium Aus0004, E. hirae ATCC 9790, E. casseliflavus EC20, and E. faecalis V583, respectively. The extent of homology relative to QU 25 is depicted using a heat map of arbitrarily chosen bins. The colour scheme and percentage identity for orthologs are as follows: red, orthologs with >90% identity; green, 70–90% identity; blue, 50–70% identity; and black, <50% identity. The 10th circle shows the positions of rRNA operons. The last two (innermost) circles represent G + C content (purple >39.5% average; green <39.5% average; range from 32 to 47%) and G + C skew, both calculated for a 10-kb window with 1-kb stepping.
The genome of QU 25 comprises a single circular chromosome of 3 022 186 bp (GC content, 38.6%) and five circular plasmids: pQY182, pQY082, pQY039, pQY024, and pQY003, with lengths of 181 920, 82 213, 38 528, 23 629, and 2584 bp, and GC contents of 36.2, 35.8, 33.8, 35.3, and 38.9%, respectively (Table 1 and Fig. 1B). Coordinates on the genome were designated as bp starting from the first nucleotide of the start codon of dnaA. We confirmed the likely location of the replication terminus by identifying a single dif sequence (ACTTTGTATAATATATATTATGTAAACT, position 1 449 088 to 1 449 115) that aligns with the shift in GC skew (Fig. 1B). A total of 2900 protein-coding sequences (CDS), 63 tRNA genes, and 6 rRNA operons were predicted in the QU 25 chromosome.
Table 1.
General features of the Enterococcus mundtii QU 25 genome
| Features | Chromosome | pQY182 | pQY082 | pQY039 | pQY024 | pQY003 |
|---|---|---|---|---|---|---|
| Size (bp) | 3 022 186 | 181 920 | 82 213 | 38 528 | 23 629 | 2584 |
| G + C content (%) | 38.6 | 36.2 | 35.8 | 33.8 | 35.3 | 38.9 |
| No. of rRNA operons | 6 | 0 | 0 | 0 | 0 | 0 |
| No. of tRNA genes | 63 | 0 | 0 | 0 | 0 | 0 |
| CDS (protein coding) | 2900 | 178 | 82 | 36 | 21 | 4 |
| Average of CDS length (bp) | 884 | 813 | 818 | 850 | 903 | 436 |
| Estimated copy number of replicon | 1 | 1 | 1 | 1 | 1 | 5 |
3.2. Genomic comparisons with other Enterococcus species
From the phylogeny based on 16S rRNA sequences, E. mundtii was closely related to E. hirae and E. faecium, while E. casseliflavus and E. faecalis were more distantly related to E. mundtii.28 To investigate the taxonomic position of E. mundtii QU 25 based on genome-wide comparisons, we first carried out ortholog analysis with the draft genome of E. mundtii ATCC 882 and five complete genomes of other Enterococcus spp., including two E. faecium strains (DO and Aus0004), E. hirae ATCC 9790, E. casseliflavus EC20, and E. faecalis V583 (Supplementary Table S1 and Fig. 1B). The results were consistent with the phylogeny based on 16S rRNA. While E. mundtii ATCC 882 showed the highest similarity to QU 25 as anticipated, two other species (E. faecium DO and Aus0004, and E. hirae ATCC 9790) showed moderate degrees of similarity, while E. casseliflavus EC20 and E. faecalis V583 were the least similar. DNA dot plot analysis showed the centre diagonal line between QU 25, the two E. faecium strains, and E. hirae (Fig. 2), indicating that not only each of the genes but also their genome structures (or gene orders) were related. There were gene regions unique to the QU 25 strain (Fig. 1B). Except for prophages, the 9-kb region (positions 1 359 588–1 368 963) includes five hypothetical proteins and a cell wall-anchored protein with a LPXTG motif.
Figure 2.
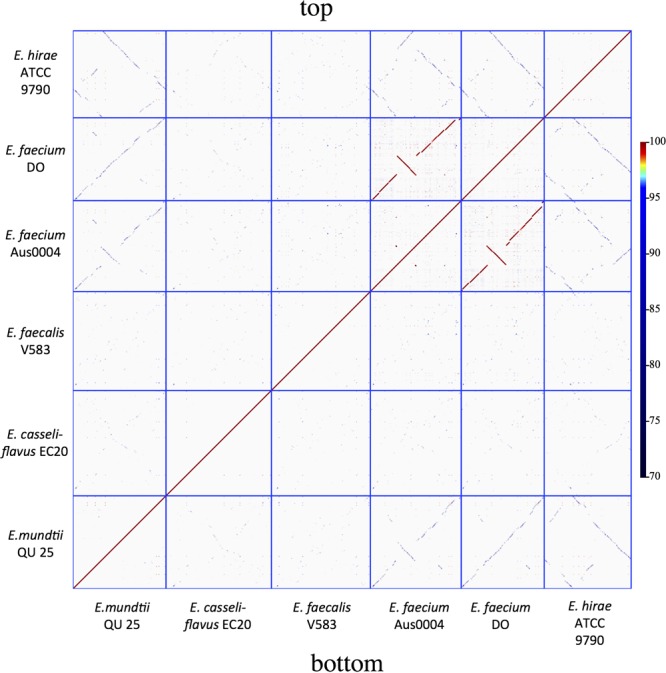
Dot plots comparing the chromosomal organization among the six Enterococcus species. Dot plots were generated using BLASTN and GenomeMatcher software. This figure appears in colour in the online version of DNA Research.
3.3. Mobile genetic elements
Three chromosomal regions were identified as prophage loci. Their positions are indicated in Fig. 1B and are named phiEmqu1 (38.7 kb; positions 806 547–845 215), phiEmqu2 (47.9 kb; positions 2 327 297–2 375 151), and phiEmqu3 (40.8 kb; positions 2 556 843–2 597 594). Sixty, 70, and 54 phage-related genes were identified in these regions, respectively (Supplementary Table S2). Prophages phiEmqu1 and phiEmqu3 contained several putative genes involved in DNA replication. However, no genes for DNA synthesis were found in the largest prophage phiEmqu2, suggesting that it is replication-defective. As shown in Fig. 1B, the locations of ori (dnaA) and ter (dif) were not exactly opposite each other. The dif motif, which is strongly associated with replication termini,29 was ∼60 kb off the exact opposite position of ori (dnaA). Phage-mediated replicore imbalance has been observed in E. faecium Aus0004.20 Thus, the replicore balance of QU 25 could be disrupted by the insertion of phiEmqu2 and/or phiEmqu3.
Clustered regularly interspaced short palindromic repeats (CRISPRs) are involved in a recently discovered interference pathway that protects cells from bacteriophages and conjugative plasmids. Approximately 40% of sequenced bacterial genomes and ∼90% of genomes from archaea contain at least one CRISPR locus.30 No CRISPR loci were detected in the QU 25 genome. Many insertion sequence elements (ISEs) have been found in enterococci. The relatively closed species, such as E. mundtii ATCC 882, E. faecium DO and Aus0004 strains, and E. hirae ATCC 9790, have 21, 180, 76, and 14 ISEs and transposase-related genes, respectively. At least 13 different ISEs were detected in the QU 25 genome, ranging in copy number from 1 to 5, representing 33 distinct copies and distributed around the chromosome and plasmids (Supplementary Table S3). The most frequently observed ISE type was the ISL3 family.
3.4. Plasmids
Enterococcus spp. have been reported to possess a number of plasmids that often confer resistance to antimicrobials and particular heavy metals, and serve to enhance virulence and/or DNA repair mechanisms.31–34 In strain QU 25, the plasmid copy number per chromosome was estimated by observing the distribution of read coverage of the Illumina sequence read, which indicated one copy of pQY182, pQY082, pQY039, and pQY024, and five copies of pQY003 (Table 1). BLASTN analyses showed that these five plasmids were similar to those of E. mundtii, E. faecium, E. hirae, or E. faecalis (Supplementary Table S4).
Plasmid pQY182 harbours genes that encode a two-component regulatory system, a cellulose 1,4-beta-cellobiosidase, a toxin–antitoxin system, and several proteins with DNA repair functions. Plasmid pQY082 harbours duplicated regions of 8.3 kb, which include the IS1675 transposase, ubiD family decarboxylase, two cell surface proteins, and two proteins with unknown functions (EMQU_3088-3094 and EMQU_3155-3160 with 99% similarity). pQY082 also harbours genes that encode several proteins with DNA repair functions and a toxin–antitoxin system. Toxin–antitoxin systems have been frequently reported in E. faecium strains,35 and the QU 25 chromosome additionally has at least four such systems. Plasmid pQY039 contains several genes for a DNA damage-inducible protein and a gene encoding a toxin–antitoxin system. Plasmid pQY024 also harbours genes for a DNA damage-inducible protein, a toxin–antitoxin system, and mundticin KS genes (see Discussion later). Plasmid pQY003 only harbours genes with unknown function, except for the replication initiation protein.
3.5. Vancomycin resistance
Because many Enterococcus isolates show vancomycin resistance which has been associated with hospital-acquired infections, we tested the sensitivity of QU 25 to this antibiotic for the safety of industrial use. The results showed that the vancomycin MIC for QU 25 was >2 µg/ml, indicating that this strain is vancomycin-sensitive. Several known genes involved in vancomycin resistance (vanA, vanB, vanX, vanH, vanR, and vanS)36 were not detected in the QU 25 genome and also in plasmids.
3.6. Bacteriocin activity and self- and cross-immunity
Bacteriocins are ribosomally synthesized bacterial peptides or proteins that show antimicrobial activity, generally against species that are closely related to bacteriocin producers.24 Mundticin is one of the bacteriocins produced by some E. mundtii strains. It is significant to clarify whether QU 25 produces mundticin or not for the resistivity to contamination in a large-scale culture. Three genes, such as munA (mundticin precursor), munB (ATP-binding cassette [ABC] transporter), and munC (mundticin KS immunity protein), are responsible for mundticin production in E. mundtii NERI 7393.37 The gene cluster containing these three genes was identified on plasmid pQY024, and munA (EMQU_3203; 100% nucleotide sequence identity), munB (EMQU_3204; 99.56% nucleotide sequence identity), and munC (EMQU_3205; 98.99% nucleotide sequence identity) showed high homology with corresponding genes in strain NERI 7393.
To examine whether the gene cluster for mundticin synthesis was functional, QU 25 was tested for bacteriocin production. QU 25 and E. mundtii QU 2 showed bacteriocin activity against three indicator strains (L. sakei JCM 1157T, E. faecalis JCM 5803T, and E. mundtii JCM 8731T), none of which showed inhibitory activity (Table 2). QU 25 and QU 2 showed no activity against each other (Table 2), which indicated that these strains have self- and cross-immunity against their bacteriocins. Bacteriocin-producing strains are known to have immunity (tolerance) to their own bacteriocins and to bacteriocins with similar structures. Collectively, these results strongly suggest that QU 25 produces a bacteriocin with similar characteristics to mundticin produced by QU 2.
Table 2.
Bacteriocin activity and immunity of Enterococcus mundtii strains
| Indicator strain | Bacteriocin producer |
||
|---|---|---|---|
| E. mundtii QU 25 | E. mundtii QU 2 | E. mundtii JCM 8731T | |
| E. mundtii QU 25 | − | − | − |
| E. mundtii QU 2 | − | − | − |
| E. mundtii JCM 8731T | + | + | − |
| E. faecalis JCM 5803T | + | + | − |
| L. sakei JCM 1157T | + | + | − |
+, growth inhibition of indicator strains; −, no inhibition of indicator strains.
3.7. Haemolysin activity
Haemolysin is one of the putative enterococcal virulence factors,20 so it is important to test the haemolysin activity for the safety of industrial use in a large-scale culture. Four putative haemolysin genes (haemolysin, haemolysin III, haemolysin A, and α-haemolysin) were identified (EMQU_0190 and EMQU_0948, EMQU_0449, EMQU_0841, and EMQU_1982, respectively). We tested haemolysin activity and no changes to the blood agar in the vicinity of the colonies were observed (data not shown), suggesting that these putative haemolysin genes in QU 25 might be inactive or silent under the tested culture conditions.
3.8. Metabolic pathways for lactic acid fermentation
QU 25 was previously reported to have two different pathways for xylose metabolism: the phosphoketolase (PK) pathway and the pentose phosphate (PP)/glycolytic pathway in low xylose concentrations.2,38 When QU 25 was grown in high xylose concentrations, PK activity was not detected. However, higher transaldolase and transketolase activities were detected2, indicating that strain QU 25 would utilize the PP/glycolytic pathway, not the PK pathway, as the main pathway for lactic acid fermentation.
Genes for xylose metabolism in the QU 25 chromosome were located in a 22-kb region (positions 2 904 895–2 926 710 bp) in two gene clusters: one involved in the initial metabolism of xylose and uptake of pentose, and the other involved in the PP/glycolytic pathway and uptake of related sugars (Fig. 3). The first gene cluster contained xylR (EMQU_2811; xylose repressor), xylA (EMQU_2810; xylose isomerase), xynB (EMQU_2809; xylan beta-1,4-xylosidase; additionally, there is another xynB gene [EMQU_2642] outside of this cluster), xylB (EMQU_2805; d-xylulose kinase), putative xylose transporter genes annotated as l-arabinose and d-ribose ABC transporter (EMQU_2806-2808), and a hypothetical protein (EMQU_2804), the N- and C-terminal regions of DNA mismatch repair protein (EMQU_2803 and EMQU_2802, respectively, which were thought as pseudogenes), ABC transporter ATP-binding protein (EMQU_2801), and its permease (EMQU_2800). Since pentose transporters have been shown to be promiscuous,39 d-xylose would likely be transported by these gene products in QU 25.
Figure 3.
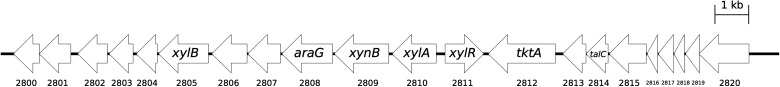
Gene clusters involved in the initial metabolism of xylose and the PP/glycolytic pathway in E. mundtii QU 25. Numbers below arrows indicate feature code (EMQU_XXXX).
The second gene cluster contains genes for the PP/glycolytic pathway, including a transketolase (EMQU_2812) and a transaldolase (EMQU_2814). Furthermore, this cluster contains an allulose-6-phosphate 3-epimerase gene (EMQU_2813), genes for a fructose-like phosphotransferase system (EMQU_2815-2819), and a putative transcriptional regulator (EMQU_2820). Transketolase is a key enzyme in the PP/glycolytic pathway, and the QU 25 chromosome additionally harbours one transketolase gene (EMQU_1275). Since allulose-6-phosphate 3-epimerase catalyses the conversion of d-allulose-6-phosphate to d-fructose-6-phosphate, this enzyme and the fructose transporters may supply various ketoses to the PP/glycolytic pathway. For genes involved in the PK pathway, PK (EMQU_1837), acetate kinase (EMQU_2620), phosphotransacetylase (EMQU_2119), acetaldehyde dehydrogenase (EMQU_2205), and alcohol dehydrogenase (EMQU_1129, EMQU_1829, and EMQU_2109) were dispersed throughout the chromosome.
To get insights into the lactic acid fermentation characteristics of strain QU 25, we performed the comparative analysis on the presence of key enzymes related to lactic acid fermentation from xylose using KO (KEGG Orthology) gene assignment (Supplementary Table S5). Two clinical isolates of E. faecium (DO and Aus0004) and E. hirae ATCC 9790 lack genes necessary for the metabolism of xylose (transaldolase, PK, xylulokinase, and xylose isomerase). Another clinical isolate, E. faecalis V583, lacks genes for transketolase, transaldolase, and PK. Thus, it is most unlikely that these strains metabolize xylose. E. casseliflavus EC20 has genes for PK, xylulokinase, and xylose isomerase, so that this strain can metabolize xylose by the PK pathway. It also has two complete genes for transketolase, but lacks transaldolase for the PP/glycolytic pathway. However, Kato et al.40 reported that Lactococcus lactis IO-1, which can utilize xylose, also lacks the gene for transaldolase and is presumed to have an alternative PP/glycolytic pathway. Therefore, EC20 might metabolize xylose also by the PP/glycolytic pathway. Two E. mundtii strains (QU 25 and ATCC 882) have two genes of full-length transketolase and all other genes necessary for the xylose metabolism by the two pathways. The results of our genomic analysis coincide with the description on their phenotype about xylose metabolism in Bergey's Manual (p. 599, Table 116).28 From these results, remarkable genomic features related to lactic acid fermentation in QU 25 still remain unclear, and the analysis of transcriptional regulation of these genes could help its clarification.
QU 25 was able to metabolize a mixture of glucose and cellobiose simultaneously without apparent CCR.1 In Gram-positive bacteria, the roles of catabolite control protein A (CcpA) and seryl-phosphorylated form of histidine-containing protein (P-Ser-HPr) in CCR have been well studied.41 Genes encoding CcpA (EMQU_1943), HPr (EMQU_0954), and HPr kinase/phosphorylase (EMQU_1951) were also found in the QU 25 genome. The mechanism by which CCR is prevented in the presence of glucose is still unknown.
Strain QU 25 shows a predominant production of l-(+)-lactate when grown at high concentrations of cellobiose and xylose, whereas d-lactic acid was not detected in the culture broth.1,2 However, two l-lactate dehydrogenase genes (l-LDH; EMQU_1380 and EMQU_2714) and one d-lactate dehydrogenase gene (d-LDH; EMQU_2453) were identified. Potentially, little or no d-LDH was expressed under these culture conditions. Lactate racemase, another gene involved in d-lactic acid formation,42 was not found in the QU 25 genome.
4. Conclusions
We have highlighted the phylogenetic relationship of E. mundtii QU 25 with related enterococcal species and characterized mobile genetic elements, including multiple prophages and ISEs, plasmids, and genes for metabolic pathways for lactic acid fermentation in this strain. In addition, we have demonstrated that QU 25 has bacteriocin activity. The complete E. mundtii QU 25 genome sequence described here may be an important resource in the genetic engineering of recombinant strains for optimized production of lactic acid.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This study was supported by the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2008-2012 (S0801025).
Supplementary Material
Acknowledgements
We thank Pacific Biosciences, Inc. (Menlo Park, CA, USA) and Tomy Digital Biology Co., Ltd (Tokyo, Japan) for PacBio sequencing and assembly, and Hitachi Solutions, Ltd (Tokyo, Japan) for optical mapping. We also thank Y. Kanesaki for helpful discussions.
References
- 1.Abdel-Rahman M.A., Tashiro Y., Zendo T., Shibata K., Sonomoto K. Isolation and characterisation of lactic acid bacterium for effective fermentation of cellobiose into optically pure homo l-(+)-lactic acid. Appl. Microbiol. Biotechnol. 2011;89:1039–49. doi: 10.1007/s00253-010-2986-4. [DOI] [PubMed] [Google Scholar]
- 2.Abdel-Rahman M.A., Tashiro Y., Zendo T., Hanada K., Shibata K., Sonomoto K. Efficient homofermentative l-(+)-lactic acid production from xylose by a novel lactic acid bacterium, Enterococcus mundtii QU 25. Appl. Environ. Microbiol. 2011;77:1892–5. doi: 10.1128/AEM.02076-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdel-Rahman M.A., Tashiro Y., Zendo T., Sonomoto K. Improved lactic acid productivity by an open repeated batch fermentation system using Enterococcus mundtii QU 25. RSC Adv. 2013;3:8437–45. [Google Scholar]
- 4.Chin C.-S., Alexander D.H., Marks P., et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods. 2013;10:563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 5.Magni C., Espeche C., Repizo G.D., et al. Draft genome sequence of Enterococcus mundtii CRL1656. J. Bacteriol. 2012;194:550. doi: 10.1128/JB.06415-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Machii M., Watanabe S., Zendo T., et al. Chemically defined media and auxotrophy of the prolific l-lactic acid producer Lactococcus lactis IO-1. J. Biosci. Bioeng. 2013;115:481–4. doi: 10.1016/j.jbiosc.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Koren S., Schatz M.C., Walenz B.P., et al. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat. Biotechnol. 2012;30:693–700. doi: 10.1038/nbt.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller J.R., Delcher A.L., Koren S., et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;24:2818–24. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagarajan N., Read T.D., Pop M. Scaffolding and validation of bacterial genome assemblies using optical restriction maps. Bioinformatics. 2008;24:1229–35. doi: 10.1093/bioinformatics/btn102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noguchi H., Taniguchi T., Itoh T. MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008;15:387–96. doi: 10.1093/dnares/dsn027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugawara H., Ohyama A., Mori H., Kurokawa K. Microbial genome annotation pipeline (MiGAP) for diverse users. The 20th International Conference on Genome Informatics (GIW2009) Poster and Software Demonstrations (Yokohama); 2009. S001-1-2. [Google Scholar]
- 12.Wheeler D.L., Barrett T., Benson D.A., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2007;35:D5–D12. doi: 10.1093/nar/gkl1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zdobnov E.M., Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–8. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 14.Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Y., Liang Y., Lynch K.H., Dennis J.J., Wishart D.S. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–52. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varani A.M., Siguier P., Gourbeyre E., Charneau V., Chandler M. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 2011;12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grissa I., Vergnaud G., Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer K.L., Carniol K., Manson J.M., et al. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 2010;192:2469–70. doi: 10.1128/JB.00153-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulsen I.T., Banerjei L., Myers G.S.A., et al. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science. 2003;299:2071–4. doi: 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 20.Lam M.M.C., Seemann T., Bulach D.M., et al. Comparative analysis of the first complete Enterococcus faecium genome. J. Bacteriol. 2012;194:2334–41. doi: 10.1128/JB.00259-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qin X., Galloway-Peña J.R., Sillanpaa J., et al. Complete genome sequence of Enterococcus faecium strain TX16 and comparative genomic analysis of Enterococcus faecium genomes. BMC Microbiol. 2012;12:135. doi: 10.1186/1471-2180-12-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaechter T., Wunderlin C., Schmidheini T., Solioz M. Genome sequence of Enterococcus hirae (Streptococcus faecalis) ATCC 9790, a model organism for the study of ion transport, bioenergetics, and copper homeostasis. J. Bacteriol. 2012;194:5126–7. doi: 10.1128/JB.01075-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohtsubo Y., Ikeda-Ohtsubo W., Nagata Y., Tsuda M. GenomeMatcher: a graphical user interface for DNA sequence comparison. BMC Bioinformatics. 2008;9:376. doi: 10.1186/1471-2105-9-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zendo T., Eungruttanagorn N., Fujioka S., et al. Identification and production of a bacteriocin from Enterococcus mundtii QU 2 isolated from soybean. J. Appl. Microbiol. 2005;99:1181–90. doi: 10.1111/j.1365-2672.2005.02704.x. [DOI] [PubMed] [Google Scholar]
- 25.Ennahar S., Asou Y., Zendo T., Sonomoto K., Ishizaki A. Biochemical and genetic evidence for production of enterocins A and B by Enterococcus faecium WHE 81. Int. J. Food Microbiol. 2001;70:291–301. doi: 10.1016/s0168-1605(01)00565-7. [DOI] [PubMed] [Google Scholar]
- 26.Shin S.C., Ahn D.H., Kim S.J., et al. Advantages of single-molecule real-time sequencing in high-GC content genomes. PLoS ONE. 2013;8:e68824. doi: 10.1371/journal.pone.0068824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Travers K.J., Chin C.-S., Rank D.R., Eid J.S., Turner S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010;38:e159. doi: 10.1093/nar/gkq543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitman W.B., Goodfellow M., Kämpfer P., et al. Bergey's Manual of Systematic Bacteriology. 2 edition. New York: Springer Publishing Company; 2012. [Google Scholar]
- 29.Hendrickson H., Lawrence J.G. Mutational bias suggests that replication termination occurs near the dif site, not at Ter Sites. Mol. Microbiol. 2007;64:42–56. doi: 10.1111/j.1365-2958.2007.05596.x. [DOI] [PubMed] [Google Scholar]
- 30.Marraffini L.A., Sontheimer E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010;11:181–90. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arias C.A., Panesso D., Singh K.V., Rice L.B., Murray B.E. Cotransfer of antibiotic resistance genes and a hylEfm-containing virulence plasmid in Enterococcus faecium. Antimicrob. Agents Chemother. 2009;53:4240–6. doi: 10.1128/AAC.00242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Migura L., Hasman H., Jensen L.B. Presence of pRI1: a small cryptic mobilizable plasmid isolated from Enterococcus faecium of human and animal origin. Curr. Microbiol. 2009;58:95–100. doi: 10.1007/s00284-008-9266-x. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Migura L., Liebana E., Jensen L.B. Transposon characterization of vancomycin-resistant Enterococcus faecium (VREF) and dissemination of resistance associated with transferable plasmids. J. Antimicrob. Chemother. 2007;60:263–8. doi: 10.1093/jac/dkm186. [DOI] [PubMed] [Google Scholar]
- 34.Hasman H., Kempf I., Chidaine B., et al. Copper resistance in Enterococcus faecium, mediated by the tcrB gene, is selected by supplementation of pig feed with copper sulfate. Appl. Environ. Microbiol. 2006;72:5784–9. doi: 10.1128/AEM.02979-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moritz E.M., Hergenrother P.J. Toxin-antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc. Natl. Acad. Sci. USA. 2007;104:311–6. doi: 10.1073/pnas.0601168104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pootoolal J., Neu J., Wright G.D. Glycopeptide antibiotic resistance. Annu. Rev. Pharmacol. Toxicol. 2002;42:381–408. doi: 10.1146/annurev.pharmtox.42.091601.142813. [DOI] [PubMed] [Google Scholar]
- 37.Kawamoto S., Shima J., Sato R., et al. Biochemical and genetic characterization of mundticin KS, an antilisterial peptide produced by Enterococcus mundtii NFRI 7393. Appl. Environ. Microbiol. 2002;68:3830–40. doi: 10.1128/AEM.68.8.3830-3840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdel-Rahman M.A., Tashiro Y., Sonomoto K. Recent advances in lactic acid production by microbial fermentation processes. Biotechnol. Adv. 2013;31:877–902. doi: 10.1016/j.biotechadv.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Song S., Park C. Utilization of d-ribose through d-xylose transporter. FEMS Microbiol. Lett. 1998;163:255–61. doi: 10.1111/j.1574-6968.1998.tb13054.x. [DOI] [PubMed] [Google Scholar]
- 40.Kato H., Shiwa Y., Oshima K., et al. Complete genome sequence of Lactococcus lactis IO-1, a lactic acid bacterium that utilizes xylose and produces high levels of l-lactic acid. J. Bacteriol. 2012;194:2102–3. doi: 10.1128/JB.00074-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita Y. Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci. Biotechnol. Biochem. 2009;73:245–59. doi: 10.1271/bbb.80479. [DOI] [PubMed] [Google Scholar]
- 42.Goffin P., Deghorain M., Mainardi J.-L., et al. Lactate racemization as a rescue pathway for supplying d-lactate to the cell wall biosynthesis machinery in Lactobacillus plantarum. J. Bacteriol. 2005;187:6750–61. doi: 10.1128/JB.187.19.6750-6761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.