

Abstract
Administration of pharmacologic doses of glucocorticoid in vivo increases renal proximal tubule apical membrane Na/H exchange and decreases Na/PO4 cotransport activity (1). Current data suggest that the NHE-3 and NaPi-2 proteins mediate significant fractions of proximal tubule apical membrane Na/H exchange and Na/PO4 cotransport, respectively. This study examines whether glucocorticoid excess or deficiency affects NHE-3 and NaPi-2 protein abundance and the intrarenal distribution of these transporters. Protein abundance of NHE-3 and NaPi-2 in control rats was compared to rats rendered glucocorticoid-deficient by bilateral adrenalectomy, and to rats receiving pharmacologic doses of dexamethasone using immunoblots and immunohistochemistry. Adrenalectomy had modest effects on NHE-3 protein abundance, but dexamethasone administration to either adrenalectomized or sham-operated rats significantly increased NHE-3 protein abundance in both the proximal tubule and thick ascending limb, but not the thin descending limb. Adrenalectomy increased NaPi-2 protein abundance in the proximal tubule, whereas dexamethasone administration dramatically suppressed NaPi-2 protein on the apical membrane in both adrenalectomized and sham-operated animals. No significant reciprocal increase in subapical NaPi-2 staining was seen in the dexamethasone-treated rats. The present study shows that glucocorticoids regulate proximal tubule apical membrane Na/H exchange and NaPi cotransport by changes in protein abundance of NHE-3 and NaPi-2, respectively.
Glucocorticoid excess increases and glucocorticoid deficiency decreases net acid excretion by the kidney (2,3). The increased net acid excretion involves increased tubular H secretion and HCO3 absorption and enhanced urinary buffer excretion due to increased ammoniagenesis and excretion, and increased phosphate excretion (3–6). Glucocorticoids exert their effects on proximal tubule HCO3 and PO4 absorption by stimulating apical membrane Na/H exchange and inhibiting Na-PO4 cotransport, respectively (1,7,8).
Immunohistochemical data suggest that proximal tubule apical membrane Na/H exchange and Na-PO4 cotransport are mediated at least in part by NHE-3 and NaPi-2 proteins, respectively (9–12). Glucocorticoid excess increases renal cortical NHE-3 mRNA in the rabbit (13), but its effect on NHE-3 protein in various nephron segments in the intact animal has not been examined. It is also unclear whether glucocorticoid deficiency affects NHE-3 protein expression. Glucocorticoid excess decreases NaPi-2 mRNA and protein in whole renal cortex (14), but the intranephron and cellular distribution of NaPi-2 has not been examined in either glucocorticoid excess or deficient states. This is particularly relevant because plasma membrane insertion has been shown to be a mechanism of regulation of apical membrane NaPi-2 abundance (15,16). In the present study, we examined the abundance of these two transporter proteins with variations in the glucocorticoid status in rats by immunohistochemistry and immunoblot. We showed that glucocorticoids increase NHE-3 and decrease NaPi-2 protein expression in the proximal tubule, and increase NHE-3 protein expression in the thick ascending limb.
Materials and Methods
Animal Models
Male Sprague Dawley rats weighing 200 to 300 g were subjected to either bilateral adrenalectomy (ADX group) or sham operations (SHM group), and were allowed free access to water and rat chow. ADX animals were supplemented with 0.9% saline in their drinking water for at least 3 d before administration of either vehicle or glucocorticoid. Both ADX and SHM animals were given either 60 μg/100 g body wt subcutaneous injections of dexamethasone (DEX) twice daily or vehicle (phosphate-buffered saline [PBS]) for 2 d (four doses), and a fifth dose was given 2 h before sacrifice. Initially, four experimental groups (ADX, SHM, ADX + DEX, SHM + DEX) with four animals in each group were used for NHE-3 and NaPi-2 staining by immunohistochemistry and immunoblots of cortical and apical membranes. Subsequently, an additional set of experiments was performed using the same four conditions (ADX, n = 5; SHM, n = 4; ADX + DEX, n = 6; SHM + DEX, n = 4) to examine NHE-3 expression in both cortical and medullary membranes. The animals were sacrificed and kidneys were harvested for either renal membrane preparation or for immunohistochemistry after perfusion fixation. Renal NHE-3 and NaPi-2 protein expression was quantified by immunoblots in all four experimental groups and by immunohistochemistry in SHM, ADX, and ADX + DEX animals.
Immunoblots
Renal cortex or the inner stripe of the outer medulla was dissected and homogenized (buffer containing in mM: 300 mannitol, 20 Hepes, pH 7.50, and 5 ethyleneglycol-bis(β-aminoethyl ether)N,N′-tetra-acetic acid; in μg/ml: 100 phenylmethylsulfonyl fluoride, 2 leupeptin, 2 aprotinin, and 2 pepstatin A; Brinkman polytron), and the membrane fraction was obtained by centrifugation (Beckman J2–21 M, JA-20 rotor, 20,000 rpm, 40 min, 4°C). Apical membrane vesicles were prepared from the cortical homogenate by exposing cortical membranes to three consecutive precipitations in 15 mM MgCl2, and the final apical membrane-enriched vesicles were pelleted from the supernatant (Beckman J2–21 M, JA-20 rotor, 20,000 rpm, 40 min, 4°C). Twenty micrograms of either cortical membranes, medullary membranes, or cortical apical membranes was fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. For immunoblotting, anti-NHE-3 antiserum (no. 1568 against epitope DSFLQADGPEEQLQ at 1:200 dilution) (9) and anti-NaPi-2 antiserum (against epitope LALPAHHNATRL at 1:1000 dilution) (11,12) were used as primary antisera. The specificity of both antisera have been characterized previously (9,12,17). Anti-β-actin antibody (1:1000 dilution) was used to ensure equal loading. After incubation with horseradish peroxidase-coupled mouse anti-rabbit secondary antibody, signals were detected by enhanced chemiluminescence (Amersham, Arlington Heights, IL) and quantified by densitometry. NHE-3 and NaPi-2 signals were normalized to β-actin signals and expressed as a percentage of the SHM group.
Immunohistochemistry
Localization and abundance of NHE-3 and NaPi-2 protein in the kidney were studied by immunohistochemistry in three experimental groups (SHAM, ADX, and ADX + DEX). The kidneys were fixed by vascular perfusion through the abdominal aorta, as described previously (9,12,15). Briefly, kidneys were perfused with 3% paraformaldehyde/0.05% picric acid in a 6:4 mixture of 0.1 M cacodylate buffer (pH 7.4, adjusted to 300 mosmol/kg water with sucrose) and 10% hydroxyethyl starch. After 5 min of fixation, the fixative was washed out by perfusion with 0.1 M cacodylate/sucrose buffer. Coronal slices of the fixed kidneys were mounted on cork disks, frozen in liquid propane cooled by liquid N2, and stored at −80°C until use. Serial sections (3 to 4 μm thick) were cut in a cryostat and placed on chrome alum gelatin-coated slides. Immunohistostaining for NHE-3 and NaPi-2 was performed as described previously (9,12,15). For NHE-3, sections were pretreated with 10% normal goat serum in PBS, and for NaPi-2, with 3% milk powder and 0.3% Triton X-100 in PBS. Sections were then incubated with anti-NHE-3 antiserum (no. 1566 YSRHELTPNEDEKQ, 1/8000 in PBS/BSA) or with anti-NaPi-2 antiserum (against epitope LALPAHHNATRL, 1:500 in PBS/milk powder) overnight in a humidified chamber at 4°C. After repeated rinsing in PBS, binding of the primary antibodies was detected with FITC-conjugated swine-anti-rabbit IgG (1:40 PBS/BSA; Dakopatts, Glostrup, Denmark). Rhodamine-conjugated phalloidin (1/100 dilution; Molecular Probes, Eugene, OR) was added to the secondary antibody for staining of actin filaments. Finally, the sections were rinsed with PBS, coverslips were applied with DAKO-Glycergel (Dakopatts) containing 2.5% 1,4-diazabicyclo-(2.2.2)-octane (DABCO; Sigma, St. Louis, MO) as a fading retardant, and the sections were studied by epifluorescence microscopy (Polyvar, Reichert-Jung, Vienna, Austria).
Statistical Analysis
Quantitative differences were assessed by ANOVA.
Results
Effect of Glucocorticoids on NHE-3
Figure 1, A and B, shows representative immunoblots examining NHE-3 protein abundance in cortical apical membranes and medullary membrane from animals from the four experimental groups. In renal cortex, NHE-3 abundance was not significantly different in the ADX and SHM animals, but was increased in both the ADX + DEX and the SHM + DEX animals (P < 0.05 for both by ANOVA). Labeling of β-actin, which served as a loading control, was not different in the four groups. Summary of four experiments showed the following relative NHE-3 signals: SHM, 100 ± 51%; ADX, 103 ± 20%; SHM + DEX, 245 ± 7%; ADX + DEX, 280 ± 16%. Immunoblots of cortical membranes showed similar results (not shown). Figure 1B shows a representative immunoblot of NHE-3 protein abundance in medullary membranes. The effect of ADX on NHE-3 abundance is more variable than in cortex, but overall ADX suppressed NHE-3 expression in the medulla (P < 0.05 by ANOVA). DEX administration to either SHM or ADX animals significantly increased NHE-3 protein abundance compared with SHM and ADX (P < 0.05 for both by ANOVA). Summary of all animals showed the following NHE-3 signals: SHM, 100 ± 25%; ADX, 54 ± 51%; SHM + DEX, 345 ± 23%; ADX + DEX, 367 ± 26%.
Figure 1.

Immunoblot of renal NHE-3 and β-actin protein abundance. Mobility in kilodaltons is indicated on the right. (A) Cortical apical membranes: ADX slightly decreased, whereas DEX significantly increased NHE-3 expression. Number of experiments: ADX, 9; SHM, 8; ADX + DEX, 10; SHM + DEX, 8. (B) Medullary membranes: ADX decreased, whereas DEX increased NHE-3 expression. Number of experiments: ADX, 5; SHM, 4; ADX + DEX, 6; SHM + DEX, 4. DEX, dexamethasone-treated; ADX, adrenalectomized; SHM, sham-operated.
NHE-3 protein was expressed in the brush border of S1 and S2 segments of proximal tubules in the cortical labyrinth (Figure 2B). NHE-3 was localized predominantly in the base of the brush border, as demonstrated by costaining of F-actin (Figure 2C). In medullary rays, there was an axial decrease of NHE-3 expression along the S2 segments. In S3 segments of medullary rays and the outer stripe, no immunoreactivity for NHE-3 was observed. Intracellular staining was not evident in these sections. NaPi staining was restricted to cortex and was evident along the entire length of the brush border (Figure 2A).
Figure 2.

Localization of NaPi-2 (A), NHE-3 (B), and F-actin (C) in proximal tubules of the cortical labyrinth. NaPi-2 is visible in small intracellular compartments and over the entire height of the brush border (A); NHE-3 immunostaining is restricted to the base of the brush border (B); F-actin (revealed by rhodamine-conjugated phalloidin) is present over the entire lengths of the brush-border microvilli (C). Bar, 20 μm.
ADX caused a very small decrease in NHE-3 expression in proximal tubules (Figure 3B) compared with sham-operated rats (Figure 3A). In ADX + DEX animals, NHE-3 abundance was profoundly increased compared to both ADX and SHM animals (Figure 3, A through C). These findings are similar to those of the immunoblots shown above. The effect of both glucocorticoid depletion or excess seemed to affect S1 and S2 proximal tubules in a similar way. The pattern described above was found consistently, with slight differences in the magnitude of the changes in all animals of the corresponding groups.
Figure 3.

Detection by immunofluorescence of NHE-3 in rat renal cortex of a sham-operated rat (A), an ADX rat (B), and an ADX + DEX rat (C). Immunostaining is seen in the brush border of proximal tubules. Adrenalectomy minimally reduces whereas administration of dexamethasone significantly increases NHE-3 abundance in the brush border of proximal tubules. Bar, 100 μm. Number of experiments: four for each group.
In the apical membrane of the thick ascending limb (TAL), ADX had modest effects on NHE-3 expression on immunohistochemistry (Figure 4, A and B). In SHM rats, TAL NHE-3 expression was similar in all animals, whereas much larger variations were seen among the individual animals of the ADX groups. This variable effect of ADX on TAL NHE-3 was also evident in the immunoblots performed on medullary membrane shown above. However, TAL NHE-3 expression was dramatically increased in the ADX + DEX animals (Figure 4C) compared with either SHM or ADX animals. NHE-3 staining in the thin descending limb (TDL), on the other hand, was not affected by ADX or ADX + DEX.
Figure 4.
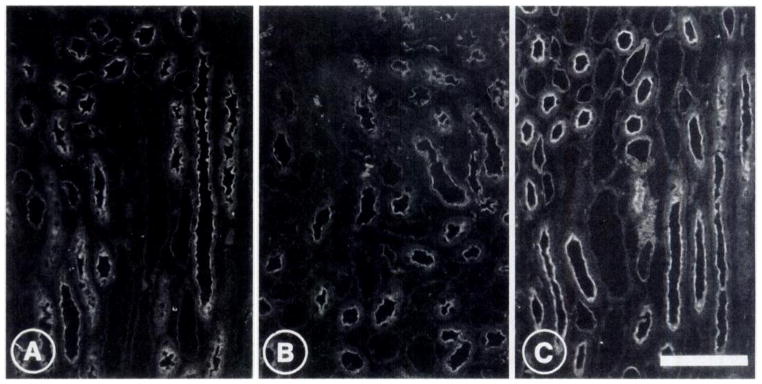
Detection by immunofluorescence of NHE-3 in the inner stripe of the rat renal medulla of a sham-operated rat (A), an ADX rat (B), and an ADX + DEX rat (C). Immunostaining is seen in the thick ascending limbs and the thin descending limbs. Adrenalectomy minimally decreases whereas administration of dexamethasone significantly increases NHE-3 abundance in the luminal membrane of thick ascending limb cells. The weak immunostaining of the luminal membrane of thin limbs is not affected by adrenalectomy or by dexamethasone treatment. Bar, 100 μm. Number of experiments: four for each group.
Effect of Glucocorticoids on NaPi-2
Figure 5 is a representative immunoblot quantifying NaPi-2 abundance in apical membrane vesicles from the four experimental groups. ADX increased NaPi-2 immunoexpression in three out of four sets of animals, with no change in one set. DEX dramatically suppressed NaPi-2 expression in all ADX or SHM animals. A summary of all four sets of animals showed the following relative NaPi-2 levels: SHM, 100 ± 23%; ADX, 217 ± 35%; SHM + DEX, 15 ± 45%; ADX + DEX, 18 ± 2% (P < 0.05 for all comparisons except between SHM + DEX and ADX + DEX; ANOVA). Immunoblots with cortical membranes showed a similar pattern (data not shown).
Figure 5.

Immunoblot of renal cortical apical membranes for NaPi-2 and β-actin protein abundance. Mobility in kilodaltons is indicated on the right. ADX increased whereas DEX decreased NaPi-2 expression. Number of experiments: four for each group.
On kidney sections (Figure 6), ADX increased NaPi-2 immunoexpression in the brush border of all nephrons examined compared with SHM (Figure 6, A and B). Intracellular staining in ADX animals was slightly more distinct, but did not appear to be more abundant than in SHM controls (Figure 6, A and B). ADX + DEX (Figure 6C) drastically reduced NaPi-2 expression in the brush border of all nephrons. Intracellular staining for NaPi-2 was more diffuse and abundant in ADX + DEX than in ADX or SHM animals.
Figure 6.

Detection by immunofluorescence of NaPi-2 in rat renal cortex of a sham-operated rat (A), an ADX rat (B), and an ADX + DEX rat (C). Immunostaining is seen in the brush border of proximal tubules and in intracellular compartments. Adrenalectomy increases NaPi-2 immunoreactivity in the brush border with little change in the intracellular staining pattern. Administration of dexamethasone drastically reduces NaPi-2 abundance in the brush border of proximal tubules and increases the intracellular staining. Bar, 100 μm. Number of experiments: four for each group.
Discussion
Increased proximal tubule apical membrane Na/H exchanger activity contributes to the increased NaHCO3 absorption and increased NH4 secretion in states of glucocorticoid excess (1,3,4). Kinsella and coworkers have demonstrated that adrenalectomy per se did not lower apical membrane Na/H activity, whereas dexamethasone administration increased the Vmax of the Na/H exchanger by 40 to 90% without altering its affinity for Na or H (1,7). This increase was observed in the background of both adrenalectomized or sham-operated animals (1). Our findings of changes in NHE-3 antigen by immunoblot and immunohistochemistry are in agreement with previous studies on apical membrane Na/H exchanger activity. Adrenalectomy per se did not have a significant effect on NHE-3 protein abundance on immunoblots performed on total cortical or brush-border membranes, although it is possible that a small decrease in proximal apical NHE-3 expression may not be detectable by immunoblot and immunohistochemistry. The increase in NHE-3 protein abundance induced by dexamethasone was profound, as assayed by both immunoblots and immunohistochemistry. This increase was independent of whether the animals were adrenalectomized. This finding is expected because 60 μg/kg represents a pharmacologic dose of glucocorticoid. The agreement between Na/H exchange activity in the previous studies (1,7) and NHE-3 abundance in the present study further confirms that NHE-3 mediates a significant portion of proximal tubule apical membrane Na/H exchange.
In the medulla, adrenalectomy seemed to have a variable suppressive effect on TAL NHE-3 expression. The suppression was demonstrable on immunoblots despite the large variation due to the more quantitative nature of immunoblots compared with immunohistochemistry. The increased TAL NHE-3 expression in DEX animals was unequivocal in immunoblots and evident even in immunohistochemistry. Immunoblots could not distinguish changes originating from TDL or TAL in the medulla, whereas immunohistochemistry clearly showed that the increased medullary NHE-3 was all due to increased expression in TAL. The lack of response in the TDL suggests that there are nephron segment-specific factors mediating the effect of glucocorticoid on NHE-3 expression.
The increased NHE-3 protein abundance is compatible with the current model of activation of NHE-3 by glucocorticoids at the level of gene transcription. In OKP cells, the glucocorticoid-induced increase in NHE-3 activity can be accounted for by an increase in steady-state NHE-3 transcript levels due to activation of transcription of the NHE-3 gene (18–20). Parallel increases in NHE-3 mRNA, protein, Na/H exchange activity, and transepithelial NaHCO3 flux have been described in the maturing nephron of the neonate (21,22). The decrease in TAL apical membrane NHE-3 expression in adrenalectomized rats is compatible with the finding that rat TAL NaHCO3 absorption was decreased by 40% in an in vivo microperfusion study by adrenalectomy and restored to normal by low-dose dexamethasone (23). The adrenalectomy-induced decrease in TAL Na-K-ATPase (24) and TAL transepithelial NaCl absorption (25,26) can be restored by aldosterone but not by dexamethasone. It is conceivable that regulation of TAL NaHCO3 transport by adrenal steroids may be mediated by effects of glucocorticoid on the apical membrane NHE-3 rather than the effect of mineralocorticoids on basolateral Na-K-ATPase. A commensurate increase in proximal tubule NHE-3 protein abundance and activity and TAL NHE-3 mRNA and protein abundance has been described in chronic metabolic acidosis in rats (27,28).
The effect of glucocorticoids on phosphate balance is complex and includes effects on gastrointestinal absorption, bone mineral balance, and renal phosphate handling. Phosphaturia and hypophosphatemia have been described in patients with and in animal models of glucocorticoid excess (29,30). The principal regulatory step of renal phosphate excretion resides in the proximal tubule apical membrane Na-PO4 cotransport system. There are two points of disparity between the current data and those of earlier transport studies. First, although dexamethasone reduced the Vmax of the Na-Pi cotransport by approximately 30%, adrenalectomy per se had little effect on apical membrane Na-Pi cotransport (1,31). Present studies show a definite increase of NaPi-2 protein abundance in adrenalectomized animals by both immunoblot and immunohistochemistry. Second, the effect of dexamethasone administration on NaPi-2 protein abundance in this study and in the report by Levi and coworkers (14) is far greater than the observed 30% decrease in apical Na-PO4 cotransport Vmax (1,31). The difference between the transport data and the protein abundance is likely due to the fact that Na-PO4 cotransport activity reflects more than one form of Na-Pi cotransporter. The relative contributions of NaPi-2 and other potential Na-PO4 cotransporter isoforms to the Na-coupled phosphate flux in vesicles is currently unknown. It has been shown that dexamethasone administration in adrenal-intact animals decreases NaPi-2 but does not affect NaPi-1 protein abundance (14). The contribution of Na-PO4 cotransporter isoforms other than NaPi-2 to apical membrane Na-PO4 cotransport can also account for the lack of detectable effect of adrenalectomy on apical membrane Na-PO4 cotransport.
The increase in intracellular staining of NaPi-2 that accompanies the decrease in apical membrane staining in dexamethasone-treated animals suggests that decreased membrane insertion and/or increased internalization likely contribute in part to the decrease in apical membrane NaPi-2 protein. Protein trafficking is a well described mechanism by which acute dietary phosphate or parathyroid hormone administration regulates apical membrane NaPi-2 protein abundance (15,16), and trafficking could well be playing a role in mediating the effect of glucocorticoids on NaPi-2. Because NaPi-2 abundance in total cortical membranes also varied with glucocorticoid status, changes in kinetics of insertion and internalization cannot be the sole mode of regulation of NaPi-2 in response to chronic changes in glucocorticoid excess. At present, one cannot conclude on the relative roles of trafficking versus protein synthesis/degradation in mediating the effect of glucocorticoid on proximal tubule apical NaPi-2 abundance. It is possible that glucocorticoids may first induce endocytosis followed by subsequent intracellular degradation of NaPi-2.
In summary, the present study demonstrates that the increase in Na/H exchange and suppressed Na-PO4 cotransport activity previously described in the proximal tubule apical membrane is due to changes in the protein abundance of NHE-3 and NaPi-2, respectively. In addition, NHE-3 protein expression in the apical membrane of the TAL is inversely proportional to the glucocorticoid status of the animal, whereas NHE-3 expression in the TDL is not regulated by glucocorticoids.
Acknowledgments
The present studies are supported by the Department of Veterans Affairs Research Service (Drs. Levi and Moe), National Institutes of Health (Grant DK39398 to Dr. Alpern, Grant DK-41612 to Dr. Baum, and Grant DK-48482 to Dr. Moe), and the Swiss National Science Foundation (Grant 31-47742.96 to Dr. Kaissling and Grant 3100-46523.96 to Dr. Murer).
References
- 1.Freiberg JM, Kinsella J, Sacktor B. Glucocorticoids increase the Na/H exchange and decrease the Na gradient-dependent phosphate-uptake systems in renal brush border membrane vesicles. Proc Natl Acad Sci USA. 1982;79:4932–4936. doi: 10.1073/pnas.79.16.4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dubrovsky AH, Nair E, Byers MK, Levine DZ. Renal net acid excretion in the adrenalectomized rat. Kidney Int. 1981;19:516–528. doi: 10.1038/ki.1981.49. [DOI] [PubMed] [Google Scholar]
- 3.Hulter HN, Licht JH, Bonner EL, Jr, Glynn RD, Sebastian A. Effects of glucocorticoid steroids on renal and systemic acid-base metabolism. Am J Physiol. 1980;239:F30–F43. doi: 10.1152/ajprenal.1980.239.1.F30. [DOI] [PubMed] [Google Scholar]
- 4.Baum M, Quigley R. Glucocorticoids stimulate rabbit proximal convoluted tubule acidification. J Clin Invest. 1993;91:110–114. doi: 10.1172/JCI116158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guntupalli J, Eby E, Lau K. Mechanism for the phosphaturia of NH4Cl: Dependence on acidemia but not on diet PO4 or PTH. Am J Physiol. 1982;242:F552–F560. doi: 10.1152/ajprenal.1982.242.5.F552. [DOI] [PubMed] [Google Scholar]
- 6.Poujeol P, Vandewalle A. Phosphate uptake by proximal cells isolated from rabbit kidney: Role of dexamethasone. Am J Physiol. 1985;249:F74–F83. doi: 10.1152/ajprenal.1985.249.1.F74. [DOI] [PubMed] [Google Scholar]
- 7.Kinsella JL, Frieberg JM, Sacktor B. Glucocorticoid activation of Na-H exchange in renal brush border membrane vesicles. Am J Physiol. 1985;248:F233–F239. doi: 10.1152/ajprenal.1985.248.2.F233. [DOI] [PubMed] [Google Scholar]
- 8.Boross M, Kinsella J, Cheng L, Sacktor B. Glucocorticoids and metabolic acidosis-induced renal transport of inorganic phosphate, calcium and NH4. Am J Physiol. 1986;250:F827–F833. doi: 10.1152/ajprenal.1986.250.5.F827. [DOI] [PubMed] [Google Scholar]
- 9.Amemiya M, Loffing J, Lötscher M, Kaissling B, Alpern RJ, Moe OW. Expression of NHE-3 in the apical membrane of rat proximal tubule and thick ascending limb. Kidney Int. 1995;48:1206–1215. doi: 10.1038/ki.1995.404. [DOI] [PubMed] [Google Scholar]
- 10.Biemesderfer D, Pizzonia JH, Exner M, Reilly RF, Igarashi P, Aronson PS. NHE3: A Na/H exchanger isoform of the renal brush border. Am J Physiol. 1993;34:F736–F742. doi: 10.1152/ajprenal.1993.265.5.F736. [DOI] [PubMed] [Google Scholar]
- 11.Magagnin S, Werner A, Markovich D, Sorribas V, Stange G, Biber J, Murer H. Expression cloning of human and rat renal cortex Na-Pi cotransport. Proc Natl Acad Sci USA. 1993;90:5979–5983. doi: 10.1073/pnas.90.13.5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Custer M, Lötscher M, Biber J, Murer H, Kaissling B. Expression of Na-Pi cotransport in rat kidney: Localization by RT-PCR and immunohistochemistry. Am J Physiol. 1994;266:F767–F774. doi: 10.1152/ajprenal.1994.266.5.F767. [DOI] [PubMed] [Google Scholar]
- 13.Baum M, Moe OW, Gentry D, Alpern RJ. Effect of glucocorticoids on renal cortical NHE-3 and NHE-1 mRNA. Am J Physiol. 1994;267:F437–F442. doi: 10.1152/ajprenal.1994.267.3.F437. [DOI] [PubMed] [Google Scholar]
- 14.Levi M, Shayman JA, Abe A, Gross SK, McCluer RH, Biber J, Murer H, Lötscher M, Cronin RE. Dexamethasone modulate rat renal brush border membrane phosphate transporter mRNA and protein abundance and glycosphingolipid composition. J Clin Invest. 1995;96:207–216. doi: 10.1172/JCI118022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lötscher M, Levi M, Biber J, Murer H, Kaissling B. Parathyroid hormone induces rapid endocytosis of renal type II Na-Pi cotransporter [Abstract] J Am Soc Nephrol. 1996;7:1804. [Google Scholar]
- 16.Lötscher M, Kaissling B, Biber J, Murer H, Levi M. Role of microtubules in the rapid regulation of renal phosphate transport in response to acute alterations in dietary phosphate content. J Clin Invest. 1997;99:1302–1312. doi: 10.1172/JCI119289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moe OW, Amemiya M, Yamaji Y. Protein kinase A activation acutely inhibits and phosphorylates Na/H exchanger NHE-3. J Clin Invest. 1995;96:2187–2194. doi: 10.1172/JCI118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baum M, Amemiya M, Dwarakanath V, Alpern RJ, Moe OW. Glucocorticoids regulate NHE-3 transcription in OKP cells. Am J Physiol. 1996;270:F164–F169. doi: 10.1152/ajprenal.1996.270.1.F164. [DOI] [PubMed] [Google Scholar]
- 19.Cano A. Characterization of the rat NHE-3 promoter. Am J Physiol. 1996;271:F629–F636. doi: 10.1152/ajprenal.1996.271.3.F629. [DOI] [PubMed] [Google Scholar]
- 20.Kandasamy RA, Orlowski J. Genomic organization and glucocorticoid transcriptional activation of the Na/H exchanger NHE-3 gene. J Biol Chem. 1996;271:10551–10559. doi: 10.1074/jbc.271.18.10551. [DOI] [PubMed] [Google Scholar]
- 21.Baum M, Biemesderfer D, Gentry D, Aronson PS. Ontogeny of rabbit renal cortical NHE3 and NHE1: Effect of glucocorticoids. Am J Physiol. 1995;268:F815–F820. doi: 10.1152/ajprenal.1995.268.5.F815. [DOI] [PubMed] [Google Scholar]
- 22.Baum M, Quigley R. Prenatal glucocorticoids stimulate neonatal juxtamedullary proximal convoluted tubule acidification. Am J Physiol. 1991;261:F746–F752. doi: 10.1152/ajprenal.1991.261.5.F746. [DOI] [PubMed] [Google Scholar]
- 23.Unwin R, Capasso G, Giebisch G. Bicarbonate transport along the loop of Henle: Effects of adrenal steroids. Am J Physiol. 1995;268:F234–F239. doi: 10.1152/ajprenal.1995.268.2.F234. [DOI] [PubMed] [Google Scholar]
- 24.Marver D. Evidence of corticosteroid action along the nephron. Am J Physiol. 1984;246:F111–F123. doi: 10.1152/ajprenal.1984.246.2.F111. [DOI] [PubMed] [Google Scholar]
- 25.Stanton BA. Regulation by adrenal corticosteroids of sodium and potassium transport in loop of Henle and distal tubule of rat kidney. J Clin Invest. 1986;78:1612–1620. doi: 10.1172/JCI112754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Work J, Jamison RL. Effect of adrenalectomy on transport in the rat medullary thick ascending limb. J Clin Invest. 1987;80:1160–1164. doi: 10.1172/JCI113174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ambühl PM, Amemiya M, Danczkay M, Lötscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ. Chronic metabolic acidosis increases NHE-3 protein abundance in rat kidney. Am J Physiol. 1996;271:F917–F925. doi: 10.1152/ajprenal.1996.271.4.F917. [DOI] [PubMed] [Google Scholar]
- 28.Laghmani K, Borenstein, Ambühl PM, Froissart M, Bichara M, Moe OW, Alpern RJ, Paillard M. Chronic metabolic acidosis enhances NHE-3 protein abundance and transport activity in the rat thick ascending limb by increasing NHE-3 mRNA. J Clin Invest. 1997;99:24–30. doi: 10.1172/JCI119128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anderson J, Forster JB. Effect of cortisone on urinary phosphate excretion in man. Clin Sci. 1959;18:437–439. [PubMed] [Google Scholar]
- 30.Laron Z, Crawford JD, Klein R. Phosphaturic effect of cortisone in normal and parathyroidectomized rats. Proc Soc Exp Biol Med. 1957;96:649–651. doi: 10.3181/00379727-96-23566. [DOI] [PubMed] [Google Scholar]
- 31.Turner ST, Kiebzak GM, Dousa TP. Mechanism of glucocorticoid effect on renal transport of phosphate. Am J Physiol. 1982;243:C227–C236. doi: 10.1152/ajpcell.1982.243.5.C227. [DOI] [PubMed] [Google Scholar]