

Abstract
Purpose
There are a number of craniosynostosis syndromes with hearing loss—including Muenke, Apert, Pfeiffer, Crouzon, Beare-Stevenson, Crouzon with acanthosis nigricans, and Jackson-Weiss syndromes—that result from mutations in the fibroblast growth factor receptor (FGFR) genes. Studies of FGFRs and their ligands, fibroblast growth factors (FGFs), have revealed clues to the precise contribution of aberrant FGFR signaling to inner ear morphogenesis and the hearing loss encountered in craniosynostoses. The purpose of this article is to review basic studies of FGFRs with emphasis on their function and expression in the inner ear and surrounding structures.
Method
A Medline search was performed to find basic science articles regarding FGFR, their ligands, and their expression and relevant mouse models. Additional items searched included clinical descriptions and studies of individuals with FGFR-related craniosynostosis syndromes.
Results
The FGF signaling pathway is essential for the morphogensis and proper function of the inner ear and auditory sensory epithelium.
Conclusion
The variable auditory phenotypes seen in individuals with Muenke syndrome may have a genetic basis, likely due to multiple interacting factors in the genetic environment or modifying factors. Further analysis and studies of mouse models of Muenke syndrome, in particular, may provide clues to the specific effects of the defining mutation in FGFR3 in the inner ear not only at birth but also into adulthood. In particular, investigations into these models may give insight into the variable expression and incomplete penetrance of this phenotype.
Keywords: Muenke syndrome, craniosynostosis, FGFR craniosynostosis, craniosynostosis hearing, craniosynostosis syndromes
Hearing loss is one of the leading causes of childhood disability in the United States (Mehra, Eavey, & Keamy, 2009) and is a common and serious problem that can have a significant impact on speech and language development, cognitive abilities, and educational performance (American Academy of Audiology, 2011). It is estimated that 1–3 in every 1,000 newborns has significant permanent hearing loss (Bielecki, Horbulewicz, & Wolan, 2011; Joseph, Pillinger, Pretorius, & Martinez-Devesa, 2010; Mehl & Thomson, 1998). When mild or greater hearing losses are considered, prevalence in children and adolescents is estimated to be 3.1% (Mehra et al., 2009).
Of all children with congenital or early-onset deafness, 50%–60% are estimated to have hereditary hearing loss. One-third of these children have syndromic hearing loss, with associated abnormalities (Marazita et al., 1993). Syndromes and physical findings associated with syndromes are important risk factors for hearing loss (Bielecki et al., 2011) and comprise four of the ten risk factors for hearing loss stipulated by the Joint Committee on Infant Hearing (JCIH) Position Statement (2007). These risk factors include craniofacial anomalies, syndromes associated with permanent hearing loss, physical findings associated with syndromes known to have hearing loss (e.g., white forelock), and neurodegenerative disorders (e.g., Charcot-Marie-Tooth syndrome).
There are more than 300 syndromic causes of hearing loss (Marazita et al., 1993), including well-known conditions such as CHARGE (coloboma, heart defects, atresia choanae, retardation of growth and/or development, genitourinary anomalies, and ear anomalies and deafness) syndrome, Waardenburg syndrome, Treacher Collins syndrome, KID (keratitis, ichthyosis, and deafness) syndrome, and syndromic craniosynostosis (Ohlms, Chen, Stewart, & Franklin, 1999). To date, there are over 180 identified forms of syndromic craniosynostosis. The most common forms are associated with mutations in the fibroblast growth factor receptor (FGFR) genes (Kimonis, Gold, Hoffman, Panchal, & Boyadjiev, 2007). Muenke syndrome—an autosomal dominant cranio-synostosis syndrome caused by a single defining c.749 C>G mutation in the FGFR3 gene, resulting in a proline to arginine amino acid substitution at codon 250 (p.Pro250Arg)—is the most common syndromic form of craniosynostosis, with an incidence of approximately 1 in 30,000 live births (Boulet, Rasmussen, & Honein, 2008; Muenke et al., 1997). In addition to craniosynostosis, or premature fusion of the cranial sutures, individuals characteristically have sensorineural hearing loss, tarsal and/or carpal bone coalition, and developmental delay (Muenke et al., 1997); see examples of carpal bone fusion and clindodactyly in Figure 1. Of all FGFR-related craniosynostosis syndromes, sensorineural hearing loss is most specific to Muenke syndrome (Doherty et al., 2007; Honnebier et al., 2008; Mansour et al., 2009).
Figure 1.
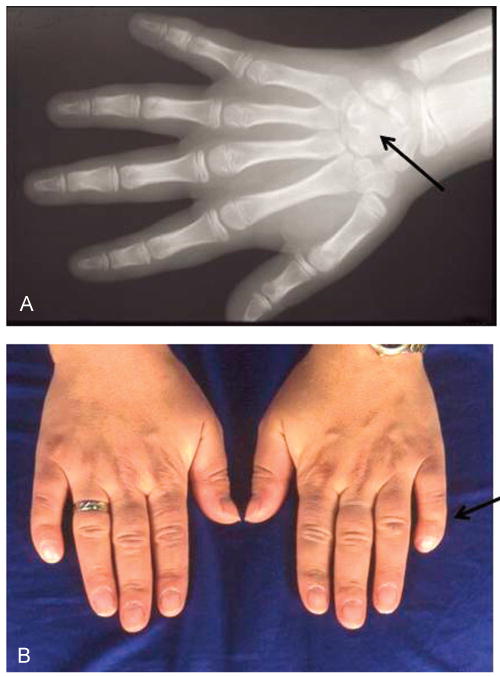
Panel A: Capal bone fusion (capitate-hamate) in an individual with Muenke syndrome (black arrow). Panel B: Broad thumbs and clindodactyly in an individual with Muenke syndrome (black arrow).
Hearing loss has been reported since the initial description of this syndrome in the 1990s, and its recognition and management in conjunction with standard clinical care is important. Initially thought to affect roughly 30%–40% of cases, more recent studies have shown that almost all individuals with Muenke syndrome have at least mild to moderate low- to mid-frequency sensorineural hearing loss (Doherty et al., 2007; Honnebier et al., 2008; Mansour et al., 2009), as exemplified in Figure 2. This hearing loss configuration is characteristically rising to normal hearing in the high frequencies, although some individuals have been reported with high-frequency hearing loss. It is important to note this predominantly low-frequency hearing loss may be missed on newborn and school hearing screenings, because these measures predominantly test hearing at mid- and high frequencies. Although the hearing loss is mild in most cases, some individuals have hearing loss that necessitates amplification (Doherty et al., 2007). Developmental delay is also a component of Muenke syndrome (Muenke et al., 1997), and there may be a yet-unidentified role that the hearing loss in these individuals plays in this developmental delay, particularly in regard to speech delay.
Figure 2.
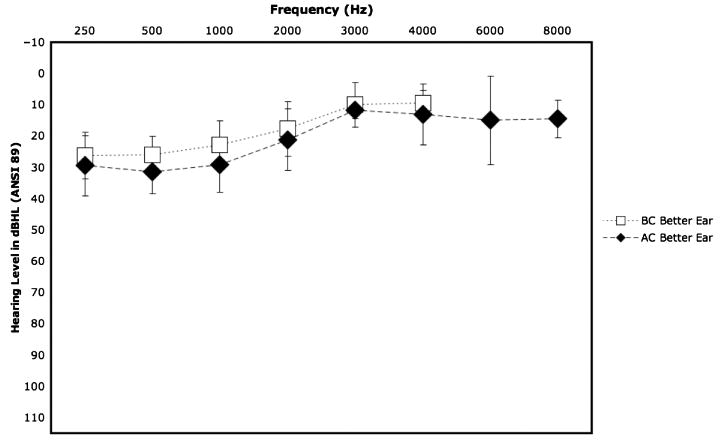
Composite audiogram showing mean (±SD) hearing threshold conduction (AC; 250 Hz–8000 Hz) and bone conduction (BC; 250 Hz–8000 Hz) for better hearing ear. Data represent averaged hearing thresholds on 18 individuals with Muenke syndrome from four facilities. Image adapted from Muenke Lab National Institutes of Health Muenke syndrome study (Doherty et al., 2007).
In this review, we describe basic studies of the expression and role of FGFRs in the inner ear and surrounding structures, with particular attention to FGFR3, the gene implicated in Muenke syndrome. Mouse models of perturbation and excessive signaling of Fgfr3 have provided much insight on the role of this protein in the morphogenesis, function, and development of the inner ear. Further, mouse models of Muenke syndrome provide compelling evidence of a plausible etiopathological nature of the hearing loss in individuals with Muenke syndrome and additional FGFR-related syndromic craniosynostoses.
Literature Review
A Medline search was conducted to find previously reported cases of Muenke syndrome from 1996 to 2011. The key words and patient terms searched included Muenke, coronal synostosis, FGFR3 craniosynostosis, P250R, Pro250Arg, syndromic craniosynostosis, FGFR3, FGFR1, FGFR2, FGFR mouse, FGFR inner ear, hearing loss, newborn hearing screen, and hearing loss epidemiology. References were also obtained from articles found through the literature search.
FGFR3 and the Inner Ear
The organ of Corti comprises the auditory sensory epithelium of the cochlea. Composed of a highly ordered array of sensory hair cells and nonsensory supporting cells that run along the length of the snail-like cochlea from base to apex, the organ of Corti represents one of the most striking examples of cellular patterning in vertebrates (Shim, Minowada, Coling, & Martin, 2005). Inner hair cells (IHCs) function primarily in the mechanoelectric transduction of sound, whereas outer hair cells (OHCs) function mainly in modulation and amplification of the cochlear response. Stereocilia, localized to the apical surface of the hair cells, detect sound-induced fluid motion with subsequent opening of ion channels, depolarization of the hair cells, and excitation of the auditory nerve. The network of supporting cells surround and couple the hair cells to the basilar membrane. These supporting cells include pillar cells, which form the triangular fluid-filled tunnel of Corti, separating the rows of IHCs and OHCs; Deiter's cells, which are found underlying each OHC; phalangeal cells, which surround the IHC; and border cells, which contact the medial surface of the IHCs (Mann & Kelley, 2011; Shim et al., 2005).
Studies in which researchers investigated the molecular mechanisms and pathways of hearing have helped demonstrate the roles of the different components in the development and maintenance of this intricate auditory network. Thus far, precise roles for two signaling pathways in patterning of the organ of Corti have been unveiled: (a) the Notch signaling pathway in hair cell specification and (b) the fibroblast growth factor (FGF) signaling pathway in the generation of hair cell progenitor pools and in the specification or differentiation of pillar cells (Colvin, Bohne, Harding, McEwen, & Ornitz, 1996; Kelley, 2003; Kiernan, Steel, & Fekete, 2002; Mueller, Jacques, & Kelley, 2002). FGFs signal via endocrine (substrate or hormone to distant target) or paracrine (cell-to-cell communication) signaling in order to mediate biological activities including developmental signaling and the regulation of metabolic processes (Belov & Mohammadi, 2013). FGF signals are carried out by the binding and subsequent dimerization of FGFRs. After dimerization, signaling may then proceed with the assistance of cofactors such as heparan sulfate and Klotho coreceptors. FGF signaling is required for the normal induction, patterning, and morphogenesis of the inner ear, as well as for the differentiation of cell types within the organ of Corti (Schimmang, 2007; Shim, 2006; Wright & Mansour, 2003a, 2003b). In particular, FGF20 and Fgfr1 are required for the differentiation of hair cells, whereas Fgf8 and Fgfr3 are required for pillar cell differentiation (Colvin et al., 1996; Domínguez-Frutos et al., 2009; Jacques, Montcouquiol, Layman, Lewandoski, & Kelley, 2007; Pirvola et al., 2002; Zelarayan et al., 2007). Fgfr2 signaling also plays a role in inner ear morphogenesis, particularly in formation of the membranous labyrinth, cochleovestibular ganglion, and nonsensory epithelium (Pirvola et al., 2000). Of these two signaling pathways, the focus from hereon will be on the FGF–FGFR signaling pathway, the pathway implicated in FGFR-related cranio-synostosis syndromes.
The hair cell has a central role in hearing as a mechanosensory receptor. Therefore, it is not surprising that mutations causing the loss or malfunction of hair cells lead to hearing deficits (Steel & Kros, 2001). An example is human deafness caused by mutations in the myosin genes (MYO7A, MYO3A, and MYO15); mouse studies have shown that these genes play a role in the organization of and are expressed in and around the hair cells and hair cell stereocilia (Liu et al., 1997; Probst et al., 1998; Self et al., 1998, 1999; Steel & Kros, 2001; Wang et al., 1998; Weil et al., 1995).
It is reasonable to predict that disturbances in the patterning of the organ of Corti via effects on supporting cells might also lead to hearing deficits. The auditory phenotype of mice lacking Fgfr3 (Fgfr3 -/-), the gene associated with Muenke syndrome, has lent much credence to this prediction. These mice are profoundly deaf; the only obvious defect in their inner ears involves the organ of Corti, where there is incomplete development of the pillar cells and the tunnel of Corti (Colvin et al., 1996). Specifically, these Fgfr3 -/-mice have no recognizable inner and outer pillar cells and lack the tunnel of Corti. In addition, the cochleae of these mice lack maturation and differentiation. Adult Fgfr3 -/- mice had cochleae resembling normal newborn mice with no differentiated pillar cells, an absent tunnel of Corti, a patent vessel below the basilar membrane (which normally involutes 2 weeks after birth), and numerous mesothelial cells below the basilar membrane. Additionally, mutant mice had reduced organ of Corti innervation. Hearing assessment via auditory brainstem responses (ABRs) revealed an absence of ABRs in the mutant mice, indicating profound deafness (Colvin et al., 1996). The inner ear defects in these mice not only suggest specific roles for Fgfr3 in pillar cell differentiation but also demonstrate that defects in cells comprising the organ of Corti that result in deafness are not limited to the sensory hair cells. All of the cellular components of the organ of Corti must function properly to achieve sound transduction.
These results are not surprising, considering earlier expression studies showed that Fgfr3 was highly expressed in the developing cochlear duct of the mouse (Peters, Ornitz, Werner, & Williams, 1993). In particular, Fgfr3 had intense expression in the differentiating hair cells and support cells. In addition, Fgfr3-mRNA was distinctly expressed in the two types of supporting cells in the rat organ of Corti, pillar cells, and Deiter's cells (Pirvola et al., 1995). It is interesting to note that in this same study, there was a noise-induced increased expression of Fgfr3 in the organ or Corti at both the mRNA and protein levels; this effect was seen in the supporting cells and adjacent OHCs but not in the IHCs (Pirvola et al., 1995). This finding suggests an additional role of FGFR3 in the protection and repair process of the organ of Corti, further supporting a main role for FGFR3 in the supporting cells—when OHC damage occurs, it is the supporting cells that prevent the progression of damage (Raphael & Altschuler, 1991).
Further, the role of FGFR3 in pillar cell development has been shown by studies in which inhibition of Fgfr3 by SU5402 (which inhibits the tyrosine kinase activity of all four FGFRs) causes a disruption in the development of pillar cells, an effect that is dose dependent (Mueller et al., 2002). This finding is consistent with the observations in the Fgfr3 mutant mice and support the hypothesis that Fgfr3 is necessary for pillar cell commitment and/or differentiation—a role that, when perturbed, results in hearing loss. Treatment of cochlear explants with exogenous FGF2, a strong activator of FGFR3c (a splice variant expressed in the developing cochlea), results in a marked increase in the number of pillar cells; this increase far outnumbered the increase in IHCs that occurred (Mueller et al., 2002). There was no increase in the number of OHCs or Deiter's cells.
A study of a mouse model of chondroplasia (Fgfr3 Y367C mutation) revealed hearing loss and, interestingly, as in the mouse that lacks Fgfr3 completely, the only inner ear defect observed in this mouse was an increased number of pillar cells and modified supporting cells in the organ of Corti (Pannier et al., 2009).
Collectively, these studies support the theory that not only is activation of FGFR3 required for pillar cell differentiation, but also that the FGF signaling pathway regulates the number of cells that will develop as pillar cells. Further, mouse models have shown that disruption of this pathway results in a lack of pillar cell differentiation, an effect that leads to hearing loss. How, exactly, pillar cell abnormalities and/or absence causes hearing loss has yet to be elucidated. Researchers speculate that changes in pillar cell number affect the properties of the basilar membrane. Pillar cells form a rigid support that is thought to couple the motion of the basilar membrane to deflection of the stereocilia on the apical surface of the hair cells (Shim, 2006). Perhaps changes in the number of pillar cells, or the absence of pillar cells, affects deflection of the stereocilia, causing an interruption in the transformation of mechanical energy of sound to electrical signals responsible for excitation of the auditory nerve.
The importance and necessity of the FGF signaling pathway for morphogenesis and proper function of the inner ear and auditory sensory epithelium is evident in many experimental systems—including zebrafish, chicken, and mouse models—where experimental manipulations have demonstrated the necessity of FGFs during different steps of inner ear induction in vitro and in vivo (Ladher, Anakwe, Gurney, Schoenwolf, & Francis-West, 2000; Ladher, Wright, Moon, Mansour, & Schoenwolf, 2005; Maroon et al., 2002; Schimmang, 2007; Wright & Mansour, 2003a; Zelarayan et al., 2007).
Inner Ear in Muenke Syndrome
All of these studies lead to a repetitive theme: the important role of FGFR3 in hearing function and in the proper formation of the organ of Corti. The next question is how this relates particularly to Muenke syndrome and what inner ear effects can be expected as a result of the specific perturbation that occurs in FGFR3 signaling in Muenke syndrome. In mice, both inhibition of Fgfr3 function and excessive Fgf signaling (via deletion of Sprouty2, which encodes a negative regulator of signaling via receptor tyrosine kinases) leads to hearing loss, with disturbances in the cryoarchitecture of the organ of Corti relating to the number and organization of supporting cells (Colvin et al., 1996; Shim et al., 2005). The model of excessive FGF signaling gives much insight into Muenke syndrome, considering the causative mutation in FGFR3 is a gain of function mutation that results in excess FGF–FGFR3 signaling due to enhanced ligand binding affinity (Ibrahimi, Zhang, Eliseenkova, Linhardt, & Mohammadi, 2004). Further, binding of the Muenke syndrome mutant FGFR3 receptor is notably enhanced for FGF9, a ligand shown to have a role in inner ear morphogenesis (Pirvola, Zhang, Mantela, Ornitz, & Ylikoski, 2004). Given the knowledge of these previous studies, it is reasonable to predict that not only would mouse models of Muenke syndrome exhibit hearing loss, but this hearing loss would likely be a result of aberrant development of the organ of Corti.
A mouse model of Muenke syndrome has provided much insight. Hearing was evaluated in the corresponding mouse model of Muenke syndrome (p.Pro244Arg; Mansour et al., 2009). Researchers measured ABR thresholds to click (broadband low frequency), 8-, 16-, and 32-kHz tone-pip stimuli in wild-type, heterozygous, and homozygous Fgfr3P244R mice bred in four different genetic backgrounds. In this study, Muenke syndrome model mice were found to be moderately hearing impaired. The Muenke syndrome model mice had hearing loss that was more severe but of a similar pattern to individuals with Muenke syndrome, with a hearing loss that was greater in the lower frequencies (Doherty et al., 2007; Honnebier et al., 2008; Mansour et al., 2009). Thus, hearing loss in Muenke syndrome model mice is qualitatively similar but more severe than the hearing loss that occurs in individuals with Muenke syndrome. The hearing loss in the Muenke syndrome model mice was dose dependent: Homozygous mice had more hearing loss than heterozygous mice. Mice with different genetic backgrounds displayed differences in the absolute and relative magnitudes of hearing loss (Mansour et al., 2009). The underlying or baseline hearing status in the four backgrounds was not described in this study. This finding proposes that the variable auditory phenotypes seen in individuals with Muenke syndrome may have a genetic basis, likely due to multiple interacting factors in the genetic environment or modifying factors.
Muenke syndrome model mice had abnormalities in organ of Corti differentiation, with excess pillar cells, too few Deiter's cells, and extra OHCs. There was no change in the amount of IHCs. These organ of Corti abnormalities were dose dependent (i.e., direct correlation with the degree of expression of mutant FGFR3/genetic burden and degree of hearing loss) and were greater at apical (low-frequency) than basal (high-frequency) regions of the cochlea, consistent with the hearing loss configuration in Muenke syndrome model mice and individuals with Muenke syndrome.
Further analysis and studies of mouse models of Muenke syndrome, in particular, may provide clues to the specific effects of the defining mutation in FGFR3 in the inner ear not only at birth but also into adulthood. Particularly, investigations into these models may give insight into the variable expression and incomplete penetrance of this phenotype—that is, why individuals with the same causative mutation present with different degrees and types of hearing loss. It is very likely that the answer to this relates to the particular histological and morphological differences present in the organ of Corti as a result of each individual's unique genetic environment in the presence of the FGFR3 mutation.
Because the changes in the inner ear of the Muenke syndrome mouse are histologically but not macroscopically evident, the defects in the inner ear of individuals with Muenke syndrome likely may not be evident on macroscopic examination—for example, via computed tomography (CT) or magnetic resonance imaging (MRI) studies. Further elucidation of the effects of the FGFR3 mutation on the structure and formation of the inner ear in humans may require more intricate studies to discern changes that are likely only microscopically apparent. Reported ear anomalies in Muenke syndrome include low-set and posteriorly rotated ears, venous dysplasia of the left petrous bone in an individual with tinnitus, and apical cartilage deformity (Kress et al., 2006; Roscioli et al., 2001). The most striking ear anomalies were found in an individual with a hypoplastic right auricle, an absent right external auditory meatus, and bony atresia of the right external auditory canal; this individual was misdiagnosed in infancy with Treacher Collins syndrome, due in part to the striking ear anomalies (Shah et al., 2006).
However, imaging studies of the outer, middle, and inner ear in individuals with Muenke syndrome—such as those done in individuals with Apert, Crouzon, and Pfeiffer syndromes—have not been performed or reported in Muenke syndrome literature to date. Thus, it is not yet clear what role each network of the ear plays in the hearing loss that occurs in individuals with Muenke syndrome. Imaging studies in individuals with Apert, Crouzon, and Pfeiffer syndromes have revealed anomalies of the middle and inner ear, including malformed and/or fused middle ear ossicles (Apert, Crouzon, and Pfeiffer syndromes), dehiscence of the semicircular canal (Apert syndrome), hypoplasia of the middle ear ossicles (Pfeiffer syndrome), ankylosis of the stapes (Pfeiffer syndrome), atresia of the external auditory canal (Crouzon and Pfeiffer syndromes), and atrophy of the tympanic membranes (Crouzon syndrome), just to name a few (Cremers, 1981; Desai et al., 2010; Orvidas, Fabry, Diacova, & McDonald, 1999; Vallino-Napoli, 1996; Zhou, Schwartz, & Gopen, 2009). Large studies such as these for Muenke syndrome will help further elucidate the precise etiology of the hearing loss in individuals with Muenke syndrome, an etiology which the Muenke syndrome mouse model has helped us get closer to understanding. CT scanning of the middle and inner ear is a reasonable starting point for evaluation of bony anomalies of the middle ear and inner ear in individuals with Muenke syndrome. As many of the changes that occur in the cochlea are likely microscopic, as inferred from the Muenke syndrome mouse model, these may be difficult to elucidate solely from imaging, whether that be via CT scanning or MRI.
This mouse model may also be useful in the research of hearing loss therapies through gene manipulation. Most of the work in the field of hearing loss and gene therapy has focused on the addition of a “missing” component by genetic methods (Atar & Avraham, 2005). However, the modulation of key components by inhibition may also be a useful approach, especially in situations where hearing loss has a known genetic component. In a study by Maeda, Fukushima, Nishizaki, and Smith (2005), researchers silenced expression of the mutant allele of the GJB2 gene, which causes the most common form of hereditary deafness (Cohen-Salmon et al., 2002; Shim, 2006). By using the RNA interference technique (a biological process in which RNA molecules inhibit gene expression) against the mutant allele in vitro and in vivo, they succeeded in restoring hearing in a genetic mouse model. The cochlea represents an ideal target for these therapies; not only is it isolated from the remainder of the body by the blood-labyrinth barrier, but it is also bathed in fluid, permitting the quick and efficient delivery of liquids (Atar & Avraham, 2005). Although there are limitations to the delivery of such therapies (i.e., delivering therapies into the inner ear without causing damage to existing or residual hearing) and to the therapies themselves (gene therapy and RNAi), there is much hope and promise for the use of these therapies in the future for the treatment of hearing loss in individuals with syndromic hearing loss, such as in Muenke syndrome.
Acknowledgments
This research was supported by the Division of Intramural Research at the National Human Genome Research Institute (National Institutes of Health [NIH], Department of Health and Human Services, USA), Protocol No. 05-HG-0131, awarded to the last author. We would like to express our gratitude to the individuals who participate in our ongoing studies of Muenke syndrome at the NIH. We are also grateful to Carmen Brewer of the National Institute on Deafness and Other Communication Disorders for her review of this article.
Footnotes
Disclosure: The authors have declared that no competing interests existed at the time of publication.
References
- American Academy of Audiology. Clinical practice guide-lines: Childhood hearing screening guidelines. 2011 Retrieved from www.audiology.org/resources/documentlibrary/Documents/ChildhoodScreeningGuidelines.pdf.
- Atar O, Avraham KB. Therapeutics of hearing loss: Expectations vs reality. Drug Discovery Today. 2005;10:1323–1330. doi: 10.1016/S1359-6446(05)03618-4. [DOI] [PubMed] [Google Scholar]
- Belov AA, Mohammadi M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harbor Perspectives in Biology. 2013;5:a015958. doi: 10.1101/cshperspect.a015958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielecki I, Horbulewicz A, Wolan T. Risk factors associated with hearing loss in infants: An analysis of 5282 referred neonates. International Journal of Pediatric Otorhinolaryngology. 2011;75:925–930. doi: 10.1016/j.ijporl.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989–2003. American Journal of Medical Genetics, Part A. 2008;146A:984–991. doi: 10.1002/ajmg.a.32208. [DOI] [PubMed] [Google Scholar]
- Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Petit C. Targeted ablation of connexin26 in the inner epithelial gap junction network causes hearing impairment and cell death. Current Biology. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nature Genetics. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Cremers CW. Hearing loss in Pfeiffer's syndrome. International Journal of Pediatric Otorhinolaryngology. 1981;3:343–353. doi: 10.1016/0165-5876(81)90059-8. [DOI] [PubMed] [Google Scholar]
- Desai U, Rosen H, Mulliken JB, Gopen Q, Meara JG, Rogers GF. Audiologic findings in Pfeiffer syndrome. Journal of Craniofacial Surgery. 2010;21:1411–1418. doi: 10.1097/SCS.0b013e3181ebcf58. [DOI] [PubMed] [Google Scholar]
- Doherty ES, Lacbawan F, Hadley DW, Brewer C, Zalewski C, Kim HJ, Muenke M. Muenke syndrome (FGFR3-related craniosynostosis): Expansion of the phenotype and review of the literature. American Journal of Medical Genetics, Part A. 2007;143A:3204–3215. doi: 10.1002/ajmg.a.32078. [DOI] [PubMed] [Google Scholar]
- Domínguez-Frutos E, Vendrell V, Alvarez Y, Zelarayan LC, López-Hernández I, Ros M, Schimmang T. Tissue-specific requirements for FGF8 during early inner ear development. Mechanisms of Development. 2009;126:873–881. doi: 10.1016/j.mod.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Honnebier MB, Cabiling DS, Hetlinger M, McDonald-McGinn DM, Zackai EH, Bartlett SP. The natural history of patients treated for FGFR3-associated (Muenke-type) craniosynostosis. Plastic and Reconstructive Surgery. 2008;121:919–931. doi: 10.1097/01.prs.0000299936.95276.24. [DOI] [PubMed] [Google Scholar]
- Ibrahimi OA, Zhang F, Eliseenkova AV, Linhardt RJ, Mohammadi M. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Human and Molecular Genetics. 2004;13:69–78. doi: 10.1093/hmg/ddh011. [DOI] [PubMed] [Google Scholar]
- Jacques BE, Montcouquiol ME, Layman EM, Lewandoski M, Kelley MW. Fgf8 induces pillar cell fate and regulates cellular patterning in the mammalian cochlea. Development. 2007;134:3021–3029. doi: 10.1242/dev.02874. [DOI] [PubMed] [Google Scholar]
- Joint Committee on Infant Hearing. Year 2007 position statement: Principles and guidelines for early hearing detection and intervention programs. Pediatrics. 2007;120:898–921. doi: 10.1542/peds.2007-2333. [DOI] [PubMed] [Google Scholar]
- Joseph JA, Pillinger T, Pretorius PM, Martinez-Devesa P. Congenital conductive hearing loss and multiple synostosis syndrome with analysis of temporal bone CT scan findings. International Journal of Pediatric Otorhinolaryngology. 2010;74:1438–1440. doi: 10.1016/j.ijporl.2010.09.001. [DOI] [PubMed] [Google Scholar]
- Kelley MW. Cell adhesion molecules during inner ear and hair cell development, including notch and its ligands. Current Topics in Developmental Biology. 2003;57:321–356. doi: 10.1016/s0070-2153(03)57011-9. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Steel KP, Fekete DM. Development of the mouse inner ear. In: Rossant J, Tam PPL, editors. Mouse development: Patterning, morphogenesis and organogenesis. San Diego, CA: Academic Press; 2002. pp. 539–566. [Google Scholar]
- Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of craniosynostosis. Seminars in Pediatric Neurology. 2007;14:150–161. doi: 10.1016/j.spen.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Kress W, Schropp C, Lieb G, Petersen B, Büsse-Ratzka M, Kunz J, Collman H. Saethre-Chotzen syndrome caused by TWIST 1 gene mutations: Functional differentiation from Muenke coronal synostosis syndrome. European Journal of Human Genetics. 2006;14:39–48. doi: 10.1038/sj.ejhg.5201507. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Anakwe KU, Gurney AL, Schoenwolf GC, Francis-West PH. Identification of synergistic signals initiating inner ear development. Science. 2000 Dec 8;290:1965–1967. doi: 10.1126/science.290.5498.1965. [DOI] [PubMed] [Google Scholar]
- Ladher RK, Wright TJ, Moon AM, Mansour SL, Schoenwolf GC. FGF8 initiates inner ear induction in chick and mouse. Genes and Development. 2005;19:603–613. doi: 10.1101/gad.1273605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene causing non-syndromic recessive deafness. Nature Genetics. 1997;16:188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Fukushima K, Nishizaki K, Smith RJ. In vitro and in vivo suppression of GJB2 expression by RNA interference. Human Molecular Genetics. 2005;14:1641–1650. doi: 10.1093/hmg/ddi172. [DOI] [PubMed] [Google Scholar]
- Mann ZF, Kelley MW. Development of tonotopy in the auditory periphery. Hearing Research. 2011;276:2–15. doi: 10.1016/j.heares.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Mansour SL, Twigg SRF, Freeland RM, Wall SA, Li C, Wilkie AOM. Hearing loss in a mouse model of Muenke syndrome. Human Molecular Genetics. 2009;18:43–50. doi: 10.1093/hmg/ddn311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML, Ploughman LM, Rawlings B, Remmington E, Arnos KS, Nance WE. Genetic epidemiologic studies of early-onset deafness in the US school age population. American Journal of Medical Genetics. 1993;43:486–491. doi: 10.1002/ajmg.1320460504. [DOI] [PubMed] [Google Scholar]
- Maroon H, Walshe J, Mahmood R, Kiefer P, Dickson C, Mason I. Fgf3 and Fgf8 are required together for formation of the otic placode and vesicle. Development. 2002;129:2099–3108. doi: 10.1242/dev.129.9.2099. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. Newborn hearing screening: The great omission. Pediatrics. 1998;101:E4. doi: 10.1542/peds.101.1.e4. [DOI] [PubMed] [Google Scholar]
- Mehra S, Eavey RD, Keamy DG. The epidemiology of hearing impairment in the United States: Newborns, children and adolescents. Otolaryngology—Head & Neck Surgery. 2009;140:461–472. doi: 10.1016/j.otohns.2008.12.022. [DOI] [PubMed] [Google Scholar]
- Mueller KL, Jacques BE, Kelley MW. Fibroblast growth factor signaling regulates pillar cell development in the organ of Corti. The Journal of Neuroscience. 2002;22:9368–9377. doi: 10.1523/JNEUROSCI.22-21-09368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenke M, Gripp KW, McDonald-McGinn DM, Gaudenz K, Whitaker LA, Bartlett SP, Zackai EH. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. American Journal of Human Genetics. 1997;60:555–564. [PMC free article] [PubMed] [Google Scholar]
- Ohlms LA, Chen AY, Stewart MG, Franklin DJ. Establishing the etiology of childhood hearing loss. Otolaryngology—Head & Neck Surgery. 1999;120:159–163. doi: 10.1016/S0194-5998(99)70400-6. [DOI] [PubMed] [Google Scholar]
- Orvidas LJ, Fabry LB, Diacova S, McDonald TJ. Hearing and otopathology in Crouzon syndrome. Laryngoscope. 1999;109:1372–1375. doi: 10.1097/00005537-199909000-00002. [DOI] [PubMed] [Google Scholar]
- Pannier S, Couloigner V, Messaddeq N, Elmaleh-Bergès M, Munnich A, Romand R, Legeai-Mallet L. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochimica et Biophysica Acta. 2009;1792:140–147. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Developmental Biology. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Cao Y, Oellig C, Suoqiang Z, Pettersson RF, Ylikoski J. The site of action of neuronal acidic fibroblast growth factor is the organ of Corti of the rat cochlea. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:9269–9273. doi: 10.1073/pnas.92.20.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Spencer-Dene B, Xing-Qun L, Kettunen P, Thesleff I, Fritzsch B, Ylikoski J. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. The Journal of Neuroscience. 2000;20:6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hébert JM, McConnell SK, Partanen J. FGFR1 is required for the development of the auditory sensory epithelium. Neuron. 2002;35:671–680. doi: 10.1016/s0896-6273(02)00824-3. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Zhang X, Mantela J, Ornitz DM, Ylikoski J. Fgf9 signaling regulates inner ear morphogenesis through epithelial-mesenchymal interactions. Developmental Biology. 2004;273:350–360. doi: 10.1016/j.ydbio.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Probst FJ, Fridell RA, Raphael Y, Saunders TL, Wang A, Liang Y, Camper SA. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science. 1998 May 29;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- Raphael Y, Altschuler RA. Scar formation after drug-induced cochlear insult. Hearing Research. 1991;51:173–183. doi: 10.1016/0378-5955(91)90034-7. [DOI] [PubMed] [Google Scholar]
- Roscioli T, Flanagan S, Mortimore RJ, Kumar P, Weedon D, Masel J, Glass IA. Premature calvarial synostosis and epidermal hyperplasia (Beare-Stevenson syndrome-like anomalies) resulting from a P250R missense mutation in the gene encoding fibroblast growth factor receptor 3. American Journal of Medical Genetics. 2001;101:187–194. doi: 10.1002/ajmg.1369. [DOI] [PubMed] [Google Scholar]
- Schimmang T. Expression and functions of FGF ligands during early otic development. International Journal of Developmental Biology. 2007;51:473–481. doi: 10.1387/ijdb.072334ts. [DOI] [PubMed] [Google Scholar]
- Self T, Mahony M, Fleming J, Walsh J, Brown SD, Steel KP. Shaker-1 mutations reveal roles for myosin VIIA in both development and function of cochlear hair cells. Development. 1998;125:557–566. doi: 10.1242/dev.125.4.557. [DOI] [PubMed] [Google Scholar]
- Self T, Sobe T, Copeland NG, Jenkins NA, Avraham KB, Steel KP. Role of myosin VI in the development of cochlear hair cells. Developmental Biology. 1999;214:331–341. doi: 10.1006/dbio.1999.9424. [DOI] [PubMed] [Google Scholar]
- Shah P, Siriwardena K, Taylor G, Steele L, Ray P, Blaser S, Chitayat D. Sudden infant death in a patient with FGFR3 P250R mutation. American Journal of Medical Genetics, Part A. 2006;140:2794–2796. doi: 10.1002/ajmg.a.31517. [DOI] [PubMed] [Google Scholar]
- Shim K. The auditory sensory epithelium: The instrument of sound perception. The International Journal of Biochemistry and Cell Biology. 2006;38:1827–1833. doi: 10.1016/j.biocel.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Shim K, Minowada G, Coling DE, Martin GR. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Developmental Cell. 2005;8:553–564. doi: 10.1016/j.devcel.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Steel KP, Kros CJ. A genetic approach to understanding auditory function. Nature Genetics. 2001;27:143–149. doi: 10.1038/84758. [DOI] [PubMed] [Google Scholar]
- Vallino-Napoli LD. Audiologic and otologic characteristics of Pfeiffer syndrome. The Cleft Palate–Craniofacial Journal. 1996;33:524–529. doi: 10.1597/1545-1569_1996_033_0524_aaocop_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Friedman TB. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998 May 29;280:1447–1451. doi: 10.1126/science.280.5368.1447. [DOI] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Petit C. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995 Mar 2;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. FGF signaling in ear development and innervation. Current Topics in Developmental Biology. 2003a;57:225–259. doi: 10.1016/s0070-2153(03)57008-9. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Mansour SL. Fgf3 and Fgf10 are required for mouse otic placode induction. Development. 2003b;130:3379–3390. doi: 10.1242/dev.00555. [DOI] [PubMed] [Google Scholar]
- Zelarayan LC, Vendrell V, Alvarez Y, Domínguez-Frutos E, Theil T, Schimmang T. Differential requirements for FGF3, FGF8 and FGF10 during inner ear development. Developmental Biology. 2007;308:379–391. doi: 10.1016/j.ydbio.2007.05.033. [DOI] [PubMed] [Google Scholar]
- Zhou G, Schwartz LT, Gopen Q. Inner ear anomalies and conductive hearing loss in children with Apert syndrome: An overlooked otologic aspect. Otology & Neurotology. 2009;30:184–189. doi: 10.1097/MAO.Ob013e318191a352. [DOI] [PubMed] [Google Scholar]